

ABSTRACT
During lytic herpes simplex virus (HSV) infections, the virion host shutoff (Vhs) (UL41) endoribonuclease degrades many cellular and viral mRNAs. In uninfected cells, spliced mRNAs emerge into the cytoplasm bound by exon junction complexes (EJCs) and are translated several times more efficiently than unspliced mRNAs that have the same sequence but lack EJCs. Notably, most cellular mRNAs are spliced, whereas most HSV mRNAs are not. To examine the effect of splicing on gene expression during HSV infection, cells were transfected with plasmids harboring an unspliced renilla luciferase (RLuc) reporter mRNA or RLuc constructs with introns near the 5′ or 3′ end of the gene. After splicing of intron-containing transcripts, all three RLuc mRNAs had the same primary sequence. Upon infection in the presence of actinomycin D, spliced mRNAs were much less sensitive to degradation by copies of Vhs from infecting virions than were unspliced mRNAs. During productive infections (in the absence of drugs), RLuc was expressed at substantially higher levels from spliced than from unspliced mRNAs. Interestingly, the stimulatory effect of splicing on RLuc expression was significantly greater in infected than in uninfected cells. The translational stimulatory effect of an intron during HSV-1 infections could be replicated by artificially tethering various EJC components to an unspliced RLuc transcript. Thus, the splicing history of an mRNA, and the consequent presence or absence of EJCs, affects its level of translation and sensitivity to Vhs cleavage during lytic HSV infections.
IMPORTANCE Most mammalian mRNAs are spliced. In contrast, of the more than 80 mRNAs harbored by herpes simplex virus 1 (HSV-1), only 5 are spliced. In addition, synthesis of the immediate early protein ICP27 causes partial inhibition of pre-mRNA splicing, with the resultant accumulation of both spliced and unspliced versions of some mRNAs in the cytoplasm. A common perception is that HSV-1 infection necessarily inhibits the expression of spliced mRNAs. In contrast, this study demonstrates two instances in which pre-mRNA splicing actually enhances the synthesis of proteins from mRNAs during HSV-1 infections. Specifically, splicing stabilized an mRNA against degradation by copies of the Vhs endoribonuclease from infecting virions and greatly enhanced the amount of protein synthesized from spliced mRNAs at late times after infection. The data suggest that splicing, and the resultant presence of exon junction complexes on an mRNA, may play an important role in gene expression during HSV-1 infections.
INTRODUCTION
During lytic herpes simplex virus 1 (HSV-1) infections, viral and cellular gene expression is regulated through the interplay of multiple transcriptional and posttranscriptional controls (1). Of the posttranscriptional mechanisms, one of the most important is the regulation of mRNA decay rates and translation by the HSV-1 virion host shutoff (Vhs) protein (2). Vhs (UL41) is an endoribonuclease (3–6) that is a minor structural component of virions (7, 8). At early times, copies of Vhs that enter the cell as components of infecting virions degrade many cellular mRNAs (9–11), thereby inhibiting the synthesis of the proteins that they encode (12). Following the onset of viral transcription, Vhs also accelerates the turnover of members of all kinetic classes of viral mRNAs (13–17). As a result, the levels and patterns of accumulation of many viral mRNAs are determined by a balance between the rates of new transcription and mRNA processing on the one hand and the rate of Vhs-mediated degradation on the other (15–18). Surprisingly, in some cells, Vhs actually stimulates the translation of specific mRNAs (19–23). This may be due to Vhs-mediated degradation of many other mRNAs, which would reduce the mRNA load in infected cells, alleviating competition between the remaining mRNAs for limiting amounts of translation factors (19–21). However, it also remains possible that Vhs helps recruit translation factors and ribosomes to some specific mRNAs.
Mutations that inactivate Vhs have only a modest effect on virus growth in many cultured cell lines (7, 12). Nevertheless, Vhs mutants are severely attenuated during animal infections (24–30). They replicate poorly in mouse corneas and other tissues and are defective in neuroinvasion (24). In addition, Vhs inhibits key components of the host's intrinsic and innate immune responses (26). Vhs inhibits the expression of ATRX, an effector of the intrinsic immune response (31). It impairs the activation of the Pkr protein kinase (32), inhibits the establishment of an interferon-mediated antiviral state (13, 33), and impedes the activation of dendritic cells (34, 35). Vhs is thus an important determinant of HSV virulence (24, 26).
Purified Vhs retains endonuclease activity in vitro in the absence of other viral or cellular proteins (3–5). Nevertheless, within infected cells, Vhs cleavage of many mRNAs appears to occur during translation initiation (36) and is dependent on the interaction of Vhs with cellular translation factors (5, 36–39). Vhs interacts with the cap-binding complex eIF4F (37) by virtue of its ability to bind eIF4AI and -4AII (38), ATP-dependent RNA helicases (40, 41) that are components of eIF4F, and eIF4H (5, 38, 39), a helicase accessory factor that binds to and stimulates the activities of eIF4AI and -4AII (40–46). eIF4H and eIF4AI/II appear to play an essential role in Vhs-mediated mRNA degradation. Mutations in Vhs that disrupt its binding to eIF4H abrogate its ability to cleave mRNAs that are translated by cap-dependent ribosome scanning (5, 38, 39), even if the mutant proteins retain endonuclease activity in vitro. Furthermore, small interfering RNA (siRNA)-mediated depletion of eIF4H from cells prior to infection prevents subsequent mRNA degradation by wild-type Vhs (47). Similarly, Vhs-mediated mRNA degradation is blocked by the depletion of eIF4AI and -4AII or by treatment of infected cells with hippuristanol (48), a specific inhibitor of the helicase activity of eIF4AI and -4AII (49).
While Vhs accelerates the decay of many mRNAs, some are refractory to Vhs-mediated degradation or at least degraded less rapidly than others (50–57). Because features of mRNAs that cause differential rates of decay could affect the balance of many host and viral gene products, with consequent effects on virus-host interactions, it is important to identify these features and elucidate their mechanisms of action. To this end, we investigated whether the splicing history of an mRNA affects its sensitivity to Vhs cleavage or translation during HSV-1 infection. The rationale for these studies was 2-fold. First, the compositions of messenger ribonucleoproteins (mRNPs) are different for spliced and unspliced mRNAs. Spliced mRNAs emerge into the cytoplasm bound by exon junction complexes (EJCs) approximately 22 nucleotides upstream of exon junctions (58–67). EJCs facilitate the recruitment of ribosomes to spliced mRNAs and translation initiation (68–75). Although EJCs are subsequently removed by elongating ribosomes (76–78), spliced mRNAs are translated to produce several times more protein per mRNA than in unspliced mRNAs that have an identical primary sequence but lack EJCs (79, 80). Since the mRNP structure could affect the rate of mRNA degradation, it was important to determine whether splicing affects the sensitivity of an mRNA to Vhs. Second, a notable difference between cellular and HSV-1 mRNAs is that most cellular mRNAs are spliced, whereas, with the exception of some immediate early and late mRNAs, viral mRNAs are not (1). An observation that spliced and unspliced mRNAs are differentially sensitive to Vhs might have important implications for gene expression during HSV-1 infections.
To examine the effect of splicing on gene expression during HSV-1 infection, cells were transfected with plasmids encoding an unspliced mRNA for renilla luciferase (RLuc) or RLuc constructs with introns near the 5′ or 3′ end of the gene. After splicing of mRNAs from the intron-containing constructs, the three RLuc mRNAs were indistinguishable. Upon infection with wild-type HSV-1 in the presence of actinomycin D (to examine mRNA degradation induced by copies of Vhs from infecting virions), spliced mRNAs were less sensitive to Vhs-mediated degradation and expressed significantly more RLuc protein than unspliced mRNAs. During productive infections (in the absence of drugs), RLuc was expressed at substantially higher levels from spliced than from unspliced mRNAs. Strikingly, HSV-1 infection actually stimulated RLuc expression from spliced mRNAs to levels that were several times higher than those from the same mRNAs in uninfected cells. The stimulatory effect of HSV-1 on the synthesis of proteins encoded by a spliced mRNA could be replicated by artificially tethering various EJC components to an unspliced RLuc transcript. The data suggest that splicing of an mRNA, and the resultant presence of an exon junction complex, can play an important role in gene expression during HSV-1 infections.
MATERIALS AND METHODS
Cells and virus.
HeLa S3 cells and Vero cells were purchased from the American Type Culture Collection and maintained in Eagle's minimum essential medium (MEM) (Gibco) supplemented with 10% (vol/vol) fetal bovine serum and antibiotics, as described previously (7, 81, 82). Wild-type HSV-1 strain KOS and mutant strain Vhs 1 (12, 15, 17), which contains a point mutation that changes threonine 214 of Vhs to isoleucine (T214I) (83), were grown, and titers were determined on Vero cells. HeLa cells were used for all transfections and assays of Vhs-mediated mRNA degradation.
Plasmids. (i) Plasmids harboring Vhs alleles.
Plasmid pKosAmp, which contains the Vhs open reading frame from wild-type HSV-1 strain KOS, cloned into the vector pCDNA1.1/Amp (Invitrogen) downstream from the cytomegalovirus (CMV) immediate early promoter, was described previously (84). Plasmids pD194N and pD215N were derived by site-directed mutagenesis of the Vhs open reading frame of pKosAmp and encode Vhs polypeptides with the point mutations D194N and D215N, respectively (84). Plasmid pVhs1 contains the UL41 open reading frame from the mutant virus Vhs 1 (12) cloned into pcDNA1.1/Amp (84). It contains the point mutation T214I (83). These plasmids are suitable for expressing wild-type and mutant forms of the Vhs protein following transfection of mammalian cells.
(ii) Plasmids harboring spliced and unspliced target mRNAs.
Plasmids harboring spliced or unspliced mRNAs for a triose phosphate isomerase (TPI)/RLuc/TPI fusion protein were described previously (79) and are diagrammed in Fig. 1A. The unspliced mRNA produced from the no-intron construct encodes a fusion protein of renilla luciferase fused to copies of exons 6 and 7 from human TPI inserted in frame at both ends of RLuc. Constructs that produce spliced mRNAs encoding the same TPI/RLuc/TPI protein contained the intron that normally separates exons 6 and 7 of TPI, inserted between the copies of exons 6 and 7 at either the 5′ or 3′ end of the gene (Fig. 1A). These constructs also had a CMV immediate early promoter and a bovine growth hormone polyadenylation site to facilitate their expression in transfected HeLa cells. Following splicing of transcripts from the intron-containing constructs, the spliced and unspliced mRNAs were indistinguishable (79).
FIG 1.

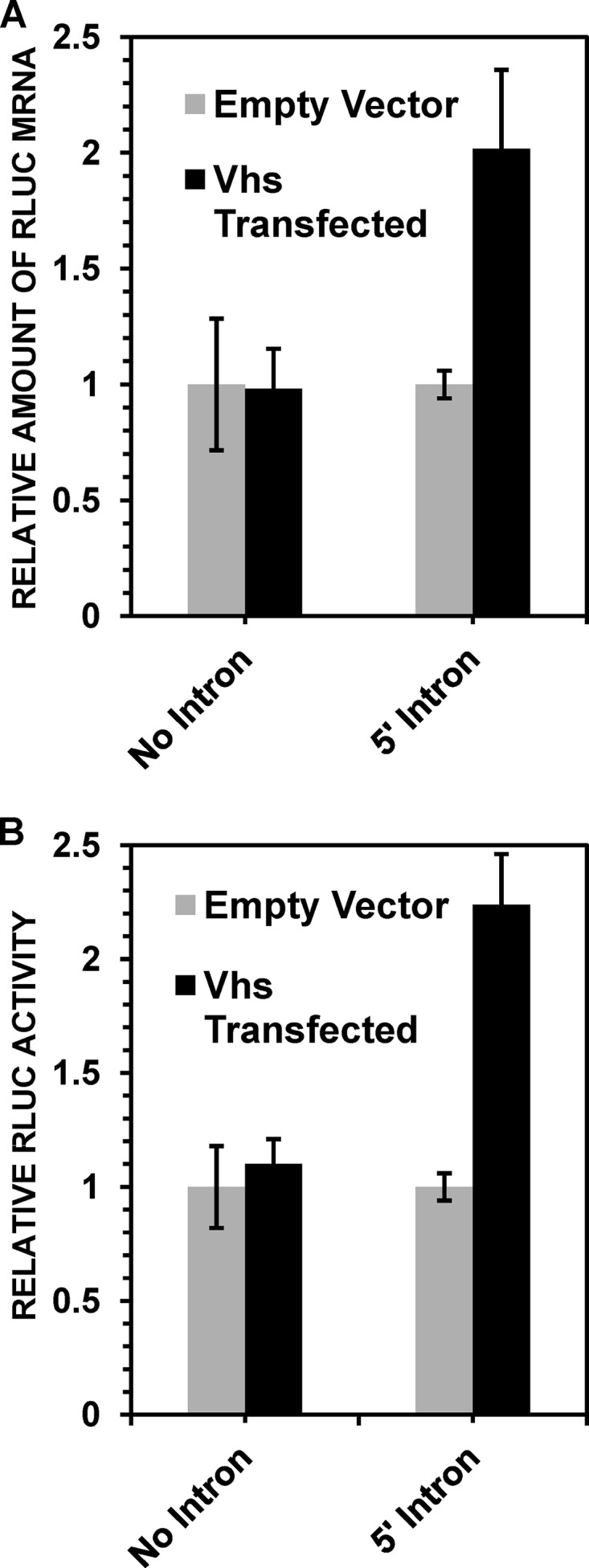
Spliced TPI/RLuc/TPI mRNAs are less sensitive to Vhs than are unspliced mRNAs. (A) Structures of intron-containing and intronless TPI/RLuc/TPI constructs. The RLuc open reading frame and TPI exons 6 and 7 are represented by white, gray, and black rectangles, respectively. The intron between TPI exons 6 and 7 is depicted by jagged lines. The transcriptional start site is shown by a rightward arrow, and the poly(A) site and start and stop codons are indicated by vertical lines. (B and C) HeLa cells were transfected with the no-intron, 5′ intron, or 3′ intron TPI/RLuc/TPI constructs. Forty-eight hours later, the cells were infected with 20 PFU/cell of wild-type (WT) HSV-1 or mock infected in the presence of 10 μg/ml of actinomycin D. Cultures were harvested 5 h after infection or mock infection and processed to determine the relative levels of TPI/RLuc/TPI mRNA (B) or RLuc activity (C), normalized to the levels in mock-infected cells. Error bars represent standard errors of the means.
T cell receptor β chain (TCR-β) minigene constructs that contain portions of three TCR-β exons and no intron, one intron, or two nonidentical introns were used to produce spliced and unspliced TCR-β mRNAs. These plasmids were described previously (79, 85) and are diagrammed in Fig. 2A. Plasmids harboring the TPI/RLuc/TPI or TCR-β mRNAs were generously provided by Melissa Moore from the University of Massachusetts. The constructs were used to examine whether the splicing history of an mRNA, or the location of an exon junction within a spliced mRNA, affects its expression during HSV-1 infections.
FIG 2.

Spliced TCR-β minigene mRNAs are less sensitive to Vhs degradation than are unspliced mRNAs. (A) Structures of TCR-β minigene constructs containing portions of three TCR-β exons and no intron, one intron, or two nonidentical introns. The three exons are depicted by light gray, dark gray, and black rectangles. Introns are represented by jagged lines. The transcriptional start site and polyadenylation site are indicated, as are the translational start and stop codons. (B) HeLa cells were transfected with the no-intron (lane a), one-intron (lane b), or two-intron (lane c) TCR-β constructs to yield approximately equal amounts of the spliced and unspliced mRNAs. Forty-eight hours after transfection, cells were infected with 20 PFU/cell of wild-type HSV-1 or mock infected, both in the presence of 10 μg/ml of actinomycin D. Cultures were harvested 5 h later and processed to determine the relative amounts of TCR-β mRNA, normalized to the levels in mock-infected cells. Error bars represent standard errors of the means.
(iii) Plasmids for tethering EJC components to an intronless mRNA.
Tethering of the exon junction complex to an unspliced mRNA was accomplished by using a target mRNA produced from a modified version of the no-intron TPI/RLuc/TPI construct described above in which the 5′ copies of TPI exons 6 and 7 were deleted and replaced with sequences containing 6 tandem copies of the binding site for the MS2 coat protein (80) (see Fig. 5A). The sequences encoding the MS2-binding sites began 49 nucleotides downstream from the start codon, in frame with the RLuc open reading frame.
FIG 5.

Effect of HSV-1 infection on unspliced mRNAs tethered to components of the exon junction complex. (A) Structures of an RLuc reporter plasmid encoding an mRNA with 6 copies of the RNA-binding site for the MS2 coat protein and a control FLuc reporter lacking MS2-binding sites. Plasmids encoding fusion proteins with the MS2 RNA-binding domain fused to various EJC components are shown at the right. (B and C) HeLa cells were transfected with equal amounts of the RLuc and FLuc reporter plasmids along with either a plasmid encoding only the MS2 RNA-binding domain or a plasmid encoding a fusion protein with the MS2 RNA-binding domain fused to the indicated EJC component. The cells were infected 48 h later with 20 PFU/cell of wild-type HSV-1 or mock infected. Cell lysates were prepared 3 h (B) or 9 h (C) after infection and analyzed for RLuc and FLuc activities, and RLuc activity was normalized to that of FLuc. (D) HeLa cells were transfected with equal amounts of the RLuc and FLuc reporter plasmids along with a plasmid encoding either the MS2 RNA-binding domain or the MS2 RNA-binding domain fused to eIF4AIII. Cell lysates were prepared 3 or 9 h after infection and analyzed for the relative amounts of RLuc mRNA. Error bars represent standard errors of the means.
Plasmids expressing N-terminal fusions of the RNA-binding domain of the MS2 coat protein and the EJC components REF2-I, DEK, RNPS1, SRm160, and eIF4AIII were described previously (85, 86), as was a plasmid encoding a C-terminal MS2 fusion to the EJC component Y14 (85, 86). Plasmids harboring target mRNAs containing MS2 coat protein-binding sites and plasmids encoding fusions of the MS2 coat protein with EJC components were provided by Melissa Moore.
DNA transfections.
To examine the effect of splicing on the sensitivity of a TPI/RLuc/TPI mRNA to copies of Vhs from infecting virions, HeLa cells were seeded into 6-well tissue culture trays at a density of 1 × 106 cells/well in MEM plus 10% (vol/vol) fetal bovine serum without antibiotics. The next day, the cells were transfected with 3.5 μg/well of the no-intron TPI/RLuc/TPI plasmid (Fig. 1A), 700 ng/well of the 5′ intron construct, or 2.5 μg/well of the 3′ intron plasmid by using Lipofectamine 2000 (Invitrogen) according to procedures recommended by the manufacturer. Enough of the plasmid pCMV-HA was added to equalize the total amount of DNA in each transfection mixture. Preliminary experiments showed that transfections with these amounts of the plasmids resulted in the accumulation of approximately equal amounts of unspliced and spliced TPI/RLuc/TPI mRNAs in the cytoplasm (data not shown). The medium was refreshed the next day. Forty-eight hours after transfection, the cells were infected with 20 PFU/cell of wild-type HSV-1 strain KOS or mock infected, both in the presence of 10 μg/ml of actinomycin D. Parallel cultures were harvested 5 h after infection or mock infection and processed for the determination of renilla luciferase activity or the relative amounts of TPI/RLuc/TPI mRNA.
Similar experiments to examine the effect of virion copies of Vhs on unspliced and spliced TCR-β minigene mRNAs were initiated by transfecting cells with 4 μg of the no-intron TCR-β minigene construct (Fig. 2A), 800 ng of the one-intron plasmid, or 400 ng of the two-intron plasmid. As was the case for transfections with the TPI/RLuc/TPI constructs, the amounts of the TCR-β minigene constructs included in the transfection mixtures were chosen because they resulted in approximately equal amounts of mature unspliced and spliced mRNAs in the cytoplasm (data not shown). Cells were infected or mock infected in the presence of actinomycin D, as described above, and harvested 5 h after infection, after which lysates were analyzed for the amounts of TCR-β minigene mRNA.
Experiments to examine the expression of unspliced and spliced TPI/RLuc/TPI mRNAs during productive HSV-1 infections were performed as described above for those analyzing virion copies of Vhs, except that cells were infected in the absence of actinomycin D and harvested 3, 6, or 9 h after infection or mock infection. To determine whether Vhs has a differential effect on the expression of spliced and unspliced mRNAs when it is the only viral protein present, cells were transfected with 3.5 μg of the no-intron TPI/RLuc/RPI plasmid or 700 ng of the 5′ intron plasmid, together with either 3.5 μg of the Vhs-expressing plasmid pKosAmp or 3.5 μg of the expression vector lacking Vhs. As described above, pCMV-HA was used to equalize the total amount of DNA in each transfection mixture. The cells were harvested 48 h after transfection and assayed for RLuc activity and the amount of TPI/RLuc/TPI mRNA.
Effect of HSV-1 on unspliced mRNAs tethered to components of the exon junction complex.
To determine whether the stimulatory effect of HSV-1 on the expression of spliced mRNAs could be replicated by artificially tethering EJC components to an unspliced mRNA, HeLa cells were transfected with 1 μg/well of a plasmid harboring an RLuc/TPI mRNA with 6 copies of the binding site for the MS2 coat protein (80) along with 1 μg/well of a control plasmid harboring an mRNA for firefly luciferase (FLuc) without any MS2-binding sites (see Fig. 5A). Transfection mixtures also contained 3 μg/well of a plasmid encoding only the RNA-binding domain of the MS2 coat protein or a plasmid encoding a fusion protein of MS2 fused to the EJC component REF2-I, DEK, SRm160, or Y14. In accord with data from a previous report (80), we found that the MS2-RNPS1 fusion protein was expressed at higher levels than the other fusion proteins (data not shown). Therefore, in these experiments, cells were transfected with 1.35 μg/well of the plasmid encoding MS2-RNPS1 along with 450 ng/well each of the plasmids harboring the RLuc/TPI and FLuc target mRNAs and 2.75 μg/well of the carrier pCMV-HA to equalize the total amounts of DNA in the transfection mixtures. Forty-eight hours after transfection, the cells were infected with 20 PFU/cell of wild-type HSV-1 or mock infected and harvested 3 or 9 h after infection. Cell lysates were analyzed for RLuc and FLuc activities, and the RLuc activity was normalized to that of FLuc.
Luciferase assays.
For cells transfected with an RLuc-expressing plasmid, luciferase activity was measured by using a renilla luciferase assay system (Promega) and a GloMax 20/20 single-tube luminometer according to protocols recommended by the manufacturers. For cells transfected with both RLuc- and FLuc-expressing plasmids, the cells were lysed in passive lysis buffer, and RLuc and FLuc activities were assayed by using a dual-luciferase assay system (Promega). For each sample, RLuc activity was normalized to FLuc activity.
Real-time quantitative reverse transcription-PCR.
The relative amounts of TPI/RLuc/TPI and TCR-β minigene mRNAs were quantified, as described previously (47), by real-time quantitative reverse transcription-PCR (RT-PCR) using an Applied Biosystems model 7500 real-time PCR system. Samples of whole-cell or total cytoplasmic RNA were extracted with TRIzol (Invitrogen) and treated with RQ1 RNase-free DNase (Promega), after which DNase activity was inactivated by stop solution and heating to 65°C. (In these experiments, whole-cell and total cytoplasmic RNAs gave similar results [data not shown].) RNA samples were reverse transcribed by using a High Capacity cDNA reverse transcription kit from Applied Biosystems. Triplicate or quadruplicate aliquots of each cDNA reaction mixture were amplified by using TaqMan primers and probes for TPI/RLuc/TPI or TCR-β minigene mRNAs, and another three or four aliquots were amplified with primers and probes for 18S rRNA. The primers and probes for TPI/RLuc/TPI and TCR-β minigene mRNAs were custom designed by Applied Biosystems. The TPI/RLuc/TPI primers and probes were designed to bind the RLuc transgene to avoid background amplification of the endogenous TPI mRNA, while the probe for TCR-β was designed to bind TCR-β mRNA after intron 2 had been excised. Primers and probes for 18S rRNA were purchased from Applied Biosystems. TPI/RLuc/TPI and TCR-β minigene mRNA levels were determined by using 18S rRNA as an internal standard, since Vhs does not affect rRNAs (15, 17, 87). For each type of transfection, the relative amounts of TPI/RLuc/TPI and TCR-β minigene mRNAs were determined by using the ΔΔCT method described in the model 7500 manual (88). The calculation and propagation of standard errors of the means were performed according to established procedures (89).
RESULTS
Spliced mRNAs are less sensitive to Vhs from infecting virions than are unspliced mRNAs.
To examine whether splicing affects the translation or half-lives of mRNAs during HSV-1 infection, we employed three plasmids (Fig. 1A) previously used by Nott and colleagues to show that in uninfected HeLa cells, splicing enhances the translation efficiency of an mRNA (79). These plasmids encode identical proteins containing the RLuc sequence flanked at both ends by copies of TPI exons 6 and 7 (79). The no-intron construct produces an unspliced TPI/RLuc/TPI mRNA, while the other two constructs contain TPI intron 6 inserted between the 5′ or 3′ copies of TPI exons 6 and 7. Previous studies showed that following transfection of HeLa cells, precursor mRNAs from the intron-containing constructs are spliced to give rise to mature mRNAs indistinguishable from those transcribed from the no-intron construct (79). The 5′-proximal AUG codon is in the 5′ copy of TPI exon 6, and, at least for transcripts with the 5′ intron, correct splicing is required to place the RLuc sequence in the same reading frame as the start codon. Therefore, for this construct, a protein with RLuc activity can be made only from mRNAs that have been correctly spliced (79).
Because splicing often affects the amount of an mRNA that accumulates in the cytoplasm (79), HeLa cells were transfected with different amounts of the no-intron, 5′ intron, or 3′ intron constructs to yield approximately equal amounts of mature TPI/RLuc/TPI mRNA in the cytoplasm. Forty-eight hours after transfection, the cells were infected with 20 PFU/cell of wild-type HSV-1 or mock infected, both in the presence of 10 μg/ml of actinomycin D. The cells were harvested 5 h later, and lysates were analyzed to determine the levels of TPI/RLuc/TPI mRNA and RLuc enzymatic activity (Fig. 1). Infection in the presence of actinomycin D precludes de novo viral and cellular transcription and allows a convenient measure of the effect of copies of Vhs from infecting virions on mRNAs transcribed prior to infection. In cells transfected with the no-intron plasmid, wild-type HSV-1 infection reduced the level of TPI/RLuc/TPI mRNA to approximately 45% of that in mock-infected cells (Fig. 1B), which is indicative of normal Vhs activity. In contrast, in cells transfected with plasmids with either a 5′ or a 3′ intron, the levels of TPI/RLuc/TPI mRNA were essentially indistinguishable in infected and mock-infected cells. Similar results were observed for the effect of HSV-1 on RLuc enzymatic activity (Fig. 1C), suggesting that the relative mRNA levels measured by real-time RT-PCR reflected the levels of translatable mRNA. The results indicate that spliced mRNAs containing either a 5′ or 3′ exon junction are considerably less sensitive to degradation by Vhs from infecting virions than are unspliced TPI/RLuc/TPI mRNAs.
To investigate whether the results with the TPI/RLuc/TPI constructs could be extended to other intron-containing constructs, HeLa cells were transfected with plasmids containing T cell receptor (TCR-β) minigenes with no introns, one intron, or two nonidentical introns (Fig. 2A). As in the above-described experiments, the cells were infected with wild-type HSV-1 or mock infected in the presence of actinomycin D and harvested 5 h later for determination of the amounts of TCR-β minigene mRNA by real-time RT-PCR using a TaqMan probe that spans the second exon junction of spliced mRNAs, hybridizing to sequences in both the second and third exons. As such, it will detect TCR-β mRNAs transcribed from the intronless minigene or mRNAs from the intron-containing constructs after the removal of the second intron by splicing but will not detect unspliced mRNAs from the intron-containing constructs. Once again, HSV-1 infection caused a significant reduction in the amount of unspliced mRNA, while mRNAs from the one-intron construct were less sensitive to Vhs-mediated degradation, and mRNAs from the two-intron construct were even less sensitive (Fig. 3B). The results show that the inclusion of an exon junction stabilizes at least two different mRNAs against Vhs-induced degradation.
FIG 3.

HSV-1 stimulates the accumulation of spliced TPI/RLuc/TPI mRNAs and RLuc activity during productive infections. HeLa cells were transfected with the no-intron (A and D), 5′ intron (B and E), or 3′ intron (C and F) TPI/RLuc/RPI constructs to yield approximately equal amounts of spliced and unspliced mRNAs. Forty-eight hours later, cells were infected with 20 PFU/cell of wild-type HSV-1 or mutant strain Vhs 1 or were mock infected. Replicate cultures were harvested 3 h, 6 h, or 9 h after infection or mock infection and processed to determine the relative amounts of RLuc enzymatic activity (A to C) or TPI/RLuc/TPI mRNA (D to F). For each type of mRNA, the levels of RLuc activity and mRNA at various times in infected and mock-infected cells were normalized to the levels at 3 h in mock-infected cells. Error bars represent the standard errors of the means.
HSV-1 stimulates expression of spliced mRNAs during productive infections.
The above-described experiments examined Vhs cleavage of spliced and unspliced mRNAs following infection of cells in the presence of actinomycin D to preclude viral gene expression and focus on mRNA cleavage by Vhs molecules that entered the cell as components of infecting virions. We next examined the accumulation and translation of spliced and unspliced mRNAs during productive HSV-1 infection. To this end, HeLa cells were transfected with the no-intron, 5′ intron, or 3′ intron TPI/RLuc/TPI constructs to yield approximately equal amounts of cytoplasmic mRNAs. Forty-eight hours after transfection, the cells were mock infected or infected with 20 PFU/cell of wild-type HSV-1 or mutant strain Vhs 1, which contains a point mutation in the UL41 open reading frame that changes threonine 214 to isoleucine and abrogates its ability to degrade mRNAs that are translated by cap-dependent ribosome scanning. The cells were harvested 3, 6, and 9 h after infection and analyzed for RLuc enzymatic activity and the amount of TPI/RLuc/TPI mRNA (Fig. 3).
Infection with wild-type HSV-1 caused a modest increase in RLuc activity encoded by the intronless construct during the first 9 h after infection (Fig. 3A). In contrast, HSV-1 infection caused a much more striking increase in RLuc activity encoded by the constructs with 5′ and 3′ introns (Fig. 3B and C), which reached levels 16 and 14 times higher than those seen in mock-infected cells by 9 h after infection. The stimulatory effect of wild-type HSV-1 infection on RLuc activity from the 5′ intron construct was accompanied at 9 h by a similar increase in mRNA levels, although at 6 h, the stimulation of RLuc activity was greater than that of TPI/RLuc/TPI mRNA (Fig. 3B and E). While wild-type HSV-1 infection caused a significant stimulation of RLuc protein and mRNA levels from the 3′ intron construct, it caused a greater stimulation levels of RLuc activity than mRNA at both 6 and 9 h (Fig. 3C and F).
Given that spliced TPI/RLuc/TPI mRNAs transcribed prior to infection are more resistant to Vhs degradation than are unspliced mRNAs (Fig. 1), it is likely that this contributes to the increased accumulation and translation of spliced relative to unspliced mRNAs during productive infection. However, if their greater resistance to Vhs was the only reason for the increased accumulation and expression of spliced mRNAs, the levels and translation of spliced and unspliced mRNAs should be similar in cells infected with a Vhs mutant lacking mRNA-degradative activity. Contrary to this expectation, infection with Vhs 1 caused significant increases in RLuc activity and mRNA levels from the intron-containing relative to the intronless constructs (Fig. 3B, C, E, and F). The results suggest that the effect of splicing is not simply to render an mRNA more resistant to Vhs degradation but also to allow positive stimulation of its accumulation and translation during HSV-1 infection.
Because this stimulatory effect was observed with Vhs 1, it could not have been dependent on the canonical mRNA-degradative activity of Vhs and may have involved additional viral proteins synthesized after infection. However, because Vhs 1 contains a single amino acid change in UL41, it remained possible that stimulation involved, at least in part, a second function of Vhs independent of its mRNA-degradative activity. To test this possibility, we examined whether Vhs could stimulate the translation and accumulation of a spliced TPI/RLuc/TPI mRNA in the absence of other viral gene products. To this end, HeLa cells were transfected with either the intronless or 5′-intron-containing TPI/RLuc/TPI constructs along with either a Vhs expression plasmid or the empty expression vector. Cells were harvested 48 h after transfection, and cytoplasmic extracts were analyzed for levels of RLuc activity and TPI/RLuc/TPI mRNA. For cells transfected with the intronless construct, the levels of RLuc protein and mRNA were indistinguishable in cells transfected with the Vhs expression plasmid and those transfected with the empty expression vector (Fig. 4). In contrast, in cells transfected with the 5′-intron-containing construct, wild-type Vhs stimulated the production of the RLuc protein and the accumulation of TPI/RLuc/TPI mRNA to levels that were approximately twice those seen in cells transfected with the empty expression vector (Fig. 4). The results indicate that Vhs caused a modest stimulation of the translation and accumulation of a spliced mRNA in the absence of other viral gene products.
FIG 4.
Vhs stimulates the accumulation and expression of spliced TPI/RLuc/TPI mRNAs in the absence of other viral gene products. HeLa cells were transfected with the no-intron or 5′ intron TPI/RLuc/TPI constructs along with either a plasmid expressing wild-type Vhs or the empty expression vector. Replicate cultures were harvested 48 h later and processed to determine the levels of TPI/RLuc/TPI mRNA (A) and RLuc activity (B), normalized to the levels in cells that received the empty expression vector. Error bars represent standard errors of the means.
Effect of tethering of EJC components to an unspliced RLuc mRNA.
To investigate whether the presence of an exon junction complex plays a role in the HSV-mediated stimulation of RLuc synthesis from spliced mRNAs, we examined whether stimulation could be replicated by artificially tethering EJC components to an unspliced RLuc mRNA. To this end, we utilized a set of plasmids used by Nott and coworkers to demonstrate that tethering of EJC components to an unspliced mRNA stimulates its translational efficiency in uninfected HeLa cells (80). Briefly, HeLa cells were transfected with an intronless RLuc reporter plasmid containing six copies of the RNA-binding site for the MS2 coat protein in frame at the 5′ end of the RLuc open reading frame along with a control reporter plasmid harboring firefly luciferase (FLuc) mRNA lacking MS2-binding sites (Fig. 5A). Transfection mixtures also contained plasmids encoding the RNA-binding domain of the MS2 coat protein or the MS2 RNA-binding domain fused to individual EJC components. As in previous studies (80), this should allow tethering of the MS2-EJC fusion proteins to RLuc mRNAs that contain MS2 RNA-binding sites but not to FLuc mRNAs, which lack them. Forty-eight hours after transfection, the cells were infected with 20 PFU/cell of wild-type HSV-1 or mock infected. Cell extracts were prepared 3 or 9 h after infection and assayed for RLuc and FLuc activities, and the RLuc activity was normalized to the level of FLuc activity (Fig. 5B and C).
Similar levels of normalized RLuc activity were seen 3 h after infection or mock infection in cells expressing the MS2 coat protein and in cells expressing any of the MS2-EJC fusion proteins, indicating that there was no detectable HSV-mediated stimulation of RLuc synthesis at that time (Fig. 5B). Nine hours after infection, similar RLuc levels were seen in infected and mock-infected cells transfected with the control plasmid encoding the MS2 coat protein as well as in cells transfected with the plasmid encoding the MS2-DEK fusion protein, indicating that in these cells, there was no HSV-1 stimulation of RLuc synthesis (Fig. 5C). However, HSV-1 infection caused a 2- to 3-fold stimulation of RLuc expression in cells transfected with plasmids encoding the MS2-SRM160, MS2-Y14, and MS2–REF2-I fusion proteins and a 5-fold stimulation in cells transfected with the MS2-RNSP1 fusion protein (Fig. 5C). The data suggest that tethering of selected EJC components to an unspliced RLuc mRNA allowed the HSV-1-mediated stimulation of RLuc synthesis.
In a similar experiment, HeLa cells were transfected with the RLuc and FLuc reporter plasmids along with a plasmid encoding either the MS2 RNA-binding domain or the MS2 RNA-binding domain fused to the EJC component eIF4AIII (Fig. 5D). The cells were infected or mock infected and then analyzed at 3 or 9 h for the relative levels of RLuc mRNA. Similar relative RLuc mRNA levels were seen 3 h after infection or mock infection in cells expressing the MS2 RNA-binding domain and in cells expressing the MS2-eIF4AIII fusion protein, indicating there was no HSV-1-mediated stimulation of RLuc expression at that time (Fig. 5D). However, by 9 h, HSV-1 infection caused a 3-fold stimulation of the relative RLuc mRNA level in cells transfected with the plasmid encoding MS2-eIF4AIII (Fig. 5D).
To examine whether this HSV-1-mediated stimulation of RLuc expression was at least partially due to Vhs, cells were transfected with the intronless RLuc reporter plasmid with the MS2 RNA-binding sites, the control FLuc reporter plasmid, and plasmids encoding either the MS2 RNA-binding domain or an MS2-eIF4AIII fusion protein. However, in this experiment, instead of infecting cells with virus, cells were transfected with plasmids encoding wild-type or mutant Vhs polypeptides, the empty expression vector, or a plasmid encoding ICP0, a multifunctional viral immediate early protein that stimulates the phosphorylation of eIF4E and the assembly of the eIF4F cap-binding complex (Fig. 6) (90). Cell lysates were prepared, and RLuc and FLuc activities were determined 24 h after transfection.
FIG 6.

Vhs stimulates eIF4AIII-dependent expression of an intronless RLuc mRNA in the absence of other HSV-1 gene products. HeLa cells were transfected with equal amounts of the RLuc and FLuc reporter plasmids along with either a plasmid encoding the MS2 RNA-binding domain (lane a) or a plasmid encoding an MS2-eIF4AIII fusion protein (lanes b to g). Transfection mixtures also contained plasmids harboring wild-type Vhs (lane c); the Vhs T214I (lane d), D194N (lane e), or D215N (lane f) mutant allele; the immediate early protein ICP0 (lane g); or the empty expression vector (lanes a and b). Cell lysates were prepared 48 h after transfection and analyzed for RLuc and FLuc activities, and RLuc activity was normalized to that of FLuc. Error bars represent standard errors of the means.
Equivalent RLuc/FLuc ratios were observed in cells transfected with the empty expression vector and plasmids encoding MS2-eIF4AIII or MS2, indicating that the MS2-eIF4AIII plasmid did not by itself stimulate RLuc activity (Fig. 6, lanes a and b). Similarly, transfection of cells with plasmids encoding ICP0 and MS2-eIF4AIII did not stimulate RLuc expression beyond the level seen in cells transfected with MS2-eIF4AIII and the empty expression vector (Fig. 6, lanes b and g). In contrast, transfection of cells with plasmids encoding wild-type Vhs and MS2-eIF4AIII resulted in a 2-fold stimulation of RLuc expression relative to control transfections involving MS2-eIF4AIII and the empty expression vector (Fig. 6, lanes b and c). This supports the results in Fig. 5D suggesting that tethering of an MS2-eIF4AIII fusion protein to an unspliced RLuc mRNA enables HSV-1-mediated stimulation of RLuc expression. Furthermore, in accord with data from the experiments shown in Fig. 3, this stimulation did not require that the Vhs protein retain mRNA degradative activity, since transfection with the Vhs T214I allele (carried by the mutant virus Vhs 1) caused an almost 3-fold stimulation of RLuc expression (Fig. 6, lane d). Similarly, transfection with the D194N allele, a Vhs allele encoding a protein that lacks nuclease activity due to a single amino acid change in the key nuclease motif (5), stimulated RLuc expression to the same extent as wild-type Vhs (Fig. 6, lane e). In contrast, transfection with the D215N allele, a Vhs allele with a change in another residue of the nuclease motif (5), did not cause an appreciable stimulation of RLuc expression above that seen with control transfections (Fig. 6, lanes b and f). These data suggest that the exon junction complex may play an important role in gene expression during HSV-1 infections.
DISCUSSION
These studies demonstrate that the splicing history of an mRNA, namely, whether or not it has been spliced, can significantly affect its level of accumulation and translation during HSV-1 infection. This conclusion was based on two types of experiments. First, during infections in the presence of actinomycin D to block viral and cellular transcription, spliced mRNAs transcribed prior to infection were more resistant to degradation by Vhs from infecting virions than were mRNAs that had not undergone splicing. Second, during productive infections, HSV-1 caused a dramatic 14- to 16-fold increase in the synthesis of proteins encoded by spliced mRNAs and an increase in the levels of mRNAs from intron-containing constructs. The magnitude of the HSV-1 stimulation of TPI/RLuc/TPI protein synthesis from spliced mRNAs was particularly striking because it represented a 14- to 16-fold stimulation above the already enhanced levels of translation seen for spliced mRNAs in uninfected cells (80).
The effects during productive infections may have been the result of several mechanisms, some of which involve Vhs and others of which do not. One would expect that if spliced TPI/RLuc/TPI mRNAs transcribed after infection are similar to those made before infection in being more resistant to Vhs degradation than unspliced mRNAs, this increased resistance should contribute to increased levels of spliced mRNAs, with increased synthesis of the proteins that they encode. However, the observation that Vhs 1, a mutant that lacks Vhs mRNA-degradative activity, still stimulates protein synthesis from spliced mRNAs (Fig. 3) suggests that splicing does not simply shield an mRNA from Vhs degradation but may also enable positive HSV-mediated stimulation of its levels and translation. These stimulatory activities may depend, at least in part, on one or more viral proteins, other than Vhs, that are synthesized after infection. In addition, because Vhs 1 encodes a UL41 protein with a single amino acid change (12, 83, 91), it is possible that the increased synthesis of proteins from spliced mRNAs depends, at least in part, on a second function of the Vhs protein, one that is independent of its canonical mRNA-degradative activity but is retained by Vhs 1. In this regard, transfection of cells with the wild-type Vhs allele or the T214I allele, the allele carried by Vhs 1, stimulates the accumulation of spliced mRNAs and their translation products in the absence of other viral gene products (Fig. 4 and 6), an observation that is consistent with the possibility that under conditions where a spliced mRNA is not degraded, either due to its inherent resistance to Vhs degradation or due to a mutation in Vhs, translation of the mRNA may be stimulated by a second function of Vhs.
Previous studies have shown that in uninfected cells, splicing enhances both the cytoplasmic accumulation and level of translation of spliced mRNAs compared to unspliced mRNAs (79, 80). However, splicing led to a greater increase in the amount of protein synthesized from an mRNA than could be explained simply by the observed increase in the amount of mRNA, leading to the conclusion that splicing increases the efficiency with which an mRNA is translated (79, 80). In the present study, HSV-1 infection caused a significant increase in the levels of spliced TPI/RLuc/TPI mRNAs and, at least at some time points, an even greater increase in the amount of RLuc protein synthesized from the intron-containing constructs (Fig. 3). Thus, for infections with wild-type HSV-1, the increase in RLuc protein synthesis from mRNAs with a 3′ exon junction was twice as great as the increase in mRNA levels 9 h after infection and almost 5 times as great as those at 6 h (Fig. 3C and F). Similarly, for mRNAs with a 5′ exon junction, the stimulation of RLuc synthesis by wild-type HSV-1 was 3 to 4 times greater at 6 h than the stimulation of TPI/RLuc/TPI mRNA levels (Fig. 3B and E), although by 9 h, the stimulations of RLuc activity and TPI/RLuc/TPI mRNA levels were more comparable. These data indicate that HSV-1 dramatically stimulates TPI/RLuc/TPI protein synthesis from spliced mRNAs and, perhaps to a lesser extent, stimulates an increase in the levels of the spliced mRNAs. This in turn suggests that HSV-1 infection may stimulate the translational efficiencies of spliced mRNAs, especially at intermediate and late times after infection, although additional experiments are required to demonstrate conclusively whether this is the case.
Recent studies, primarily from the Smiley laboratory, have reported instances in which Vhs stimulates the translation of specific mRNAs instead of degrading them (20–23). These include mRNAs containing several cellular internal ribosome entry sites (IRESs) (23) as well as specific viral mRNAs that are translated by cap-dependent ribosome scanning at late times after infection (20–22). However, data from those previous studies differed from our current results in several important respects. First, none of the previous reports linked translational stimulation by Vhs to splicing. Second, Dauber and coworkers provided evidence suggesting that one way in which Vhs stimulates the cap-dependent translation of some late viral mRNAs is by preventing mRNA overload (20), a function that probably is dependent upon the mRNA-degradative activity of Vhs. In contrast, data from the present studies suggest that Vhs enhances the accumulation of spliced mRNAs and their translation products through a mechanism that, at least partially, is independent of its mRNA-degradative activity. The present results are not necessarily incompatible with those from previous studies, and both processes may occur during wild-type HSV-1 infections. An interesting feature of those previous studies is that translational stimulation by Vhs was observed in HeLa cells but not in Vero cells or several other commonly used cell lines (20, 22, 23). We used HeLa cells in the present experiments because previous studies had shown that mRNAs transcribed from the TPI/RLuc/TPI and TCR-β minigene constructs are correctly spliced in these cells (79, 80). It will be interesting to determine if HSV-1 infection stimulates the translation and accumulation of spliced mRNAs in the same way in Vero cells as it does in HeLa cells.
The mechanism(s) by which splicing stabilizes an mRNA against degradation by virion copies of Vhs and by which HSV-1 stimulates the accumulation of spliced mRNAs and their translation products during productive infection remains to be determined. At least the latter effect appears to involve the presence of exon junction complexes on spliced mRNAs. This is suggested by the fact that artificial tethering of selected EJC components to an unspliced RLuc mRNA allowed HSV-1-mediated stimulation of RLuc synthesis during productive infection (Fig. 5). Whether HSV-1 somehow amplifies a normal cellular pathway that enhances the expression of spliced mRNAs or acts through a novel mechanism that exists only in virus-infected cells remains to be determined.
Interestingly, these experiments did not identify just one EJC component that, when part of an MS2 fusion protein, enabled HSV-1-mediated stimulation of the expression of unspliced mRNAs containing MS2-binding sites. Instead, fusion proteins involving several different EJC components enabled stimulation although to different extents (Fig. 5). The reason for this is unclear, although a similar result was observed by Nott and coworkers in their experiments showing that tethering of fusion proteins involving MS2 and selected EJC components to an unspliced mRNA stimulated its translation in uninfected cells (80). Those authors speculated that the binding of more than one kind of MS2-EJC fusion protein to the MS2-binding sites on an mRNA might nucleate the assembly of a complex involving one or more other EJC components on the mRNA and that such a complex or partial EJC might enable the stimulation of translation. Whether such a mechanism was responsible for the HSV-1-stimulated expression of RLuc from unspliced mRNAs with MS2-binding sites remains to be determined.
At first, our observation that spliced TPI/RLuc/TPI mRNAs, synthesized prior to the addition of actinomycin D at the time of infection, are less sensitive to degradation by Vhs from infecting virions than are unspliced mRNAs seemed surprising, because Vhs has long been known to degrade many cellular mRNAs (2, 5, 7, 9–11, 15, 16, 92), although most cellular mRNAs are spliced. A potential answer to this conundrum comes from the observation that although exon junction complexes facilitate translation initiation, they are removed from an mRNA during the pioneer round of translation. Almost all studies of host shutoff by Vhs have focused on the degradation of cellular mRNAs that were transcribed prior to infection and have been translated multiple times. These mRNAs should no longer be bound by EJCs and, therefore, should be equally as sensitive to degradation by Vhs as unspliced mRNAs.
Whether the relative resistance of spliced mRNAs to Vhs from incoming virions plays a significant role during HSV-1 infections is unknown. In contrast to most cellular mRNAs, all but five HSV-1 mRNAs are unspliced (1). Three of the spliced viral transcripts (ICP0, ICP22, and ICP47) are immediate early mRNAs, and two (UL15 and gC) are late mRNAs. If viral mRNAs transcribed and spliced immediately after infection are similar to newly synthesized spliced mRNAs from uninfected cells in being bound by exon junction complexes, the presence of EJCs on spliced immediate early mRNAs might temporarily shield them from Vhs degradation at the same time that Vhs is degrading many older spliced cellular mRNAs that have already been translated multiple times and from which the EJCs have been removed. This might increase the synthesis of these immediate early proteins and be beneficial in order to jump-start the infection. Subsequently, once the immediate early mRNAs have been translated and the EJCs have been removed, they would become as sensitive to Vhs degradation as unspliced viral mRNAs, allowing Vhs to play its role in the sequential expression of different classes of viral mRNAs. In addition, while Vhs accelerates the decay of many cellular mRNAs, HSV-1 infection actually stimulates the accumulation of some host mRNAs (55–57). If these newly induced cellular mRNAs are spliced, the presence of EJCs might transiently protect them from Vhs degradation and increase the production of the proteins that they encode. This could be beneficial if the induced mRNAs encode host proteins that play a role in virus replication.
Similarly, our observation that productive HSV-1 infection stimulates protein synthesis from spliced TPI/RLuc/TPI mRNAs (Fig. 3) was surprising because of the body of data indicating that the viral immediate early protein ICP27 inhibits splicing (93–98). This inhibition plays a significant role, along with Vhs, in the shutoff of host protein synthesis during HSV-1 infections (99, 100), and ICP27 is required for the transport of unspliced viral mRNAs from the nucleus to the cytoplasm (93, 101). UL15 is unique among the spliced HSV-1 mRNAs in that the splice site is in the open reading frame, meaning that the UL15 protein can be synthesized from spliced mRNAs only (98). The ICP27-mediated inhibition of splicing is incomplete, leading, at late times, to the accumulation of the mature spliced UL15 mRNA and its unspliced precursor in the cytoplasm (98). One possibility is that the stimulation of translation of spliced mRNAs seen in the present study facilitates the translation of the spliced UL15 mRNA to ensure sufficient synthesis of this structural viral protein in the face of the overall ICP27-mediated downturn in splicing. Similarly, spliced and unspliced gC mRNAs are found at late times during HSV-1 infection (102). The unspliced mRNA encodes the membrane glycoprotein that is an abundant component of the virion envelope (1), while the spliced mRNAs encode a secreted form of gC (102). It is possible that the stimulation of translation of spliced mRNAs plays a role in facilitating the synthesis of secreted gC.
It is worth noting that while three of the viral immediate early mRNAs (ICP0, ICP22, and ICP47) are spliced, two immediate early mRNAs (ICP4 and ICP27) are not (1). In light of the present results, it will be interesting to determine whether there are any splicing-dependent differences in the fates and expression of the different immediate early mRNAs at late times after infection. In addition, in light of the observation by Smiley et al. that knockout mutations in Vhs have a more pronounced effect on late protein synthesis and viral growth in HeLa cells than in some other cell lines (20, 22), it will be interesting to determine whether any effects of mRNA splicing on gene expression during HSV-1 infections are dependent on the type of cells that are infected.
The results of the present study suggest that splicing may stabilize mRNAs against degradation by Vhs from incoming virions and increase the amount of protein made from spliced mRNAs during productive HSV-1 infections. While very suggestive, the present experiments focused on artificially constructed TPI/RLuc/TPI and TCR-β minigene mRNAs. A key test of these results will be to determine the effect of introns on the Vhs sensitivity of naturally occurring cellular mRNAs, expressed constitutively prior to infection or induced at the time of infection or shortly thereafter, and on the Vhs sensitivity of viral immediate early mRNAs. Another test will be to examine the effect of introns on the translation of naturally occurring viral late mRNAs or on cellular mRNAs induced at late times after infection. These experiments are under way.
ACKNOWLEDGMENTS
We thank Melissa Moore from the University of Massachusetts Medical School for her generosity in providing plasmids harboring spliced and unspliced reporter mRNAs and for tethering EJC components to an unspliced reporter. We also thank our other colleagues Lora Shiflett and Emma Hayes at the University of Missouri—Kansas City School of Biological Sciences for many helpful discussions.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Read GS. 2013. Virus-encoded endonucleases: expected and novel functions. Wiley Interdiscip Rev RNA 4:693–708. doi: 10.1002/wrna.1188. [DOI] [PubMed] [Google Scholar]
- 3.Taddeo B, Zhang W, Roizman B. 2006. The U(L)41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc Natl Acad Sci U S A 103:2827–2832. doi: 10.1073/pnas.0510712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J Virol 80:9341–9345. doi: 10.1128/JVI.01008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Everly DN Jr, Feng P, Mian IS, Read GS. 2002. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J Virol 76:8560–8571. doi: 10.1128/JVI.76.17.8560-8571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elgadi MM, Hayes CE, Smiley JR. 1999. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J Virol 73:7153–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Read GS, Karr BM, Knight K. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J Virol 67:7149–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smibert CA, Johnson DC, Smiley JR. 1992. Identification and characterization of the virion-induced host shutoff product of herpes simplex virus gene UL41. J Gen Virol 73(Part 2):467–470. [DOI] [PubMed] [Google Scholar]
- 9.Strom T, Frenkel N. 1987. Effects of herpes simplex virus on mRNA stability. J Virol 61:2198–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schek N, Bachenheimer SL. 1985. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J Virol 55:601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fenwick ML, McMenamin MM. 1984. Early virion-associated suppression of cellular protein synthesis by herpes simplex virus is accompanied by inactivation of mRNA. J Gen Virol 65(Part 7):1225–1228. [DOI] [PubMed] [Google Scholar]
- 12.Read GS, Frenkel N. 1983. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J Virol 46:498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasieka TJ, Lu B, Crosby SD, Wylie KM, Morrison LA, Alexander DE, Menachery VD, Leib DA. 2008. Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J Virol 82:5527–5535. doi: 10.1128/JVI.02047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lam Q, Smibert CA, Koop KE, Lavery C, Capone JP, Weinheimer SP, Smiley JR. 1996. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J 15:2575–2581. [PMC free article] [PubMed] [Google Scholar]
- 15.Oroskar AA, Read GS. 1989. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J Virol 63:1897–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwong AD, Frenkel N. 1987. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc Natl Acad Sci U S A 84:1926–1930. doi: 10.1073/pnas.84.7.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oroskar AA, Read GS. 1987. A mutant of herpes simplex virus type 1 exhibits increased stability of immediate-early (alpha) mRNAs. J Virol 61:604–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Read GS. 1997. Control of mRNA stability during herpes simplex virus infections, p 311–321. In Harford JB, Morris DR (ed), mRNA metabolism and post-transcriptional gene regulation, 1st ed, vol 17 Wiley-Liss, Inc, New York, NY. [Google Scholar]
- 19.Dauber B, Poon D, dos Santos T, Duguay BA, Mehta N, Saffran HA, Smiley JR. 20 April 2016. The herpes simplex virus virion host shutoff protein enhances translation of viral true late mRNAs independently of suppressing protein kinase R and stress granule formation. J Virol doi: 10.1128/JVI.03180-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dauber B, Saffran HA, Smiley JR. 2014. The herpes simplex virus 1 virion host shutoff protein enhances translation of viral late mRNAs by preventing mRNA overload. J Virol 88:9624–9632. doi: 10.1128/JVI.01350-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finnen RL, Hay TJ, Dauber B, Smiley JR, Banfield BW. 2014. The herpes simplex virus 2 virion-associated ribonuclease vhs interferes with stress granule formation. J Virol 88:12727–12739. doi: 10.1128/JVI.01554-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dauber B, Pelletier J, Smiley JR. 2011. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J Virol 85:5363–5373. doi: 10.1128/JVI.00115-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saffran HA, Read GS, Smiley JR. 2010. Evidence for translational regulation by the herpes simplex virus virion host shutoff protein. J Virol 84:6041–6049. doi: 10.1128/JVI.01819-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasieka TJ, Menachery VD, Rosato PC, Leib DA. 2012. Corneal replication is an interferon response-independent bottleneck for virulence of herpes simplex virus 1 in the absence of virion host shutoff. J Virol 86:7692–7695. doi: 10.1128/JVI.00761-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korom M, Wylie KM, Morrison LA. 2008. Selective ablation of virion host shutoff protein RNase activity attenuates herpes simplex virus 2 in mice. J Virol 82:3642–3653. doi: 10.1128/JVI.02409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smiley JR. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J Virol 78:1063–1068. doi: 10.1128/JVI.78.3.1063-1068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith TJ, Morrison LA, Leib DA. 2002. Pathogenesis of herpes simplex virus type 2 virion host shutoff (vhs) mutants. J Virol 76:2054–2061. doi: 10.1128/jvi.76.5.2054-2061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith TJ, Ackland-Berglund CE, Leib DA. 2000. Herpes simplex virus virion host shutoff (vhs) activity alters periocular disease in mice. J Virol 74:3598–3604. doi: 10.1128/JVI.74.8.3598-3604.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strelow LI, Leib DA. 1996. Analysis of conserved domains of UL41 of herpes simplex virus type 1 in virion host shutoff and pathogenesis. J Virol 70:5665–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strelow LI, Leib DA. 1995. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J Virol 69:6779–6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurak I, Silverstein LB, Sharma M, Coen DM. 2012. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J Virol 86:10093–10102. doi: 10.1128/JVI.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sciortino MT, Parisi T, Siracusano G, Mastino A, Taddeo B, Roizman B. 2013. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase R in cells infected with herpes simplex virus 1. J Virol 87:3271–3276. doi: 10.1128/JVI.03049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy JA, Duerst RJ, Smith TJ, Morrison LA. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol 77:9337–9345. doi: 10.1128/JVI.77.17.9337-9345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cotter CR, Kim WK, Nguyen ML, Yount JS, Lopez CB, Blaho JA, Moran TM. 2011. The virion host shutoff protein of herpes simplex virus 1 blocks the replication-independent activation of NF-kappaB in dendritic cells in the absence of type I interferon signaling. J Virol 85:12662–12672. doi: 10.1128/JVI.05557-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cotter CR, Nguyen ML, Yount JS, Lopez CB, Blaho JA, Moran TM. 2010. The virion host shut-off (vhs) protein blocks a TLR-independent pathway of herpes simplex virus type 1 recognition in human and mouse dendritic cells. PLoS One 5:e8684. doi: 10.1371/journal.pone.0008684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiflett LA, Read GS. 2013. mRNA decay during herpes simplex virus (HSV) infections: mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (Vhs) protein. J Virol 87:94–109. doi: 10.1128/JVI.01557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Page HG, Read GS. 2010. The virion host shutoff endonuclease (UL41) of herpes simplex virus interacts with the cellular cap-binding complex eIF4F. J Virol 84:6886–6890. doi: 10.1128/JVI.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng P, Everly DN Jr, Read GS. 2005. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J Virol 79:9651–9664. doi: 10.1128/JVI.79.15.9651-9664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng P, Everly DN Jr, Read GS. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J Virol 75:10272–10280. doi: 10.1128/JVI.75.21.10272-10280.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parsyan A, Svitkin Y, Shahbazian D, Gkogkas C, Lasko P, Merrick WC, Sonenberg N. 2011. mRNA helicases: the tacticians of translational control. Nat Rev Mol Cell Biol 12:235–245. doi: 10.1038/nrm3083. [DOI] [PubMed] [Google Scholar]
- 41.Rogers GW Jr, Komar AA, Merrick WC. 2002. eIF4A: the godfather of the DEAD box helicases. Prog Nucleic Acid Res Mol Biol 72:307–331. doi: 10.1016/S0079-6603(02)72073-4. [DOI] [PubMed] [Google Scholar]
- 42.Richter-Cook NJ, Dever TE, Hensold JO, Merrick WC. 1998. Purification and characterization of a new eukaryotic protein translation factor. Eukaryotic initiation factor 4H. J Biol Chem 273:7579–7587. [DOI] [PubMed] [Google Scholar]
- 43.Richter NJ, Rogers GW Jr, Hensold JO, Merrick WC. 1999. Further biochemical and kinetic characterization of human eukaryotic initiation factor 4H. J Biol Chem 274:35415–35424. doi: 10.1074/jbc.274.50.35415. [DOI] [PubMed] [Google Scholar]
- 44.Rogers GW Jr, Richter NJ, Merrick WC. 1999. Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J Biol Chem 274:12236–12244. doi: 10.1074/jbc.274.18.12236. [DOI] [PubMed] [Google Scholar]
- 45.Rogers GW Jr, Lima WF, Merrick WC. 2001. Further characterization of the helicase activity of eIF4A. Substrate specificity. J Biol Chem 276:12598–12608. doi: 10.1074/jbc.M007560200. [DOI] [PubMed] [Google Scholar]
- 46.Rogers GW Jr, Richter NJ, Lima WF, Merrick WC. 2001. Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J Biol Chem 276:30914–30922. doi: 10.1074/jbc.M100157200. [DOI] [PubMed] [Google Scholar]
- 47.Sarma N, Agarwal D, Shiflett LA, Read GS. 2008. Small interfering RNAs that deplete the cellular translation factor eIF4H impede mRNA degradation by the virion host shutoff protein of herpes simplex virus. J Virol 82:6600–6609. doi: 10.1128/JVI.00137-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sadek JS. 2013. PhD thesis University of Missouri—Kansas City, Kansas City, MO. [Google Scholar]
- 49.Lindqvist L, Oberer M, Reibarkh M, Cencic R, Bordeleau ME, Vogt E, Marintchev A, Tanaka J, Fagotto F, Altmann M, Wagner G, Pelletier J. 2008. Selective pharmacological targeting of a DEAD box RNA helicase. PLoS One 3:e1583. doi: 10.1371/journal.pone.0001583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corcoran JA, Hsu WL, Smiley JR. 2006. Herpes simplex virus ICP27 is required for virus-induced stabilization of the ARE-containing IEX-1 mRNA encoded by the human IER3 gene. J Virol 80:9720–9729. doi: 10.1128/JVI.01216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu WL, Saffran HA, Smiley JR. 2005. Herpes simplex virus infection stabilizes cellular IEX-1 mRNA. J Virol 79:4090–4098. doi: 10.1128/JVI.79.7.4090-4098.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Esclatine A, Taddeo B, Roizman B. 2004. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J Virol 78:8582–8592. doi: 10.1128/JVI.78.16.8582-8592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Esclatine A, Taddeo B, Evans L, Roizman B. 2004. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc Natl Acad Sci U S A 101:3603–3608. doi: 10.1073/pnas.0400354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Esclatine A, Taddeo B, Roizman B. 2004. The UL41 protein of herpes simplex virus mediates selective stabilization or degradation of cellular mRNAs. Proc Natl Acad Sci U S A 101:18165–18170. doi: 10.1073/pnas.0408272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ray N, Enquist LW. 2004. Transcriptional response of a common permissive cell type to infection by two diverse alphaherpesviruses. J Virol 78:3489–3501. doi: 10.1128/JVI.78.7.3489-3501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hobbs WE, DeLuca NA. 1999. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J Virol 73:8245–8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khodarev NN, Advani SJ, Gupta N, Roizman B, Weichselbaum RR. 1999. Accumulation of specific RNAs encoding transcriptional factors and stress response proteins against a background of severe depletion of cellular RNAs in cells infected with herpes simplex virus 1. Proc Natl Acad Sci U S A 96:12062–12067. doi: 10.1073/pnas.96.21.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ. 2012. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151:750–764. doi: 10.1016/j.cell.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stroupe ME, Tange TO, Thomas DR, Moore MJ, Grigorieff N. 2006. The three-dimensional arcitecture [sic] of the EJC core. J Mol Biol 360:743–749. doi: 10.1016/j.jmb.2006.05.049. [DOI] [PubMed] [Google Scholar]
- 60.Chan CC, Dostie J, Diem MD, Feng W, Mann M, Rappsilber J, Dreyfuss G. 2004. eIF4A3 is a novel component of the exon junction complex. RNA 10:200–209. doi: 10.1261/rna.5230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shibuya T, Tange TO, Sonenberg N, Moore MJ. 2004. eIF4AIII binds spliced mRNA in the exon junction complex and is essential for nonsense-mediated decay. Nat Struct Mol Biol 11:346–351. doi: 10.1038/nsmb750. [DOI] [PubMed] [Google Scholar]
- 62.Tange TO, Nott A, Moore MJ. 2004. The ever-increasing complexities of the exon junction complex. Curr Opin Cell Biol 16:279–284. doi: 10.1016/j.ceb.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 63.Kim VN, Yong J, Kataoka N, Abel L, Diem MD, Dreyfuss G. 2001. The Y14 protein communicates to the cytoplasm the position of exon-exon junctions. EMBO J 20:2062–2068. doi: 10.1093/emboj/20.8.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le Hir H, Gatfield D, Izaurralde E, Moore MJ. 2001. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J 20:4987–4997. doi: 10.1093/emboj/20.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kataoka N, Yong J, Kim VN, Velazquez F, Perkinson RA, Wang F, Dreyfuss G. 2000. Pre-mRNA splicing imprints mRNA in the nucleus with a novel RNA-binding protein that persists in the cytoplasm. Mol Cell 6:673–682. doi: 10.1016/S1097-2765(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 66.Le Hir H, Izaurralde E, Maquat LE, Moore MJ. 2000. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J 19:6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Le Hir H, Moore MJ, Maquat LE. 2000. Pre-mRNA splicing alters mRNP composition: evidence for stable association of proteins at exon-exon junctions. Genes Dev 14:1098–1108. [PMC free article] [PubMed] [Google Scholar]
- 68.Chazal PE, Daguenet E, Wendling C, Ulryck N, Tomasetto C, Sargueil B, Le Hir H. 2013. EJC core component MLN51 interacts with eIF3 and activates translation. Proc Natl Acad Sci U S A 110:5903–5908. doi: 10.1073/pnas.1218732110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le Hir H, Seraphin B. 2008. EJCs at the heart of translational control. Cell 133:213–216. doi: 10.1016/j.cell.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 70.Ma XM, Yoon SO, Richardson CJ, Julich K, Blenis J. 2008. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 133:303–313. doi: 10.1016/j.cell.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 71.Diem MD, Chan CC, Younis I, Dreyfuss G. 2007. PYM binds the cytoplasmic exon-junction complex and ribosomes to enhance translation of spliced mRNAs. Nat Struct Mol Biol 14:1173–1179. doi: 10.1038/nsmb1321. [DOI] [PubMed] [Google Scholar]
- 72.Lu S, Cullen BR. 2003. Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA 9:618–630. doi: 10.1261/rna.5260303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wiegand HL, Lu S, Cullen BR. 2003. Exon junction complexes mediate the enhancing effect of splicing on mRNA expression. Proc Natl Acad Sci U S A 100:11327–11332. doi: 10.1073/pnas.1934877100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matsumoto K, Wassarman KM, Wolffe AP. 1998. Nuclear history of a pre-mRNA determines the translational activity of cytoplasmic mRNA. EMBO J 17:2107–2121. doi: 10.1093/emboj/17.7.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Braddock M, Muckenthaler M, White MR, Thorburn AM, Sommerville J, Kingsman AJ, Kingsman SM. 1994. Intron-less RNA injected into the nucleus of Xenopus oocytes accesses a regulated translation control pathway. Nucleic Acids Res 22:5255–5264. doi: 10.1093/nar/22.24.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gehring NH, Lamprinaki S, Kulozik AE, Hentze MW. 2009. Disassembly of exon junction complexes by PYM. Cell 137:536–548. doi: 10.1016/j.cell.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 77.Sato H, Maquat LE. 2009. Remodeling of the pioneer translation initiation complex involves translation and the karyopherin importin beta. Genes Dev 23:2537–2550. doi: 10.1101/gad.1817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dostie J, Dreyfuss G. 2002. Translation is required to remove Y14 from mRNAs in the cytoplasm. Curr Biol 12:1060–1067. doi: 10.1016/S0960-9822(02)00902-8. [DOI] [PubMed] [Google Scholar]
- 79.Nott A, Meislin SH, Moore MJ. 2003. A quantitative analysis of intron effects on mammalian gene expression. RNA 9:607–617. doi: 10.1261/rna.5250403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nott A, Le Hir H, Moore MJ. 2004. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev 18:210–222. doi: 10.1101/gad.1163204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Read GS, Patterson M. 2007. Packaging of the virion host shutoff (Vhs) protein of herpes simplex virus: two forms of the Vhs polypeptide are associated with intranuclear B and C capsids, but only one is associated with enveloped virions. J Virol 81:1148–1161. doi: 10.1128/JVI.01812-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Everly DN Jr, Read GS. 1997. Mutational analysis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): characterization of HSV type 1 (HSV-1)/HSV-2 chimeras. J Virol 71:7157–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kwong AD. 1988. The herpes simplex virus virion host shutoff and mRNA destabilization function, abstr 48. Abstr 13th Int Herpesvirus Workshop, Irvine, CA. [Google Scholar]
- 84.Everly DN Jr, Read GS. 1999. Site-directed mutagenesis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): analysis of functional differences between HSV type 1 (HSV-1) and HSV-2 alleles. J Virol 73:9117–9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lykke-Andersen J, Shu MD, Steitz JA. 2000. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103:1121–1131. doi: 10.1016/S0092-8674(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 86.Lykke-Andersen J, Shu MD, Steitz JA. 2001. Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science 293:1836–1839. doi: 10.1126/science.1062786. [DOI] [PubMed] [Google Scholar]
- 87.Krikorian CR, Read GS. 1991. In vitro mRNA degradation system to study the virion host shutoff function of herpes simplex virus. J Virol 65:112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. 2006. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol Chapter 15:Unit 15.8. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- 89.Bevington PR. 1969. Data reduction and error analysis for the physical sciences. McGraw-Hill Book Company, New York, NY. [Google Scholar]
- 90.Walsh D, Mohr I. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev 18:660–672. doi: 10.1101/gad.1185304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kwong AD, Kruper JA, Frenkel N. 1988. Herpes simplex virus virion host shutoff function. J Virol 62:912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fenwick ML, Everett RD. 1990. Transfer of UL41, the gene controlling virion-associated host cell shutoff, between different strains of herpes simplex virus. J Gen Virol 71(Part 2):411–418. [DOI] [PubMed] [Google Scholar]
- 93.Sandri-Goldin RM. 2008. The many roles of the regulatory protein ICP27 during herpes simplex virus infection. Front Biosci 13:5241–5256. [DOI] [PubMed] [Google Scholar]
- 94.Sciabica KS, Dai QJ, Sandri-Goldin RM. 2003. ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBO J 22:1608–1619. doi: 10.1093/emboj/cdg166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lindberg A, Kreivi JP. 2002. Splicing inhibition at the level of spliceosome assembly in the presence of herpes simplex virus protein ICP27. Virology 294:189–198. doi: 10.1006/viro.2001.1301. [DOI] [PubMed] [Google Scholar]
- 96.Bryant HE, Wadd SE, Lamond AI, Silverstein SJ, Clements JB. 2001. Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome-associated protein 145 and inhibits splicing prior to the first catalytic step. J Virol 75:4376–4385. doi: 10.1128/JVI.75.9.4376-4385.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sandri-Goldin RM, Hibbard MK, Hardwicke MA. 1995. The C-terminal repressor region of herpes simplex virus type 1 ICP27 is required for the redistribution of small nuclear ribonucleoprotein particles and splicing factor SC35; however, these alterations are not sufficient to inhibit host cell splicing. J Virol 69:6063–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hardy WR, Sandri-Goldin RM. 1994. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J Virol 68:7790–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soliman TM, Sandri-Goldin RM, Silverstein SJ. 1997. Shuttling of the herpes simplex virus type 1 regulatory protein ICP27 between the nucleus and cytoplasm mediates the expression of late proteins. J Virol 71:9188–9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hardwicke MA, Sandri-Goldin RM. 1994. The herpes simplex virus regulatory protein ICP27 contributes to the decrease in cellular mRNA levels during infection. J Virol 68:4797–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen IH, Sciabica KS, Sandri-Goldin RM. 2002. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J Virol 76:12877–12889. doi: 10.1128/JVI.76.24.12877-12889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sedlackova L, Perkins KD, Lengyel J, Strain AK, van Santen VL, Rice SA. 2008. Herpes simplex virus type 1 ICP27 regulates expression of a variant, secreted form of glycoprotein C by an intron retention mechanism. J Virol 82:7443–7455. doi: 10.1128/JVI.00388-08. [DOI] [PMC free article] [PubMed] [Google Scholar]