

ABSTRACT
Virus-like particles (VLPs) are attractive as a vaccine concept. For human respiratory syncytial virus (hRSV), VLP assembly is poorly understood and appears inefficient. Hence, hRSV antigens are often incorporated into foreign VLP systems to generate anti-RSV vaccine candidates. To better understand the assembly, and ultimately to enable efficient production, of authentic hRSV VLPs, we examined the associated requirements and mechanisms. In a previous analysis in HEp-2 cells, the nucleoprotein (N), phosphoprotein (P), matrix protein (M), and fusion protein (F) were required for formation of filamentous VLPs, which, similar to those of wild-type virus, were associated with the cell surface. Using fluorescence and electron microscopy combined with immunogold labeling, we examined the surfaces of transfected HEp-2 cells and further dissected the process of filamentous VLP formation. Our results show that N is not required. Coexpression of P plus M plus F, but not P plus M, M plus F, or P plus F, induced both viral protein coalescence and formation of filamentous VLPs that resembled wild-type virions. Despite suboptimal coalescence in the absence of P, the M and F proteins, when coexpressed, formed cell surface-associated filaments with abnormal morphology, appearing longer and thinner than wild-type virions. For F, only the carboxy terminus (Fstem) was required, and addition of foreign protein sequences to Fstem allowed incorporation into VLPs. Together, the data show that P, M, and the F carboxy terminus are sufficient for robust viral protein coalescence and filamentous VLP formation and suggest that M-F interaction drives viral filament formation, with P acting as a type of cofactor facilitating the process and exerting control over particle morphology.
IMPORTANCE hRSV is responsible for >100,000 deaths in children worldwide, and a vaccine is not available. Among the potential anti-hRSV approaches are virus-like particle (VLP) vaccines, which, based on resemblance to virus or viral components, can induce protective immunity. For hRSV, few reports are available concerning authentic VLP production or testing, in large part because VLP production is inefficient and the mechanisms underlying particle assembly are poorly understood. Here, we took advantage of the cell-associated nature of RSV particles and used high-resolution microscopy analyses to examine the viral proteins required for formation of wild-type-virus-resembling VLPs, the contributions of these proteins to morphology, and the domains involved in incorporation of the antigenically important viral F protein. The results provide new insights that will facilitate future production of hRSV VLPs with defined shapes and compositions and may translate into improved manufacture of live-attenuated hRSV vaccines.
INTRODUCTION
Human respiratory syncytial virus (hRSV) is an important viral agent of respiratory tract disease in infants, children, immunosuppressed individuals, and the elderly (1–3). In pursuit of a vaccine to prevent hRSV disease, a virus-like particle (VLP) approach is among the potential strategies. Generation of hRSV VLPs from plasmids was reported previously but was inefficient (4, 5). The vast majority of the reports of tests of vaccine potential concern heterologous VLPs or nanoparticles carrying the hRSV F and/or G protein, in part because these systems are established or efficient and because hRSV particle assembly is poorly understood. The heterologous systems include Newcastle disease virus-, Sendai virus-, or baculovirus-based VLPs; nanoparticles; and gold-based nanorods and have shown encouraging results in the BALB/c mouse model (6–12) and humans (13, 14). In comparison, authentic hRSV VLPs structurally resemble wild-type (wt) virions and also incorporate some of the internal hRSV proteins (5), features that may be advantageous for vaccine purposes. To enable efficient hRSV VLP production and allow future testing of the vaccine potential of hRSV VLPs, a better understanding of the processes that govern particle assembly, morphology, and protein incorporation is needed.
hRSV is a pleiomorphic virus. Whereas highly purified or freeze-thawed preparations can exhibit a relative high proportion of spherical particles, hRSV infectivity appears to be predominantly associated with a filamentous virion form (15–20). This was further supported by recent findings in which hRSV particles or regions of particles containing regularly spaced fusion (F) protein in the prefusion conformation had matrix (M) protein underlying the membrane and were often filamentous in nature (21). hRSV VLPs also appear to be predominantly filamentous, as M mutations that prevented M dimerization simultaneously blocked the formation of cell surface hRSV filaments and budding/release of VLPs (22). In the hRSV life cycle, filamentous virions emerge at the cell surface at a late stage, and the majority remain associated with the infected cell surface by an unknown mechanism (19, 23–25). No morphological differences were reported between secreted and cell-associated virions (21). The cell-associated nature of virions is problematic for virus purification but, on the other hand, allows visualization at the infected cell surface by immunofluorescence microscopy (IFM) or electron microscopy (EM). Moreover, observing morphology directly at the cell surface avoids potential changes in hRSV morphology associated with particle purification (21) and thus is well suited to studying the contributions of viral proteins to virion or VLP structure. Filamentous virions range in length from 2 to 8 μm and, by scanning EM (SEM), often appear as interconnected filaments with a branched appearance (15, 19, 25–29). For many enveloped viruses, production of VLPs can be accomplished to some degree by coexpressing select structural proteins in cell culture (for a review, see references 30 and 31). In the case of hRSV, an early study examined the requirements for passage of an hRSV minigenome between HEp-2 cells in a helper virus-dependent assay. The results showed that the nucleoprotein (N), the phosphoprotein (P), the matrix protein (M), and the fusion protein (F) were each necessary for packaging and passaging of a minigenome (32). More recent studies reported the same minimal requirements for VLP production in HEp-2 cell culture: the N, P, M, and F proteins were both necessary and sufficient to form cell surface filamentous structures that carried the viral proteins (5, 22). Few studies have addressed the assembly-related functions of these four proteins, especially N and P, and hence, we have little understanding of their individual contributions and the architecture of the assembly process in general.
We know from previous work that the M protein and the F protein cytoplasmic tail (CT) are important in the organization of virion assembly (5, 24, 33–35). M is a nonglycosylated phosphorylated protein of 256 amino acids (36–38). M is thought, in part by analogy to the roles of matrix proteins from other viruses, to play a key role in virion assembly by inhibiting viral replication and by forming a bridge between the viral RNP and envelope (30, 37, 39–42). F resembles the prototypic paramyxovirus fusion protein and plays a critical role in virus entry (43, 44). F, however, has also been shown to play a role in virus assembly, for which its small CT was essential (5, 24, 33). In the absence of the CT, or with mutations near the carboxy-terminal end of the CT, virion formation was abrogated (5, 33) and the F protein was found to accumulate around cytoplasmic virus-induced bodies (termed inclusion bodies [IBs] in the hRSV field), while M protein accumulated inside the IBs (33). This indicated that M and F may interact at or near the IBs and implicated these structures in the cascade of late-stage events that lead to virions. IBs are multifunctional bodies (45, 46) and are believed to represent the sites of viral replication. Coexpression of N and P is sufficient to induce formation of IBs (47). Besides critical involvement in the formation of IBs and in the hRSV polymerase complex, the roles of the N and P proteins in particle morphogenesis are not clear. However, a role for P and P-M interaction in virus entry, and potentially virus assembly, was recently proposed (48, 49).
To be able to test the potential of authentic hRSV VLPs as a vaccine strategy and to address the need to improve cell culture-based large-scale production of hRSV and hRSV VLPs, we are interested in understanding and controlling how virus particles are formed. For that purpose, we further dissected the reported requirements for VLP formation. Taking advantage of the cell-associated and predominantly filamentous nature of infectious hRSV, we examined the surfaces of transiently transfected HEp-2 cells at high resolution to document the formation and morphology of filamentous VLPs. Our data show that neither N nor IBs are required and that P, M, and the F carboxy terminus (Fstem) are sufficient for robust filamentous VLP formation. Whereas M and F together could drive the formation of filamentous VLPs with abnormal morphology, indicative of M-F interaction, all three proteins (P, M, and F) were required for major viral protein coalescence and the formation of filamentous particles resembling wt virus. This suggests an unexpected role for P in controlling hRSV particle morphology.
MATERIALS AND METHODS
Cells and primary antibodies.
HEp-2 cells were acquired from the American Type Culture Collection and grown in standard growth medium containing 10% fetal bovine serum (FBS). Monoclonal antibody (MAb) A5 (anti-F) was provided by Edward Walsh (University of Rochester School of Medicine, Rochester, NY). Synagis (anti-F) was provided by MedImmune, Inc. Anti-N antibody was acquired from AbD Serotec. Anti-P antibody was acquired from Abcam. The anti-myc antibody was acquired from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH. The AcV5 antibody (for Mep [M protein epitope tagged at the N terminus] detection) was provided by Gary Blissard (Boyce Thompson Institute at Cornell University). A rabbit polyclonal anti-M peptide serum was reported previously (34).
Construction of plasmids.
The M- and F-expressing plasmids were previously reported (33, 34). For N and P, the open reading frames (ORFs) (A2 strain) were codon optimized according to the method of Haas et al. (50), assembled from primers, and cloned into a pcDNA3-derived expression plasmid, as previously described (33). To generate Mep, the AcV5 epitope (SWKDASGWS) from the baculovirus GP64 protein plus two additional amino acids (alanine and leucine) were inserted at amino acid position 2 of the M ORF. Fstem plasmid constructs were generated by deleting the F ectodomain using standard molecular biology techniques. The myc epitope (EQKLISEEDL) was inserted by site-directed mutagenesis. Note that the following F plasmid constructs were all generated by standard techniques, such as site-directed mutagenesis, overlapping PCR, or direct subcloning. All modifications involved plasmids, but the numbers given are amino acid numbers from the original, full-length viral proteins unless otherwise indicated. For Fstem-VSV-CT, F CT amino acids 554 to 574 were replaced by amino acids 492 to 511 of the vesicular stomatitis virus (VSV) Indiana strain G protein using overlapping PCR. In Fstem-VSV-TMD, F transmembrane domain (TMD) amino acids 525 to 548 were replaced by amino acids 465 to 488 of the VSV Indiana strain G protein. In constructs labeled TMDn and TMDc, which contain either the N-terminal or C-terminal half of the VSV G TMD, respectively, F amino acids 525 to 536 or 537 to 548 were replaced, respectively, with VSV G amino acids 465 to 476 or 477 to 488. For Fstem constructs labeled ΔGL, the Asn residues at positions 27 and 50, which correspond to positions 27 and 500 in the full-length F protein, were mutated to Ala and Thr, respectively. The Fstem-green fluorescent protein (GFP) construct was generated by cloning the entire GFP ORF (without start and stop codons) into a unique EcoRI site within Fstem496, located 10 to 15 nucleotides upstream of the myc tag sequence. To make Fstem-EboGP, the Ebola virus Zaire glycoprotein ORF ectodomain was codon optimized and synthesized with GenScript. The ectodomain sequence encoding amino acids 42 to 615 was cloned into the EcoRI site of Fstem496.
Detection of viral surface filaments by fluorescence microscopy.
Infected (1.5 h) or transfected (4 h) HEp-2 cells on glass coverslips were fixed at various times, as indicated, with freshly dissolved 4% paraformaldehyde (20 min); permeabilized with 0.1% SDS or 0.2% Triton X-100 (5 min); and incubated with anti-P, anti-M, anti-F (A5 or Synagis), anti-AcV5, or anti-myc epitope antibodies or phalloidin-488 (Life Technologies). Following incubation with secondary antibodies conjugated with Alexa 488, 568, or 594, the cells were stained with DAPI (4′,6-diamidino-2-phenylindole), washed, and photographed at ×600 magnification on a Nikon TE2000 inverted fluorescence microscope with DS-Qi1 and DS-U2 camera controllers or at approximately ×1,500 magnification (using a 60× objective with ∼2.5× zoom) on a Leica TCS SP2 inverted scanning confocal microscope system. Images were processed using Nikon NIS-Elements basic software (IFM) or Adobe Photoshop CS5.1 (confocal).
Detection of viral surface filaments by field emission SEM.
HEp-2 cells on plastic coverslips were infected or transfected as described above. At 26 hours postinfection (hpi), the cells were fixed with 4% freshly dissolved paraformaldehyde for 15 min. The fixed cells were incubated with anti-F antibody (A5) or anti-myc antibody. The cells were washed and incubated with goat-anti-mouse antibodies conjugated to 15-nm colloidal gold (Aurion). The cells were washed three times and fixed in 2.5% glutaraldehyde for 120 min at room temperature. After fixation, the cells were washed once, incubated in 1% osmium tetroxide for 1 h, and washed three additional times. The cells were then dehydrated in ethanol in stepwise fashion and incubated in hexamethyldisilazane for 1 min. Following hexamethyldisilazane treatment, samples were air dried, carbon coated, and examined in an FEI Quanta 600 field emission gun scanning electron microscope. Samples were examined using secondary electron (SE) and backscattered electron (BSE) modes at various magnifications and photographed at ×20,000, ×50,000, or ×100,000 magnifications. Images at ×50,000 and ×100,000 magnifications were overlaid to determine the locations of gold particles relative to identified VLPs. Overlaid images were processed with Photoshop CS5.1 to ensure the gold particles were visible.
Calculation of the lengths and diameters of filamentous VLPs.
Ten independent digital photographs per condition were used to calculate the mean diameter and length of filamentous VLPs. Depending on the suitability of the image, 1 or 2 filaments were measured per image. For M- and F-induced filaments, accurate length measurements could not be obtained (for reasons discussed in Results below). As a consequence, the observed length differences in the absence versus the presence of P could not be statistically validated. For diameter comparisons in the absence versus the presence of P, an unpaired two-tailed Student t test was applied to the data to determine statistical significance.
Measuring cell surface levels of Fstem proteins by cell ELISA.
Relative surface levels of Fstem proteins were quantitated by whole-cell enzyme-linked immunosorbent assay (ELISA) as previously described (33). In short, transfected cells were briefly fixed with 4% freshly dissolved paraformaldehyde, blocked in 1% bovine serum albumin (BSA), and incubated with anti-myc antibody. Following incubation with a horseradish peroxidase-conjugated secondary antibody, the wells were incubated in O-phenylenediamine-based substrate. At short time intervals, aliquots were collected and added to 3 M sulfuric acid in a 96-well plate to stop the reaction. The optical density at 490 nm was measured in a Spectramax M2E plate reader (Molecular Devices). The final reported values (relative surface levels with the mean of Fstem496-ΔGL set at 100%) represent the means and standard deviations (SD) of the results of 3 independent experiments. Statistical significance was determined by an unpaired two-tailed Student t test.
VLP (Western blot) assay.
The VLP (Western blot) assay was based on a previously published protocol (46) with minor modifications. The medium of transfected cells was replaced at 16 h posttransfection (hpt) with reduced-serum medium (MEM-RS; HyClone). At 24 hpt, cells were washed gently with MEM-RS and collected by scraping with a rubber cell scraper. The collected cells were agitated by pipetting up and down with a pipetman, after which cell debris was removed by centrifugation for 5 min at 800 × g. The supernatants were transferred to new vials and centrifuged for 45 min at 21,000 × g. The supernatant was discarded, and pellets containing VLPs were resuspended and boiled in 1× Laemli buffer. Negative controls that did not form filamentous VLPs by IFM were included. Samples were electrophoresed on 12% reducing SDS-PAGE gels and transferred to Immobilon blots using a semidry apparatus (Bio-Rad). The blots were incubated with anti-M (peptide serum), anti-F (MAb19), anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase), or anti-myc antibodies, followed by a horseradish peroxidase-conjugated secondary antibody. The blots were developed using enhanced chemiluminescence (ECL) (Pierce) and scanned on a C-DiGit blot scanner (Li-Cor Biosciences). Relative protein levels were determined using C-DiGit Image Studio Software (Li-Cor Biosciences) (see below). The GAPDH content of purified VLPs was compared to the GAPDH levels in transfected cells to gauge the level of cell protein contamination (see below). Note that the results presented were obtained without the use of a sucrose cushion. However, the assays were repeated in the absence and presence of a 20% sucrose cushion in the VLP centrifugation step, with similar results. In some experiments, we also collected the supernatant of transfected cells, removed the cell debris, and pelleted VLPs as described above to determine if VLPs were secreted into the medium (see Fig. 3).
FIG 3.
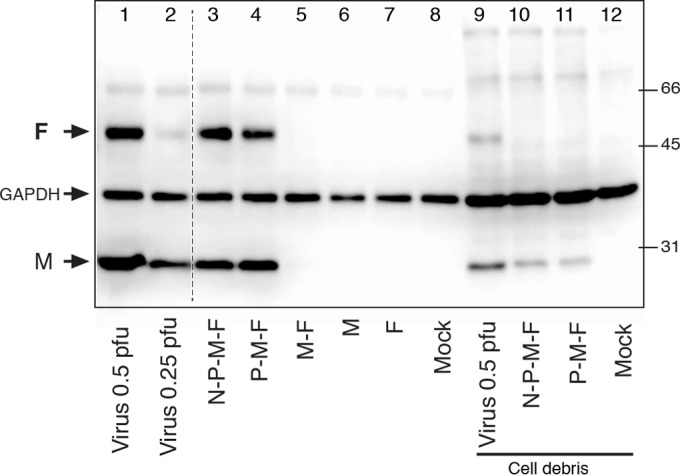
Relative F protein levels in VLPs versus wt virions. HEp-2 cells were infected with wt virus at 0.5 or 0.25 PFU/cell or transfected as for Fig. 2 with various plasmid combinations. Cell surface-associated VLPs and wt viruses were harvested as described in the text at 24 hpt or 22 hpi, respectively. Western blots were generated and incubated with anti-M, anti-F, and anti-GAPDH antibodies. Protein levels were quantitated using a Li-Cor scanner and Image Studio software. Lanes 1 to 8, particle yields from ∼300,000 cells; lanes 9 to 12, non-VLP fraction (cell debris) at ∼12,000 cells/lane. The relative protein amounts were determined, and the F/M ratio was calculated (see Materials and Methods). The positions of M, F, and GAPDH and the molecular weights of biotinylated protein markers (on the right) are indicated in thousands. The dashed line between lanes 2 and 3 indicates where two lanes containing samples not relevant to the experiment were removed.
Viral protein quantitation (Western blotting). (i) F/M ratios.
Relative amounts of F and M on Western blots were measured using C-DiGit Image Studio Software (Li-Cor). F/M ratios were calculated (as an approximation of the relative amount of F protein per filamentous VLP), and the fold increase was determined by dividing the F/M ratios of N-P-M-F- or P-M-F-induced VLPs by the ratio calculated for wt virus. The final reported values (fold increase over the F/M ratio of wt virus) represent the means and standard deviations of the results of 3 independent experiments. Statistical significance was determined by unpaired two-tailed Student t tests.
(ii) Cell protein levels.
To estimate the levels of cell-derived proteins in the VLP fraction, we included on the Western blots the cell debris fraction that was cleared from each VLP sample by low-speed centrifugation. Anti-GAPDH antibody was included during primary antibody blot incubations, and GAPDH bands were quantitated using C-DiGit Image Studio software. From the quantitations, we calculated the total amount of GAPDH in each sample by adding the amounts of GAPDH from the VLP and cell debris fractions. The percent GAPDH in each VLP fraction was calculated as the amount of GAPDH associated with VLPs divided by the amount of GAPDH in the total sample times 100%.
(iii) VLP production after VSV G TMD exchanges.
M bands on Western blots were quantitated using C-DiGit Image Studio software. As an indicator of the numbers of VLPs produced, the relative amounts of M in the VLP fractions were calculated as the amount of M in each sample divided by the amount of M in a control sample of Fstem-ΔGL times 100%. The final reported values (relative VLP production) represent the means and standard deviations of the results of 3 independent experiments. Statistical significance was determined by unpaired two-tailed Student t tests.
RESULTS
The P, M, and F proteins are sufficient for filamentous VLP formation.
In a previous report based on immunofluorescence, coexpression of the N, P, M, and F proteins in HEp-2 cells was both required and sufficient to generate filamentous VLPs (5, 22). To elucidate the roles of these four hRSV proteins in virion assembly and morphogenesis, we coexpressed various combinations of N, P, M, and F proteins from plasmids with codon-optimized ORFs in HEp-2 cells and examined VLP formation. In cell culture, a large majority of infectious progeny virus remains cell associated and is filamentous in nature (15, 16, 19, 21, 26, 28, 51), allowing virion morphogenesis to be readily assessed by examining the cell surface. First, we compared cells coexpressing N, P, M, and F to cells infected with hRSV. HEp-2 cells transfected with N-, P-, M-, and F-expressing plasmids were fixed and permeabilized at various times posttransfection, stained for viral proteins, and examined by IFM. The results of fixation at 22 hpt are shown in Fig. 1A. At the transfected cell surface, numerous viral-protein-containing rod-like structures (VLPs) formed that were similar in appearance to virions formed at the surface of a wt-virus-infected cell (Fig. 1A, 1 to 4). The efficiency of formation appeared to be higher than previously reported, as by 22 hpt, the cell surfaces were already densely covered with filamentous VLPs. In contrast to nonviral membrane extrusions, VLPs did not appear to contain polymerized actin (see below), in agreement with previous work (5). Note that we do not use the term “F-actin” to indicate the polymerization status of actin to avoid confusion when discussing the F protein and polymerized (F) actin. Transient expression of individual viral proteins revealed that the N, P, and M proteins were each distributed relatively evenly throughout the cell and that none of the four proteins alone induced filamentous VLP formation (Fig. 1A, 5 to 8).
FIG 1.

Viral protein requirements for filamentous VLP formation. The N protein and IBs are dispensable. HEp-2 cells on glass coverslips were infected with wt hRSV or transfected with various combinations of plasmids expressing hRSV N, P, M, or F protein. The distribution of viral proteins was examined by IFM or confocal microscopy at 22 hpt, after paraformaldehyde fixation, detergent permeabilization, and incubation with anti-N, -P, -M, or -F antibodies and Alexa-conjugated secondary antibodies. (A) N-P-M-F-induced VLP formation in transfected cells by IFM. hRSV-infected cells (1 and 2) and cells transfected with N-P-M-F-encoding plasmids (3 and 4) were compared to cells expressing N, P, M, or F alone (5 to 8). (B) Viral protein requirements for filamentous VLP formation. Cells transfected with various combinations of three plasmids were examined by IFM as for panel A. (C) Confocal analysis of P-M-F-induced VLPs. Cells transfected with P-M-F-encoding plasmids were processed as for panel A; labeled with P, M, or F antibody or phalloidin-488 to visualize polymerized actin; and examined by confocal microscopy. (A, B, and C) The plasmids used are indicated above or beside the images. The primary antibodies used and the color of the secondary conjugate are indicated in the upper right corner of each image. IFM images, ×600 magnification; confocal images, approximately ×1,600 magnification. Maximum projections are represented (merge of all z-stacks).
FIG 4.

Analysis of VLP formation by coexpression of combinations of two viral proteins. M and F proteins form VLPs of aberrant length and diameter. HEp-2 cells were transfected with combinations of two hRSV protein-encoding plasmids, and the distribution of viral proteins was examined by epifluorescence or confocal microscopy at 22 hpt as for Fig. 1. (A) VLP formation in the presence of three different two-protein combinations. Cells transfected with P-plus-M, P-plus-F, or M-plus-F plasmids were compared by epifluorescence (1 to 6) or by confocal scanning microscopy (7 and 8). Images 1, 3, 5, and 7 were detergent permeabilized; images 2, 4, 6, and 8 were left unpermeabilized. (B) Analysis of M-F-induced VLPs. A matrix protein carrying an N-terminal epitope tag (to improve detection under mild-detergent conditions) was coexpressed with the F protein. Cells were fixed and then permeabilized with Triton X-100 and processed as for Fig. 1 by epifluorescence (1 to 4) or by confocal microscopy (5 and 6). (A and B) The plasmids used are indicated above or in the upper left corner of each image. The primary antibodies used and the color of the secondary conjugate are indicated in the upper right corners. The detergent used for permeabilization after fixation is indicated in the lower left corners. Magnifications: ×600 (A, 1 to 6, and B, 1 to 4) and approximately ×1,600 (A, 7 and 8, and B, 5 and 6). The confocal images are maximum projections (merge of all z-stacks).
FIG 5.

High-resolution analysis of M-F-induced filamentous structures. HEp-2 cells were transfected with plasmids expressing M and F. The cells were fixed and processed for SEM at 22 hpt as for Fig. 2. Prior to SEM processing, the cells were labeled with anti-F antibodies, followed by secondary antibodies conjugated with 15-nm gold particles. For the ×50,000 and ×100,000 analyses, SE and BSE images were overlaid to show the locations of gold particles. The boxed areas with an asterisk in the left panels are shown on the right at higher magnification. Individual images of the same sample are shown.
FIG 8.

Incorporation of foreign proteins into hRSV VLPs. Fstem496 was modified to include GFP or the Ebola virus GP ectodomain. (A) Schematic of constructs. Sequences encoding the entire GFP (without start and stop codons) or Ebola virus Zaire GP ectodomain residues 42 to 615 were cloned into Fstem496 as shown and named Fstem-GFP and Fstem-EboGP, respectively. Fstem-GFP and Fstem-EboGP were transiently expressed in HEp-2 cells, and their distribution was examined by IFM. (Images 1 and 2) Both modified Fstem proteins were detected via the included myc tag. (B) Fstem-GFP and Fstem-EboGP were coexpressed with P and M proteins and examined by IFM at 22 hpt. The primary antibodies used and the color of the secondary conjugate are indicated in the upper right corners of each image. Magnification, ×600. (C) Cells were transfected as for panel B, labeled with myc antibody and 15 nm-gold particles at 22 hpt, and subjected to SEM. For all the images, SE and BSE pictures were overlaid as for Fig. 2 to show the locations of gold particles.
To further examine viral protein requirements, we sequentially omitted one of the four plasmids and observed the impact on VLP formation by IFM (Fig. 1B). In the absence of F (Fig. 1B, 1), no filamentous VLPs were formed and M accumulated in IBs. This is consistent with our previous work in the context of a replicating virus lacking F (F null) (33). Coexpression of N-P-F or N-M-F proteins also failed to yield VLPs (Fig. 1B, 2 and 3). In contrast, numerous filamentous VLPs were observed extruding from the plasma membrane after coexpression of P-M-F (Fig. 1B4). The P-M-F-induced VLPs were abundant and contained high levels of viral proteins. Whereas M was evenly distributed throughout the cells after N-M-F coexpression, upon P-M-F coexpression, M underwent a redistribution event and accumulated in many cells almost exclusively in VLPs (Fig. 1B, 3 versus 4). Thus, efficient targeting of M to VLPs was dependent on the combined presence of P and F and independent of N. Note also that IBs were absent (Fig. 1B, 3 and 4) due to the omission of P or N, both of which are required for IB formation (47).
FIG 2.

High-resolution analysis of PMF-induced VLPs. HEp-2 cells were infected with wt hRSV (1 and 2) or transfected with plasmids expressing N-P-M-F (3 and 4) or P-M-F (5 and 6). The cells were fixed and processed at 22 hpt and examined by field emission SEM. Prior to SEM processing, the cells were fixed in paraformaldehyde and labeled with anti-F antibodies, followed by secondary antibodies conjugated with 15-nm gold particles. The ×100,000-magnification samples (2, 4, and 6) were scanned using both SE and BSE modes to visualize gold particles, and the SE and BSE photographs were overlaid to show the locations of the gold particles, as described previously (34). The boxed areas in the ×20,000 images (1, 3, and 5) are shown in detail on the right.
It was previously reported that N-P-M-F-induced VLPs and hRSV virions could be distinguished from nonviral membrane extrusions, such as microvilli, based on the respective absence or presence of polymerized actin (5). To assess the localization of polymerized actin relative to VLPs, cells transfected with P, M, and F plasmids were examined with confocal microscopy (Fig. 1C); the status of the P protein under P-M-F coexpression conditions was also examined. Polymerized actin was occasionally observed at the base of the viral filamentous structures. Although we could not quantitatively assess the amount of VLP-associated polymerized actin, in the majority of cases, polymerized actin did not accumulate in the filamentous VLPs, consistent with a previous report using complete virus (20) (Fig. 1C, 3). The results indicate that under these conditions, F protein targeted to filamentous VLPs and not to nonviral filamentous cell extrusions. In contrast to actin, the P protein strongly colocalized with F and M in VLPs (Fig. 1C, 1 and 2). Thus, coexpression of P, M, and F proteins led to viral protein coalescence and was sufficient for abundant, IB-independent formation of VLPs. These VLPs contained high concentrations of viral proteins and appeared to be distinct from polymerized-actin-containing cell extrusions.
High-resolution analysis: P-M-F-induced VLPs are morphologically similar to wt virions and contain high levels of F protein.
Due to the limited resolution of IFM, we examined the morphology of P-M-F-induced filamentous VLPs at higher magnification. Cells transfected with P-, M-, and F-expressing plasmids were examined by field emission SEM and compared to N-P-M-F-induced VLPs and virions formed at the surfaces of hRSV-infected cells (Fig. 2). To distinguish VLPs from nonviral membrane extrusions (which do not contain the F protein, as shown in Fig. 1 and previously by Shaikh et al. [5]), we labeled the viral F protein with 15-nm gold particles prior to processing for SEM. At the surfaces of hRSV-infected cells, high numbers of filamentous virions were detected, in which anti-F gold label was present, albeit at low levels (Fig. 2, 1 and 2). N-P-M-F- and P-M-F-transfected cells also carried large numbers of filamentous VLPs (Fig. 2, 3 to 6), which were morphologically similar to hRSV virions. The VLPs consistently contained higher levels of gold particles than the wt virions. To quantitate the levels of F in cell surface-associated VLPs relative to wt virions, we harvested the VLPs and viruses using a previously described protocol (52) with minor modifications. Briefly, washed cells were collected by scraping at 24 hpt, and VLPs were isolated by gentle pipetting, removal of cells and cell debris by low-speed centrifugation, and pelleting of particles at high g force. Note that at a later stage we repeated the experiments using a 20% sucrose cushion in the pelleting step, with identical results (data not shown). Viral protein levels were determined using quantitative Western blotting (Fig. 3). As negative controls, we included parallel samples in which no filamentous VLPs were observed (Fig. 1) (transfection with M alone or F alone), as well as a sample in which M and F were coexpressed. In samples expressing M, F, or M plus F, the M and F proteins were generally undetectable (Fig. 3, lanes 5, 6, and 7), indicating the protocol does not isolate these viral proteins when expressed alone. Occasionally, a low level of M protein was observed in the harvested fraction from cells transfected with M alone and from M-F-transfected cells, which never exceeded 5% of the level of M from N-P-M-F- or P-M-F-induced VLPs. We also compared GAPDH levels within the VLP fraction (Fig. 3, lanes 1 to 8) to those of cell debris lysates, a small portion of which was loaded on the same gels (Fig. 3, lanes 9 to 12) as a measure of cell protein contamination. Although GAPDH was detected in all the fractions, quantitation showed that VLP fraction-associated GAPDH was ≤3% of the total GAPDH present, suggesting a minor level of host cell protein contamination. In contrast to transfections with M and F alone, the M and F proteins were readily observed in N-P-M-F- and P-M-F-transfected cells and virus-infected cells. The relative amounts of F and M were quantitated, and the F/M ratios were calculated and compared as an indicator of relative F levels in filamentous VLPs. In support of the SEM data, the average F/M ratios in N-P-M-F- and P-M-F-induced VLPs from three independent experiments were higher than those of wt virions: 1.98 times (SD, 0.424; P < 0.05) and 1.94 times (SD, 0.69; not statistically significant), respectively. These data confirm that, at least for N-P-M-F-induced VLPs, higher levels of F protein were detected in VLPs than in wt virions; this may in part be due to available space, as the wt virions also have to accommodate the G and SH proteins.
FIG 6.

VLP formation by F proteins lacking the ectodomain (Fstem). (A) Four F variants were constructed, each lacking the majority of the F ectodomain. The deleted residues are indicated. A myc epitope tag was inserted in place of the ectodomain. The two remaining native N-linked glycosylation sites (residues 27 and 500 based on full-length F) are also indicated. Each construct was expressed individually in HEp-2 cells, fixed and permeabilized at 24 hpt, and examined by IFM (images 1 to 4). The antibody used is indicated. Magnification, ×600. (B) Fstem constructs were coexpressed with Mep in HEp-2 cells. The cells were fixed at 22 hpt, permeabilized, and examined by confocal microscopy. The Fstem constructs used are indicated in the upper left corners of the images. The primary antibodies used and the color of the secondary conjugate are indicated in the upper right corners. Magnification, approximately ×1,500. All the images are maximum projections (merge of all z-stacks). The insets provide higher-magnification detail. (C) HEp-2 cells were transfected with plasmids expressing M and Fstem496 (1 and 2) or P, M, and Fstem496 (3 and 4). The cells were fixed and processed for SEM at 22 hpt. Prior to SEM processing, the cells were labeled with anti-myc antibody, followed by secondary antibodies conjugated with 15-nm gold particles. For images 2 and 4, the SE and BSE pictures were overlaid, as for Fig. 2, to show the locations of gold particles.
We have not yet made efforts to optimize VLP production. However, a crude comparison of the amounts of matrix protein associated with VLPs and wt virus in Fig. 3 indicates that the numbers of filamentous particles harvested from N-P-M-F- and P-M-F-transfected cells were comparable to the number of particles from cells infected with wt virus at low multiplicity (between 0.25 and 0.5 PFU/cell). Given that these VLPs were obtained without optimization and at a relatively early time point (24 h posttransfection), VLP formation was not inefficient. Combined, Fig. 1 to 3 show that coexpression of P, M, and F proteins was sufficient for strong coalescence of viral proteins and for detection of VLPs by microscopy and Western blotting and that these VLPs were morphologically similar to wt virions but contained higher levels of F protein.
In the absence of P, the M and F proteins form long filamentous structures.
To examine the contributions of the P, M, and F proteins to VLP formation, we sequentially omitted one of the three plasmids and examined the distribution of the remaining viral proteins by IFM (Fig. 4). Transfected cells were processed as shown in Fig. 1 and detergent permeabilized or left unpermeabilized to focus on surface-associated proteins. Note that in the absence of detergent, the P and M proteins were minimally detected, consistent with their predicted topologies (inside the plasma or virion membrane). When P and F proteins were coexpressed, no filamentous VLPs were detected (Fig. 4A, 3). Small patches containing P and F proteins were occasionally observed in nonpermeabilized cells (Fig. 4A, 4). However, the majority of P and F proteins did not appear to influence each other's subcellular distribution; rather, each was distributed as if expressed alone (Fig. 4A, 3). The same was true when P and M or M and F were coexpressed (Fig. 4A, 1 and 5): in both cases, the proteins remained evenly distributed and did not noticeably alter each other's subcellular localization. At the surfaces of nonpermeabilized M-F-transfected cells, however, rod-shaped structures were present, which appeared to be unusually long (occasionally >20 μm) and contained the F protein (Fig. 4A, 6). In general, these structures were less abundant than P-M-F-induced VLPs, appeared to be thinner, and were difficult to document by epifluorescence. Samples coexpressing M and F were therefore examined with confocal microscopy, with similar results (Fig. 4A, 7 and 8). The fact that M-F-induced filaments were more readily observed without detergent permeabilization (two distinct anti-F Abs gave similar results) suggests that these structures were disrupted by SDS treatment and thus were more fragile than P-M-F- and N-P-M-F-induced VLPs.
FIG 7.

Dissecting the role of Fstem domains in VLP formation. Fstem496 was modified to determine the relative importance of the remaining ectodomain residues, the TMD, and the CT. (A) Overview of constructs. Within Fstem496, ectodomain residues with a potential role in assembly were individually mutated to Ala or Gly (collectively named Fstem-Ecto-ala), and the F TMD and CT were exchanged with VSV G TMD or CT residues, as depicted. (B) IFM analysis of cells coexpressing P, M, and Fstem-VSVG-CT or Fstem-VSVG-TMD at 22 hpt. The primary antibodies used and the color of the secondary conjugate are indicated in the upper right corners of the images (modified Fstem proteins were detected via the myc tag). Magnification, ×600. Note that in the presence of Fstem-VSVG-TMD, filaments were largely absent, and cells displaying filaments as depicted were only occasionally observed. (C to E) Quantitation of TMD impact. (C) Overview of constructs. To improve quantification on Western blots, the remaining glycosylation sites in Fstem496 were mutated (Fstem-ΔGL). Within construct Fstem-ΔGL, either the full TMD or the N-terminal half or the C-terminal half of the TMD was exchanged with the TMD counterpart of VSV G (named Fstem-ΔGL-VSVG-TMD, -TMDn, or -TMDc). (D) Relative surface levels of TMD-modified Fstem proteins. Transfected HEp-2 cells were fixed with fresh paraformaldehyde at 24 hpt, and Fstem levels were determined by cell ELISA as previously described (33), using an anti-myc antibody. NGFR-myc is a surface marker with a myc tag at the N terminus and served as a positive control. The mean signal of Fstem-ΔGL was set at 100%. The error bars represent standard deviations from the means of triplicate samples. (E) Quantitative analysis of the role of the F TMD in VLP formation. Plasmids expressing Fstem496, Fstem-ΔGL, or Fstem-ΔGL with full or partial TMD exchanges were cotransfected with P and M plasmids, and VLPs were harvested as for Fig. 2 at 24 hpt. The relative levels of M were quantitated by Western blotting (Li-Cor) as an indicator of VLPs generated, with the mean value of Fstem-ΔGL set at 100%. Due to overlap of the M and Fstem bands, lane 1 was not quantitated. The values represent the means and standard deviations derived from the results of three independent experiments. Statistical significance is indicated: *, P < 0.01; **, P < 0.05 (unpaired two-tailed t test).
To determine whether M was present in the M-F-induced filaments, we repeated the experiment using an M protein epitope tagged at the N terminus (Mep). This was done because the M antibody utilized previously requires SDS treatment for M recognition by IFM, and SDS appeared to disrupt the M-F-induced structures. A plasmid expressing Mep was cotransfected with F-expressing plasmid, and samples were fixed and treated with a milder detergent (Triton X-100) in an attempt to permeabilize the membrane without disrupting the filament structures (Fig. 4B). As a control, cells were left unpermeabilized to verify whether Mep could recapitulate the phenotype of untagged M (Fig. 4B, 1). As observed in Fig. 4A, long rod-like structures were detected at the surface when Mep2 and F were coexpressed. When identically transfected cells were permeabilized with Triton X-100 instead of SDS, the Mep protein was observed colocalizing with F in the surface filaments by IFM and confocal microscopy (Fig. 4B, 3 and 5). In contrast, expression of Mep alone resembled expression of untagged M (Fig. 4B, 2). Costaining for polymerized actin revealed that polymerized actin was occasionally present at the bases of the structures near the plasma membrane but was not consistently present in the M-F-induced filaments (Fig. 4B, 4 and 6). The results suggest that the M-F-induced filaments were not generic plasma membrane extrusions but likely represent a “rudimentary” filamentous VLP form specifically induced by the viral M and F proteins.
High-resolution analysis of M-F-induced surface filaments.
To ask whether the M-F-induced filaments differed in length and/or diameter relative to wt virions, as suggested by Fig. 4, we examined the cell surface at higher resolution (Fig. 5). Cells transfected with M- and F-expressing plasmids were processed for field emission SEM as for Fig. 2, and the viral F protein was again labeled with 15-nm gold particles. Consistent with IFM, SEM showed rod-like structures that contained the F protein. Statistically significant length comparisons could not be obtained because the fragile M-F-induced filaments were often not intact and appeared to collapse and become entangled during SEM preparation. In addition, gold label was not easily detected at the low magnification needed to see their entire lengths. Nevertheless, an approximate average length of M-F-induced filamentous VLPs was derived from multiple independent images and found to be 8 μm. For the reasons stated above, this number is likely an underestimate of the true average length. A similarly derived length average of P-M-F-induced VLPs was 4 to 5 μm, suggesting that viral protein-induced filaments are longer in the absence of P.
With regard to diameter, measurements from 10 independent SEM images showed a statistically significant difference (P < 0.05): M-F-induced filaments ranged from approximately 50 to 110 nm, with an average diameter of 87 nm, whereas P-M-F- and N-P-M-F-induced VLPs were similar to wt virions and had average diameters of 144 and 160 nm, respectively. M-F-induced filaments were sometimes found amid membrane extrusions of larger diameter that completely lacked F (presumably microvilli). Combined, Fig. 4 and 5 show that coexpression of M and F led to VLPs that (i) were longer than particles formed in the presence of P, (ii) carried high concentrations of M and F, (iii) appeared to lack polymerized actin and thus were likely distinct from nonviral membrane extrusions, and (iv) had a reduced diameter. These results suggest that M and F proteins can interact in the absence of P and form rudimentary filamentous VLP structures at the cell surface. However, a significant proportion of M and F proteins were not in VLPs and remained distributed throughout the cell, presumably due to a failure to significantly coalesce in the absence of P.
The F ectodomain is not required for VLP formation.
The CT is essential for the role of F in virus assembly (5, 24, 33). With the aim of mapping the minimal RSV protein domains required for VLP production, we next asked whether the F carboxy-terminal domain, when coexpressed with M, would be sufficient to allow formation of rudimentary VLPs, as observed in Fig. 5. In anticipation of a potential impact on the protein expression level or targeting, four different F variants lacking the majority of the F ectodomain were generated. This was done by removing F amino acids 36 to 485 (Fstem486), 36 to 495 (Fstem496), 36 to 505 (Fstem506), or 36 to 515 (Fstem516) (Fig. 6A). These deletions left the authentic signal peptide (SP), signal peptide cleavage site, TMD, and CT intact and maintained a small but varying portion of the ectodomain near the TMD. A myc epitope tag was inserted in place of the deleted F ectodomain section to track the truncated F proteins. To examine expression, plasmids encoding the various Fstem constructs were transfected into HEp-2 cells. The cells were fixed and permeabilized at 24 hpt and stained with anti-myc antibody for IFM analysis (Fig. 6A). The Fstem constructs displayed a staining pattern consistent with the secretory pathway and were detected at similar levels (Fig. 6A, 1 to 4).
Next, each of the Fstem constructs was coexpressed with the Mep protein in HEp-2 cells to test by confocal microscopy its ability to support formation of rudimentary VLPs. Transfected cells were permeabilized with Triton X-100 and processed as for Fig. 4B. All the Fstem constructs supported the formation of filamentous structures, which appeared to be thinner than wt virions, and contained Fstem and Mep proteins (Fig. 6B, 1 to 4). When we expressed the Fstem proteins in parallel experiments in the presence of P and M (not shown), significant proportions of Fstem506 and Fstem516 were found to be present in the cytoplasm, indicative of aberrant processing (which was not observed when the proteins were expressed alone [Fig. 6A]). For this reason, we focused our studies on Fstem496, which was the minimum construct to support VLP formation without an aberrant expression profile. Cells transfected with M and Fstem496 were examined by SEM using an anti-myc antibody to label Fstem with 15-nm gold particles (Fig. 6C). Consistent with the confocal results, surface filaments that had a reduced diameter and contained the Fstem protein were detected (Fig. 6C, 1 and 2). High levels of gold particles indicated that Fstem496 was incorporated efficiently into VLPs. We similarly examined cells coexpressing P, M, and Fstem496 to ask if the Fstem construct could also support VLP formation in the presence of both M and P (Fig. 6C, 3 and 4). Coexpression of P, M, and Fstem496 induced VLPs similar in appearance and diameter to VLPs induced by P-M-F or N-P-M-F and to wt virions. Combined, the results show that the F ectodomain was not required for formation of filamentous VLPs and that the F carboxy-terminal domain was sufficient for incorporation of F into particles.
Dissecting the role of the F carboxy terminus in VLP formation.
Fstem496 was the minimum construct that both supported efficient VLP formation and lacked aberrant cytoplasmic expression in the presence of M and P (see above) and was further modified to identify relevant domains. The TMD and the CT domain were separately exchanged with the equivalent domains of another type 1 viral transmembrane glycoprotein, the VSV G protein (Fig. 7A). Remaining F ectodomain residues near the TMD were also examined. The results from the smallest construct (Fstem516), which still supported VLP formation (Fig. 6), left a potential role for F ectodomain residues 516 to 524. These residues, as well as the first few residues of the TMD (525 to 529), were individually mutated within Fstem496 to alanine or glycine. Each of the above-mentioned Fstem496 modifications, as well as unchanged Fstem496 as a control, were coexpressed with the P and M proteins in HEp-2 cells, and the potential to form VLPs was documented by IFM as described above (Fig. 7B). None of the individual point mutations of residues 517 to 529 had an impact on the level or appearance of VLPs (data not shown). In contrast, when the F CT was replaced with that of VSV G, filamentous VLPs were absent (Fig. 7B, 1), consistent with previous work in the N-P-M-F context (5) and the viral context (24, 33). The absence of VLPs was not due to changes in Fstem expression levels, as replacement of the F CT with the VSV G CT was previously shown not to impact surface expression (24, 33). When the F TMD was exchanged, the level of filamentous VLPs by IFM was strongly reduced, but we could occasionally find cells displaying VLPs. An example of this is shown in Fig. 7B, 2.
To quantitate the impact of TMD exchange, we subjected modified TMD proteins to the VLP assay shown in Fig. 3 (Fig. 7C to E). Preliminary data showed that Fstem496 appeared on Western blots as a diffuse band that overlapped the M band, precluding quantitation. Therefore, we mutated the two remaining glycosylation sites of Fstem496 and termed the resulting protein Fstem-ΔGL. The latter protein still supported VLP formation and resulted in a quantifiable band (see below). To measure the impact of TMD exchange and at the same time further dissect the TMD, we generated Fstem-ΔGL proteins carrying either the entire VSV G TMD (VSVG-TMD), the N-terminal half (VSVG-TMDn), or the C-terminal half (VSVG-TMDc). Each construct was examined for surface expression by cell ELISA (Fig. 7D) and coexpressed with P and M to harvest and quantitate VLPs, as described above (Fig. 7E). Since the M protein was unaltered and is a core element of a filamentous particle, the M protein level was used as an indicator of VLPs produced. Fstem496 and Fstem-ΔGL, expressed alone, were included as controls. The diffuse nature of the Fstem496 band (Fig. 7E, lane 1) did not allow accurate comparison of M levels before and after removal of Fstem glycosylation sites. However, Fstem-ΔGL supported VLP formation at a level sufficient to assess the impact of TMD exchange (Fig. 7E, lane 2). Exchange of the N-terminal half of the TMD had a minor impact on VLP production (an average reduction of 21%) (Fig. 7E, lane 3) and surface expression (an average reduction of 22%) (Fig. 7D). Exchange of the C-terminal half of the TMD and the full TMD reduced VLP formation by 62 and 76%, respectively (Fig. 7E, lanes 4 and 5). However, the last two modifications also reduced relative Fstem surface levels by 51 and 64%, respectively (Fig. 7D). Due to the concomitant decrease in Fstem surface levels and numbers of VLPs produced, a potential role for the C-terminal half of the TMD in VLP assembly could not be assessed. However, the reduced surface levels indicate that the C-terminal half of the TMD is required for proper Fstem expression and/or surface transport. Combined, the results shown in Fig. 7 indicate that (i) the CT plays a direct and important role in VLP formation, (ii) the C-terminal half of the TMD plays a role in F expression and/or surface transport and thereby an indirect role in VLP formation, and (iii) neither the N-terminal half of the TMD nor the individual F ectodomain residues were important for VLP formation. The ectodomain residues near the TMD, however, may have a role in proper Fstem expression or targeting, since the shorter constructs Fstem506 and Fstem516 were present in the cytoplasm under some conditions (see above).
Utilizing the Fstem to incorporate foreign proteins into VLPs.
Efficient VLP formation and incorporation of the Fstem into VLPs led us to examine the ability to incorporate foreign proteins into VLPs to explore the potential for future hRSV VLP-based multivalent vaccines. The full coding sequence of GFP and the sequence encoding the Ebola virus Zaire envelope glycoprotein ectodomain (residues Thr 42 to Trp 615) were individually cloned into the Fstem496 construct (Fig. 8A, 1). Both constructs maintained the myc epitope in the position near the TMD, as indicated. To verify expression, Fstem-GFP and Fstem-EboGP were transfected into HEp-2 cells, labeled with anti-myc Ab at 22 hpt, and examined with IFM (Fig. 8A, 2 and 3). Both constructs were readily detected, although levels of Fstem-EboGP were lower than those of Fstem-GFP. Both proteins displayed an expression pattern consistent with a transmembrane glycoprotein and were detected in the secretory pathway, as well as at the cell surface. Next, both proteins were coexpressed with the hRSV P and M proteins and compared to cells coexpressing P, M, and Fstem496 proteins by confocal microscopy (Fig. 8B). Fstem-GFP and Fstem-EboGP were labeled via the included myc tag and were found to concentrate in filamentous VLPs, which were similar to VLPs induced by P-M-Fstem496. Incorporation of Fstem-GFP and Fstem-EboGP was also examined with SEM, using anti-myc antibodies and 15-nm gold particles to detect the chimeric Fstem proteins (Fig. 8C). Both proteins were detected in VLPs that were morphologically similar to VLPs formed by P, M, and Fstem496 (Fig. 6C). Fstem-EboGP VLPs contained fewer gold particles than VLPs induced by Fstem-GFP. This may be caused by lower expression levels of Fstem-EboGP, as seen in Fig. 8A (inclusion of the Ebola virus GP protein ectodomain in Fstem496 was not optimized for expression and relied on the F protein SP and SP cleavage site) or by other features specific to the Ebola virus glycoprotein that might hinder optimal incorporation into hRSV VLPs. Nevertheless, high levels of incorporation of Fstem-GFP detected by IFM and SEM show that foreign sequences can be efficiently incorporated into hRSV VLPs by grafting onto an Fstem construct.
DISCUSSION
Previous work has shown that only a few of the hRSV proteins are required to form a VLP. This process, however, was inefficient (4, 5, 32), and very little is known about the mechanisms of VLP and virus assembly and the individual contributions of the viral proteins to particle morphology and composition. As a consequence, it remains challenging to produce VLPs or virus stocks with defined and consistent content and characteristics. The long-term aim of our studies is to better understand the contributions of viral proteins that form the shell of the hRSV particle, both to learn to generate authentic hRSV VLPs with defined morphology and composition and (in the long term) to improve the production process of live or inactivated viruses. Because both infectious virus and purified particles carrying prefusion F appear to be predominantly filamentous, we began our studies by dissecting the viral protein requirements for filamentous VLP formation and examined the roles that the individual viral proteins play. This was done by investigating the cell surfaces of transfected cells, as cell culture is an important platform for production of vaccine candidates and because the majority of wt infectious virions remain associated with the cell surface in different cell types (28). As harvesting and purification can lead to significant structural and morphological changes (21), examining assembly directly at the cell surface is well suited to assessing the impact of viral proteins on particle morphology. For this study, we have not thoroughly examined the medium overlying the cells. However, under the conditions used, we detected at most trace amounts of VLPs in the medium (not shown), suggesting that the vast majority of filamentous VLPs are associated with the cell surface, similar to our previous findings with infectious virus. In addition, no differences were reported between cell-associated and secreted hRSV virions in a recent cryo-EM study (21). Most reports of the viral N, P, M, and F proteins concern functions not related to particle assembly. A putative interaction between M and F as a component of the assembly process has long been suspected based on analogies with other viruses and circumstantial evidence. However, such interaction has never been directly demonstrated, which suggested it may be fragile or short-lived. Our findings show that M-F coexpression induces rudimentary filament formation, indicative of interaction. Moreover, they show that P (but not N) is required for efficient redistribution of F and major viral protein coalescence and for formation of filamentous VLPs. The data thus establish P, M, and F as the minimal components for formation of filamentous VLPs that resemble wt virus and point to an important role for P in filament structure and morphology.
The N protein and cytoplasmic IBs are not required.
In a previous study in HEp-2 cells, the N protein was required to form VLPs, whereas in our studies, VLPs were formed efficiently in the absence of N. We do not yet understand this discrepancy, but potential explanations include differences in expression efficiencies (different codon-optimized plasmids were used), differences in transfection efficiencies, and differences in amino acid contents (our viral proteins were also derived from strain A2, but minor differences exist between A2 isolates). Timing is unlikely to be responsible for the difference, as our studies were done at earlier time points (22 hpt for microscopy and 24 hpt for Western blotting-based VLP assays) than the previous studies. In line with the dispensable nature of N, we found that IBs are dispensable for filamentous VLP formation. This is somewhat surprising, as previous work from several groups, including our previous observation that M and F localize in and around IBs when present in the viral context (33), suggests that IBs are a type of scaffold from which infectious filamentous progeny viruses form in a genuine hRSV infection cycle (23, 33, 34, 53, 54). The data presented here, however, do not exclude a role for IBs in virus assembly, and we believe it is likely that IBs represent a body or mechanism that brings together all of the viral components required for formation of an infectious particle. In N-P-M-F-transfected cells, IBs sequestered at least a proportion of the P protein. Under these conditions, IBs may contribute to filamentous VLP production. However, the present work does show that the filamentous particle shell itself can be generated by the P, M, and F proteins in the absence of all other viral proteins and IBs.
F facilitates M self-assembly, but P is required for optimal viral protein coalescence and morphology.
The M protein by itself was previously shown to form helical structures when expressed in the presence of select lipids (55), suggesting that M self-association is the main engine behind hRSV filament formation. This is consistent with recent work in which blocking of M dimerization also blocked viral filament formation and VLP production (22). Our work shows that M, when expressed alone in cell culture, does not form filamentous surface structures. However, when coexpressed with F, filament formation occurred, albeit with abnormal morphology. Initially, we did not detect the M-F-induced filaments because the SDS permeabilization required for successful recognition of M by our peptide serum destroyed the filaments. Thus, these small-diameter, rudimentary VLPs are more fragile than P-M-F- and N-P-M-F-induced VLPs. M-F-induced filament formation was also less abundant than P-M-F-induced filament formation. The data suggest that this is due to the absence of P, under which condition M-F coalescence is not optimal. Thus, the F protein carboxy terminus appears to act as a cofactor for M self-assembly into rod-like structures and can do so independently of P. This F cofactor function then likely serves two purposes, namely, to facilitate M self-association (filament formation) and to ensure F protein incorporation, which is essential to generate an infectious unit.
The formation of thin filaments by M plus F, or M plus Fstem (and not by M or F alone), and the fact that both proteins are concentrated in these filaments suggest specific interaction between M and F. However, the data show that P plays an important role in the efficiency of bringing M and F together. When M and F or Fstem were coexpressed with P, we observed a drastic change in protein distribution, with all three proteins concentrating within numerous viral surface filaments. Thus, P, M, and F are each required to achieve major viral protein coalescence. Coexpression of P also changed the morphology of filaments from thin and unusually long to a morphology that closely resembles wt virions by SEM. Thus, P plays a role both in the efficiency of VLP formation (through its ability to coalesce the three essential proteins P, M, and F) and in actual VLP morphology. How P influences the diameter and length of the VLP remains to be determined.
The roles of the CT and TMD.
Only the F carboxy terminus was required for F's role in VLP formation, as Fstem constructs supported VLP formation. Although we could detect all Fstem variants at the cell surface by cell ELISA (not shown), the shortest versions were present in the cytoplasm at relatively high levels under some conditions. This indicates that the presence of a small part of the ectodomain may facilitate proper expression and targeting of Fstem, independent of Fstem's function in assembly. Within the carboxy terminus of F, we found that both the CT and TMD play a role in VLP formation. For the TMD, however, this role (carried out primarily via its C-terminal half) may be indirect and may concern an unknown function related to optimal expression and/or surface transport. The TMD was previously shown to be an apical sorting signal (56), a function that involved the apical recycling endosome. Although HEp-2 cells do not polarize into separate apical and basolateral domains, the decrease in surface expression (and indirectly the decrease in VLP formation) after exchanging F TMD residues 537 to 548 with the equivalent of the VSV G TMD may thus be caused by aberrant protein sorting. In contrast to the TMD, the CT is more likely to have a direct role, as VLPs were not formed in the absence of the F CT despite strong surface expression. This finding matches earlier work in the viral context (5, 33). With regard to the CT, we showed previously that amino acids 569 to 572 are essential for infectious-progeny production (33). It is therefore possible that these amino acids represent the residues critical for CT assembly functions.
Efficiency of VLP formation and foreign-protein incorporation.
Our studies aimed to identify requirements and mechanisms, and we have not yet worked on optimization of the process. However, relatively high levels of surface-associated VLPs were observed at an early time (22 to 24 hpt), and we could readily harvest partially purified VLPs equivalent to particles generated in cells infected at low multiplicity (0.25 to 0.5 PFU/cell). We anticipate that with efforts to optimize VLP production, these levels could be further increased. Western blot quantitation indicated that 2 times more F protein is present in N-P-M-F- or P-M-F-induced VLPs than in wt virions. This increase in F may be due to the absence of SH and G proteins in the VLPs and might be advantageous when used for vaccine purposes. As for foreign-protein incorporation, we were able to incorporate the Ebola virus GP ectodomain by grafting it onto the Fstem. SEM indicated only moderate levels of Fstem-EboGP incorporation (but on par with the level of F in wt virions), which may be due to low expression levels of Fstem-EboGP relative to Fstem-GFP. Fstem-GFP was incorporated into VLPs as efficiently as the Fstem alone (Fstem496), indicating strong potential for incorporating foreign sequences into hRSV VLPs by using the Fstem as a vehicle.
A role for M2-1 in virus assembly?
Previous work has shown by cryo-EM that the M2-1 protein is present in the virion as a layer between the ribonucleoprotein complex and M (17) and is required for targeting of M to IBs (57). In identifying the minimal players involved in assembly, we based our work on previous reports showing that M2-1 is not required for VLP formation (5, 22) or for packaging and passaging of hRSV minigenomes (32). As such, we have not examined M2-1. In our experiments, VLPs formed efficiently in the absence of M2-1, suggesting that M2-1 does not play a major role. We have also observed on many occasions, including this study (Fig. 1B, 1), that M targets to IBs efficiently without M2-1. Based on the results presented here, we suspect that M2-1 is included in infectious virions due to its role within the polymerase complex packaged inside the virion (required for productive entry) and does not have a key role in the organization of particle assembly.
Morphology.
We have focused our studies so far on filamentous virions, because evidence suggests that the filamentous morphology is the form that represents the majority of infectious virus and contains prefusion F. We have not yet investigated whether different morphologies, such as spherical VLPs, can form under the conditions used in this study. However, under the conditions used, our high-resolution SEM analyses have not detected any significant numbers of nonfilamentous particles associated with the cell surface, and we have not been able to detect significant numbers of VLPs from the medium overlying the cells. Previous work has described “membranous vesicles” that formed after coexpression of P and M (49). We have noticed that coexpression of P and M in the absence of F appears to increase cell damage (lysis or apoptosis), which could be partly responsible for the previous observations. We currently lack an antibody that properly detects P on Western blots, but we plan to develop methods to further examine the reported observations.
Cell type.
To further narrow the viral protein requirements for VLP assembly, we built on previous work carried out in HEp-2 cells, a cell type in which wt hRSV replicates efficiently. However, we have obtained the same results in HEK293 cells (data not shown), suggesting the findings are not cell specific. Future production of hRSV particles for vaccine purposes, be it VLPs or viruses, may be possible in different cell types. As multiple host factors are likely involved in hRSV VLP assembly, host species and/or cell-type-specific differences for optimal VLP production may come into play and will have to be analyzed and optimized, depending on the cell culture system of choice. A recent paper reported that immunogenic VLPs were isolated from HEK293 cells coexpressing the M, G, and F proteins (58). Our present work would predict that in the absence of P, coalescence of M, F, and G proteins and VLP formation are not optimal and that the efficiency could be enhanced by including P. However, the systems are not readily compared, as the VLPs produced by M-G-F coexpression were spherical and were harvested at a much later time point from suspensions of HEK293 cells at ultrahigh g force, whereas we focused on cell-associated filamentous VLPs at an earlier time point to avoid potential morphological changes associated with purification, as well as excessive cell lysis and/or death. Although we have so far been unable to collect VLPs from the supernatant of transfected cells, we have not yet done an exhaustive analysis and cannot rule out the possibility that round particles with a very high sedimentation coefficient are formed and secreted after coexpression of P, M, and F proteins.
In conclusion.
We have investigated the requirements and mechanisms underlying hRSV VLP formation. Understanding hRSV assembly in cell culture is relevant, as production of virus or VLPs for vaccine purposes will most likely depend on a cell culture platform. The data show that the P, M, and F proteins are sufficient for optimal viral protein coalescence and formation of a particle resembling wt virus and provide novel insights into the contributions of individual proteins, with the F carboxy terminus appearing to act as a type of cofactor for M self-assembly and P having an impact on particle length and diameter. A better understanding of the proteins and protein domains that organize assembly will facilitate needed improvements in manufacturing RSV or RSV-like particles with defined composition and characteristics for vaccine purposes.
ACKNOWLEDGMENTS
We thank the members of the Oomens laboratory for helpful discussions during the preparation of the manuscript; Edward Walsh, Geraldine Taylor, and Gary Blissard for providing antibodies; and Lisa Whitworth of the Oklahoma State University Microscopy Laboratory for technical expertise.
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Couch RB, Englund JA, Whimbey E. 1997. Respiratory viral infections in immunocompetent and immunocompromised persons. Am J Med 102:2–9, 25–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han LL, Alexander JP, Anderson LJ. 1999. Respiratory syncytial virus pneumonia among the elderly: an assessment of disease burden. J Infect Dis 179:25–30. doi: 10.1086/314567. [DOI] [PubMed] [Google Scholar]
- 3.Prince GM, Mathews A, Curtis SJ, Porter D. 2000. Treatment of respiratory syncytial virus bronchiolitis and pneumonia in a cotton rat model with systemically administered monoclonal antibody (palivizumab) and glucocorticosteroid. J Infect Dis 182:1326–1330. doi: 10.1086/315894. [DOI] [Google Scholar]
- 4.McGinnes LW, Gravel KA, Finberg RW, Kurt-Jones EA, Massare MJ, Smith G, Schmidt MR, Morrison TG. 2011. Assembly and immunological properties of Newcastle disease virus-like particles containing the respiratory syncytial virus F and G proteins. J Virol 85:366–377. doi: 10.1128/JVI.01861-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaikh FY, Cox RG, Lifland AW, Hotard AL, Williams JV, Moore ML, Santangelo PJ, Crowe JE Jr. 2012. A critical phenylalanine residue in the respiratory syncytial virus fusion protein cytoplasmic tail mediates assembly of internal viral proteins into viral filaments and particles. mBio 3:e00270–11. doi: 10.1128/mBio.00270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang HS, Kwon YM, Lee JS, Yoo SE, Lee YN, Ko EJ, Kim MC, Cho MK, Lee YT, Jung YJ, Lee JY, Li JD, Kang SM. 2014. Co-immunization with virus-like particle and DNA vaccines induces protection against respiratory syncytial virus infection and bronchiolitis. Antiviral Res 110:115–123. doi: 10.1016/j.antiviral.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorquera PA, Choi Y, Oakley KE, Powell TJ, Boyd JG, Palath N, Haynes LM, Anderson LJ, Tripp RA. 2013. Nanoparticle vaccines encompassing the respiratory syncytial virus (RSV) G protein CX3C chemokine motif induce robust immunity protecting from challenge and disease. PLoS One 8:e74905. doi: 10.1371/journal.pone.0074905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JS, Kwon YM, Hwang HS, Lee YN, Ko EJ, Yoo SE, Kim MC, Kim KH, Cho MK, Lee YT, Lee YR, Quan FS, Kang SM. 2014. Baculovirus-expressed virus-like particle vaccine in combination with DNA encoding the fusion protein confers protection against respiratory syncytial virus. Vaccine 32:5866–5874. doi: 10.1016/j.vaccine.2014.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee S, Quan FS, Kwon Y, Sakamoto K, Kang SM, Compans RW, Moore ML. 2014. Additive protection induced by mixed virus-like particles presenting respiratory syncytial virus fusion or attachment glycoproteins. Antiviral Res 111:129–135. doi: 10.1016/j.antiviral.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murawski MR, McGinnes LW, Finberg RW, Kurt-Jones EA, Massare MJ, Smith G, Heaton PM, Fraire AE, Morrison TG. 2010. Newcastle disease virus-like particles containing respiratory syncytial virus G protein induced protection in BALB/c mice, with no evidence of immunopathology. J Virol 84:1110–1123. doi: 10.1128/JVI.01709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt MR, McGinnes LW, Kenward SA, Willems KN, Woodland RT, Morrison TG. 2012. Long-term and memory immune responses in mice against Newcastle disease virus-like particles containing respiratory syncytial virus glycoprotein ectodomains. J Virol 86:11654–11662. doi: 10.1128/JVI.01510-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raghunandan R, Lu H, Zhou B, Xabier MG, Massare MJ, Flyer DC, Fries LF, Smith GE, Glenn GM. 2014. An insect cell derived respiratory syncytial virus (RSV) F nanoparticle vaccine induces antigenic site II antibodies and protects against RSV challenge in cotton rats by active and passive immunization. Vaccine 32:6485–6492. doi: 10.1016/j.vaccine.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glenn GM, Smith G, Fries L, Raghunandan R, Lu H, Zhou B, Thomas DN, Hickman SP, Kpamegan E, Boddapati S, Piedra PA. 2013. Safety and immunogenicity of a Sf9 insect cell-derived respiratory syncytial virus fusion protein nanoparticle vaccine. Vaccine 31:524–532. doi: 10.1016/j.vaccine.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Stone JW, Thornburg NJ, Blum DL, Kuhn SJ, Wright DW, Crowe JE Jr. 2013. Gold nanorod vaccine for respiratory syncytial virus. Nanotechnology 24:295102. doi: 10.1088/0957-4484/24/29/295102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachi T, Howe C. 1973. Morphogenesis and ultrastructure of respiratory syncytial virus. J Virol 12:1173–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gower TL, Pastey MK, Peeples ME, Collins PL, McCurdy LH, Hart TK, Guth A, Johnson TR, Graham BS. 2005. RhoA signaling is required for respiratory syncytial virus-induced syncytium formation and filamentous virion morphology. J Virol 79:5326–5336. doi: 10.1128/JVI.79.9.5326-5336.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiss G, Holl JM, Williams GM, Alonas E, Vanover D, Lifland AW, Gudheti M, Guerrero-Ferreira RC, Nair V, Yi H, Graham BS, Santangelo PJ, Wright ER. 2014. Structural analysis of respiratory syncytial virus reveals the position of M2-1 between the matrix protein and the ribonucleoprotein complex. J Virol 88:7602–7617. doi: 10.1128/JVI.00256-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norrby E, Marusyk H, Orvell C. 1970. Morphogenesis of respiratory syncytial virus in a green monkey kidney cell line (Vero). J Virol 6:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts SR, Compans RW, Wertz GW. 1995. Respiratory syncytial virus matures at the apical surfaces of polarized epithelial cells. J Virol 69:2667–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, Goldenring JR, Crowe JE Jr. 2008. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc Natl Acad Sci U S A 105:10209–10214. doi: 10.1073/pnas.0712144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liljeroos L, Krzyzaniak MA, Helenius A, Butcher SJ. 2013. Architecture of respiratory syncytial virus revealed by electron cryotomography. Proc Natl Acad Sci U S A 110:11133–11138. doi: 10.1073/pnas.1309070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forster A, Maertens GN, Farrell PJ, Bajorek M. 2015. Dimerization of matrix protein is required for budding of respiratory syncytial virus. J Virol 89:4624–4635. doi: 10.1128/JVI.03500-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeffree CE, Brown G, Aitken J, Su-Yin DY, Tan BH, Sugrue RJ. 2007. Ultrastructural analysis of the interaction between F-actin and respiratory syncytial virus during virus assembly. Virology 369:309–323. doi: 10.1016/j.virol.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Oomens AG, Bevis KP, Wertz GW. 2006. The cytoplasmic tail of the human respiratory syncytial virus F protein plays critical roles in cellular localization of the F protein and infectious progeny production. J Virol 80:10465–10477. doi: 10.1128/JVI.01439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santangelo PJ, Bao G. 2007. Dynamics of filamentous viral RNPs prior to egress. Nucleic Acids Res 35:3602–3611. doi: 10.1093/nar/gkm246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuchs H, Bachi T. 1975. Scanning electron microscopical demonstration of respiratory syncytial virus antigens by immunological markers. J Ultrastruct Res 52:114–119. doi: 10.1016/S0022-5320(75)80026-8. [DOI] [PubMed] [Google Scholar]
- 27.Jeffree CE, Rixon HW, Brown G, Aitken J, Sugrue RJ. 2003. Distribution of the attachment (G) glycoprotein and GM1 within the envelope of mature respiratory syncytial virus filaments revealed using field emission scanning electron microscopy. Virology 306:254–267. doi: 10.1016/S0042-6822(02)00016-8. [DOI] [PubMed] [Google Scholar]
- 28.Oomens AG, Megaw AG, Wertz GW. 2003. Infectivity of a human respiratory syncytial virus lacking the SH, G, and F proteins is efficiently mediated by the vesicular stomatitis virus G protein. J Virol 77:3785–3798. doi: 10.1128/JVI.77.6.3785-3798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stope MB, Karger A, Schmidt U, Buchholz UJ. 2001. Chimeric bovine respiratory syncytial virus with attachment and fusion glycoproteins replaced by bovine parainfluenza virus type 3 hemagglutinin-neuraminidase and fusion proteins. J Virol 75:9367–9377. doi: 10.1128/JVI.75.19.9367-9377.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen BJ, Lamb RA. 2008. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372:221–232. doi: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El Najjar F, Schmitt AP, Dutch RE. 2014. Paramyxovirus glycoprotein incorporation, assembly and budding: a three way dance for infectious particle production. Viruses 6:3019–3054. doi: 10.3390/v6083019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teng MN, Collins PL. 1998. Identification of the respiratory syncytial virus proteins required for formation and passage of helper-dependent infectious particles. J Virol 72:5707–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baviskar PS, Hotard AL, Moore ML, Oomens AG. 2013. The respiratory syncytial virus fusion protein targets to the perimeter of inclusion bodies and facilitates filament formation by a cytoplasmic tail-dependent mechanism. J Virol 87:10730–10741. doi: 10.1128/JVI.03086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitra R, Baviskar P, Duncan-Decocq RR, Patel D, Oomens AG. 2012. The human respiratory syncytial virus matrix protein is required for maturation of viral filaments. J Virol 86:4432–4443. doi: 10.1128/JVI.06744-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaikh FY, Utley TJ, Craven RE, Rogers MC, Lapierre LA, Goldenring JR, Crowe JE Jr. 2012. Respiratory syncytial virus assembles into structured filamentous virion particles independently of host cytoskeleton and related proteins. PLoS One 7:e40826. doi: 10.1371/journal.pone.0040826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghildyal R, Ho A, Jans DA. 2006. Central role of the respiratory syncytial virus matrix protein in infection. FEMS Microbiol Rev 30:692–705. doi: 10.1111/j.1574-6976.2006.00025.x. [DOI] [PubMed] [Google Scholar]
- 37.Henderson G, Murray J, Yeo R. 2002. Sorting of the respiratory syncytial virus matrix protein into detergent-resistant structures is dependent on cell-surface expression of the glycoproteins. Virology 300:244. doi: 10.1006/viro.2002.1540. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez L, Cuesta I, Asenjo A, Villanueva N. 2004. Human respiratory syncytial virus matrix protein is an RNA-binding protein: binding properties, location and identity of the RNA contact residues. J Gen Virol 85:709–719. doi: 10.1099/vir.0.19707-0. [DOI] [PubMed] [Google Scholar]
- 39.Ghildyal R, Mills J, Murray M, Vardaxis N, Meanger J. 2002. Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J Gen Virol 83:753–757. doi: 10.1099/0022-1317-83-4-753. [DOI] [PubMed] [Google Scholar]
- 40.Harrison MS, Sakaguchi T, Schmitt AP. 2010. Paramyxovirus assembly and budding: building particles that transmit infections. Int J Biochem Cell Biol 42:1416–1429. doi: 10.1016/j.biocel.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitt AP, Lamb RA. 2004. Escaping from the cell: assembly and budding of negative-strand RNA viruses. Curr Top Microbiol Immunol 283:145–196. [DOI] [PubMed] [Google Scholar]
- 42.Takimoto T, Portner A. 2004. Molecular mechanism of paramyxovirus budding. Virus Res 106:133–145. doi: 10.1016/j.virusres.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 43.Heminway BR, Yu Y, Tanaka Y, Perrine KG, Gustafson E, Bernstein JM, Galinski MS. 1994. Analysis of respiratory syncytial virus F, G, and SH proteins in cell fusion. Virology 200:801–805. doi: 10.1006/viro.1994.1245. [DOI] [PubMed] [Google Scholar]
- 44.Morton CJ, Cameron R, Lawrence LJ, Lin B, Lowe M, Luttick A, Mason A, McKimm-Breschkin J, Parker MW, Ryan J, Smout M, Sullivan J, Tucker SP, Young PR. 2003. Structural characterization of respiratory syncytial virus fusion inhibitor escape mutants: homology model of the F protein and a syncytium formation assay. Virology 311:275–288. doi: 10.1016/S0042-6822(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 45.Fricke J, Koo LY, Brown CR, Collins PL. 2013. p38 and OGT sequestration into viral inclusion bodies in cells infected with human respiratory syncytial virus suppresses MK2 activities and stress granule assembly. J Virol 87:1333–1347. doi: 10.1128/JVI.02263-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE Jr, Santangelo PJ. 2012. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol 86:8245–8258. doi: 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia J, Garcia-Barreno B, Vivo A, Melero JA. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195:243–247. doi: 10.1006/viro.1993.1366. [DOI] [PubMed] [Google Scholar]
- 48.Asenjo A, Calvo E, Villanueva N. 2006. Phosphorylation of human respiratory syncytial virus P protein at threonine 108 controls its interaction with the M2-1 protein in the viral RNA polymerase complex. J Gen Virol 87:3637–3642. doi: 10.1099/vir.0.82165-0. [DOI] [PubMed] [Google Scholar]
- 49.Asenjo A, Rodriguez L, Villanueva N. 2005. Determination of phosphorylated residues from human respiratory syncytial virus P protein that are dynamically dephosphorylated by cellular phosphatases: a possible role for serine 54. J Gen Virol 86:1109–1120. doi: 10.1099/vir.0.80692-0. [DOI] [PubMed] [Google Scholar]
- 50.Haas J, Park EC, Seed B. 1996. Codon usage limitation in the expression of HIV-1 envelope glycoprotein. Curr Biol 6:315–324. doi: 10.1016/S0960-9822(02)00482-7. [DOI] [PubMed] [Google Scholar]
- 51.Buchholz UJ, Granzow H, Schuldt K, Whitehead SS, Murphy BR, Collins PL. 2000. Chimeric bovine respiratory syncytial virus with glycoprotein gene substitutions from human respiratory syncytial virus (HRSV): effects on host range and evaluation as a live-attenuated HRSV vaccine. J Virol 74:1187–1199. doi: 10.1128/JVI.74.3.1187-1199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Batonick M, Oomens AG, Wertz GW. 2008. Human respiratory syncytial virus glycoproteins are not required for apical targeting and release from polarized epithelial cells. J Virol 82:8664–8672. doi: 10.1128/JVI.00827-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown G, Rixon HW, Sugrue RJ. 2002. Respiratory syncytial virus assembly occurs in GM1-rich regions of the host-cell membrane and alters the cellular distribution of tyrosine phosphorylated caveolin-1. J Gen Virol 83:1841–1850. doi: 10.1099/0022-1317-83-8-1841. [DOI] [PubMed] [Google Scholar]
- 54.Radhakrishnan A, Yeo D, Brown G, Myaing MZ, Iyer LR, Fleck R, Tan BH, Aitken J, Sanmun D, Tang K, Yarwood A, Brink J, Sugrue RJ. 2010. Protein analysis of purified respiratory syncytial virus particles reveals an important role for heat shock protein 90 in virus particle assembly. Mol Cell Proteomics 9:1829–1848. doi: 10.1074/mcp.M110.001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McPhee HK, Carlisle JL, Beeby A, Money VA, Watson SM, Yeo RP, Sanderson JM. 2011. Influence of lipids on the interfacial disposition of respiratory syncytial virus matrix protein. Langmuir 27:304–311. doi: 10.1021/la104041n. [DOI] [PubMed] [Google Scholar]
- 56.Brock SC, Heck JM, McGraw PA, Crowe JE Jr. 2005. The transmembrane domain of the respiratory syncytial virus F protein is an orientation-independent apical plasma membrane sorting sequence. J Virol 79:12528–12535. doi: 10.1128/JVI.79.19.12528-12535.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li D, Jans DA, Bardin PG, Meanger J, Mills J, Ghildyal R. 2008. Association of respiratory syncytial virus M protein with viral nucleocapsids is mediated by the M2-1 protein. J Virol 82:8863–8870. doi: 10.1128/JVI.00343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walpita P, Johns LM, Tandon R, Moore ML. 2015. Mammalian cell-derived respiratory syncytial virus-like particles protect the lower as well as the upper respiratory tract. PLoS One 10:e0130755. doi: 10.1371/journal.pone.0130755. [DOI] [PMC free article] [PubMed] [Google Scholar]