

Abstract
In vitro assays of compartment mixing have been key tools in the biochemical dissection of organelle docking and fusion. Many such assays measure compartment mixing through the enzymatic modification of reporter proteins. Homotypic fusion of yeast vacuoles is measured with a coupled assay of proteolytic maturation of pro-alkaline phosphatase (pro-ALP). A kinetic lag is observed between the end of docking, marked by the acquisition of resistance to anti-SNARE reagents, and ALP maturation. We therefore asked whether the time taken for pro-ALP maturation adds a kinetic lag to the measured fusion signal. Prb1p promotes ALP maturation; overproduction of Prb1p accelerates ALP activation in detergent lysates but does not alter the measured kinetics of docking or fusion. Thus, the lag between docking and ALP activation reflects a lag between docking and fusion. Many vacuoles in the population undergo multiple rounds of fusion; methods are presented for distinguishing the first round of fusion from ongoing rounds of fusion. A simple kinetic model distinguishes between two rates, the rate of fusion and the rate at which fusion competence is lost, and allows estimation of the number of rounds of fusion completed.
Keywords: Pho8p, protease B, SNARE, Rab/Ypt, tethering
Membrane docking and fusion events have been measured by many methods. Among the most commonly used are in vitro, coupled enzymatic assays of compartment mixing between two vesicle populations (1). One vesicle population contains a reporter substrate, and the other vesicle population contains an enzyme that can covalently modify the reporter substrate. We call vesicles containing the modifying enzyme “effector” vesicles and vesicles containing the substrate “reporter” vesicles. Fusion causes content mixing between effector and reporter vesicles, allowing the enzyme access to the reporter substrate. The amount of reporter modified thereby indicates the extent of fusion.
Coupled transport and fusion assays have made possible the identification of molecules that mediate vesicle budding, docking and fusion, and the dissection of the mechanisms by which these molecules act. However, coupled assays have potential limitations. Treatments that inhibit the reporter system may be interpreted incorrectly as fusion inhibitors. Moreover, if the enzymatic processing of the reporter substrate is slow relative to the docking or fusion reaction of interest, the kinetics of the processing event will dominate, and thus obscure, the kinetics of fusion.
We study yeast vacuole/vacuole homotypic fusion. A key tool in our work is a cell-free, coupled enzymatic assay of vacuole fusion (2). In this assay, the reporter substrate is the enzyme alkaline phosphatase (ALP), encoded by the PHO8 gene. ALP is synthesized as an inactive precursor, pro-ALP, and is processed into an active form by proteolytic cleavage (3, 4). The reporter vacuoles contain pro-ALP but are protease deficient. The effector vacuoles are protease replete but lack pro-ALP. Content mixing between effector and reporter vacuoles results in processing of pro-ALP to ALP. In addition to effector–reporter fusion, effector–effector and reporter–reporter fusion also occurs; the latter events do not yield a signal in the coupled assay. ALP maturation is monitored by a change of Pho8p mobility in immunoblots or a colorimetric ALP activity assay (2). The colorimetric version of the vacuole fusion assay is rapid and quantitative and will be amenable to high-throughput experimentation.
Inhibitors have been used in kinetic and staging experiments to divide the overall vacuole fusion reaction into three major subreactions: priming, docking, and fusion. In priming, which can occur before vacuole–vacuole contact, Sec18/17p (yeast NSF/α-SNAP) consumes ATP to diassemble cis-SNARE complexes on the vacuole membrane (5, 6). The docking step of the reaction begins with membrane tethering, which depends on Ypt7p, a Rab-family GTPase (7–9). Upon tethering, Ypt7p:GTP governs the assembly of a lipid- and protein-enriched subdomain called the vertex (10, 11). Docking culminates with the formation of trans-SNARE complexes that span the docking junction and the SNARE-dependent release of Ca2+ from the vacuole lumen (12). Ca2+ release may trigger the final subreaction, fusion (13). Fusion inhibitors generally exhibit one of three distinct kinetic profiles, corresponding to action by their target molecules during priming, docking, or fusion. This kinetics-based taxonomy of fusion factors and reaction stages is largely, but not completely, in accord with staging based on block/release protocols. However, several factors may complicate the interpretation of these experiments. As we show here, vacuoles can undergo multiple rounds of fusion in vitro, but it has not been possible to discriminate between the first round and subsequent rounds of fusion. Moreover, it has not been clear whether pro-ALP processing is fast or slow relative to the steps of fusion. Slow pro-ALP processing could distort measurements of fusion kinetics; the apparent delay after docking and before fusion might instead reflect a lag between fusion (content mixing) and pro-ALP activation.
In this report, we present a detailed analysis of the vacuole fusion assay system. We show that Prb1p, a vacuolar serine protease, activates the reporter substrate ALP; this reaction is fast relative to the fusion reaction. Methods are introduced to distinguish treatments that block fusion from those that inhibit the activation of pro-ALP. Finally, we show that modifications of the fusion assay allow discrimination between a single round of fusion and ongoing rounds of fusion.
Materials and Methods
Strains, Plasmids, and Growth Conditions. The standard tester strains used for the vacuole fusion reaction are the reporter/pro-ALP donor BJ3505 [MATα pep4::HIS3 prb1-Δ1.6R his3–200 lys2–801 trp1-Δ101 (gal3) ura3–52 gal2 can1] (14) and the effector/protease donor DKY6281 (MATα leu2–3 leu2–112 ura3–52 his3-Δ200 trp1-Δ901 lys2–801 suc2-9 pho8Δ::TRP1) (15). For some experiments, pho8Δ::neo and pep4Δ::neo derivatives of BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) (16) were used in place of BJ3505 and DKY6281. pCM189 (CEN TetO tTA URA3) and pCM185 (CEN ARS TetO tTA TRP1) are low-copy vectors designed for doxycycline-repressible gene expression (17). pAJM41 and pAJM43 were constructed by ligating the coding region of PRB1 into pCM189 or pCM185, immediately 3′ of the tTA promoters. pAJM44 and pAJM46 were constructed by ligating the coding region of PEP4 into pCM189 or pCM185, immediately 3′ of the tTA promoter. BJ2168 (MATa leu2 trp1 ura3–52 prb1–1122 pep4–3 prc1–407 gal2) (14) was used as a recipient strain for PEP4 and PRB1 complementation experiments: AMY51 carries pCM189, AMY52 carries pAJM41, AMY53 carries pCM189 and pCM185, AMY54 carries pCM189 and pAJM46, AMY55 carries pAJM41 and pCM185, and AMY56 carries pAJM41 and pAJM46. DKY6281 was used as the background for PEP4 and PRB1 overexpression: AMY45 carries pCM189, AMY47 carries pAJM41, and AMY49 carries pAJM44. The PRB1 overexpresser AMY47 loses viability after extended growth on selective plates without doxycycline; for this reason, AMY45, AMY47, and AMY49 were grown in the presence of 5 μg/ml doxycycline to prevent protease overproduction during routine passage. These strains were grown for vacuole isolation by first growing 100-ml broth cultures overnight in synthetic defined-uracil medium without doxycycline. Cells were harvested by centrifugation, used to inoculate 1-liter cultures in yeast extract peptone dextrose broth, and grown ≈16 h at 30°C. In some experiments, recombinant IB2 (150 μg per 1.0 OD600 liter of cells) was added at the DEAE dextran treatment step of vacuole isolation (2) to reduce proteolysis caused by Prb1p released from ruptured vacuoles, a procedure that significantly improved the fusion competence of vacuoles derived from Prb1p overexpressers. Density gradient floatation of the vacuoles during isolation left most of the added IB2 activity at the bottom of the Ficoll gradient, along with cell debris.
Fusion and ALP Activity Measurement. A standard 30-μl fusion reaction contained a total of 6 μg of vacuoles (3 μg of effector and 3 μg of reporter vacuoles), isolated as described (2), 125 mM KCl, 20 mM K-Pipes (pH 6.8), 6 mM MgCl2, 10 μM CoA, 4.5 μM recombinant IB2 (Pbi2p), and 10 nM recombinant his6-Sec18p. ATP-regenerating system (2) was added from a 10× stock to 1× final. Reactions were incubated at 27°C. For assays containing different proportions of effectors and reporters, a total of 6 μg of vacuoles per 30-μl reaction was always used. ALP maturation reactions with dilute detergent lysates contained a total of 6 μg of vacuoles (typically 3 μg of effector and 3 μg of reporter vacuoles), 125 mM KCl, 20 mM K-Pipes, pH 6.8, 6 mM MgCl2, and 0.3% (vol/vol) Triton X-100 detergent.
ALP-dependent formation of nitrophenolate from p-nitrophenylphosphate was measured by absorbance at 400 nm, as described (2), except that the assay buffer contained 250 mM Tris·Cl (pH 8.5), 0.4% (vol/vol) Triton X-100, 10 mM MgCl2, 1 mM CaCl2, and 1 mM p-nitrophenylphosphate. In this buffer at 30°C, crude vacuolar ALP has an apparent KM for p-nitrophenylphosphate of 260 ± 20 μM (mean ± 95% confidence interval; data not shown). The ALP assay was used within its linear range (i.e., it was not substrate limited) under the conditions used in this study. One unit of ALP activation is defined as an amount of ALP that catalyzes formation of 1 μmol of nitrophenolate product per minute. Vacuole fusion-mediated ALP activation is normalized to ALP unit per microgram of effector vacuoles (2). In experiments initiated with different proportions of effector and reporter vacuoles, the use of this normalization corrects for the initial amount of reporter vacuoles present. ALP activation in detergent lysates is reported as the increase in active ALP per time (ALP activation unit per minute). The fusion and ALP maturation data shown are representative examples from three to five independent experiments, except for the experiment shown in Fig. 2B, which was done twice.
Fig. 2.

Apparent kinetics of fusion are not changed by Prb1p overproduction in effector vacuoles. Fusion or detergent lysate (Insets) reactions contained reporter vacuoles obtained from strain BJ3505 and effector vacuoles from either strain AMY45 (control strain, circles) or AMY47 (Prb1p overproducer, triangles). (Insets) Graphs show the kinetics of ALP processing in dilute detergent lysates, with the same vacuole preparations used for the fusion experiments in the main graphs. (A) Kinetics of inhibition experiment. Master reactions were incubated at 27°C. At the intervals shown, 30-μl aliquots were removed from the master reactions and placed on ice (open symbols) or mixed with anti-Vam3p antibody and incubated at 27°C for the remainder of the 80 min (filled symbols). (B) Kinetics after synchronous docking. Master reactions were initiated as in A but contained anti-Sec17p IgG added from the start. After incubation for 20 min at 27°C, recombinant Vam7p (5 μM) was added to the master reactions, and at intervals, 30-μl aliquots were removed from the master reactions and placed on ice (open symbols) or mixed with anti-Vam3p antibody and incubated at 27°C for the remainder of the 80 min (filled symbols). The 0 time point in this experiment is the signal obtained with a reaction not rescued by Vam7p addition and was equivalent to the signal given in a reaction in which anti-Vam3p was added immediately before Vam7p. The ALP values are plotted in normalized form; maximum ALP activity values at 80 min were 3.6 units for the reactions containing control effector vacuoles (circles) and 4.5 units for the reactions containing Prb1p overproducer vacuoles (triangles).
Microscopy. Microscopy and morphometry were as described (12). The images shown in Fig. 4 were acquired by wide-field microscopy, then subjected to high-pass (16-pixel threshold) and sharpening filters (5 × 5 hat) to emphasize fine structure in the fields. Microscopic image analysis was done with image/j 1.32j (http://rsb.info.nih.gov/ij), and data were analyzed with jmp 5.0.1a (SAS Institute, Cary, NC).
Fig. 4.

Quantitative analyses of single and multiple rounds of fusion in the homotypic vacuole fusion system. (A) Cartoon depicting effects of initiating fusion reactions with different proportions of effector and reporter vacuoles. (B) Modeling of the quantitative consequences of varying effector and reporter vacuole fractions. Details of the model are given in Materials and Methods. For these simulations, the fusion rate constant was set at 0.03 min–1. In both panels, the darker traces indicate reactions containing equal fractions of effector and reporter vacuoles, the standard reaction condition. (Left) The fraction of total vacuoles that are effectors. Because any vacuole that contains proteases is an effector, content mixing between effectors and reporters causes the fraction of effectors to increase over time. When reactions are initiated with large fractions of effectors, reporter vacuoles will, when they fuse, almost always fuse with an effector, yielding an ALP signal (Right). Under these conditions, only the first round of fusion contributes substantially to the ALP signal. At lower effector fractions, the fraction of effectors increases dramatically over time (Left), with the consequence that the ALP signal (Right) increases most rapidly during later fusion events. (C) Raw data from an experiment in which the initial effector and reporter were varied over a wide range. Background (ice incubation) values are subtracted from all signals; these values were 0.16, 0.27, and 1.0 units for reactions containing initial effector vacuole fractions of 0.025, 0.5, and 0.975, respectively. (D) Modeling as in B but with the incorporation of a rate constant for irreversible decay of fusion competence. The downward-sloping trace (Left) shows the decrease in proportion of fusion-competent vacuoles over time. For these simulations, the fusion rate constant k1 was set at 0.03 min–1, and the rate constant for loss of fusion competence k2 was set at 0.01 min–1.(E) Fits of the model described in D to the experimental data shown in C. Details are given in Materials and Methods.
Modeling and Data Analysis. Modeling and data analysis were done by using igorpro 4.08 (WaveMetrics, Lake Oswego, OR) and berkeley madonna 8.1β9a. We model the coupled fusion assay with a system of differential equations:
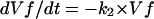 |
[1] |
 |
[2] |
 |
[3] |
Vf is the fraction of fusion-competent vacuoles, Vp is the fraction of effectors (vacuoles containing protease), and Va is the fraction of reporters (vacuoles containing immature pro-ALP). k1 is the forward fusion rate constant. k2 is the rate constant for loss of fusion competence. Both processes are controlled by mass action and are irreversible. For the traces in Fig. 4B, Vf was set at a constant value of 1 (k2 = 0).
A, the amount of activated ALP at time t, is
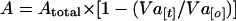 |
[4] |
where Atotal is the amount of activatable pro-ALP in a reaction, determined in an in vitro ALP maturation experiment with detergent lysates. Typically, Atotal = 10 units. Fits of experimental data to A were done in two steps. An initial fit was done with simultaneous optimization of k1, k2, and Vf[0], the initial fusogenic fraction of vacuoles. A second fit was then performed with Vf clamped to its mean value from the initial set of fits and with optimization of k1 and k2.
S, the fold-increase in vacuole surface area (assuming conservation of membrane), increases as
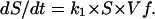 |
[5] |
R, the number of rounds of fusion, is defined as the average of fusion events per vacuole in the population, and (again, assuming conservation of membrane) is given by
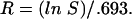 |
[6] |
Results
Prb1p Mediates ALP Activation. The vacuole fusion assay relies on Pep4p (protease A)-dependent proteolytic maturation of pro-ALP (3, 4). We now report that a second protease, Prb1p, is rate limiting for ALP maturation. Because Pep4p is required for Prb1p activation, pep4 mutant strains lack both Pep4p and Prb1p activities (4, 14, 18). Vacuoles isolated from any of several pep4 strains contain pro-ALP but lack significant ALP activity. The pro-ALP in these vacuoles can be activated in vitro, either by fusion with protease-replete vacuoles that lack ALP (Fig. 1A) or by mixing detergent lysates of these vacuole populations (Fig. 1B). ALP activation in detergent lysates is potently inhibited by inhibitors of protease B (PMSF and IB2) (14), but maturation is not inhibited by an inhibitor of protease A (pepstatin A) (14). In fusion experiments with intact vacuoles, the membrane-permeant inhibitor PMSF blocked ALP maturation, but the membrane-impermeant inhibitor IB2 had no effect on maturation. Thus, fusion-dependent ALP maturation occurs within the vacuole lumen as expected and is Prb1p dependent. However, vacuoles isolated from BY4742 prb1Δ cells exhibit significant ALP activity, typically 25% of the activity observed in vacuoles isolated from the BY4742 parental strain, and overexpression of plasmid-borne PEP4 complements the ALP maturation defect in the pep4 prb1 prc1 strain BJ2168 (data not shown). These results suggest that in vivo ALP activation occurs through two pathways, both of which depend on Pep4p: a fast pathway that requires Prb1p and a slow pathway that does not. To test this idea, Pep4p or Prb1p were overproduced in the PEP4 PRB1 pho8Δ strain DKY6281 by introducing episomal copies of PEP4 or PRB1 driven by a strong promoter (see Materials and Methods). Prb1p overproduction increased the rate of pro-ALP activation in detergent lysates (0.064 ± 0.018 units per min versus 0.020 ± 0.004 units per min for vector control; means ± SD; n = 5 experiments). In contrast, Pep4p overproduction did not alter the rate of pro-ALP activation in detergent lysates (0.021 ± 0.005 units per min; n = 3 experiments). We conclude that when Pep4p and Prb1p are both present at normal levels, Prb1p is rate limiting for ALP activation.
Fig. 1.

Inhibitor sensitivity of in vitro ALP activation. (A) Fusion-dependent ALP activation. (B) ALP activation in dilute detergent lysates. Standard reactions were initiated as described in Materials and Methods. Inhibitors were added either at the start or the end of a 60-min incubation at 27°C. Inhibitor concentrations were 25 μM for IB2, 1 mM for PMSF, 50 μM for pepstatin, and 110 mM for anti-Vam3p.
ALP Activation Is Fast Relative to the Docking–Fusion Transition. Does the time taken by Prb1p to activate pro-ALP add a significant kinetic lag to the in vitro vacuole fusion assay? If such a lag exists, we might expect that increasing the rate of pro-ALP activation should increase the apparent rate of fusion, but it should not influence docking kinetics. To measure the relative rates of docking and fusion, a kinetics of inhibition assay was used. Two master fusion reactions were initiated, one containing effector vacuoles from a control (vector alone) strain and the other containing effector vacuoles from the Prb1p overproducer. Protease-deficient reporter vacuoles containing pro-ALP were present in both fusion reactions. At intervals, aliquots were withdrawn from the master reactions and placed either in tubes with anti-Vam3p antibody (which inhibits during docking by interfering with SNARE protein function) or in tubes held on ice, which inhibits fusion. ALP activation can occur only after content mixing, the final step of fusion. If ALP activation is slow relative to the transition between docking and fusion, accelerated ALP activation should increase the apparent rate of fusion without changing the kinetics of docking. In other words, the ice curve should shift left, toward the docking curve. As shown in Fig. 2 A, PRB1 overexpression accelerated ALP activation in detergent extracts but did not change the observed kinetics of either docking or fusion. Similarly, PEP4 overexpression had no detectable effect on the kinetics of fusion-dependent ALP maturation (data not shown). An assumption central to this analysis is that most of the Prb1p-containing effector vacuoles are in fact derived from overexpresser cells. Because PRB1 is overexpressed from a plasmid and the cells are grown in nonselective medium for ≈9 doublings, and because PRB1 overexpressers are relatively slow-growing (doubling time 130 min vs. 105 min for vector control), we measured the rate of plasmid loss by replica-plating onto selective and nonselective media. At harvest, >90% of the cells retained the plasmid. These results suggest that the time taken for ALP activation by Prb1p does not add a substantial component to the apparent kinetics of vacuole fusion in vitro.
To increase the sensitivity of this analysis, we synchronized docking by using priming bypass conditions (Fig. 2B; refs. 12 and 19). A standard reaction is incubated for 20 min with anti-Sec17p antibody, which prevents priming; the anti-Sec17p block is then bypassed by addition of a soluble recombinant SNARE, rVam7p. Upon rVam7p addition, the reaction rapidly loses sensitivity to anti-Vam3p antibody (Fig. 2B, •; half-time <2 min), and fusion events begin to occur within 1 min (12, 19). However, ALP is activated more slowly, with a half-time of 24 min (Fig. 2B, ○). Again, we reasoned that Prb1p overproduction should accelerate ALP activation, and thereby increase the apparent rate of fusion, if postfusion ALP activation is a slow step. Under priming bypass conditions (Fig. 2B), neither the acquisition of insensitivity to anti-Vam3p (Fig. 2B, ▴) nor the activation of ALP (Fig. 2B, ▵) was affected by Prb1p overexpression. Thus, postfusion ALP activation by Prb1p is fast relative to the transition from docking to fusion, and ALP activation is a faithful kinetic reporter of fusion events.
Assay of Single or Ongoing Rounds of Fusion. Microscopic observations of vacuole size during in vitro fusion indicate that very large vacuoles appear in the population (Fig. 3), indicating that many vacuoles fuse more than once. It would be useful to discriminate between processes required for a single round of fusion and for ongoing rounds of fusion. However, light-microscopic measurement of vacuole size distributions (Fig. 3C) will systematically underestimate the amount of fusion. First, vacuolar membrane is not conserved during fusion. In most fusion events, membrane from the docking junction is lost into the vacuole lumen (10); as a consequence, the product vacuole has less membrane surface than its precursors do. Second, many vacuoles present in the starting population are too small to accurately measure by light microscopy; these vacuoles can be observed by electron microscopy (A.J.M., L. Wang, L. Orci, and W.T.W., unpublished data). If small vacuoles fuse, their fusion products will repopulate the smallest size classes that can be measured by light microscopy. The formation of very large vacuoles provides strong evidence that many vacuoles undergo multiple rounds of vacuole fusion in the cell-free reaction. However, for the reasons just mentioned, it is difficult to quantify how many rounds of fusion have occurred in the population as a whole by using light-microscopic size measurements.
Fig. 3.

Formation of large vacuoles as a consequence of in vitro fusion. (A and B) Standard fusion reactions were initiated in the absence or presence of anti-Vam3p antibody, as indicated, incubated for 120 min at 27°C, stained with MDY-64 fluorophore, and mounted for microscopy. (C) Vacuole diameters were measured from micrographs, and surface areas were estimated and plotted as histograms.
One round of fusion has occurred when, on average, each vacuole in the starting population has fused once. Upon fusion with a protease-replete effector vacuole, the pro-ALP in a reporter vacuole should be completely activated. The consequences of this are diagrammed in Fig. 4A. If protease-replete effector vacuoles are in excess over pro-ALP-containing reporter vacuoles (Fig. 4A, effectors > reporters), the first fusion event that a reporter vacuole undergoes will likely result in ALP activation. Conversely, if ALP-containing reporter vacuoles are in excess over protease-replete effector vacuoles, a reporter vacuole may undergo multiple rounds of fusion with other reporter vacuoles before encountering a Prb1p-replete effector vacuole (Fig. 4A, effectors < reporters). By varying the proportions of effector and reporter vacuoles, we bias the assay toward detection of either the first round of fusion or ongoing rounds of fusion.
The effects of varying the proportions of the two vacuole populations are modeled in Fig. 4B (details are provided in Materials and Methods). In this simulation, fusion events are ongoing at a constant rate, with one round of fusion per 33 min of elapsed time. (Note that after one round of fusion, some vacuoles have not yet fused, whereas others have fused more than once.) When a reaction is initiated with protease-replete effector vacuoles in excess over pro-ALP-containing reporters, the fraction of protease-replete effectors remains high and relatively stable (Fig. 4B Left). Under these conditions (e.g., Fig. 4B Right, trace 0.87), only the first round of fusion contributes substantially to the ALP signal because the first time a reporter vacuole fuses, it will fuse with an effector. If the reaction is instead initiated with pro-ALP-containing reporter vacuoles in excess over protease-replete effectors, repeated rounds of fusion (and thus content mixing) cause the fraction of protease-replete effector vacuoles to increase over time (Fig. 4B Left). Under these conditions, early fusion events contribute to the ALP signal less than later fusion events (e.g., Fig. 4B Right, trace 0.07).
The kinetics of ALP maturation were measured in fusion reactions initiated with various fractions of effector and reporter vacuoles (Fig. 4C). As expected, faster ALP maturation was observed with increasing fractions of effector vacuoles, consistent with a first-round bias under these conditions. However, ALP maturation was even slower than expected in reactions initiated with low fractions of effector vacuoles (compare Fig. 4 C with B Right). The simplest explanation of this result is that vacuoles lose fusion competence over time, and indeed, a kinetic model that incorporates a rate constant for the loss of fusion competence (Fig. 4D) fits the experimental data reasonably well (Fig. 4E). We typically find that the decay rate constant (k2) is smaller than the forward rate of fusion (k1) but is still substantial. For the experiment shown in Fig. 4 C and D, decay was somewhat faster than usual, with k1 = 0.027 ± 0.007 min–1 and k2 = 0.025 ± 0.009 min–1, and 48% of the starting vacuole population was fusion active (see Materials and Methods). The fusion capacity of a given vacuole preparation is determined by the initial fraction of fusion-competent vacuoles and by k1 and k2, the rate constants for fusion and for decay of fusion competence. In this experiment, 0.7 rounds of fusion occurred in the whole vacuole population at 120 min; at this time, the reaction was essentially complete. The vacuoles that were actively fusing (the population that yields a signal in the ALP assay) underwent ≈1.5 rounds of fusion during the 120-min interval.
Discussion
The coupled cell-free assay of vacuole docking and fusion (2) offers significant experimental advantages, including ease of genetic manipulation, relatively high throughput, and large organelle size, which facilitates microscopic analyses. Although the vacuole fusion assay is most often used in an endpoint format, kinetic experiments have been used extensively in the functional characterization of docking and fusion factors. In this study, we characterized the kinetic response of the ALP assay and developed methods for discriminating between the first round of fusion and later fusion events.
The coupled assay of vacuole fusion-mediated content mixing relies on the proteolytic activation of vacuolar ALP (Pho8p). Protease inhibitor sensitivities and protease overproduction experiments indicate that Prb1p, vacuolar protease B, is rate limiting for ALP maturation in vitro, in both detergent lysates and intact vacuoles. However, at least some PEP4-dependent ALP maturation occurs in vivo in prb1 mutants. Together, these observations suggest that ALP can be activated through two pathways, both of which require protease A (Pep4p). A fast pathway, probably the dominant pathway in wild-type cells, requires Prb1p; a slower pathway is Prb1p independent. Overproduction of Prb1p, but not Pep4p, accelerates ALP activation in vacuole detergent lysates, confirming that Prb1p is rate limiting for ALP maturation. In contrast to these results, Prb1p overproduction has no detectable effect on fusion-dependent ALP maturation. This strongly suggests that ALP maturation in the vacuole lumen, upon content mixing between effector and reporter vacuoles, is fast compared to the interval between docking and fusion. In accord with this conclusion, mature ALP appears in vivo with a half-time of 6 min (3). This time includes transport through the endoplasmic reticulum and Golgi complex to the vacuole, so the proteolytic maturation of pro-ALP takes <6 min. The coupled colorimetric assay is therefore a useful reporter of fusion kinetics. There is a substantial lag between the end of sensitivity to inhibitors of Ypt7p and SNARE proteins and fusion. The reasons for this delay are unclear, but as discussed below, similar delays are seen in other in vitro fusion systems, including systems that do not rely on a coupled assay.
Our experiments provide specific methods for evaluating reagents, especially small-molecule inhibitors, that modulate the amount of fusion signal observed. ALP processing in detergent extracts can be used to test whether inhibitors slow or prevent ALP maturation. For example, the protein phosphatase 1 inhibitor microcystin-LR is a putative inhibitor of vacuole fusion (20, 21). Microcystin-LR inhibition of fusion-dependant ALP activation has a kinetic profile consistent with inhibition of the fusion (as opposed to priming or docking) subreaction. However, microcystin-LR also prevents ALP maturation in dilute detergent lysates and has no apparent effect on fusion-mediated vacuole size increase or content mixing assayed with fluorescent probes (unpublished results). Results obtained with microcystin-LR must therefore be interpreted with caution. Other experiments with inhibitors and glc7 mutants have provided independent evidence of a role for protein phosphatase 1 in vacuole fusion (21).
The kinetics of fusion in the vacuole system are comparable to the kinetics in other in vitro fusion systems. Fusion of predocked and primed secretory granules in cracked PC12 cell preparations, for example, takes several minutes (22). In our system, the first round of fusion occurs with a half-time of <20 min, including priming and docking (Fig. 4), and many vacuoles undergo multiple fusion events (Fig. 3). The mechanisms that cause the loss of fusion competence over time (Fig. 4 C–E) are unclear. We have begun to screen fusion promoting and inhibiting reagents for differential effects on early and late rounds of fusion. The soluble SNARE Vam7p both accelerates the forward rate of fusion and reduces the rate of inactivation. In contrast, when GTP is not added to the in vitro reaction, GTP hydrolysis by the Rab protein Ypt7p appears to increase the rate at which fusion competence is lost (unpublished data). We inferred values for k2 by curve fitting in this study, but it is also possible to directly determine the rate at which fusion competence is lost. Fusion reactions containing only target or effector vacuoles are first incubated in separate tubes; at intervals the effector and reporter reactions are mixed together, and the remaining fusion capacity is measured. This approach gives decay rates (e.g., k2 = 0.02 for figure 8 in ref. 5) similar to the values inferred from fusion kinetics in the present study.
The events responsible for the observed lag between the end of sensitivity to Rab- and SNARE-targeted inhibitors, and fusion (e.g., Fig. 2), are not well understood. Several factors have been suggested to participate in late stages of vacuole fusion, including protein phosphatase 1 (20), actin (23), subunits of the V0 complex of the vacuolar proton ATPase (24), and the armadillo-repeat protein Vac8p (25, 26). We find that protein and peptide ligands of phosphoinositides inhibit fusion-dependent ALP maturation with postdocking kinetics, inhibit vacuole size increase, and do not inhibit ALP maturation in dilute detergent lysates (R. Fratti, A.J.M., N. Thorngren, and W.T.W., unpublished results). Thus, phosphoinositides are also implicated in postdocking steps of fusion, as proposed previously (27).
Acknowledgments
We thank C. Barlowe and H. Higgs for essential discussions and criticism throughout these studies. This work was supported by the National Institute of General Medical Sciences and fellowships from the National Institutes of Health and the Damon Runyon Cancer Research Foundation (to A.J.M.).
Abbreviation: ALP, alkaline phosphatase.
References
- 1.Cook, N. R. & Davidson, H. W. (2001) Traffic 2, 19–25. [DOI] [PubMed] [Google Scholar]
- 2.Haas, A. (1995) Methods Cell Sci. 17, 283–294. [Google Scholar]
- 3.Klionsky, D. J. & Emr, S. D. (1989) EMBO J. 8, 2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones, E. W., Zubenko, G. S. & Parker, R. R. (1982) Genetics 102, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayer, A., Wickner, W. & Haas, A. (1996) Cell 85, 83–94. [DOI] [PubMed] [Google Scholar]
- 6.Ungermann, C., Nichols, B. J., Pelham, H. R. & Wickner, W. (1998) J. Cell Biol. 140, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eitzen, G., Will, E., Gallwitz, D., Haas, A. & Wickner, W. (2000) EMBO J. 19, 6713–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haas, A., Scheglmann, D., Lazar, T., Gallwitz, D. & Wickner, W. (1995) EMBO J. 14, 5258–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayer, A. & Wickner, W. (1997) J. Cell Biol. 136, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, L., Seeley, E. S., Wickner, W. & Merz, A. J. (2002) Cell 108, 357–369. [DOI] [PubMed] [Google Scholar]
- 11.Wang, L., Merz, A. J., Collins, K. M. & Wickner, W. T. (2003) J. Cell Biol. 160, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merz, A. J. & Wickner, W. T. (2004) J. Cell Biol. 164, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters, C. & Mayer, A. (1998) Nature 396, 575–580. [DOI] [PubMed] [Google Scholar]
- 14.Jones, E. W. (2002) Methods Enzymol. 351, 127–150. [DOI] [PubMed] [Google Scholar]
- 15.Haas, A., Conradt, B. & Wickner, W. (1994) J. Cell Biol. 126, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brachmann, C. B., Davies, A., Cost, G. J., Caputo, E., Li, J., Hieter, P. & Boeke, J. D. (1998) Yeast 14, 115–132. [DOI] [PubMed] [Google Scholar]
- 17.Gari, E., Piedrafita, L., Aldea, M. & Herrero, E. (1997) Yeast 13, 837–848. [DOI] [PubMed] [Google Scholar]
- 18.Moehle, C. M., Dixon, C. K. & Jones, E. W. (1989) J. Cell Biol. 108, 309–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thorngren, N., Collins, K. M., Fratti, R. A., Wickner, W. & Merz, A. J. (2004) EMBO J., in press. [DOI] [PMC free article] [PubMed]
- 20.Peters, C., Andrews, P. D., Stark, M. J., Cesaro-Tadic, S., Glatz, A., Podtelejnikov, A., Mann, M. & Mayer, A. (1999) Science 285, 1084–1087. [DOI] [PubMed] [Google Scholar]
- 21.Conradt, B., Shaw, J., Vida, T., Emr, S. & Wickner, W. (1992) J. Cell Biol. 119, 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang, X., Kim-Miller, M. J., Fukuda, M., Kowalchyk, J. A. & Martin, T. F. (2002) Neuron 34, 599–611. [DOI] [PubMed] [Google Scholar]
- 23.Eitzen, G., Wang, L., Thorngren, N. & Wickner, W. (2002) J. Cell Biol. 158, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayer, M. J., Reese, C., Buhler, S., Peters, C. & Mayer, A. (2003) J. Cell Biol. 162, 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, Y. X., Kauffman, E. J., Duex, J. E. & Weisman, L. S. (2001) J. Biol. Chem. 276, 35133–35140. [DOI] [PubMed] [Google Scholar]
- 26.Veit, M., Laage, R., Dietrich, L., Wang, L. & Ungermann, C. (2001) EMBO J. 20, 3145–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayer, A., Scheglmann, D., Dove, S., Glatz, A., Wickner, W. & Haas, A. (2000) Mol. Biol. Cell 11, 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]