

ABSTRACT
Although Nef is the viral gene product used by most simian immunodeficiency viruses to overcome restriction by tetherin, this activity was acquired by the Vpu protein of HIV-1 group M due to the absence of sequences in human tetherin that confer susceptibility to Nef. Thus, it is widely accepted that HIV-1 group M uses Vpu instead of Nef to counteract tetherin. Challenging this paradigm, we identified Nef alleles of HIV-1 group M isolates with significant activity against human tetherin. These Nef proteins promoted virus release and tetherin downmodulation from the cell surface and, in the context of vpu-deleted HIV-1 recombinants, enhanced virus replication and resistance to antibody-dependent cell-mediated cytotoxicity (ADCC). Further analysis revealed that the Vpu proteins from several of these viruses lack antitetherin activity, suggesting that under certain circumstances, HIV-1 group M Nef may acquire the ability to counteract tetherin to compensate for the loss of this function by Vpu. These observations illustrate the remarkable plasticity of HIV-1 in overcoming restriction by tetherin and challenge the prevailing view that all HIV-1 group M isolates use Vpu to counteract tetherin.
IMPORTANCE Most viruses of HIV-1 group M, the main group of HIV-1 responsible for the global AIDS pandemic, use their Vpu proteins to overcome restriction by tetherin (BST-2 or CD317), which is a transmembrane protein that inhibits virus release from infected cells. Here we show that the Nef proteins of certain HIV-1 group M isolates can acquire the ability to counteract tetherin. These results challenge the current paradigm that HIV-1 group M exclusively uses Vpu to counteract tetherin and underscore the importance of tetherin antagonism for efficient viral replication.
INTRODUCTION
Tetherin, also known as BST-2 or CD317, is an interferon-inducible transmembrane protein that impedes the detachment of enveloped viruses from infected cells (1–3). Although initially identified as the cellular factor responsible for a late-stage defect in the release of vpu-deleted human immunodeficiency virus type 1 (HIV-1) from restrictive cells (4, 5), tetherin is now understood to have antiviral activity against a broad spectrum of enveloped viruses (6–10). This antiviral activity is a function of its unusual topology, which enables opposite ends of the protein to simultaneously embed in viral and cellular membranes and to thereby tether budding virions to the cell surface (11). In addition to directly inhibiting virus replication, tetherin also serves as a sensor of viral infection and a link between innate and adaptive immunity. Virion-induced clustering of tetherin on the surface of infected cells triggers NF-κB activation and the release of proinflammatory cytokines (12, 13), and the accumulation of captured virions on the plasma membrane increases the sensitivity of infected cells to elimination by antibody-dependent cell-mediated cytotoxicity (ADCC) (14–17). Thus, the impact of tetherin on limiting virus replication in vivo may be amplified by coordinated innate and adaptive immune responses.
Tetherin has had a profound effect on the course of lentiviral evolution in primates. Over 40 species of African apes and Old World monkeys are naturally infected with simian immunodeficiency viruses (SIVs) (18), and viruses of apes (chimpanzee SIV [SIVcpz] and gorilla SIV [SIVgor]) and sooty mangabeys (SIVsmm) have been transmitted to humans on multiple occasions, resulting in at least four phylogenetically distinct groups of HIV-1 (groups M, N, O, and P) and eight groups of HIV-2 (groups A to H). Whereas HIV-1 group M, the main HIV-1 group responsible for the global AIDS pandemic, and HIV-1 group N, which has so far been found in fewer than 20 individuals (19), are the result of the cross-species transmission of SIVcpz from chimpanzees to humans (20), HIV-1 groups O and P appear to have originated from gorillas as a result of the zoonotic transmission of SIVgor (21). Although most SIVs, including SIVcpz and SIVgor, use Nef to counteract the tetherin proteins of their hosts, HIV-1 group M evolved to use Vpu as a tetherin antagonist because of an ancestral deletion of 5 amino acids from the N terminus of human tetherin required for susceptibility to Nef (22–24). The absence of sequences in human tetherin that confer susceptibility to Nef, together with the lack of a vpu gene, also explains why this activity was acquired by the envelope glycoproteins of certain HIV-2 isolates (24, 25).
HIV-1 group O appears to have taken yet another path to overcome restriction by tetherin. In contrast to the potent antitetherin activity of most HIV-1 group M Vpu proteins, this function was not acquired by the Vpu proteins of HIV-1 group O isolates (22); however, the Nef proteins of these viruses were found to have significant activity against human tetherin (26). Although tetherin antagonism by HIV-1 group O Nef proteins is less efficient than typically observed for most SIV Nefs (26), this activity may have contributed to the spread of this virus in central Africa, where it is estimated to have infected approximately 100,000 people (27). These observations provide an explanation for how HIV-1 group O counteracts tetherin and reveal that it is possible for Nef to serve this function in humans.
Here we show that the Nef proteins of certain HIV-1 group M isolates can also acquire the ability to counteract human tetherin. HIV-1 group M Nefs that downmodulate human tetherin on the cell surface, enhance virus replication in cells expressing tetherin, and provide resistance to ADCC were identified. Several of these HIV-1 Nef alleles were associated with Vpu alleles that lack antitetherin activity, suggesting that under certain circumstances, Nef may acquire the ability to counteract human tetherin to compensate for the loss of this function by Vpu. These findings reveal a previously unappreciated role for the Nef proteins of HIV-1 group M in tetherin antagonism and challenge the prevailing view that these viruses exclusively use Vpu to overcome restriction by tetherin.
MATERIALS AND METHODS
Cell lines.
HEK293T cells and Jurkat cells were obtained from the ATCC and cultured under standard conditions. Natural killer (NK) effector cells and CEM.NKR-CCR5 target cells were maintained as described previously (28). Jurkat-TAg parental (JTAg), Jurkat-TAg L-tetherin, and Jurkat-TAg S-tetherin cells were kindly provided by Stuart J. Neil, King's College London, and maintained in RPMI medium supplemented with 10% fetal calf serum and gentamicin, as previously described (29).
Sequence analyses.
Nef and Vpu amino acid sequences were retrieved from the Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov/content/index). Nef sequences were aligned to the NL4-3 Nef sequence. Sequences at the 5′ and 3′ ends and insertions relative to the NL4-3 reference sequence were removed by using the bioinformatics software platform Geneious version 9.0.2 (30). This normalized the amino acid positions to the NL4-3 reference sequence. Sequence data were then analyzed by writing analysis parameters in the programming language Python version 2.7. Specifically, the modified sequences described above were queried for any combination of the following amino acids at the specified position: T4, M5, E28, A33, Y40, L90, and Y103. The script then output fasta files containing nucleotide sequences for Nef and a matching Vpu sequence, if it existed, for each of the amino acid signatures identified (e.g., a fasta file containing the Nef sequences that have T4 and E28 would be one output, while a second file would be generated for sequences containing T4, E28, and Y40).
Plasmid DNA constructs.
Selected HIV-1 group M nef and vpu alleles (Tables 1 and 2) were cloned into a bicistronic expression vector (pCGCG) by standard cloning techniques, as previously described (24, 31). Selected nef alleles were also cloned into a vpu-deleted HIV-1 NL4-3 proviral vector designed to coexpress Nef and enhanced green fluorescent protein (eGFP) from a downstream internal ribosomal entry site (IRES) (pBR-NL43-IRES-eGFP-Nef, obtained through the NIH AIDS Reagent Program [ARP] from Frank Kirchhoff), as described previously (32, 33). The vpu-deleted variant of pBR-NL43-IRES-eGFP-Nef (HIV-1.Δvpu.IeG) was generated by introducing multiple stop codons after the fifth codon of vpu as described previously (34). HIV-1NL4-3 (pNL4-3) was obtained through the AIDS Reagent Program from Malcolm Martin (35). The vpu-deleted variant of HIV-1NL4-3 (pNL4-3.Δvpu) was generated by deleting nucleotide 6076, resulting in multiple stop codons after the fifth codon of vpu (34). All plasmid DNA expression constructs were sequence confirmed.
TABLE 1.
HIV-1 isolates and GenBank accession numbers for Nef sequences
| Nef allele | Subtype | GenBank accession no. | Isolate or clone | Country | Yr of isolation |
|---|---|---|---|---|---|
| N1 | C | DQ093607 | 03ZASK213B1 | South Africa | 2003 |
| N2 | C | AF286223 | 94IN476 | India | 1994 |
| N3 | C | DQ396366 | 04ZASK052MB2 | South Africa | 2004 |
| N4 | CRF02_AG | AB286863 | 03GH197AG | Ghana | 2003 |
| N5 | C | AY727526 | 04BR137 | Brazil | 2004 |
| N6 | C | AB485644 | pPRD320-07C28 | Djibouti | 2009 |
| N7 | B | HM369928 | 08YJN3-5037 | South Korea | 2008 |
| N8 | C | DQ056411 | 03ZASK110B1 | South Africa | 2003 |
| N9 | C | AF286233 | 98IS002 | Israel | 1998 |
| N10 | C | AY713415 | 89SM_145 | Somalia | 1989 |
| N11 | C | AB485643 | pPRD320-07C10 | Djibouti | 2009 |
| N12 | C | AF286224 | 96ZM651 | Zambia | 1996 |
| N13 | C | AF286234 | 98TZ013 | Tanzania | 1998 |
| N14 | C | AY253322 | 01TZBD9_11 | Tanzania | 2001 |
| N15 | C | AY736823 | 02ET_14 | Ethiopia | 2002 |
| C1 | C | U46016 | ETH2220 | Ethiopia | 1996 |
| A1 | A | M62320 | U455_U455A | Uganda | 1985 |
| AM | A | AM000053 | 97CD.KCC2 | Democratic Republic of Congo | 1997 |
| EF | B | EF514706 | CTL 018 | Denmark | 2007 |
TABLE 2.
HIV-1 isolates and GenBank accession numbers for Vpu sequences
| Vpu allele | Subtype | GenBank accession no. | Isolate or clone | Country | Yr of isolation |
|---|---|---|---|---|---|
| N1 | C | DQ093607 | 03ZASK213B1 | South Africa | 2003 |
| N2 | C | AF286223 | 94IN476 | India | 1994 |
| N6 | C | AB485644 | pPRD320-07C28 | Djibouti | 2009 |
| N8 | C | DQ056411 | 03ZASK110B1 | South Africa | 2003 |
| N11 | C | AB485643 | pPRD320-07C10 | Djibouti | 2009 |
| N12 | C | AF286224 | 96ZM651 | Zambia | 1996 |
| C1 | C | U46016 | ETH2220 | Ethiopia | 1996 |
| AM | A | AM000053 | 97CD.KCC2 | Democratic Republic of Congo | 1997 |
Virus release assay.
To assess the ability of HIV-1 Nef and Vpu alleles to rescue virus release in the presence of human tetherin, HEK293T cells were cotransfected with (i) HIV-1 NL4-3 Δvpu Δnef proviral DNA (100 ng); (ii) pcDNA3-based expression constructs for human tetherin (pcDNA3-hBST-2) or an empty vector (pcDNA3); and (iii) pCGCG-based expression constructs for Nef or Vpu or the empty vector (pCGCG). Transfections were performed by using GenJet Lipid verII transfection reagents (SignaGen Laboratories, Gaithersburg, MD) in duplicate in 6-well plates seeded the day before at 1.2 × 106 cells per well. At 48 h posttransfection, the amount of virus released into the cell culture supernatant was measured by an HIV p24 antigen capture enzyme-linked immunosorbent assay (ELISA) (Advanced Bioscience Laboratories, Inc., Kensington, MD). The percentage of maximal virus release was calculated by dividing the mean p24 release in the presence of tetherin by the mean virus release in the absence tetherin and multiplying this value by 100, as previously described (24, 31).
ADCC assay.
ADCC was measured as previously described (28, 36). An NK cell line transduced with a retroviral vector to stably express human CD16 (FCGR3A) served as effector cells (36). Target cells (CEM.NKR-CCR5-sLTR-Luc) were derived from CEM.NKR-CCR5 CD4+ T cells obtained through the ARP (Division of AIDS, NIAID, NIH) from Alexandra Trkola (37) and modified to express firefly luciferase upon HIV-1 infection (36). Target cells were infected with HIV-1 by spinoculation. At 4 days postinfection, NK cells and HIV-1-infected target cells were incubated for 8 h at an effector-to-target cell (E:T) ratio of 10:1 in triplicate wells at each antibody concentration. Background and maximal luciferase activities were determined, respectively, from six wells containing uninfected target cells and six wells containing HIV-infected target cells plus NK cells but without antibody. ADCC activity (percent relative light units [RLU]) was calculated as (mean RLU at a given antibody concentration − mean background RLU)/(mean maximal RLU − mean background RLU) × 100.
Virus stock production and T-cell line infections.
Viral stocks of HIV-1 NL4-3 wild-type, HIV-1 NL4-3 ΔVpu, and selected HIV-1.Δvpu.IeG-Nef constructs were generated in HEK293T cells by transient transfection as described previously (38). Briefly, plasmids were transfected into HEK293T cells, and culture supernatants were harvested at 48 and 72 h posttransfection. Subsequently, virus concentrations were determined by an anti-p24 ELISA (Advanced Bioscience Laboratories, Inc., Kensington, MD). Virus release assays with HIV target cells were performed as described previously (12), with minor modifications. HIV-1 envelope surface staining to confirm infection of target cells was performed by using an established protocol (14).
Immunoblotting.
Cell lysates were prepared in ice-cold radioimmunoprecipitation assay (RIPA) buffer (Pierce Biotechnology) containing EDTA and a protease inhibitor cocktail (Pierce Biotechnology), cleared by centrifugation at 2,500 × g at 4°C for 5 min, and suspended in 2× Laemmli buffer (Sigma-Aldrich). Proteins were subsequently separated on either 12% polyacrylamide gels or Mini-Protean TGX Any kd gradient gels (Bio-Rad), transferred onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare), blocked with phosphate-buffered saline (PBS) plus 2% bovine serum albumin (BSA), and probed with commercially available monoclonal antibodies to Nef (clone 2H12), β-actin (clone ACTN05), and HIV-1 p55 (clone HIV.OT11) or a rabbit polyclonal antibody to Vpu obtained through the ARP from Klaus Strebel (39). Blots were then washed with PBS plus 0.05% Tween 20 and probed with a horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (Pierce) or an HRP-conjugated goat anti-rabbit antibody (Bio-Rad). For the analysis of virion release, culture supernatants were filtered and prepared for SDS-PAGE as described above. PVDF membranes were subsequently probed with a monoclonal antibody to HIV-1 p24 CA (clone 183-H12). Immunoblots were developed with enhanced chemiluminescence (GE Healthcare) and imaged by using a Fujifilm LAS-4000 Image Reader (Fujifilm) (31).
Coimmunoprecipitation.
HEK293T cells were seeded into 6-well plates (1.2 × 106 cells/well) and cotransfected the following day with the pCGCG-C1-Nef or pCGCG vector (2 μg) and the pcDNA3-hBST-2 or pcDNA3 vector (2 μg) by using the GenJet transfection reagent (SignaGen). Forty-eight hours later, the cells were washed in ice-cold PBS and lysed in 300 μl lysis buffer (Thermo Scientific) containing protease and phosphatase inhibitors (Sigma and Roche). A portion of the cell lysate (50 μl) was set aside to confirm Nef and tetherin expression by Western blot analysis, and the remaining sample (250 μl) was incubated with 25 μl magnetic protein G beads (New England BioLabs) at 4°C on a rotating platform for 1 h. After magnetic separation, the beads were discarded, and the cell lysates were incubated with 1 μg of IgG2a monoclonal antibody to BST-2 (Sigma) for 1 h at 4°C with rotation. Fresh protein G magnetic beads (25 μl) were then added, and incubation was continued overnight at 4°C with continuous rotation. The following day, the beads were washed five times, suspended in 35 μl of 2× SDS sample buffer (Sigma), and denatured at 95°C for 5 min. After a magnet was applied to remove the beads, samples were separated by SDS-PAGE using 8% polyacrylamide gels, transferred onto a PVDF membrane, probed with an anti-Myc antibody (Abcam), and developed with an HRP-conjugated anti-mouse IgG1 secondary antibody (Abcam) to minimize cross-reactivity with the anti-BST-2 IgG2a antibody used for immunoprecipitation. The blots were visualized by using a Li-Cor Odyssey Fc imaging system.
Flow cytometry.
Cells were stained at room temperature in PBS plus 2% fetal bovine serum (FBS) and 1% NaN3 with fluorochrome-conjugated antibodies specific for tetherin (allophycocyanin [APC]; clone RS38E), CD4 (Alexa Fluor 700; clone RPA-T4), and CD45 (peridinin chlorophyll protein [PerCP]; clone 2D1). For Env staining, an indirect method was used: cells were first incubated with HIV immunoglobulin (HIVIG) obtained through the ARP from Luba Vujcic (40) or purified human IgG from HIV-negative donors (Invitrogen), followed by incubation with a fluorochrome-conjugated isotype-specific mouse anti-human IgG antibody (phycoerythrin [PE]-Cy7; clone G18-145). For intracellular staining of Gag, cells were fixed and permeabilized with the Cytofix/Cytoperm kit (BD Biosciences), followed by staining with monoclonal antibody KC57 (PE; clone FH190-1-1) (41). Samples were then washed, fixed in 2% paraformaldehyde, and analyzed by using an LSR-II flow cytometer (Becton, Dickinson). Dead cells were excluded by using the Live/Dead fixable dead cell aqua stain (Invitrogen). After gating on viable, CD45+ cells, the surface expression of Env, tetherin, and CD4 was analyzed by using FlowJo 9.6.4 software (TreeStar).
Statistical methods.
Differences in mean virus release in the presence of all nef alleles were calculated by one-way analysis of variance (ANOVA), adjusting significance levels for multiple comparisons by using a Holm-Sidak test. Differences in the surface expression levels of tetherin and Env were calculated by one-way ANOVA with adjustment for multiple comparisons. Correlations between tetherin and Env staining on the surface of infected cells and susceptibility to ADCC were analyzed by using the Pearson correlation test. Kruskal-Wallis tests were used to compare median differences in ADCC activity (percent RLU), and one-way ANOVA was used to compare mean differences in area under the concentration-time curve (AUC) values. Differences in mean virus release between infected JTAg L-tetherin and JTAg S-tetherin cells were calculated with unpaired t tests. Differences in mean virus release for the vpu alleles were calculated by a Kruskal-Wallis test with adjustment for multiple comparisons using Dunn's multiple-comparison test.
RESULTS
Antagonism of human tetherin by HIV-1 group M Nefs.
We previously noticed that HIV-1 NA7 Nef was unusually effective at counteracting sooty mangabey tetherin and was also able to partially rescue virus release in the presence of human tetherin (24). By testing recombinants between HIV-1 NA7 and NL4-3 Nefs, we identified a tyrosine-versus-histidine polymorphism at position 40 (Y40H) that appeared to be important for this activity (data not shown). To determine if other HIV-1 Nef alleles with this polymorphism were able to counteract human tetherin, cDNA sequences for additional nef alleles encoding Y40 were synthesized and tested for the ability to rescue virus release for HIV-1 Δvpu Δnef in the presence of human tetherin. Two Nef alleles with antitetherin activity were identified (A1 and C1 Nefs), one of which (C1 Nef) rescued virus release to an extent comparable to that of NL4-3 Vpu (Fig. 1A and B). In contrast, the corresponding Vpu proteins expressed by these viruses were ineffective tetherin antagonists (Fig. 1A and B). Consistent with a role for the Y40 polymorphism in tetherin antagonism, replacement of this residue with the corresponding histidine residue of NL4-3 Nef (Y40H) eliminated the antitetherin activity of A1 and C1 Nefs (Fig. 1A); however, the introduction of Y40 into NL4-3 Nef (H40Y) was not sufficient to confer activity against human tetherin (Fig. 1C), suggesting that other amino acid substitutions are also required.
FIG 1.

Identification of residues in an HIV-1 group M Nef protein required for tetherin antagonism. (A to F) The Vpu and Nef proteins expressed by two primary HIV-1 isolates (A and B), NL4-3 Nef recombinants containing the indicated domains of C1 Nef (C and D), and C1 Nef mutants containing the indicated amino acid residues of NL4-3 Nef (E and F) were tested for their ability to rescue virus release in HEK293T cells cotransfected with HIV-1 NL4-3 Δvpu Δnef proviral DNA, a vector expressing human tetherin, and Vpu or Nef expression constructs. As controls, additional transfections were performed with constructs expressing NL4-3 Vpu, NL4-3 Nef, and an empty vector (pCGCG). At 48 h posttransfection, the accumulation of HIV-1 p24 in the cell culture supernatant was measured by an antigen capture ELISA (A, C, and E), and differences in the amounts of p24 in supernatants versus p55 Gag and Nef or Vpu in cell lysates were compared by Western blot analysis (B, D, and F). The error bars represent the standard deviations of the means for replicate assays. Differences in mean virus release compared to vector controls (A and C) or NL4-3 Vpu (E) were estimated by ordinary one-way ANOVA with adjustment for multiple comparisons (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001). (G) Amino acid sequence alignment of the HIV-1 NL4-3 and C1 Nef proteins. Dots indicate amino acid identity, dashes indicate gaps, and residues involved in tetherin antagonism are shaded. Color-coded sequences correspond to the N-terminal (yellow), globular core (blue), flexible loop (orange), and C-terminal (green) domains.
To identify the residues of C1 Nef that contribute to tetherin antagonism, recombinants between C1 Nef and NL4-3 Nef H40Y were constructed and tested in virus release assays. Domain exchange mutants were first tested to determine which regions of C1 Nef are required for this activity. Whereas none of the individual domains of C1 Nef were sufficient for tetherin antagonism, a chimera containing the N-terminal and globular core domains of C1 Nef rescued virus release as efficiently as wild-type C1 Nef (Fig. 1C and D). Similar activity was observed for a chimera containing the N-terminal, globular core, and flexible loop domains of C1 Nef (Fig. 1C and D). Thus, amino acid differences in the N-terminal and globular core domains are sufficient to account for this activity. To identify the specific residues of C1 Nef required for activity against human tetherin, C1 Nef mutants in which individual amino acids in these regions were replaced with the corresponding residues of NL4-3 Nef were also tested. This analysis revealed that substitutions at positions 4, 5, 28, 33, 40, 90, 103, and 109 resulted in a significant loss of antitetherin activity for C1 Nef (Fig. 1E to G).
Identification of other HIV-1 group M Nefs with antitetherin activity.
To identify other HIV-1 group M Nefs with antitetherin activity, the HIV-1 Sequence Database (http://www.hiv.lanl.gov/content/index) was searched for Nef alleles matching C1 Nef at one or more of the residues highlighted in Fig. 1G. Although this analysis did not reveal any other Nef alleles that were a perfect match at all eight positions, 14,157 sequences shared at least one of these residues with C1 Nef. To narrow our search to the most promising candidates, we focused on Nef alleles that matched C1 Nef at four or more positions, which were found in sequences from a total of 705 primary isolates. Of these, cDNA sequences were synthesized for 20 nef alleles encoding different combinations of four to six C1 Nef residues and screened for tetherin antagonism in virus release assays. The 15 Nef alleles with the highest antitetherin activity were further characterized for their ability to rescue virus release and downmodulate tetherin from the surface of transfected cells (Fig. 2). As a reflection of the derivation of the query sequences from subtype C Nef, most of these nef alleles belong to subtype C viruses (Table 1).
FIG 2.

HIV-1 group M Nef antagonism of human tetherin. The indicated HIV-1 group M Nef proteins were tested for their ability to rescue virus release for HIV-1 NL4-3 Δvpu Δnef in HEK293T cells expressing human tetherin, as described in the legend of Fig. 1. (A and B) The accumulation of HIV-1 p24 in the cell culture supernatant was measured by an antigen capture ELISA (A), and the amounts of p24 in the culture supernatants versus Nef and p55 Gag in cell lysates were compared by Western blot analysis (B). (C) Tetherin downmodulation was assessed by transfecting a HEK293T cell line that stably expresses human tetherin (293T-hBST-2 cells) with bicistronic pCGCG constructs that coexpress Nef and eGFP. Histograms show tetherin (BST-2) staining on the surface of viable, eGFP-positive cells relative to cells transfected with the empty pCGCG vector (dark blue) or stained with an isotype control antibody (shaded). (D) Differences in the surface expression levels of tetherin are plotted as a percentage of tetherin staining on 293T-hBST-2 cells transfected with an empty vector. The error bars represent the standard deviations of the means for replicate assays, and differences in mean virus release (A) and tetherin downmodulation (D) compared to empty vector controls were estimated by ordinary one-way ANOVA (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001). The data shown in panels A to D are representative of results from three independent experiments.
Although none of these Nef alleles counteracted restriction as efficiently as NL4-3 Vpu or C1 Nef, several exhibited partial activity against human tetherin. Compared to NL4-3 Nef and two other Nef controls that lack antitetherin activity (AM Nef and EF Nef), a significant increase in virus release was observed for N1, N2, N6, N8, N11, and N15 by p24 ELISAs (Fig. 2A). These results are supported by Western blot analyses showing similar qualitative differences in virus release (Fig. 2B). Moreover, each of the Nef alleles that significantly enhanced virus release also downmodulated tetherin from the surface of transfected cells (Fig. 2C and D). Hence, these results identified six additional HIV-1 group M Nef alleles with partial activity against human tetherin.
The short isoform of human tetherin is insensitive to antagonism by HIV-1 Nef.
As a result of alternative translational initiation, tetherin is expressed as two different isoforms that differ by 12 amino acids at the N terminus of the protein (42). Whereas the long isoform (L-tetherin) is sensitive to antagonism by HIV-1 group M Vpu and group O Nef proteins, the short isoform (S-tetherin) is resistant to these viral antagonists (26, 42). To determine if L- and S-tetherins differ in their susceptibility to HIV-1 group M Nefs, Jurkat-TAg cells expressing long and short isoforms of human tetherin (JTAg L-tetherin and JTAg S-tetherin cells, respectively) (29) were infected with vpu-deleted HIV-1 NL4-3-IRES-eGFP-Nef (HIV-1.Δvpu.IeG-Nef) recombinants engineered to express selected group M Nef alleles, and virus release and tetherin downmodulation were compared on day 3 postinfection. Whereas virus release did not differ for JTAg L- and S-tetherin cells infected with HIV-1.Δvpu.IeG expressing NL4-3 Nef, virus release was significantly higher in JTAg L-tetherin cells than in JTAg S-tetherin cells infected with recombinant viruses expressing C1, N1, N6, or N8 Nef (Fig. 3A). These results were corroborated by data from Western blot analyses comparing the accumulation of HIV-1 p24 capsid in cell culture supernatants to p55 Gag and Nef expression in cell lysates (Fig. 3B). The effects of these Nef proteins on virus release also corresponded to their effects on surface levels of tetherin, since C1, N1, N6, and N8 Nefs downmodulated L-tetherin, but not S-tetherin, on the surface of infected cells (Fig. 3C). These results reveal differential sensitivities of L- versus S-tetherin to HIV-1 group M Nefs, indicating that this activity depends on the first 12 amino acids of the long isoform of human tetherin.
FIG 3.
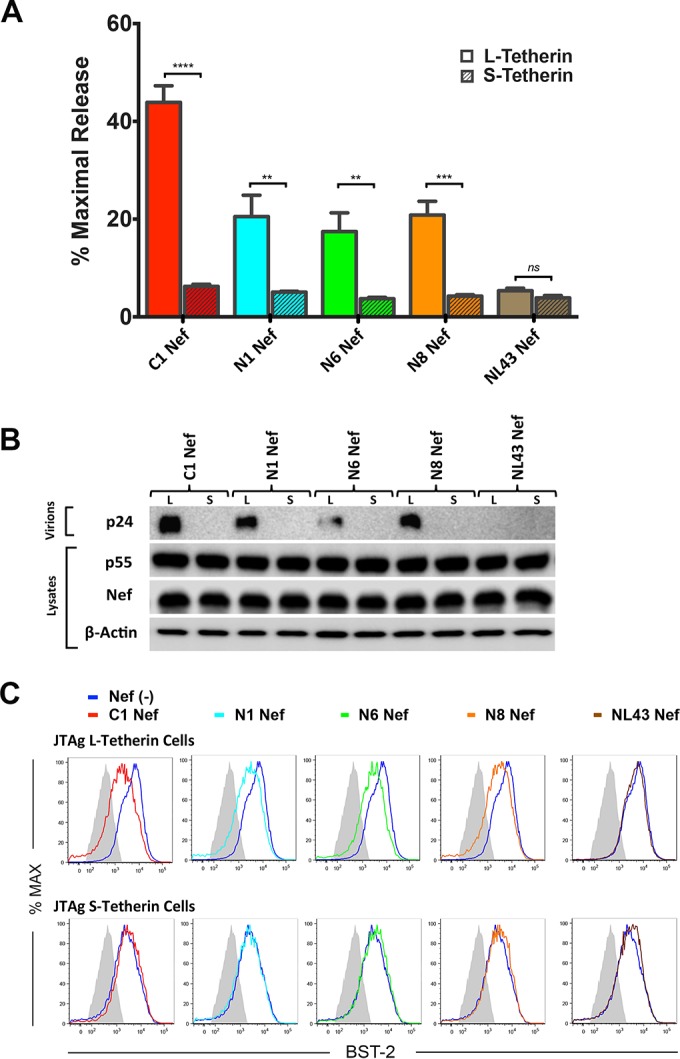
HIV-1 group M Nefs antagonize the long isoform, but not the short isoform, of human tetherin. (A and B) Jurkat-TAg cells expressing long versus short isoforms of human tetherin (JTAg L- versus S-tetherin cells) were infected with HIV-1.Δvpu.IeG-Nef recombinants expressing the indicated Nef alleles, and virus release was measured by a p24 antigen capture ELISA (A) and by Western blot analysis of culture supernatants and cell lysates (B). To maximize infection, cells were infected by spinoculation for 2 h with concentrated virus (2 μg/ml p24) in the presence of Polybrene (8 μg/ml). The error bars represent the standard deviations of the means for independent infections, and mean virus release was compared to that in JTAg L-tetherin cells infected with a virus expressing NL4-3 Nef by unpaired t tests (**, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001; ns, not significant). (C) Histograms show tetherin (BST-2) staining on the surface of cells infected with recombinant viruses expressing the indicated Nef alleles relative to cells infected with a nef-minus control virus or stained with an isotype control antibody (shaded). Virus-infected cells were identified by gating on viable, p55+ CD4low cells. The data in panels A to C are representative of results from three independent experiments.
HIV-1 C1 Nef binds to residues at the N terminus of human tetherin.
Consistent with a role for the first 12 amino acids of the long isoform of human tetherin in determining susceptibility to antagonism by HIV-1 group M Nefs, substitutions in this region disrupted binding to C1 Nef. As previously observed for SIV Nef and macaque tetherin (43), a physical interaction between C1 Nef and human tetherin was detectable by coimmunoprecipitation assays (Fig. 4A). This interaction was greatly diminished, however, by alanine substitutions at positions 5-6, 7-8, and 11-12 in L-tetherin (Fig. 4A). These residues, which include the dual-tyrosine motif (Y6Y8) required for tetherin endocytosis and signaling (44, 45), correspond to the same region previously shown to confer sensitivity to HIV-1 group O Nefs (26), suggesting that these HIV-1 group M Nef proteins counteract human tetherin by a similar mechanism.
FIG 4.
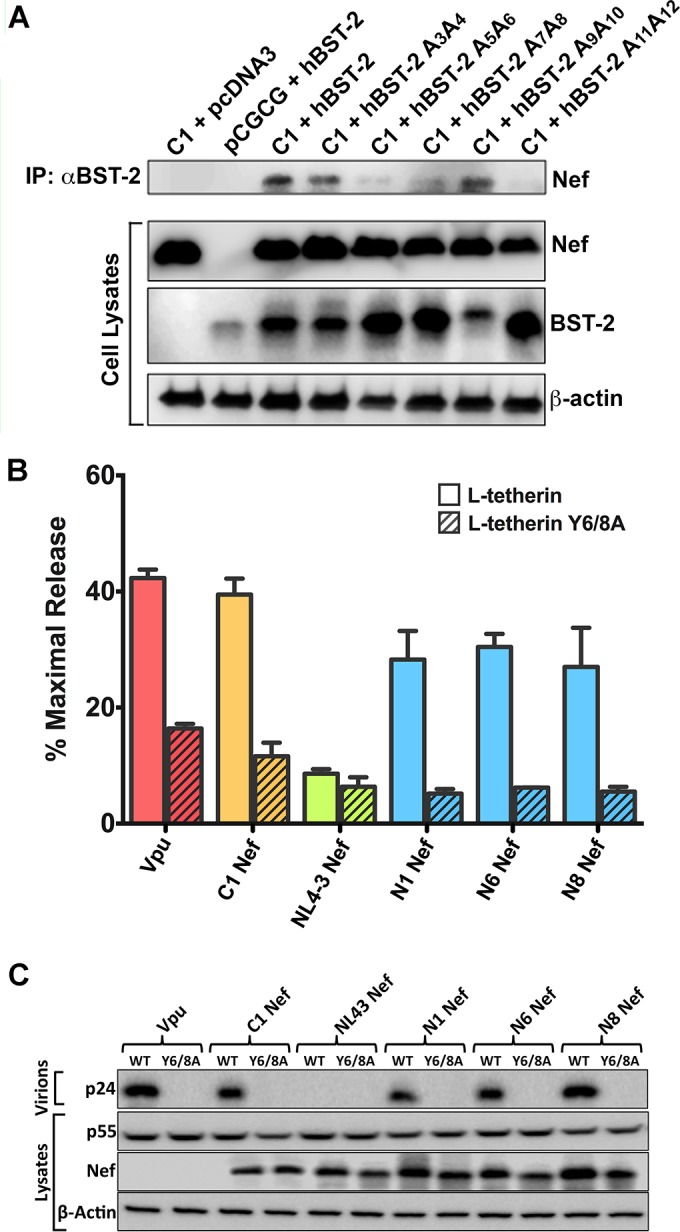
C1 Nef physically interacts with residues at the N terminus of human tetherin. (A) HEK293T cells were cotransfected with constructs expressing C1 Nef and either wild-type (WT) human tetherin (hBST-2) or tetherin mutants with alanine substitutions at the indicated positions in the N terminus of the protein. As controls, cells were also transfected with constructs expressing C1 Nef and an empty vector control for tetherin (pcDNA3) or human tetherin and an empty vector control for Nef (pCGCG). Whole-cell lysates and proteins immunoprecipitated (IP) by using an antibody to tetherin were separated by SDS-PAGE on 8% polyacrylamide gels and transferred onto PVDF membranes. Membranes were probed with antibodies to tetherin and to a Myc epitope tag appended to the C terminus of Nef. As a control for sample loading, the cell lysate membranes were also stripped and reprobed with an antibody to β-actin. (B and C) The indicted HIV-1 group M Nef proteins and HIV-1 NL4-3 Vpu were tested for their ability to rescue virus release for HIV-1 NL4-3 Δvpu Δnef in HEK293T cells cotransfected with constructs expressing L-tetherin or L-tetherin Y6/8A. The accumulation of HIV-1 p24 in the cell culture supernatant was measured by antigen capture ELISA (B), and the amounts of p24 in culture supernatants versus Nef and p55 Gag in cell lysates were compared by Western blot analysis (C).
To further corroborate HIV-1 Nef interactions with tetherin sequences spanning the dual-tyrosine motif, group M Nefs were tested for their ability to rescue virus release from cells expressing wild-type L-tetherin versus an L-tetherin mutant with tyrosine-to-alanine substitutions at positions 6 and 8 (Y6/8A L-tetherin). In accordance with their activity against L- versus S-tetherin (Fig. 3), C1, N1, N6, and N8 Nefs rescued virus release in the presence of wild-type L-tetherin but not Y6/8A L-tetherin, and NL4-3 Nef was unable to counteract restriction by either wild-type or Y6/8A L-tetherin (Fig. 4B and C). As recently reported, the Y6/8A substitutions also impaired tetherin antagonism by HIV-1 NL4-3 Vpu (Fig. 4B and C) (46, 47). These results are therefore consistent with a role for sequences at the N terminus of L-tetherin spanning the dual-tyrosine motif in interactions with group M Nef proteins that have acquired activity against human tetherin.
HIV-1 group M Nefs with antitetherin activity enhance virus replication in cells expressing human tetherin.
We next sought to determine the effects of tetherin antagonism by group M Nefs on virus replication. Parental Jurkat-TAg cells and JTAg L- and S-tetherin cells were infected with HIV-1.Δvpu.IeG-Nef recombinants expressing selected Nef proteins. These recombinants included viruses expressing the C1, N1, N6, and N8 Nef proteins with activity against human tetherin and viruses expressing the NL4-3, AM, and EF Nef proteins that lack detectable antitetherin activity. Whereas all of these viruses replicated with similar kinetics in parental Jurkat-TAg cells (Fig. 5A), virus replication in JTAg L-tetherin cells corresponded with the antitetherin activity of Nef (Fig. 5B). Virus replication was most efficient for HIV-1.Δvpu.IeG expressing C1 Nef, which counteracts human tetherin to a similar extent as NL4-3 Vpu, followed by recombinant viruses expressing N1, N6, and N8 Nefs with partial antitetherin activity, and least efficient for viruses expressing NL4-3, AM, and EF Nefs, which lack activity against human tetherin (Fig. 5B). In accordance with the inability of these Nef proteins to overcome restriction by the short isoform of tetherin, virus replication was strongly impaired for all of the recombinants in JTAg S-tetherin cells (Fig. 5C).
FIG 5.

Tetherin antagonism by Nef facilitates HIV-1 replication in cells expressing human tetherin. (A to E, G, and H) JTAg parental (A), JTAg L-tetherin (B), JTAg S-tetherin (C), and activated CD4+ T lymphocytes from two different donors (D, E, G, and H) were infected with HIV-1.Δvpu.IeG-Nef recombinants expressing the indicated Nef alleles, and virus replication was monitored by measuring the accumulation of p24 in cell culture supernatants by an antigen capture ELISA. For primary CD4+ T lymphocytes, the cultures were divided 48 h after infection and maintained in medium with (E and H) and without (D and G) 100 U/ml IFN-α. (F and I) Tetherin upregulation in response to IFN-α was verified by surface staining on day 3 postinfection. Histograms show the fluorescence intensity of staining with an antibody to tetherin (BST-2) compared to staining with an isotype control antibody (shaded). The data are representative of results from at least two independent experiments.
Similar differences in virus replication were observed under conditions of interferon-induced upregulation of tetherin in primary CD4+ lymphocytes. Activated CD4+ T cells from two different donors were infected with HIV-1.Δvpu.IeG-Nef recombinants, divided into separate cultures 2 days later, and maintained in medium with or without 100 U/ml alpha interferon (IFN-α). Tetherin upregulation was verified by surface staining, and virus replication was monitored by the accumulation of HIV-1 p24 in culture supernatants (Fig. 5D to I). The effects of each of the Nef alleles on virus replication varied between donors. Although C1 Nef facilitated virus replication in the presence or absence of IFN-α, expression of this Nef allele further enhanced virus replication relative to controls in IFN-α-treated cultures from donor 1 (Fig. 5D and E). The Nef alleles with intermediate antitetherin activity (N1, N6, and N8 Nefs) also enhanced virus replication in the presence, but not in the absence, of IFN-α in lymphocytes from this donor (Fig. 5D and E). These differences were not observed, however, in lymphocytes from donor 2. Although recombinant viruses expressing Nef alleles with antitetherin activity replicated better than viruses expressing inactive Nefs, they did not have an advantage in the presence of IFN-α (Fig. 5G and H). The reasons for these differences are presently unclear but may reflect donor-to-donor variation in basal levels of tetherin expression and/or responsiveness to interferon.
Tetherin antagonism by Nef protects HIV-infected cells from ADCC.
We and others recently reported that tetherin antagonism by Vpu protects HIV-1-infected cells from ADCC (14–17). To determine if HIV-1 Nefs with antitetherin activity can also protect virus-infected cells from ADCC, CEM.NKR-CCR5-sLTR-Luc cells were infected with HIV-1.Δvpu.IeG recombinants expressing group M Nef alleles and tested for sensitivity to NK cell-mediated lysis in the presence of purified immunoglobulin from HIV-1-positive donors (HIVIG). Consistent with previously reported results showing that tetherin antagonism by Vpu protects HIV-1-infected cells from ADCC (14), cells infected with wild-type HIV-1 NL4-3 were significantly more resistant to ADCC than cells infected with HIV-1 NL4-3.Δvpu (Fig. 6A). Among the recombinant viruses, C1 Nef, which consistently exhibited the highest antitetherin activity, afforded the greatest resistance to ADCC (Fig. 6A). Indeed, cells infected with HIV-1.Δvpu.IeG expressing C1 Nef were as resistant to ADCC as wild-type HIV-1-infected cells (Fig. 6A) and were also significantly more resistant to ADCC than cells infected with HIV-1 NL4-3.Δvpu. Accordingly, cells infected with viruses expressing Nef alleles with partial antitetherin activity (N1, N6, and N8 Nefs) were partially resistant to ADCC (Fig. 6A); however, these differences did not reach statistical significance. As expected, cells infected with viruses expressing Nef alleles that lack antitetherin activity (NL4-3, AM, and EF Nefs) were similar to HIV-1 NL4-3.Δvpu-infected cells in their susceptibility to ADCC (Fig. 6A). These differences in sensitivity to ADCC are not a result of differences in Nef expression (Fig. 6B) but instead reflect differences in the efficiency of tetherin downmodulation (Fig. 6C) and concomitant changes in Env exposure on the surface of virus-infected cells (Fig. 6D). Moreover, sensitivity to ADCC strongly correlated with surface expression of tetherin (Fig. 6E) and Env (Fig. 6F). Thus, similarly to tetherin antagonism by Vpu, tetherin antagonism by Nef protects HIV-1-infected cells from ADCC.
FIG 6.

Tetherin antagonism by Nef protects HIV-1-infected cells from ADCC. (A) CEM.NKR-CCR5-sLTR-Luc cells were infected with wild-type HIV-1 NL4-3 (Vpu+), HIV-1 NL4-3 Δvpu, or HIV-1.Δvpu.IeG-Nef recombinants expressing the indicated Nef alleles and incubated with a CD16+ NK cell line at an effector-to-target cell ratio of 10:1 in the presence of serial dilutions of purified IgG from HIV-1-positive donors (HIVIG). ADCC was calculated from the luciferase activity (RLU) after an 8-h incubation. Error bars indicate the standard deviations of the means for triplicate wells at each antibody concentration, and the dotted line indicates 50% killing of HIV-1-infected cells. (B) Nef and p55 Gag expression in virus-infected cells was confirmed by Western blot analysis of cell lysates with staining for β-actin as a control for sample loading. (C and D) Histograms showing the fluorescence intensity of tetherin (BST-2) (C) and Env (D) staining on the surface of viable, HIV-1-infected (p55+ CD4low) cells as described above for panel A. Tetherin and Env staining on cells infected with virus expressing NL4-3 Nef is shown as a reference (red histograms). The shaded histograms indicate nonspecific staining with either an isotype control for the BST-2-specific antibody (C) or IgG from HIV-negative donors (D). (E and F) Susceptibility to ADCC correlates with the fluorescence intensity of tetherin (E) and Env (F) staining on the surface of virus-infected cells (Pearson correlation test). The data are representative of results from three independent experiments.
Tetherin antagonism by Nef is associated with a loss of antitetherin activity by Vpu.
To better understand the circumstances under which HIV-1 Nef may acquire the ability to counteract tetherin, cDNA sequences coding for Vpu proteins of virus isolates expressing Nef alleles with partial antitetherin activity were synthesized and tested in virus release assays. Additional constructs expressing NL4-3, AM, and C1 Vpu proteins were also included as controls. Whereas the NL4-3 and AM Vpu proteins rescued virus release for HIV-1 Δvpu Δnef in the presence of human tetherin as expected, the N1, N2, N6, N8, N11, and N12 Vpu proteins all failed to restore virus release (Fig. 7A). These results were corroborated by data from Western blot analyses comparing the accumulation of p24 capsid in cell culture supernatants to p55 Gag expression in cell lysates (Fig. 7B).
FIG 7.
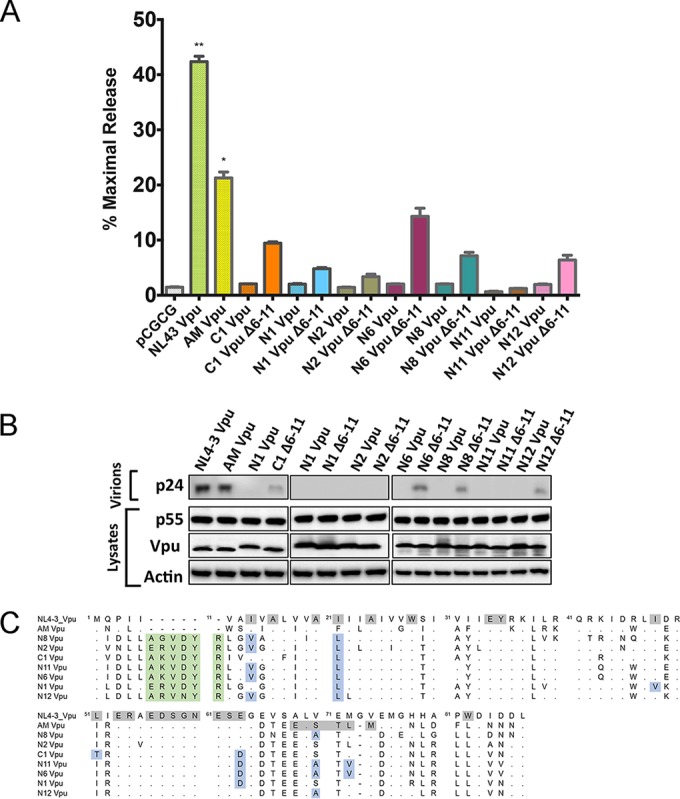
Tetherin antagonism by Nef is associated with a loss of antitetherin activity by Vpu. (A and B) The Vpu proteins of HIV-1 group M isolates with tetherin-antagonizing Nefs were tested for their ability to rescue virus release for HIV-1 NL4-3 Δvpu Δnef in HEK293T cells expressing human tetherin by measuring p24 levels in culture supernatants by an antigen capture ELISA (A) and by comparing the amount of p24 in supernatants to the amounts of Vpu and p55 Gag in cell lysates by Western blot analysis (B). Deletion mutants of these Vpu alleles (Δ6-11), removing a 6-amino-acid insertion common among subtype C isolates, were also tested. The error bars represent the standard deviations of the means for replicate assays, and differences in mean virus release compared to the empty vector control were significant for NL4-3 Vpu (**, P ≤ 0.01) and AM Vpu (*, P ≤ 0.05) by the Kruskal-Wallis test with adjustment for multiple comparisons. (C) Alignment of the amino acid sequences of the Vpu proteins in panel A to NL4-3 Vpu. Dots indicate amino acid identity, dashes indicate gaps, and residues involved in tetherin antagonism are shaded. Vpu polymorphisms corresponding to positions previously shown to be important for tetherin antagonism are highlighted in blue, and residues corresponding to a 6-amino-acid insertion common among subtype C Vpu alleles are shaded in green.
Comparison of the amino acid sequences of Vpu proteins that lack antitetherin activity to the sequences for NL4-3 and AM Vpu proteins revealed a number of differences that may account for their inability to counteract tetherin. The Vpu proteins that do not rescue virus release contain six additional amino acids in the N terminus of the protein and multiple amino acid differences at positions previously shown to affect tetherin antagonism (Fig. 7C) (48, 49). The additional sequences in the N terminus of Vpu (residues 6 to 11) correspond to a polymorphism that is present in ∼84% of the Vpu proteins expressed by subtype C isolates but rare among other HIV-1 subtypes (0.001% of subtype B and 0.02% of subtype G Vpu alleles) (http://www.hiv.lanl.gov/content/index). To determine if these sequences account for the inability to counteract tetherin, deletion mutants lacking residues 6 to 11 were also tested. Deletion of residues 6 to 11 partially increased the antitetherin activity of C1, N1, N6, N8, and N12 Vpu proteins (Fig. 7A and B), suggesting that these residues impair tetherin antagonism; however, this deletion did not fully restore virus release for any of the Vpu alleles tested, indicating that amino acid polymorphisms at other positions must impair this function by Vpu. Accordingly, amino acid differences at positions previously shown to be important for tetherin antagonism (48), including the transmembrane domain and the beta-transducin repeat-containing protein (β-TrCP) binding site, were present in each of the inactive Vpu alleles (Fig. 7C).
To further investigate the possibility that the loss of tetherin antagonism by Vpu and the gain of this function by Nef may be common features of subtype C viruses, we compared the antitetherin activities of Vpu and Nef consensus sequences representing six different HIV-1 subtypes. Vpu and Nef consensus sequences for HIV-1 subtypes A, B, C, D, G, and CRF_02 AG were assessed for their ability to rescue virus release for HIV-1 Δvpu Δnef in the presence of human tetherin. Each of the Vpu consensus sequences, including a consensus sequence for subtype C Vpu containing residues 6 to 11, efficiently rescued virus release (Fig. 8). In contrast, although virus release was somewhat higher for the subtype C Nef consensus, none of the Nef consensus sequences had significant antitetherin activity (Fig. 8). Tetherin antagonism by Nef therefore does not appear to be a common activity of any particular HIV-1 subtype, and the prevalence of subtype C Nef alleles found to have antitetherin activity probably reflects the derivation of the sequence signature used to identify these Nef alleles from a subtype C virus.
FIG 8.
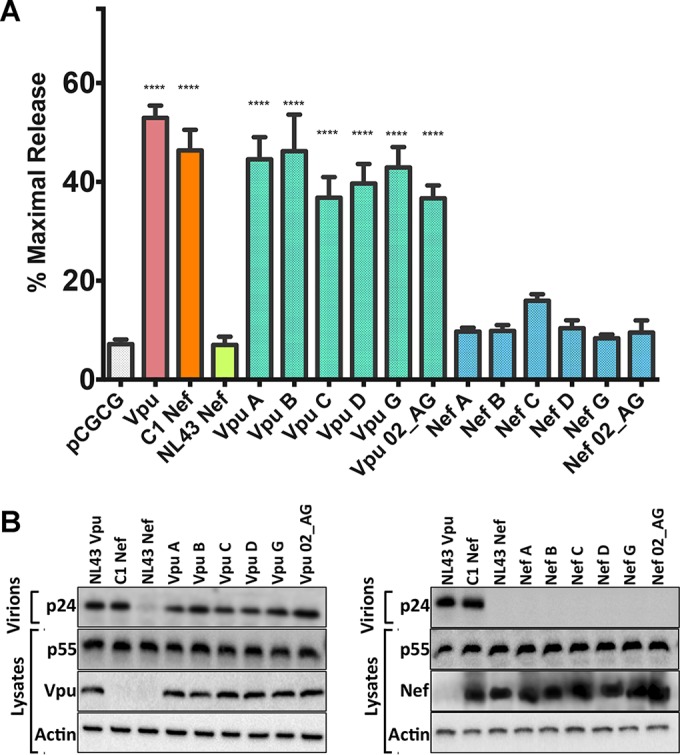
Tetherin antagonism by Vpu and Nef consensus sequences for six different HIV-1 subtypes. Vpu and Nef consensus sequences for HIV-1 subtypes A, B, C, D, G, and CRF_02 AG were compared for their abilities to rescue virus release from HEK293T cells cotransfected with proviral DNA for HIV-1 NL4-3 Δvpu Δnef and a construct for human tetherin. Virus release was assessed by measuring the accumulation of p24 capsid in cell culture supernatants by an antigen capture ELISA (A) and by comparing the amount of p24 in supernatants to the amount of p55 Gag in cell lysates by Western blot analysis (B). The error bars represent the standard deviations of the means for three independent assays, and differences in mean virus release compared to the empty vector control (pCGCG) were significant for all Vpu constructs and C1 Nef by ordinary one-way ANOVA with adjustment for multiple comparisons (****, P ≤ 0.0001).
DISCUSSION
It is widely held that HIV-1 group M isolates use Vpu rather than Nef to overcome restriction by tetherin due to the absence of a 5-amino-acid sequence in the cytoplasmic domain of human tetherin that confers susceptibility to Nef (22). The resistance of human tetherin to Nef does not appear to be absolute, however, as this function was retained by the Nef proteins of HIV-1 group O isolates following the transmission and adaptation of SIVgor to humans (26). In the present study, we identify several HIV-1 group M Nef alleles with significant activity against human tetherin, demonstrating that under certain circumstances, HIV-1 group M Nef may acquire the ability to counteract human tetherin. These group M Nefs downmodulate human tetherin on the cell surface and enhance virus release and virus replication in cells expressing human tetherin. The lack of antitetherin activity for the corresponding Vpu proteins of these viruses suggests that group M Nefs may acquire activity against human tetherin to compensate for the loss of this function by Vpu.
Although a subtype C Nef that antagonized human tetherin nearly as efficiently as NL4-3 Vpu was initially identified, subsequently identified Nef alleles exhibited only partial activity against human tetherin. Whereas minimal effects of these Nefs on virus release and tetherin downmodulation were observed in transfected HEK293T cells, more striking differences were observed in JTAg L-tetherin cells infected with vpu-deleted HIV-1 recombinants expressing selected Nef alleles. These results are similar to previous findings for HIV-1 group O Nefs, which poorly enhance virus release in transfected HEK293T cells but exhibit greater activity in primary CD4+ T cells infected with recombinant viruses (26). While efficient antagonism of human tetherin therefore appears to be rare, HIV-1 group M Nef proteins may acquire moderate levels of antitetherin activity more frequently.
The Nef alleles with partial antitetherin activity were selected on the basis of a mutational analysis of C1 Nef, which defined 8 amino acid differences from NL4-3 Nef required for tetherin antagonism. While no other Nef alleles were a perfect match with C1 Nef at all eight positions, 15 alleles with different combinations of four or more of these residues were selected for further analysis. Of these alleles, six were found to have significant activity against human tetherin. Amino acid sequence comparisons of these Nef proteins did not, however, reveal common residues that would account for their antitetherin activity. Whereas all of these Nefs had an alanine at position 33 and a tyrosine at position 103, these residues were also found in Nef proteins that lack activity against human tetherin. Thus, the sequence determinants for the antagonism of human tetherin appear to be complex and context dependent.
Although a majority of group M Nef alleles found to have activity against human tetherin belong to subtype C isolates, most of the subtype C Nef alleles selected for screening did not have significant antitetherin activity. Furthermore, a consensus sequence for subtype C Nef also lacked activity against human tetherin. Therefore, the predominance of subtype C Nefs with antitetherin activity identified in this study probably reflects the derivation of the query sequence used to select these alleles from a subtype C virus rather than a common or ancestral function of subtype C Nef proteins.
Tetherin antagonism by the group M Nef proteins identified in this study depends on N-terminal residues that differentiate long from short isoforms of human tetherin. Whereas each of the group M Nef proteins with antitetherin activity downmodulated tetherin from the cell surface and enhanced virus replication in JTAg L-tetherin cells, none of these Nef proteins promoted virus release or tetherin downmodulation in JTAg S-tetherin cells. Moreover, substitutions at positions 5-6, 7-8, and 11-12 of L-tetherin impaired a physical interaction with C1 Nef that was detectable by coimmunoprecipitation assays, and substitutions at positions 6 and 8, which correspond to a dual-tyrosine motif in L-tetherin required for intracellular trafficking and signal transduction (13, 44, 45), abrogated sensitivity to antagonism by each of the group M Nef proteins altogether. Thus, tetherin antagonism by group M Nefs depends on residues at the N terminus of the long isoform of human tetherin that are missing from the short isoform of the protein, reminiscent of the antitetherin activity HIV-1 group O Nefs, which maps to sequences spanning the dual-tyrosine motif at the N terminus of L-tetherin (26).
The recognition of the same N-terminal region of tetherin by HIV-1 group M and O Nef proteins suggests that they may use similar mechanisms to counteract restriction. In contrast to SIV Nefs, which downmodulate the tetherin proteins of their nonhuman primate hosts by AP-2-dependent endocytosis (43, 50), HIV-1 group O Nefs impair the anterograde transport of nascent tetherin molecules to the cell surface (26). It is therefore conceivable that group M Nefs that acquire antitetherin activity do so by preventing the trafficking of newly synthesized tetherin to sites of virus assembly and release at the plasma membrane. An alternative, but perhaps not mutually exclusive, possibility is that Nef may displace tetherin from sites of virus assembly independent of tetherin downmodulation. It is becoming increasingly clear that the ability of HIV-1 Vpu to reduce steady-state levels of tetherin on the cell surface is not sufficient to account for its ability to counteract restriction (51, 52). This has been attributed to the lateral displacement of tetherin from sites of virus assembly at the plasma membrane (53–55). Although displacement is not an activity that has been attributed to Nef, the relatively modest effects of group M Nefs on cell surface levels of tetherin suggest that a similar mechanism may contribute to the enhancement of virus release.
Consistent with the possibility that HIV-1 Nef may gain activity against human tetherin to compensate for the loss of this function by Vpu, the Vpu proteins of virus isolates expressing tetherin-antagonizing Nefs were found to lack antitetherin activity. Sequence comparisons revealed that these Vpus contain 6 amino acids at the N terminus of the protein that are not present in NL4-3 Vpu or other Vpu proteins previously shown to antagonize tetherin (22, 48). These sequences are more common among subtype C isolates than among isolates of other HIV-1 subtypes and include charged residues adjacent to the transmembrane domain that could potentially alter the orientation of the tetherin binding interface (49, 56); however, while the deletion of these residues partially increased antitetherin activity for some of the Vpu alleles, their elimination did not fully restore tetherin antagonism. Moreover, a subtype C Vpu consensus sequence containing these residues retained potent antitetherin activity. These results indicate that one or more of the amino acid polymorphisms scattered throughout these Vpu alleles, rather than this 6-amino-acid insertion, impair tetherin antagonism. The circumstances leading to the loss of tetherin antagonism by Vpu are presently unknown but may include the selection of escape mutations by CD8+ T cells and/or founder effects during mucosal transmission resulting in the establishment of infection by vpu variants that have lost the ability to counteract tetherin.
Similarly to recent studies by our group and others showing that tetherin antagonism by Vpu protects HIV-1-infected cells from ADCC (14–17), we found that the antitetherin activity of group M Nefs also affords resistance to ADCC. Indeed, resistance to ADCC corresponded with the antitetherin activity of Nef. Whereas cells infected with vpu-deleted HIV-1 expressing C1 Nef were as resistant to ADCC as cells infected with wild-type HIV-1, cells infected with viruses expressing Nef alleles with partial antitetherin activity exhibited intermediate resistance to ADCC. These observations are further supported by correlations between the efficiency of tetherin downmodulation, concomitant reductions in Env levels on the surface of virus-infected cells, and resistance to ADCC. Thus, similar to the antitetherin activity of Vpu, tetherin antagonism by Nef can protect HIV-1-infected cells from antibody-dependent lysis. Given the potential for tetherin to amplify the antiviral effects of antibodies in vivo, ADCC or other Fcγ receptor-dependent functions of antibodies may play an important role in the selective pressure for Nef to acquire activity against human tetherin.
The identification of HIV-1 group M Nef proteins with activity against human tetherin challenges the prevailing view that the main group of viruses responsible for the global AIDS pandemic exclusively uses Vpu to counteract this factor. While most HIV-1 group M isolates use Vpu to counteract tetherin, the Nef proteins of certain viruses were found to have significant activity against human tetherin. Moreover, the Vpu proteins expressed by several of these viruses were unable to oppose tetherin, suggesting that this function may be acquired by Nef under circumstances where it is lost by Vpu. One implication of these observations is that efforts to develop antiretroviral drugs that interfere with the ability of Vpu to counteract tetherin may lead to the selection of Nef variants that gain this function (57, 58). Thus, understanding the conditions that lead to tetherin antagonism by Nef will be important for the development of therapies to increase the susceptibility of HIV-1 to this restriction factor.
ACKNOWLEDGMENTS
We thank Stuart Neil, King's College School of Medicine, for providing the Jurkat-TAg L-tetherin and Jurkat-TAg S-tetherin cell lines and Reiko Nishihara, Dana-Farber Cancer Institute, for her guidance with the statistical methods used for this study. We also thank Kim L. Weisgrau and Eva G. Rakasz, Wisconsin National Primate Research Center, for flow cytometry services.
D.T.E. is an Elizabeth Glaser Scientist of the Elizabeth Glaser Pediatric AIDS Foundation.
This work was supported by National Institutes of Health grants AI098485, AI095098, AI121135, and P51OD011106.
REFERENCES
- 1.Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. 2009. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139:499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammonds J, Wang JJ, Yi H, Spearman P. 2010. Immunoelectron microscopic evidence for tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog 6:e1000749. doi: 10.1371/journal.ppat.1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitzpatrick K, Skasko M, Deerinck TJ, Crum J, Ellisman MH, Guatelli J. 2010. Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles. PLoS Pathog 6:e1000701. doi: 10.1371/journal.ppat.1000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 6.Mangeat B, Cavagliotti L, Lehmann M, Gers-Huber G, Kaur I, Thomas Y, Kaiser L, Piguet V. 2012. Influenza virus partially counteracts restriction imposed by tetherin/BST-2. J Biol Chem 287:22015–22029. doi: 10.1074/jbc.M111.319996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weidner JM, Jiang D, Pan XB, Chang J, Block TM, Guo JT. 2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J Virol 84:12646–12657. doi: 10.1128/JVI.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J. 2009. Inhibition of Lassa and Marburg virus production by tetherin. J Virol 83:2382–2385. doi: 10.1128/JVI.01607-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K. 2009. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma-associated herpesvirus. J Virol 83:9672–9681. doi: 10.1128/JVI.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. 2009. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol 83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. 2003. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4:694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- 12.Galao RP, Le Tortorec A, Pickering S, Kueck T, Neil SJ. 2012. Innate sensing of HIV-1 assembly by tetherin induces NFkappaB-dependent proinflammatory responses. Cell Host Microbe 12:633–644. doi: 10.1016/j.chom.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tokarev A, Suarez M, Kwan W, Fitzpatrick K, Singh R, Guatelli J. 2013. Stimulation of NF-kappaB activity by the HIV restriction factor BST2. J Virol 87:2046–2057. doi: 10.1128/JVI.02272-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, Evans DT. 2014. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A 111:6425–6430. doi: 10.1073/pnas.1321507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alvarez RA, Hamlin RE, Monroe A, Moldt B, Hotta MT, Rodriguez Caprio G, Fierer DS, Simon V, Chen BK. 2014. HIV-1 Vpu antagonism of tetherin inhibits antibody-dependent cellular cytotoxic responses by natural killer cells. J Virol 88:6031–6046. doi: 10.1128/JVI.00449-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pham TN, Lukhele S, Hajjar F, Routy JP, Cohen EA. 2014. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 11:15. doi: 10.1186/1742-4690-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol 88:2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans DT, Elder JH, Desrosiers RC. 2013. Nonhuman lentiviruses, p 1584–1612. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 19.Delaugerre C, De Oliveira F, Lascoux-Combe C, Plantier JC, Simon F. 2011. HIV-1 group N: travelling beyond Cameroon. Lancet 378:1894. doi: 10.1016/S0140-6736(11)61457-8. [DOI] [PubMed] [Google Scholar]
- 20.Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JF, Sharp PM, Shaw GM, Peeters M, Hahn BH. 2006. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D'Arc M, Ayouba A, Esteban A, Learn GH, Boue V, Liegeois F, Etienne L, Tagg N, Leendertz FH, Boesch C, Madinda NF, Robbins MM, Gray M, Cournil A, Ooms M, Letko M, Simon VA, Sharp PM, Hahn BH, Delaporte E, Mpoudi Ngole E, Peeters M. 2015. Origin of the HIV-1 group O epidemic in western lowland gorillas. Proc Natl Acad Sci U S A 112:E1343–E1352. doi: 10.1073/pnas.1502022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogando Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6:409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johsnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6:54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT. 2009. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog 5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Tortorec A, Neil SJD. 2009. Antagonism and intracellular sequestration of human tetherin by the HIV-2 envelope glycoprotein. J Virol 83:11966–11978. doi: 10.1128/JVI.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kluge SF, Mack K, Iyer SS, Pujol FM, Heigele A, Learn GH, Usmani SM, Sauter D, Joas S, Hotter D, Bibollet-Ruche F, Plenderleith LJ, Peeters M, Geyer M, Sharp PM, Fackler OT, Hahn BH, Kirchhoff F. 2014. Nef proteins of epidemic HIV-1 group O strains antagonize human tetherin. Cell Host Microbe 16:639–650. doi: 10.1016/j.chom.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mourez T, Simon F, Plantier JC. 2013. Non-M variants of human immunodeficiency virus type 1. Clin Microbiol Rev 26:448–461. doi: 10.1128/CMR.00012-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alpert MD, Harvey JD, Lauer WA, Reeves RK, Piatak M Jr, Carville A, Mansfield KG, Lifson JD, Li W, Desrosiers RC, Johnson RP, Evans DT. 2012. ADCC develops over time during persistent infection with live-attenuated SIV and is associated with complete protection against SIV(mac)251 challenge. PLoS Pathog 8:e1002890. doi: 10.1371/journal.ppat.1002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinelt J, Neil SJ. 2014. Differential sensitivities of tetherin isoforms to counteraction by primate lentiviruses. J Virol 88:5845–5858. doi: 10.1128/JVI.03818-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serra-Moreno R, Jia B, Breed M, Alvarez X, Evans DT. 2011. Compensatory changes in the cytoplasmic tail of gp41 confer resistance to tetherin/BST-2 in a pathogenic nef-deleted SIV. Cell Host Microbe 9:46–57. doi: 10.1016/j.chom.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schindler M, Wurfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Munch J, Kirchhoff F. 2003. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol 77:10548–10556. doi: 10.1128/JVI.77.19.10548-10556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schindler M, Munch J, Kirchhoff F. 2005. Human immunodeficiency virus type 1 inhibits DNA damage-triggered apoptosis by a Nef-independent mechanism. J Virol 79:5489–5498. doi: 10.1128/JVI.79.9.5489-5498.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong SK, Connole M, Sullivan JS, Choe H, Carville A, Farzan M. 2009. A New World primate deficient in tetherin-mediated restriction of human immunodeficiency virus type 1. J Virol 83:8771–8780. doi: 10.1128/JVI.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency virus syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alpert MD, Heyer LN, Williams DE, Harvey JD, Greenough T, Allhorn M, Evans DT. 2012. A novel assay for antibody-dependent cell-mediated cytotoxicity against HIV-1- or SIV-infected cells reveals incomplete overlap with antibodies measured by neutralization and binding assays. J Virol 86:12039–12052. doi: 10.1128/JVI.01650-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trkola A, Matthews J, Gordon C, Ketas T, Moore JP. 1999. A cell line-based neutralization assay for primary human immunodeficiency virus type 1 isolates that use either the CCR5 or the CXCR4 coreceptor. J Virol 73:8966–8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Bredow B, Arias JF, Heyer LN, Gardner MR, Farzan M, Rakasz EG, Evans DT. 2015. Envelope glycoprotein internalization protects human and simian immunodeficiency virus-infected cells from antibody-dependent cell-mediated cytotoxicity. J Virol 89:10648–10655. doi: 10.1128/JVI.01911-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maldarelli F, Chen MY, Willey RL, Strebel K. 1993. Human immunodeficiency virus type 1 Vpu protein is an oligomeric type I integral membrane protein. J Virol 67:5056–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vujcic LK, Quinnan GV Jr. 1995. Preparation and characterization of human HIV type 1 neutralizing reference sera. AIDS Res Hum Retroviruses 11:783–787. doi: 10.1089/aid.1995.11.783. [DOI] [PubMed] [Google Scholar]
- 41.Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell J IV, Bixby D, Savona MR, Collins KL. 2010. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med 16:446–451. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cocka LJ, Bates P. 2012. Identification of alternatively translated tetherin isoforms with differing antiviral and signaling activities. PLoS Pathog 8:e1002931. doi: 10.1371/journal.ppat.1002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serra-Moreno R, Zimmermann K, Stern LJ, Evans DT. 2013. Tetherin/BST-2 antagonism by Nef depends on a direct physical interaction between Nef and tetherin, and on clathrin-mediated endocytosis. PLoS Pathog 9:e1003487. doi: 10.1371/journal.ppat.1003487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rollason R, Korolchuk V, Hamilton C, Schu P, Banting G. 2007. Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J Cell Sci 120:3850–3858. doi: 10.1242/jcs.003343. [DOI] [PubMed] [Google Scholar]
- 45.Galão Rui P, Pickering S, Curnock R, Neil SJD. 2014. Retroviral retention activates a Syk-dependent HemITAM in human tetherin. Cell Host Microbe 16:291–303. doi: 10.1016/j.chom.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia X, Weber E, Tokarev A, Lewinski M, Rizk M, Suarez M, Guatelli J, Xiong Y. 2014. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife 3:e02362. doi: 10.7554/eLife.02362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lau D, Kwan W, Guatelli J. 2011. Role of the endocytic pathway in the counteraction of BST-2 by human lentiviral pathogens. J Virol 85:9834–9846. doi: 10.1128/JVI.02633-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pickering S, Hue S, Kim EY, Reddy S, Wolinsky SM, Neil SJ. 2014. Preservation of tetherin and CD4 counter-activities in circulating Vpu alleles despite extensive sequence variation within HIV-1 infected individuals. PLoS Pathog 10:e1003895. doi: 10.1371/journal.ppat.1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vigan R, Neil SJ. 2010. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J Virol 84:12958–12970. doi: 10.1128/JVI.01699-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang F, Landford WN, Ng M, McNatt MW, Bieniasz PD, Hatziioannou T. 2011. SIV Nef proteins recruit the AP-2 complex to antagonize tetherin and facilitate virion release. PLoS Pathog 7:e1002039. doi: 10.1371/journal.ppat.1002039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goffinet C, Homann S, Ambiel I, Tibroni N, Rupp D, Keppler OT, Fackler OT. 2010. Antagonism of CD317 restriction of human immunodeficiency virus type 1 (HIV-1) particle release and depletion of CD317 are separable activities of HIV-1 Vpu. J Virol 84:4089–4094. doi: 10.1128/JVI.01549-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jafari M, Guatelli J, Lewinski MK. 2014. Activities of transmitted/founder and chronic clade B HIV-1 Vpu and a C-terminal polymorphism specifically affecting virion release. J Virol 88:5062–5078. doi: 10.1128/JVI.03472-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McNatt MW, Zang T, Bieniasz PD. 2013. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog 9:e1003299. doi: 10.1371/journal.ppat.1003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewinski MK, Jafari M, Zhang H, Opella SJ, Guatelli J. 2015. Membrane anchoring by a C-terminal tryptophan enables HIV-1 Vpu to displace bone marrow stromal antigen 2 (BST2) from sites of viral assembly. J Biol Chem 290:10919–10933. doi: 10.1074/jbc.M114.630095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pujol FM, Laketa V, Schmidt F, Mukenhirn M, Muller B, Boulant S, Grimm D, Keppler OT, Fackler OT. 2016. HIV-1 Vpu antagonizes CD317/tetherin by adaptor protein-1-mediated exclusion from virus assembly sites. J Virol 90:6709–6723. doi: 10.1128/JVI.00504-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skasko M, Wang Y, Tian Y, Tokarev A, Munguia J, Ruiz A, Stephens EB, Opella SJ, Guatelli J. 2012. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J Biol Chem 287:58–67. doi: 10.1074/jbc.M111.296772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Q, Liu Z, Mi Z, Li X, Jia P, Zhou J, Yin X, You X, Yu L, Guo F, Ma J, Liang C, Cen S. 2011. High-throughput assay to identify inhibitors of Vpu-mediated down-regulation of cell surface BST-2. Antiviral Res 91:321–329. doi: 10.1016/j.antiviral.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 58.Mi Z, Ding J, Zhang Q, Zhao J, Ma L, Yu H, Liu Z, Shan G, Li X, Zhou J, Wei T, Zhang L, Guo F, Liang C, Cen S. 2015. A small molecule compound IMB-LA inhibits HIV-1 infection by preventing viral Vpu from antagonizing the host restriction factor BST-2. Sci Rep 5:18499. doi: 10.1038/srep18499. [DOI] [PMC free article] [PubMed] [Google Scholar]