

ABSTRACT
Although a highly effective vaccine is available, the number of yellow fever cases has increased over the past 2 decades, which highlights the pressing need for antiviral therapeutics. In a high-throughput screening campaign, we identified an acetic acid benzodiazepine (BDAA) compound which potently inhibits yellow fever virus (YFV). Interestingly, while treatment of YFV-infected cultures with 2 μM BDAA reduced the virion production by greater than 2 logs, the compound was not active against 21 other viruses from 14 different viral families. Selection and genetic analysis of drug-resistant viruses revealed that replacement of the proline at amino acid 219 (P219) of the nonstructural protein 4B (NS4B) with serine, threonine, or alanine conferred YFV with resistance to BDAA without apparent loss of replication fitness in cultured mammalian cells. However, replacement of P219 with glycine conferred BDAA resistance with significant loss of replication ability. Bioinformatics analysis predicts that the P219 amino acid is localized at the endoplasmic reticulum lumen side of the fifth putative transmembrane domain of NS4B, and the mutation may render the viral protein incapable of interacting with BDAA. Our studies thus revealed an important role and the structural basis for the NS4B protein in supporting YFV replication. Moreover, in YFV-infected hamsters, oral administration of BDAA protected 90% of the animals from death, significantly reduced viral load by greater than 2 logs, and attenuated virus infection-induced liver injury and body weight loss. The encouraging preclinical results thus warrant further development of BDAA or its derivatives as antiviral agents to treat yellow fever.
IMPORTANCE Yellow fever is an acute viral hemorrhagic disease which threatens approximately 1 billion people living in tropical areas of Africa and Latin America. Although a highly effective yellow fever vaccine has been available for more than 7 decades, the low vaccination rate fails to prevent outbreaks in at-risk regions. It has been estimated that up to 1.7 million YFV infections occur in Africa each year, resulting in 29,000 to 60,000 deaths. Thus far, there is no specific antiviral treatment for yellow fever. To cope with this medical challenge, we identified a benzodiazepine compound that selectively inhibits YFV by targeting the viral NS4B protein. To our knowledge, this is the first report demonstrating in vivo safety and antiviral efficacy of a YFV NS4B inhibitor in an animal model. We have thus reached a critical milestone toward the development of specific antiviral therapeutics for clinical management of yellow fever.
INTRODUCTION
Yellow fever is a mosquito-borne hemorrhagic disease that is frequently associated with jaundice and is caused by yellow fever virus (YFV) infection (1). Vaccination with attenuated YFV-17D vaccine is the most important measurement to prevent yellow fever. The vaccine is safe, affordable, and highly effective, and a single dose of the vaccine is sufficient to confer sustained immunity and lifelong protection against yellow fever disease (2, 3). However, the decline in herd immunity and discontinuation of mosquito control measures in many parts of Africa are considered the major causes for the resurgence of yellow fever and periodic epidemics in East and West African countries during the last 2 decades (4). In addition, recent clinical studies also showed that an activated immune microenvironment with other microbial infections prior to vaccination impeded efficacy of the YFV-17D vaccine in an African cohort, suggesting that vaccine regimens may need to be boosted in African populations to achieve efficient protective immunity (5–7). It has been estimated that up to 1.7 million YFV infections may occur in Africa each year, resulting in 29,000 to 60,000 deaths (8, 9) (http://www.who.int/csr/disease/yellowfev/yellowfeverburdenestimation_summary2013.pdf). Once an outbreak starts, vaccination is less effective, and other intervention strategies, such as antiviral therapies, are needed. However, there are no specific antiviral therapeutics to treat the life-threatening disease, and development of antiviral agents that inhibit YFV replication would meet a pressing medical need (8).
YFV is the prototypic member of the Flavivirus genus in the family Flaviviridae (10). The virus has a single-stranded, positive-sense, approximately 11-kb RNA genome. The viral genomic RNA contains a single open reading frame that encodes a long polyprotein flanked by 5′- and 3′-untranslated regions (UTR) harboring essential cis-elements for regulation of viral protein translation and RNA replication. The viral polyprotein is cotranslationally and posttranslationally processed into three structural and seven nonstructural (NS) proteins by host cellular and viral proteases. While the three structural proteins, capsid (C), premembrane (prM), and envelope (E), are involved in virion assembly and infectious entry into host cells, the seven NS proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5, form membrane-bound replication complexes for replication of the viral RNA genome. Specifically, the three small hydrophobic proteins NS2A, NS4A, and NS4B anchor the viral replication complex to the endoplasmic reticulum (ER) membrane, and the NS5 protein functions as a methyltransferase and RNA-dependent RNA polymerase to catalyze viral RNA synthesis and facilitate viral RNA translation (11). In addition to their distinct roles in viral replication, viral proteins are also involved in activation and/or evasion of host innate and adaptive immune responses and thus play important roles in pathogenesis (12–14).
In the last decades, tremendous industrial and academic efforts have been made in discovery and development of antiviral agents against flaviviruses, most notably for dengue viruses (DENV) (15, 16). These efforts have resulted in identification of a variety of small molecules that inhibit viral replication through targeting distinct viral and host cellular functions (17–19). Small molecules that specifically target viral capsid assembly (20), NS3/2A proteases (21–23), NS3 RNA helicase (24), NS4B protein (25, 26), NS5 methyltransferase (27), and RNA-dependent RNA polymerase (28, 29) have been discovered, and the in vivo antiviral efficacies for four of these direct-acting antiviral (DAA) agents have been demonstrated in animal models (24, 30–32); however, none of them has reached human clinical trials. Balapiravir, a tri-isobutyrate ester prodrug of 4′-azidocytidine (R1479) that was originally developed for treatment of chronic hepatitis C virus (HCV) infection by Hoffmann-La Roche, is active against DENV in cultured cells. However, a phase II clinical trial of Balapiravir for treatment of dengue fever failed to improve the clinical and virological parameters in patients (33), most likely due to a DENV-induced cytokine response that decreased the efficiency of conversion of R1479 to its triphosphate form in mononuclear cells (34).
Compared to dengue virus and other flaviviruses, development of antiviral agents against YFV has been less robust, perhaps because of the availability of an effective vaccine and the difficulty of conducting clinical trials during sporadic outbreaks of yellow fever. Thus far, alpha interferon (IFN-α) and a few broad-spectrum antiviral nucleoside analogues, including ribavirin, T-705, and BCX4430, have been demonstrated to inhibit YFV replication in vivo in hamsters (35–39). In addition, a high-throughput screening effort using a subgenomic replicon cell-based phenotypic assay identified two small molecular compounds that inhibited YFV, and their resistance mutation was mapped to the NS4B gene (40). However, the antiviral efficacies of these compounds in vivo in animal models have not been reported.
In a high-throughput screening with a HEK293 cell-based IFN-β promoter reporter assay, we identified a benzodiazepine compound that potently inhibits YFV. Selection and genetic analysis of drug-resistant viruses revealed that replacement of the proline at amino acid 219 of the NS4B protein with serine conferred YFV resistance to the compound. Pharmacological studies showed that the compound was well-tolerated in mice and hamsters, and oral administration of the compound protected 90% of YFV-infected hamsters from death, significantly reduced the viral load, and normalized liver function. The results thus strongly support the further development of the benzodiazepine compound or its derivatives as yellow fever therapeutics.
MATERIALS AND METHODS
Cell lines, virus stocks, and compounds.
Huh7.5 (human hepatoma cells; a gift of Charles M. Rice, Rockefeller University), HEK293 (human embryonic kidney cells; ATCC), and Vero (green monkey kidney cells; ATCC) cells were maintained in Dulbecco's modified minimal essential medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum at 37°C (41). The C6/C36 mosquito cell line was purchased from ATCC and maintained in Eagle's minimum essential medium (EMEM) supplemented with 10% fetal bovine serum at 28°C.
A plasmid containing the YFV 17D complete genomic cDNA, pACNR/FLYF-17Dx, was a gift of Charles M. Rice (42, 43). YFV 17D virus stock was produced by electroporation of Huh7.5 cells with in vitro-transcribed RNA from pACNR/FLYF-17Dx (44). The medium was harvested at 48 h posttransfection, clarified by low-speed centrifugation, and stored at −80°C until use. The virus titers were determined by a plaque assay on Vero cells as described previously (45).
BDAA and its enantiomers were synthesized in-house with greater than 95% purity. Scale-up synthesis of BDAA for animal efficacy studies was done by SRI International via the NIAID Preclinical Service Program. Clinical psychoactive benzodiazepines (alprazolam, estazolam, nordiazepam, temazepam, prazepam, and lorazepam) were obtained from a certified reference material provider (Cerilliant). Ribavirin was obtained from ICN Pharmaceuticals, Inc. For in vivo studies, BDAA was prepared in a solution of Solutol HS15 (Sigma-Aldrich) and methyl-2-pyrrolidinone (NMP; Sigma-Aldrich) in a 1:1 (wt/vol) ratio. Ribavirin was prepared in sterile saline.
In-Cell Western assay.
An In-Cell Western assay to simultaneously detect YFV envelope protein and cell viability was performed essentially as described previously (46). Briefly, Huh7.5 cells in a 96-well plate were infected with YFV at a multiplicity of infection (MOI) of 0.1 and either mock treated (1% dimethyl sulfoxide [DMSO]) or treated with a serial dilution of BDAA for 48 h. Cells were fixed and incubated with a mouse monoclonal antibody against flavivirus envelope proteins (4G2; Millipore), followed by incubation with anti-mouse IRDye 800CW-labeled secondary antibody together with two reagents for cell staining (DRAQ5 [Biostatus] and Sapphire700 [LI-COR]). YFV envelope protein expression and cell viability were visualized by using the LI-COR Odyssey system.
qRT-PCR.
Total cellular RNA was extracted from Huh7.5 or C6/C36 cells at 2 days postinfection (dpi) with YFV at the indicated MOI by using the NucleoSpin 96 RNA kit (Macherey-Nagel). The amounts of YFV RNA were determined in a quantitative one-step reverse transcription-PCR (qRT-PCR) assay by using a LightCycler 480II apparatus (Roche) and the primers 5′-ATGCTGTCCCTTTTGGTTTG-3′ and 5′-GCCACTGTGAGTTTCAGCAA-3′. β-Actin mRNA was quantified with the primers 5′-CCAACCGCGAGAAGATGA-3′ and 5′-CCAGAGGCGTACAGGGATAG-3′ to normalize the levels of viral RNA in Huh7.5 cells. Aedes albopictus S7 ribosomal mRNA was quantified with the primers 5′-GTCCACGATCCCGCACTCT-3′ and 5′-GTGGTCTGCTGGTTCTTGTCC-3′ to normalize the levels of viral RNA in C6/C36 cells.
Virus yield reduction assay.
Monolayers of Vero or Huh7.5 cells in 24-well plates were infected for 1 h with serial 10-fold dilutions of the culture medium harvested from YFV-infected cells with or without treatment, followed by overlay with medium containing 0.75% methylcellulose and incubation at 37°C for 3 to 5 days. Plaques were counted after crystal violet staining (47).
Cytotoxicity assay.
To determine the cytotoxicity of compounds, a 3-(4,5-dimethylthiazol)-2-(4-sulfophenyl)-2H-tetrazolium (MTT) assay (Sigma) was performed. Uninfected Huh7.5 cells were mock treated or treated with the indicated concentrations of testing compounds under conditions that were identical to those used for the antiviral assays. The dose-dependency curves were generated to determine the inhibitory concentration required to inhibit cell viability by 50% (CC50) by using Prism 5 (GraphPad Software, Inc.).
Selection and sequence analysis of resistant viruses.
The YFV 17D strain rescued from an infectious cDNA clone was used to infect Huh7.5 cells. Drug-resistant viruses were selected by multiple rounds of passaging of the viruses in cultures treated with increasing concentrations of BDAA. Briefly, Huh7.5 cells grown in a 12-well plate were infected either with wild-type YFV 17D at an MOI of 0.1 or in medium from a previous passage that contained virus and the compound. After incubation at 37°C for 1 h, the inocula were removed and replaced with fresh medium containing BDAA. For each passage, the cell culture was incubated for 3 days, during which time the cytopathic effect (CPE) was monitored as one of the readouts for the development of resistance. The viruses were passaged 6 times in the presence of 3 μM BDAA, followed by passage 6 times in the presence of 6 μM BDAA and 6 times in the presence of 8 μM BDAA. As a control, wild-type YFV 17D was passaged in the presence of 0.5% DMSO in parallel. The viruses in the supernatants of the 12th passage in the presence of 6 μM BDAA and 18th passage in the presence of 8 μM BDAA were evaluated for compound sensitivity in a yield reduction assay. To map drug resistance mutations, virus in the 18th passage was used to infect Huh7.5 cells. Total cellular RNAs were extracted on day 3 postinfection. YFV cDNA was synthesized and amplified by RT-PCR. Purified PCR products were used for direct DNA sequencing (Genewiz). Mutations were identified by sequence alignment with parental YFV 17D cDNA.
Recombinant virus cDNA construction.
To construct full-length YFV 17D cDNA clones containing the desired NS4B P219 mutations, 995-bp fragments flanking the NS4B P219 site with the desired nucleotide substitutions to confer P219S, P219T, P219A, or P219G mutations were synthesized and confirmed by DNA sequencing (GenScript). The corresponding fragment in pACNR/FLYF-17Dx was then replaced with the synthesized fragments to yield plasmids containing full-length YFV 17D cDNA clones with the desired point mutations. Mutant viruses were rescued by electroporation of Huh7.5 cells with in vitro-transcribed RNA from each of the recombinant YFV cDNA clones. The medium and cells were harvested at 4 to 6 days posttransfection. Virus titers were determined by a plaque assay in Vero or Huh7.5 cells.
Benzodiazepine receptor binding assay.
The binding activities of BDAA to benzodiazepine receptors were assessed in a radioactive-labeled ligand competition binding assay (48, 49). The central nervous system benzodiazepine receptor (GABAAR) was prepared from rat cerebral cortex, and the peripheral nervous system benzodiazepine receptor (PBR) was prepared from rat heart. [3H]flunitrazepam and [3H]PK11195 were used as the labeled agonists, and diazepam and unlabeled PK11195 were used as reference agonists for the central nervous system and peripheral nervous system benzodiazepine receptor binding assays, respectively. The bound and unbound labeled ligands were separated by centrifugation, and bound labeled ligands were quantified by scintillation counting.
Pharmacokinetics of BDAA.
Single-dose pharmacokinetics (PK) studies were performed for BDAA in mice. Intravenous (i.v.), oral (per os [p.o.]), intramuscular (i.m.), or intraperitoneal (i.p.) administration routes were included with dose levels of either 2 mg/kg of body weight (i.v.) or 10 mg/kg for all other routes (IAS, Berkeley, CA). Following each administration, at various time points postadministration blood samples were collected to determine the plasma concentration of the test compound. Three female CD-1 mice per time point were used. The single-compartment PK parameters were calculated using PK functions within Excel.
In vivo toxicity analysis in mice and hamsters.
A repeated-dose oral study and i.p. maximum tolerated dose (MTD) study (MB Research Laboratories, Spinnerstown, PA) for BDAA were performed in CD-1 mice at 5, 10, 25, 50, and 100 mg/kg every 8 h for 7 days. Each treatment group included three mice. The animals were observed once daily for mortality and body weight changes as well as any sign of illness.
A repeated-dose oral MTD study for BDAA was performed in female Syrian golden hamsters at 50, 100, and 150 mg/kg twice daily for 7 days. Each treatment group included 5 animals. The animals were observed once daily for mortality and body weight changes as well as any sign of illness.
Antiviral efficacy in YFV-infected hamsters.
The in vivo efficacy experiments were performed using lethal YFV infection of hamsters as reported previously (36, 37). Female Syrian golden hamsters (Charles River Laboratories) with an average weight of 110 g were used after a quarantine period of greater than 48 h. Randomly grouped animals were infected with Jimenez, a hamster-adapted YFV strain, via i.p. inoculation, at 10 50% tissue culture infective doses (TCID50)/animal. The infected hamsters (n = 10 per group) were treated twice daily with 100 mg/kg, 50 mg/kg, or 25 mg/kg of BDAA for 7 days beginning at 4 h prior to infection via oral administration. Ribavirin was included as a positive control and was administered orally at 25 mg/kg with the same treatment schedule. A placebo-treated infection control group was included, and these animals were treated with the same vehicle used for BDAA. Sham-infected controls (n = 5 per group) were either treated with the highest dose of BDAA using the same schedule and route of administration to monitor for signs of toxicity, and negative controls were treated with vehicle. Uninfected and untreated normal controls (n = 5) were also included as controls for weight changes and mortality. The survival rate was observed until day 21 postinfection. Body weights were measured on 3 and 6 days postinfection. Serum was collected via ocular sinus bleed on 4 and 6 days postinfection. An alanine aminotransferase (ALT) assay was performed with serum samples obtained 6 days postinfection and added into a commercial reagent (Teco Diagonostic, Anaheim, CA). The virus titers were determined using serum samples from 4 days postinfection in a CPE-based infectious cell culture assay in Vero 76 cells to calculate the endpoint of infection (35).
Ethics statement.
The in vivo efficacy study, including veterinary care and experimental procedures, was reviewed and approved by the Institutional Animal Care and Use Committee of Utah State University (identification number 2400). The study was carried out in accordance with the recommendations and guidelines of the National Institutes of Health for the care and use of laboratory animals.
Statistical analyses.
In the in vivo efficacy study, survival data were analyzed using the Wilcoxon log-rank survival analysis, and all other statistical analyses were performed using a one-way analysis of variance (ANOVA) with a Bonferroni group comparison (Prism 5; GraphPad Software, Inc.).
RESULTS
Discovery of a benzodiazepine compound that specifically inhibits YFV.
We reported previously that infection of a HEK293 cell-based reporter cell line expressing firefly luciferase under the control of a human IFN-β promoter with several RNA viruses, including DENV and YFV, activated reporter gene expression that correlated quantitatively with the levels of virus replication and progeny virus production. This assay had been used for high-throughput screening of compounds that inhibited either viral replication or the virus-induced proinflammatory cytokine response (45). Through screening of an in-house small-molecule library, we identified a benzodiazepine compound, 2-(S)-3-[(S)-sec-butyl]-7-chloro-2-oxo-5-phenyl-2,3-dihydro-1H-benzo[e] (1,4)diazepin-1-ylacetic acid, designated BDAA (Fig. 1A), that inhibited DENV serotype 2 (DENV-2) and YFV infection. Its antiviral activity against YFV was demonstrated in three independent assays in human hepatoma cell line Huh7.5. As shown in Fig. 1B to D, BDAA caused dose-dependent inhibition of YFV envelope protein expression, RNA replication, and progeny virion production, as demonstrated in In-Cell Western, qRT-PCR, and plaque assays, respectively. The 50% effective concentrations (EC50s) of the compound in the three assays were between 0.21 and 0.9 μM. Moreover, treatment of YFV-infected cultures with 2 or 10 μM BDAA reduced progeny virion production by >2 to 4 logs. Cytotoxicity of the compounds was determined in an MTT assay in Huh7.5 cells, and CC50 values were greater than 100 μM. In addition to Huh7.5 cells, similar antiviral results against YFV were also obtained in HEK293 and Vero cells in a qRT-PCR assay (Table 1). Investigation of the antiviral spectrum through the National Institutes of Health (NIH) in vitro antiviral screening program showed that BDAA is a potent YFV inhibitor which inhibited YFV-induced CPE, with an EC50 of 0.05 μM, and reduced YFV yields, with an EC90 of 0.03 μM in Vero cells. In addition, BDAA also modestly reduced DENV-2 yield, with an EC90 of 8.6 μM. However, the compound did not apparently inhibit the infection/replication of 21 other viruses from 14 different virus families, including four additional representative human-pathogenic flaviviruses (Table 1). The narrow antiviral spectrum suggests that BDAA most likely targets a YFV-specific function.
FIG 1.
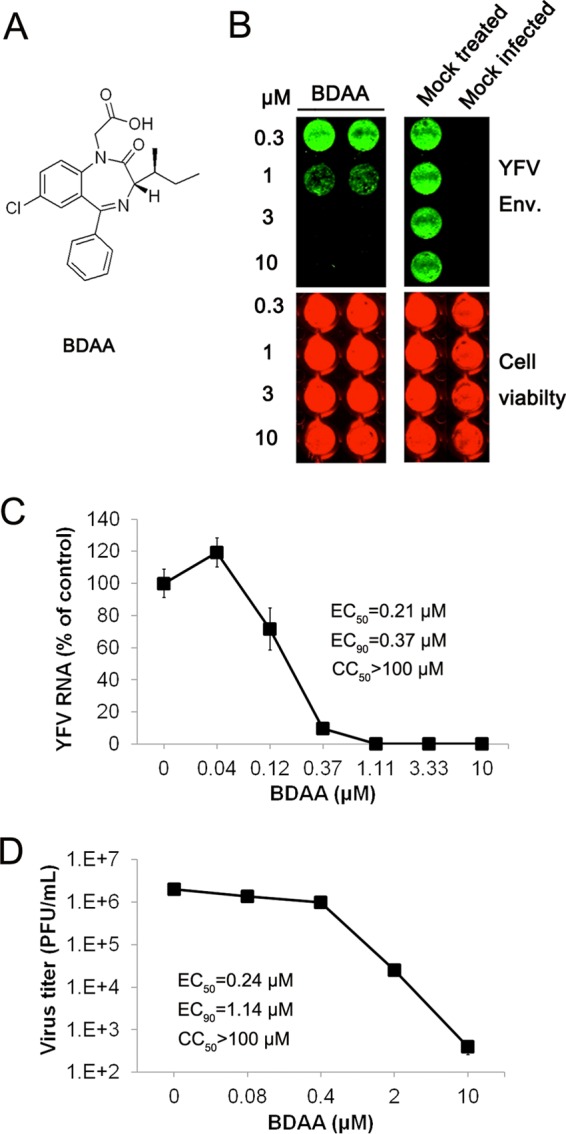
In vitro antiviral activity of BDAA against YFV. (A) Structure of BDAA. (B) Huh7.5 cells seeded in 96-well plates were infected with YFV strain 17D at an MOI of 0.1 for 1 h, followed by treatment with the indicated concentrations of BDAA for 48 h. YFV E protein was visualized by In-Cell immunostaining (green). Cell viability was determined using DRAQ 5 and Sapphire 700 stains (red). (C and D) Huh7.5 cells seeded in 96-well plates were infected with YFV at an MOI of 0.01 for 1 h, followed by treatment with the indicated concentrations of BDAA for 48 h. Total intracellular RNAs were extracted to determine the amount of YFV RNA by qRT-PCR. YFV RNAs were normalized to β-actin mRNA and presented as the percentage of mock treated control (C). Data represented the mean results from four independent replicates (± standard deviations). Culture media were harvested from infected cells treated with various concentrations of BDAA to determine the virus titer in a plaque assay in Vero cells (D). Values represent average results from a triplicate experiment (± standard deviations). EC50, EC90, and CC50 values were calculated using Prism 5 (GraphPad Software, Inc.).
TABLE 1.
Antiviral spectrum of BDAAa
| Virus | Family | Strain | Cell type | Assay | EC50 | EC90 | CC50 |
|---|---|---|---|---|---|---|---|
| Herpes simplex virus 1 | Herpesviridae | E-377 | HFF | CPE | >100 | ND | >100 |
| Human cytomegalovirus | AD169 | HFF | CPE | >100 | ND | >100 | |
| Rift Valley fever virus | Bunyaviridae | MP-12 | Vero 76 | CPE | 18 | ND | 43 |
| Tacaribe virus | Arenaviridae | TRVL11573 | Vero | CPE | 18 | ND | 40 |
| Venezuelan equine encephalitis virus | Togaviridae | TC-83 | Vero | CPE | 32 | ND | 32 |
| BK virus | Papovaviridae | Gardner | HFF | CPE | 62.4 | ND | >100 |
| JC virus | MAD-1 | Cos7 | qPCR | 95.5 | ND | >100 | |
| Vaccinia virus | Poxviridae | Copenhagen | HFF | CPE | >100 | ND | >100 |
| SARSb coronavirus | Coronaviridae | Toronto-2 | Vero E6 | CPE | >100 | ND | >100 |
| Influenza A H1N1 virus | Othomyxoviridae | Influenza A/California/7/2009 | MDCK | CPE | >100 | ND | >100 |
| Hepatitis B virus | Hepadnaviridae | ayw | HepG2.215 | Hybridization | >100 | ND | >100 |
| Respiratory syncytial virus | Paramyxoviridae | A | Hep-2 | CPE | >100 | ND | >100 |
| Human papillomavirus | Papillomaviridae | HPV-11 | HEK293 | qPCR | 3 | 71 | >100 |
| Norovirus | Caliciviridae | GI | HG23 | Hybridization | >100 | ND | >100 |
| Encephalomyocarditis virus | Picornaviridae | Huh7.5 | Yield reduction | >10 | ND | >100 | |
| Hepatitis C virus | Flaviviridae | Con-1 | Huh-luc/NeoET | Luciferase reporter | >20 | ND | >20 |
| HCVcc JC1 | Huh7.5 | qPCR | >10 | ND | >100 | ||
| Bovine viral diarrhea virus | NADL | MDBK | Yield reduction | >10 | ND | >100 | |
| West Nile virus | KERN515 WN02 | Vero 76 | CPE | 32 | ND | 32 | |
| Japanese encephalitis virus | SA-14 | Vero 76 | CPE | >57 | ND | 57 | |
| Powassen virus | LB | BHK-21 | CPE | 32 | ND | 32 | |
| Zika virus | Zika-016 | Vero 76 | CPE | >100 | ND | >100 | |
| Dengue virus | DENV-2 New guinea C | Vero | Yield reduction | ND | 8.6 | >50 | |
| Yellow fever virus | 17D | Vero 76 | CPE | 0.05 | ND | >50 | |
| Vero 76 | Yield reduction | ND | 0.03 | >50 | |||
| Huh7.5 | qPCR# | 0.21 | 0.37 | >100 | |||
| HEK293 | qPCR# | 0.17 | 0.30 | >50 | |||
| Vero | qPCR# | 0.13 | 0.27 | >100 |
All the assays were performed by the NIAID Pre-clinical Service Program, except for those marked with a #, which were conducted in-house at Baruch S. Blumberg Institute. The EC50, EC90, and CC50 values reported are the micromolar concentrations. ND, not determined.
SARS, severe acute respiratory syndrome.
Antiviral activity of BDAA enantiomers against YFV.
BDAA is a benzodiazepine compound with two chiral centers. We therefore synthesized and compared the antiviral activity of the four enantiomers against YFV (Fig. 2A). As shown in Fig. 2B, the rank order of the potency of the enantiomers to inhibit YFV RNA replication in Huh7.5 cells was (S,S) > (S,R) > (R,S) > (R,R), with EC50s of 0.2, 0.5, 2.4, and >10 μM, respectively. We note that the BDAA compound in our chemical library is an (S,S) enantiomer. These results suggest a critical role of the S chiral configuration at the alpha carbon atom in inhibition of YFV replication and establish a basis for further lead optimization efforts toward identification of benzodiazepine derivatives with superior pharmacological properties and antiviral activities against YFV.
FIG 2.

Antiviral activity of BDAA enantiomers. (A) Structures of BDAA and its enantiomers. (B) Determination of the antiviral activities of BDAA and its enantiomers from a qRT-PCR assay. Huh7.5 cells were cultured and treated as described for Fig. 1C. YFV RNAs were normalized to β-actin mRNA levels and are presented as percentages of mock-treated control results. Values represent the mean results from four independent replicates (± standard deviations). EC50 and EC90 values were calculated using Prism 5 (GraphPad Software, Inc.).
Antiviral and benzodiazepine receptor binding activities depend on distinct structural features.
Benzodiazepines are the most common antianxiety drugs administered around the world (50). This class of drugs works by allosterically modulating the binding of the neurotransmitter gamma-aminobutyric acid (GABA) to the GABAA receptor (GABAAR), an ionotropic receptor and ligand-gated ion channel, which results in sedative, hypnotic (sleep-inducing), anxiolytic (antianxiety), anticonvulsant, muscle relaxant, and other pharmacological properties (51). Benzodiazepine drugs can also interact with peripheral benzodiazepine receptors (PBR), which are structurally unrelated to GABAAR and are present in the peripheral nervous system and other tissues. Binding of benzodiazepines to PBRs modulates immune responses and other host functions (52).
Benzodiazepine is a chemical name for compounds with a core structure of fused benzene and diazepine rings. Benzodiazepine drugs are substituted 1,4-benzodiazepines with different side groups attached to this core structure. The different side groups affect the binding of the molecule to GABAAR and thus modulate the pharmacological properties. Interestingly, BDAA is structurally similar to benzodiazepine drugs used in the clinic. Therefore, it is very important to know whether BDAA binds GABAAR and/or peripheral benzodiazepine receptors. Obviously, our goal is to develop benzodiazepines with potent anti-YFV activity, but without psychiatric or other pharmacological/toxicological activities. Accordingly, the goals of our lead optimization efforts are not only to improve the antiviral activity, but also, more importantly, to focus on identifying lead compounds with favorable pharmacological properties and without GABAAR and/or PBR binding activities.
To investigate the structure-activity relationship (SAR) of benzodiazepines in antiviral and receptor binding activities, we first tested six representative clinical psychoactive benzodiazepine drugs (Fig. 3A) for their antiviral activities against YFV. As shown in Fig. 3B, while BDAA potently inhibited YFV RNA replication in Huh7.5 cells, none of the six clinically used benzodiazepine drugs demonstrated any detectable inhibitory effects on YFV infection. Next, we tested if BDAA competitively inhibited the binding of [3H]flunitrazepam and [3H]PK11195, representative benzodiazepine drugs, to their cognate receptors, GABAAR and PBR, respectively. In this assay, an inhibition of more than 50% of the labeled ligand binding by a test compound indicates a strong benzodiazepine receptor binding activity. On the contrary, an inhibition of less than 20% of the labeled ligand binding is interpreted as no specific binding activity of the test compound. As shown in Fig. 3C, while 0.3 μM diazepam and PK11195 completely inhibited the labeled ligands binding to GABAAR and PBR, respectively, 10 μM BDAA reduced the labeled ligand binding to their cognate receptors by less than 15%. The results thus indicate that the anti-YFV and psychiatric activities of benzodiazepines rely on distinct structural features, and development of benzodiazepine anti-YFV drugs without psychiatric activities is possible.
FIG 3.

The anti-YFV and psychiatric activities of benzodiazepines require distinct structural features. (A) Structures of six representative clinically psychoactive benzodiazepines. (B) Determination of the antiviral activities of clinically used benzodiazepines based on a qRT-PCR assay. Huh7.5 cells were cultured and treated as described for Fig. 1C. YFV RNA levels were normalized to β-actin mRNA levels and are presented as the percentage of the mock-treated control values. Values represent the mean results from four independent replicates (± standard deviations). (C) The affinities of BDAA to central and peripheral nervous system benzodiazepine receptors were measured in radioligand competitive binding assays. The binding of [3H]flunitrazepam and [3H]PK11195 to the corresponding benzodiazepine receptors was measured in the presence or absence of 10 μM BDAA, and the effects of BDAA on radioligand banding are expressed as the percentage of the mock-treatment control. The reference compounds diazepam and nonlabeled PK11195 with known benzodiazepine receptor binding activities were tested at 0.3 μM. Values represent the mean results from duplicate experiments (± standard deviations).
BDAA does not inhibit the early steps of YFV replication.
As the first step toward understanding the antiviral mechanism of BDAA, we performed a time-of-addition experiment to map the viral replication step(s) inhibited by the compound during a synchronized YFV infection of Huh7.5 cells. As shown in Fig. 4A, pretreatment as well as treatment of Huh7.5 cells during the virus inoculation period did not inhibit YFV infection, as indicated by a normal onset of viral RNA replication in the infected cultures. Those results imply that the compound neither inactivates the infectivity of YFV nor inhibits the infectious entry of YFV. Interestingly, addition of BDAA as late as 12 h postinfection was still able to potently inhibit viral RNA replication, and treatment starting at 24 h postinhibition reduced viral RNA by approximately 40%. A similar result was obtained in a parallel study that measured the YFV titers in the culture medium (Fig. 4B). These observations indicated that BDAA most likely inhibits a postentry step of YFV replication.
FIG 4.

Time-of-addition analysis of BDAA antiviral activity against YFV. Huh7.5 cells were infected with YFV at an MOI of 10 for 1 h. BDAA (5 μM) was present either transiently for 2 h before infection (−2 to 0), during infection (0, 1), or added at the indicated time points until 48 h postinfection. For each time point, 1% DMSO was added to mock-treated controls. Total intracellular RNAs were extracted, and culture media samples were collected at 48 h postinfection to determine the amounts of YFV RNA in a qRT-PCR assay (A) and virus titers were determinedin a plaque assay (B). YFV RNAs were normalized to β-actin mRNA levels. The graph denotes the percentages of YFV RNA relative to that of the mock-treated control added at the time of infection (time point 0). Virus titers in the culture media were measured by plaque assay in Vero cells. Each data point is the mean results from four independent replicates (± standard deviation).
Selection and genetic analysis of BDAA-resistant YFV.
In order to determine the viral target of BDAA, we selected drug-resistant YFV strains via a procedure depicted in Fig. 5A. Briefly, Huh7.5 cells were infected by YFV 17D and cultured in the presence of the indicated concentrations of BDAA. After 3 days, culture medium was harvested and inoculated into a fresh Huh7.5 culture in the presence of BDAA. Extensive CPE was observed at passage 18 in cells treated with 8 μM BDAA, suggesting the outgrowth of BDAA-resistant YFV. Resistance of the selected YFV strain to BDAA was validated in Huh7.5 cells with a virus yield reduction assay. As shown in Fig. 5B, BDAA reduced the yield of parental YFV 17D, with an EC50 of 0.1 μM, but reduced the yield of BDAA-selected YFV, with an EC50 of 3.2 μM. More strikingly, the EC90s of BDAA to reduce the yields of wild-type (WT) YFV 17D and BDAA-selected YFV were 0.9 and 28.5 μM, respectively, suggesting successful selection of highly BDAA-resistant YFV.
FIG 5.

Selection and characterization of BDAA-resistant YFV. (A) Schematic representation of the procedure to select BDAA-resistant YFV. At the 18th passage, CPE was observed in the culture treated with BDAA. The culture medium was harvested as a BDAA-resistant YFV stock. (B) Effects of BDAA on parental YFV 17D and BDAA-resistant virus were determined in a virus yield reduction assay. Values represent average results from two independent wells with duplication for virus titer titration. EC50 and EC90 values were calculated using Prism 5 (GraphPad Software, Inc.). (C) Alignment of the amino acid sequence flanking the putative NS4B protein transmembrane domain 5 (pTMD5) of YFV and other flaviviruses. The predicted membrane topology of YFV NS4B is shown at the top of amino acid sequence. Amino acid P219 of YFV is highlighted. GenBank accession numbers for the listed viruses are as follows: YFV strain 17D (X03700.1), YFV strain BeH655417 (JF912190.1), DENV-1 Western Pacific strain (U88536.1), DENV-2 New Guinea C strain (AF038403.1), DENV-3 H87 strain (KU050695.1), DENV-4 rDEN4 clone (AF326825.1), WNV NY99 (NC_009942.1), JEV JaOArS982 strain (M18370.1), and ZIKV strain BeH819966 (KU365779.1). The amino acid position of NS4B is numbered according to that for YFV 17D.
In order to map the BDAA resistance mutations, six overlapped cDNA segments covering the entire genomic RNA of the drug-resistant YFV as well as parental YFV 17D were amplified by RT-PCR. The PCR products were subjected for direct DNA sequencing. Sequence alignments between the parental and drug-resistant viruses revealed only a single-nucleotide mutation, from C to U, at nucleotide 7423 in BDAA-resistant YFV, which resulted in the replacement of proline at amino acid 219 (P219) of the NS4B protein with a serine (Fig. 5C). Sequence alignment of NS4B indicated that P219 is absolutely conserved in all the 38 full-length YFV sequences deposited in GenBank, but it is replaced with a threonine at the position in DENV, West Nile virus (WNV), and Japanese Encephalitis virus (JEV) or with a serine in Zika virus (ZIKV). Such a difference may account for the selective inhibition of BDAA for YFV but not other flaviviruses.
The flaviviral NS4B are integral membrane proteins and are primarily localized in cytoplasmic foci originating from the endoplasmic reticulum (ER) (53). Studies of DENV NS4B suggest that the protein is an essential component of the viral RNA replication complex (54). It has also been demonstrated that the DENV NS4B protein antagonizes the virus-induced type I interferon response and may thus play a role in viral pathogenesis (55). Bioinformatics analysis predicts that the YFV NS4B protein contains five putative intra- or transmembrane domains (pTMD1 to pTMD5), with a membrane topology in which the N-terminal part resides in the ER lumen, while the C-terminal part is composed of three transmembrane domains with the C-terminal tail localized in the cytoplasm. This model is generally consistent with experimentally determined topology of the DENV NS4B protein (53). P219 resides at the N terminus of the fifth putative transmembrane domain, pTMD5 (Fig. 5C).
Effects of P219 substitutions on YFV replication and resistance.
To confirm that P219S mutation does confer BDAA resistance and to investigate the role of P219 in NS4B function and BDAA inhibition of YFV replication, we produced YFV 17D viruses with a P219S, P219T, P219A, or P219G mutation by engineering the desired mutations in the infectious YFV 17D cDNA clone and rescuing the mutant viruses by transfection of in vitro-transcribed RNA into Huh7.5 cells. While parental YFV 17D, P219S, or P219T RNA induced CPE at day 4 posttransfection, P219G and P219A RNA caused CPE at day 5 and 6 posttransfection, respectively. At the day of CPE appearance, culture media were harvested. The cells were lysed in serum-free DMEM by three freeze-thaw cycles. The cell lysates were cleared by centrifugation at 1,000 × g and 4°C for 10 min. The virus titers in culture media were determined in Vero cells via a plaque assay. While the parental YFV 17D, P219S, P219T, and P219A RNA-transfected cells produced similar amounts of viruses, ranging from 8 × 104 to 1.2 × 105 PFU/ml, and caused similar sizes of plaques in Vero cell monolayers, no infectious virus could be detected from the culture medium of P219G RNA-transfected cells. We then compared the yields of cell-free (medium) and cell-associated (lysate) viruses between YFV 17D and P219G RNA-transfected cells in a plaque assay in Huh7.5 cell monolayers. We found that while the yields of cell-associated viruses were similar between YFV 17D and P219G RNA-transfected cells, P219G RNA-transfected cells produced approximately 60-fold less cell-free viruses than did YFV 17D RNA-transfected cells. Moreover, P219G virus produced smaller plaques. These results indicated that the NS4B P219G mutation may compromise virus replication and/or secretion of infectious virions.
To investigate the biological properties of the mutant viruses, we first examined the cell-free viruses rescued from Huh7.5 cells transfected with parental YFV 17D, P219S, P219T, or P219A RNA. Infection of the three mutant viruses caused an accumulation of viral RNA and produced infectious virions with similar kinetics in Huh7.5 cells as did parental YFV 17D (Fig. 6A and B). We next determined the sensitivity of the three mutant viruses to BDAA. As shown in Fig. 6C and D, while BDAA efficiently inhibited wild-type YFV 17D RNA replication and reduced viral yields, all three mutant viruses were resistant to BDAA, and the P219S mutant virus demonstrated the highest level of resistance to BDAA inhibition of viral RNA replication and infectious virion production.
FIG 6.

Antiviral activity of BDAA against replication-competent recombinant YFV with P219 mutations in Huh7.5 cells. (A and B) Replication kinetics of recombinant viruses with different P219 mutations. Huh7.5 cells were infected at an MOI of 0.1 for 1 h. Total cellular RNAs were extracted, and the culture media were collected from cells at the indicated time points postinfection. YFV RNA was detected by a qRT-PCR assay and normalized to β-actin mRNA levels. The YFV RNA arbitrary units are the amount relative to that of the RNA level in uninfected cells (A). Supernatants from the cultures were used to determine the virus yields in a plaque assay (right panel). Data presented are mean results from two independent replicates with duplication in the plaque assay (± standard deviations) (B). (C and D) Sensitivity of recombinant viruses to BDAA. Huh7.5 cells were infected with YFV 17D or mutant virus at an MOI of 0.1 and mock treated or treated with indicated concentrations of BDAA for 48 h. Total cellular RNAs were extracted to detect YFV RNA in a qRT-PCR assay, using β-actin as an internal control. Values are the percentage relative to mock-treated controls (C). Supernatants from the cultures were harvested to determine the virus yields in a plaque assay in Vero cells (D). Data are mean results from four independent replicate experiments (± standard deviation).
In order to determine the effects of the P219G substitution on viral replication and virion secretion, we first infected Huh7.5 cells with cell-associated viruses harvested from YFV 17D and P219G RNA-transfected Huh7.5 cells to compare the kinetics of viral RNA replication (Fig. 7A) and production of cell-associated (Fig. 7B) and secreted (Fig. 7C) viruses. The results clearly demonstrated that the P219G mutant YFV has approximately 10-fold lower viral RNA replication as well as production of both cell-associated and cell-free viruses, suggesting that the mutation compromises viral RNA replication but does not directly affect virion assembly and secretion. Moreover, despite lower replication and progeny virion production, the P219G virus is highly resistant to BDAA in Huh7.5 cells (Fig. 7D and E).
FIG 7.

Antiviral activity of BDAA against recombinant YFV with a P219G mutation in Huh7.5 cells. (A to C) Replication kinetics of P219G recombinant YFV. Huh7.5 cells were infected with cell-associated viruses harvested from YFV 17D or P219G RNA-transfected Huh7.5 cells at an MOI of 0.1 for 1 h. Intracellular viral RNA (A) and yields of cell-associated virions (B) and secreted infectious virions (C) at the indicated times postinfection were quantified in a qRT-PCR assay and plaque assay, respectively. YFV RNA was normalized as described for Fig. 6A. (D and E) Sensitivity of YFV P219G to BDAA. Huh7.5 cells were infected with cell-associated YFV 17D or P219G YFV at an MOI of 0.1 and mock treated or treated with the indicated concentrations of BDAA for 48 h. Total cellular RNA was extracted to detect YFV RNA by qRT-PCR using β-actin as an internal control. Values are percentages relative to mock-treated controls (D). Supernatants from the cultures were harvested to determine the virus yields in a plaque assay in Vero cells (E). Data presented are means from four independent replicate experiments (± standard deviation).
The antiviral effect of BDAA against wild-type 17D and mutant recombinant YFV was further tested in the mosquito cell line C6/C36. Figure 8 shows that the resistance profile in C6/C36 cells was similar to that in Huh7.5 cells. While BDAA efficiently inhibited wild-type YFV 17D RNA replication, with an EC50 of 0.08 μM, all three mutant viruses were resistant to BDAA, with EC50s ranging from 0.4 to 4.7 μM, among which the P219S mutant virus demonstrated the highest level of resistance to BDAA inhibition.
FIG 8.
Sensitivity of YFV to BDAA in C6/C36 cells. C6/C36 cells were infected with YFV 17D or mutant virus at an MOI of 0.1 and mock treated or treated with indicated concentrations of BDAA for 48 h. Total cellular RNAs were extracted to detect YFV RNA in a qRT-PCR assay, using Aedes albopictus S7 ribosomal mRNA as the internal control. Values are percentages relative to mock-treated controls. Data presented are means from four independent replicates (± standard deviation).
Taken together, our results imply that while proline 219 is critical to confer sensitivity to BDAA, it is not essential for YFV replication, because it can be replaced with amino acids containing a small nonpolar hydrophobic side chain (A) or polar uncharged side chain (S or T) without apparent loss of replication fitness in both human and mosquito cells. Mechanistically, the similar resistant profiles of NS4B mutants in both the human and mosquito cells imply that the amino acid P219 residues may mediate the direct binding of BDAA to the YFV NS4B protein. Nevertheless, the high conservation of P219 in all the clinical/field YFV isolates suggests that P219 is important for viral replication fitness in human and mosquitoes and thus favors the development of BDAA or its derivatives as antiviral agents for the treatment of yellow fever.
Evaluation of pharmacological and toxicological properties of BDAA.
In order to determine the dosing schedule and route of administration for the in vivo antiviral efficacy study in animals, we first determined the pharmacokinetics of BDAA in mice. A single dose of BDAA was administered i.v., p.o., s.c., i.m., or i.p. routes, and blood samples were obtained at various time points postadministration to determine the plasma drug concentrations. The pharmacokinetic studies of BDAA indicated that BDAA demonstrated good oral bioavailability (35%) and good retention of drug. For instance, at 8 h post-oral administration of 10 mg/kg BDAA, the plasma concentration of BDAA was maintained above 0.2 μM. Assuming a linear relationship was maintained with increased dosages, at a dose of 50 mg/kg, BDAA could be maintained in the plasma at the level that is above the EC90 for inhibition of YFV in culture, for at least 8 h post-single oral dosing (data not shown).
In order to find the MTD of BDAA in animals, a repeated-dose oral and i.p. MTD study was performed in mice. The results showed that oral administration of BDAA at the highest dose tested (100 mg/kg three times daily for 7 days) was very well tolerated. However, administration of BDAA with a dosing schedule of 100 mg/kg three times daily via the i.p. route caused significant toxicity to all the animals, which were euthanized on day 4 or 5. A repeated-dose oral MTD study was also performed in hamsters and showed that BDAA was well tolerated at the highest dose tested (150 mg/kg twice daily for 7 days).
Antiviral efficacy of BDAA in YFV-infected hamsters.
The above in vivo toxicology studies showed that BDAA is better tolerated with oral administration. The highest equilibrium solubility of BDAA in solutol:cremaphore:saline diluent was 100 mg/ml. Due to its relatively low aqueous solubility, it was difficult to keep BDAA in solution in order to dose the animals at the 150-mg/kg dose. Hence, a 100-mg/kg/dose level was selected as the highest dose for the efficacy study. Grouping and treatment schedules for YFV-infected hamsters are described in the legend of Fig. 9. The infected animal survival rate, body weight changes between 3 to 6 dpi, levels of serum ALT at 6 dpi, and also virus titers at 4 dpi in serum were measured to determine the activities of the test compound in suppressing viral replication and improving viral disease management.
FIG 9.

In vivo efficacy of BDAA in a hamster model of lethal YFV infection. Female Syrian golden hamsters were challenged with Jimenez, a hamster-adapted YFV strain, via i.p. injection (10 CC50/animal). Three groups of infected animals were treated with escalated doses of BDAA twice daily (BID) via oral administration for 7 days, beginning 4 h prior to infection. Animals treated with ribavirin served as positive controls, and a placebo-treated group served as a negative control. Each infected group had 10 animals. Sham-infected controls were either treated with the highest dose of BDAA to monitor toxicity or placebo treated (n = 5). A group of 5 animals without any treatment served as normal controls. (A) The effect of treatment on mortality of hamsters infected with YFV. *, P < 0.05 compared with placebo. (B) Weight change between 3 and 6 dpi from hamsters infected with YFV and treated with various doses of BDAA or with ribavirin. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (compared with placebo). (C) The effect of BDAA at various doses or ribavirin on serum ALT at 6 dpi from hamsters infected with YFV. ***, P < 0.001; **, P < 0.01. (D) Virus titers in serum on day 4 of YFV-infected hamsters treated with various doses of BDAA or with ribavirin. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
As shown in Fig. 9A, improvement of YFV-infected animal survival was observed in groups treated with 100 mg/kg or 50 mg/kg of BDAA twice daily orally, although this was only statistically significant (P < 0.05 [compared with the placebo control group]) for the group treated with 50 mg/kg of BDAA. This was likely due to the relatively higher survival rate in the placebo-treated group. Both of these two dosing groups performed slightly better than the ribavirin-treated controls (70% survival). The lowest dose of 25 mg/kg BDAA twice daily resulted in a survival curve that was similar to that for placebo treatment.
Comparing the weight changes between 3 and 6 dpi among the treatment groups, significant improvements in animals treated with either 100 or 50 mg/kg (P < 0.01 and P < 0.05, respectively) were observed. However, treatment with 25 mg/kg of BDAA resulted in a weight change curve similar to that for the placebo control group. The weight change curve of animals treated with ribavirin was similar to that of normal control animals and was significantly improved compared with placebo (P < 0.001) (Fig. 9B).
Serum levels of ALT, a biomarker for hepatocyte injury, at 6 dpi were significantly reduced in animals treated with 100 mg/kg of BDAA (P < 0.01). A trend toward reduced ALT was also observed in the 50-mg/kg/dose group, although this reduction was not significant. As with survival and weight change, serum ALT levels in animals treated with 25 mg/kg of BDAA were similar to those in placebo-treated animals, further confirming the inactivity of the compound at this dose. Ribavirin treatment also significantly reduced serum ALT, as expected (P < 0.001) (Fig. 9C).
Finally, dosing with 100 mg/kg of BDAA beginning just prior to virus challenge resulted in an approximate 2-log10 reduction in average virus titer in serum at 4 dpi compared with placebo treatment, which was statistically significant (P < 0.001). A reduction in virus titer was also observed in animals treated with 50 mg/kg of BDAA (P < 0.05) or in those treated with ribavirin (P < 0.01) (Fig. 9D).
In the efficacy studies, sham-infected animals were also included as controls. In sham-infected animals, treatment with 100 mg/kg BDAA twice daily for 7 days did not produce any signs of toxicity, as judged by weight changes and ALT levels, compared with placebo-treated animals and normal animals without sham infection or treatment.
DISCUSSION
We report herein the discovery of a benzodiazepine compound, BDAA, that potently inhibits YFV replication by specifically targeting NS4B protein. Because benzodiazepines are well-known as antianxiety drugs that bind GABAA receptor and PBRs, intensive attention has been paid to the potential neurologic and psychiatric activities of BDAA in our studies. Fortunately, we obtained several lines of evidence suggesting that the anti-YFV and psychiatric activities of benzodiazepines rely on distinct structural features. Specifically, while all six psychoactive benzodiazepine drugs do not inhibit YFV replication, BDAA had negligible GABAAR and PBR binding activities (Fig. 3). Furthermore, in our repeated-dose maximum tolerated dose studies, central nervous system effects, including tremors and convulsions, changes in the level of activity, gait, and posture, reactivity to handling or sensory stimuli, altered strength, and stereotypic or bizarre behavior (e.g., self-mutilation, walking backwards), were not observed in all the BDAA-treated mice. In addition, compounds with the benzodiazepine scaffold have been reported to have antiviral activities against respiratory syncytial virus (56), hepatitis B virus (57), filoviruses (58), and human immunodeficiency virus (59). Hence, development of benzodiazepine derivatives with potent anti-YFV activity, but without psychiatric side effects, is plausible.
Concerning the antiviral mechanism of BDAA, a single amino acid substitution in NS4B, P219S, was identified to confer YFV resistance to BDAA. NS4B is a nonenzymatic integral membrane protein (53). Flaviviral NS4B proteins participate in viral RNA replication and evasion of host innate immune response (55). The critical roles of this protein in viral replication and pathogenesis are well illustrated by the fact that many cell culture and animal adaptive mutations map to NS4B (60). Although the exact functional involvement of flavivirus NS4B in the viral replication cycle and the molecular mechanism to evade host innate immunity remain to be determined, prior studies have already demonstrated that NS4B dimerization and interaction with other viral nonstructural proteins, including NS1, NS2A, NS3, and NS4A, as well as many host cellular proteins, are important for its function in viral RNA replication and antagonism of the immune response (14, 31, 61, 62). It is thus conceivable that blocking the NS4B protein interaction with other viral and host cellular proteins could lead to direct inhibition of viral RNA replication or, alternatively, could act through activation of the host innate antiviral response to restrict viral replication.
Interestingly, several structurally distinct compounds have been identified that inhibit DENV or YFV replication through targeting NS4B (40, 63–67). In addition, multiple inhibitors of the NS4B protein of hepatitis C virus, a distantly related member of the Flaviviridae family, are currently under preclinical and clinical development for treatment of hepatitis C infection (68). Intriguingly, as summarized in Fig. 10, except for F164L, which confers DENV resistance to SDM25N and AM404 and localizes at the cytosolic loop between TMD3 and TMD4 (65, 67), all other drug resistance mutations are mapped in the transmembrane domains. Furthermore, except for DENV V63 and YFV P219 mutations, which specifically confer resistance to spiropyrazolopyridone compound-14a and BDAA, respectively, all other DENV and YFV drug resistance mutations identified thus far confer resistance to multiple drugs (25). Particularly, while each of the DENV P104, A119, and F164 mutations confers resistance to AM404 and/or SDM25N (65, 67), the DENV T108 mutation confers resistance to AZD0530 and dasatinib, which disrupt DENV RNA replication by inhibition of cellular Fyn kinase (64). Accordingly, we speculate that while spiropyrazolopyridone and BDAA, the only two NS4B inhibitors with a very narrow antiviral spectrum and demonstrated in vivo antiviral efficacy, may directly target NS4B protein; other NS4B inhibitors may disrupt NS4B function via targeting host cellular enzymes or other proteins that interact with NS4B or regulate NS4B function. The fact that the majority of drug resistance mutations map within the transmembrane domain of NS4B strongly suggests that interruption of the “intramembrane” interaction of NS4B with other viral and host membrane proteins might be the most important targets of the small molecular antiviral compounds. However, because the DENV TMD5, spanning the membrane from the ER lumen to the cytoplasm, may flip back to the ER lumen upon cleavage at its C terminus by NS2B/NS3 protease during polyprotein processing (53), it is also possible that BDAA interacts with YFV NS4B to alter its membrane topology. In fact, proline is frequently observed as the first residue of a transmembrane α helix due to its conformational rigidity, whereas replacement with a glycine tends to disrupt the helical structure due to its high flexibility. Hence, the observed reduction of replication fitness of P219G YFV could be due to an altered membrane topology of pTMD5. Moreover, the strict requirement of P219 for the susceptibility of YFV to BDAA strongly suggests that YFV NS4B structural features conferred by this specific amino acid residue may favor BDAA binding. This hypothesis will be investigated in our future studies.
FIG 10.

Flavivirus NS4B membrane topology and locations of drug-resistant amino acid substitutions.
Traditionally, virus-encoded enzymes are primary antiviral targets (17, 69). However, nonenzymatic viral structural and nonstructural proteins have been shown to be the targets of highly selective and potent antiviral agents over the last decade, as highlighted by FDA-approved HCV NS5A inhibitor as well as HCV NS4B inhibitors, and also HIV and HBV capsid inhibitors that are currently under development (70–72). Among the flaviviral NS4B inhibitors, only spiropyrazolopyridone compound 14a was recently reported to significantly reduce the viremia of DENV-2 in an AG129 mice model when mice were treated with the inhibitor immediately after infection or when treatment was delayed for 48 h (63). Herein, we showed, for the first time, that an YFV NS4B inhibitor, BDAA, protected 90% of YFV-infected hamsters from death, significantly reduced viral load by greater than 2 logs, and attenuated virus infection-induced liver injury and body weight loss. We have thus proved the concept that NS4B is a viable antiviral target against YFV and we have reached a critical milestone toward the development of specific antiviral therapeutics for clinical management of yellow fever. In addition, potent and safe anti-YFV agents may also be used as prophylaxis means for nonimmunized residents and travelers during outbreaks in regions where YFV is endemic.
ACKNOWLEDGMENTS
We acknowledge support from the Nonclinical and Preclinical Services Program of NIAID, National Institutes of Health, Department of Health and Human Services, under contracts HHSN2722011000221 (scale-up synthesis), HHSN272201100019I (in vitro antiviral spectrum screening), and HHSN272201000039I (efficacy study with hamsters).
Funding Statement
This work was supported by the National Institutes of Health (AI104636), a sponsored research program from Unither Virology, LLC, and the Hepatitis B Foundation (through an appropriation from the Commonwealth of Pennsylvania). The founders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Monath TP, Vasconcelos PF. 2015. Yellow fever. J Clin Virol 64:160–173. doi: 10.1016/j.jcv.2014.08.030. [DOI] [PubMed] [Google Scholar]
- 2.Poland JD, Calisher CH, Monath TP, Downs WG, Murphy K. 1981. Persistence of neutralizing antibody 30-35 years after immunization with 17D yellow fever vaccine. Bull World Health Organ 59:895–900. [PMC free article] [PubMed] [Google Scholar]
- 3.Reinhardt B, Jaspert R, Niedrig M, Kostner C, L'Age-Stehr J. 1998. Development of viremia and humoral and cellular parameters of immune activation after vaccination with yellow fever virus strain 17D: a model of human flavivirus infection. J Med Virol 56:159–167. doi: 10.1002/(SICI)1096-9071(199810)56:2<159::AID-JMV10>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 4.Jentes ES, Poumerol G, Gershman MD, Hill DR, Lemarchand J, Lewis RF, Staples JE, Tomori O, Wilder-Smith A, Monath TP, Informal WHO Working Group on Geographic Risk for Yellow Fever . 2011. The revised global yellow fever risk map and recommendations for vaccination, 2010: consensus of the Informal WHO Working Group on Geographic Risk for Yellow Fever. Lancet Infect Dis 11:622–632. doi: 10.1016/S1473-3099(11)70147-5. [DOI] [PubMed] [Google Scholar]
- 5.Maciel M Jr, Cruz Fda S, Cordeiro MT, da Motta MA, Cassemiro KM, Maia Rde C, de Figueiredo RC, Galler R, Freire Mda S, August JT, Marques ET, Dhalia R. 2015. A DNA vaccine against yellow fever virus: development and evaluation. PLoS Negl Trop Dis 9:e0003693. doi: 10.1371/journal.pntd.0003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muyanja E, Ssemaganda A, Ngauv P, Cubas R, Perrin H, Srinivasan D, Canderan G, Lawson B, Kopycinski J, Graham AS, Rowe DK, Smith MJ, Isern S, Michael S, Silvestri G, Vanderford TH, Castro E, Pantaleo G, Singer J, Gillmour J, Kiwanuka N, Nanvubya A, Schmidt C, Birungi J, Cox J, Haddad EK, Kaleebu P, Fast P, Sekaly RP, Trautmann L, Gaucher D. 2014. Immune activation alters cellular and humoral responses to yellow fever 17D vaccine. J Clin Invest 124:3147–3158. doi: 10.1172/JCI75429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akondy RS, Johnson PL, Nakaya HI, Edupuganti S, Mulligan MJ, Lawson B, Miller JD, Pulendran B, Antia R, Ahmed R. 2015. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc Natl Acad Sci U S A 112:3050–3055. doi: 10.1073/pnas.1500475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Julander JG. 2013. Experimental therapies for yellow fever. Antiviral Res 97:169–179. doi: 10.1016/j.antiviral.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beasley DW, McAuley AJ, Bente DA. 2015. Yellow fever virus: genetic and phenotypic diversity and implications for detection, prevention and therapy. Antiviral Res 115:48–70. doi: 10.1016/j.antiviral.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Harris E, Holden KL, Edgil D, Polacek C, Clyde K. 2006. Molecular biology of flaviviruses. Novartis Found Symp 277:23–39. doi: 10.1002/0470058005.ch3. [DOI] [PubMed] [Google Scholar]
- 11.Selisko B, Wang C, Harris E, Canard B. 2014. Regulation of flavivirus RNA synthesis and replication. Curr Opin Virol 9:74–83. doi: 10.1016/j.coviro.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diamond MS, Pierson TC. 2015. Molecular insight into dengue virus pathogenesis and its implications for disease control. Cell 162:488–492. doi: 10.1016/j.cell.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laurent-Rolle M, Morrison J, Rajsbaum R, Macleod JM, Pisanelli G, Pham A, Ayllon J, Miorin L, Martinez-Romero C, ten Oever BR, Garcia-Sastre A. 2014. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 16:314–327. doi: 10.1016/j.chom.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munoz-Jordan JL, Laurent-Rolle M, Ashour J, Martinez-Sobrido L, Ashok M, Lipkin WI, Garcia-Sastre A. 2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J Virol 79:8004–8013. doi: 10.1128/JVI.79.13.8004-8013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim SP, Wang QY, Noble CG, Chen YL, Dong H, Zou B, Yokokawa F, Nilar S, Smith P, Beer D, Lescar J, Shi PY. 2013. Ten years of dengue drug discovery: progress and prospects. Antiviral Res 100:500–519. doi: 10.1016/j.antiviral.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Behnam MA, Nitsche C, Boldescu V, Klein CD. 2016. The medicinal chemistry of dengue virus. J Med Chem 59:5622–5649. doi: 10.1021/acs.jmedchem.5b01653. [DOI] [PubMed] [Google Scholar]
- 17.Chang J, Block TM, Guo JT. 2013. Antiviral therapies targeting host ER alpha-glucosidases: current status and future directions. Antiviral Res 99:251–260. doi: 10.1016/j.antiviral.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noble CG, Chen YL, Dong H, Gu F, Lim SP, Schul W, Wang QY, Shi PY. 2010. Strategies for development of dengue virus inhibitors. Antiviral Res 85:450–462. doi: 10.1016/j.antiviral.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 19.Noble CG, Shi PY. 2012. Structural biology of dengue virus enzymes: towards rational design of therapeutics. Antiviral Res 96:115–126. doi: 10.1016/j.antiviral.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Scaturro P, Trist IM, Paul D, Kumar A, Acosta EG, Byrd CM, Jordan R, Brancale A, Bartenschlager R. 2014. Characterization of the mode of action of a potent dengue virus capsid inhibitor. J Virol 88:11540–11555. doi: 10.1128/JVI.01745-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu H, Bock S, Snitko M, Berger T, Weidner T, Holloway S, Kanitz M, Diederich WE, Steuber H, Walter C, Hofmann D, Weissbrich B, Spannaus R, Acosta EG, Bartenschlager R, Engels B, Schirmeister T, Bodem J. 2015. Novel dengue virus NS2B/NS3 protease inhibitors. Antimicrob Agents Chemother 59:1100–1109. doi: 10.1128/AAC.03543-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timiri AK, Sinha BN, Jayaprakash V. 2016. Progress and prospects on DENV protease inhibitors. Eur J Med Chem 117:125–143. doi: 10.1016/j.ejmech.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 23.Behnam MA, Graf D, Bartenschlager R, Zlotos DP, Klein CD. 2015. Discovery of nanomolar dengue and West Nile virus protease inhibitors containing a 4-benzyloxyphenylglycine residue. J Med Chem 58:9354–9370. doi: 10.1021/acs.jmedchem.5b01441. [DOI] [PubMed] [Google Scholar]
- 24.Byrd CM, Grosenbach DW, Berhanu A, Dai D, Jones KF, Cardwell KB, Schneider C, Yang G, Tyavanagimatt S, Harver C, Wineinger KA, Page J, Stavale E, Stone MA, Fuller KP, Lovejoy C, Leeds JM, Hruby DE, Jordan R. 2013. Novel benzoxazole inhibitor of dengue virus replication that targets the NS3 helicase. Antimicrob Agents Chemother 57:1902–1912. doi: 10.1128/AAC.02251-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie X, Zou J, Wang QY, Shi PY. 2015. Targeting dengue virus NS4B protein for drug discovery. Antiviral Res 118:39–45. doi: 10.1016/j.antiviral.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Zou B, Chan WL, Ding M, Leong SY, Nilar S, Seah PG, Liu W, Karuna R, Blasco F, Yip A, Chao A, Susila A, Dong H, Wang QY, Xu HY, Chan K, Wan KF, Gu F, Diagana TT, Wagner T, Dix I, Shi PY, Smith PW. 2015. Lead optimization of spiropyrazolopyridones: a new and potent class of dengue virus inhibitors. ACS Med Chem Lett 6:344–348. doi: 10.1021/ml500521r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim SP, Sonntag LS, Noble C, Nilar SH, Ng RH, Zou G, Monaghan P, Chung KY, Dong H, Liu B, Bodenreider C, Lee G, Ding M, Chan WL, Wang G, Jian YL, Chao AT, Lescar J, Yin Z, Vedananda TR, Keller TH, Shi PY. 2011. Small molecule inhibitors that selectively block dengue virus methyltransferase. J Biol Chem 286:6233–6240. doi: 10.1074/jbc.M110.179184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim SP, Noble CG, Shi PY. 2015. The dengue virus NS5 protein as a target for drug discovery. Antiviral Res 119:57–67. doi: 10.1016/j.antiviral.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 29.Chen YL, Yokokawa F, Shi PY. 2015. The search for nucleoside/nucleotide analog inhibitors of dengue virus. Antiviral Res 122:12–19. doi: 10.1016/j.antiviral.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 30.Byrd CM, Dai D, Grosenbach DW, Berhanu A, Jones KF, Cardwell KB, Schneider C, Wineinger KA, Page JM, Harver C, Stavale E, Tyavanagimatt S, Stone MA, Bartenschlager R, Scaturro P, Hruby DE, Jordan R. 2013. A novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob Agents Chemother 57:15–25. doi: 10.1128/AAC.01429-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou J, Xie X, Wang QY, Dong H, Lee MY, Kang C, Yuan Z, Shi PY. 2015. Characterization of dengue virus NS4A and NS4B protein interaction. J Virol 89:3455–3470. doi: 10.1128/JVI.03453-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin Z, Chen YL, Schul W, Wang QY, Gu F, Duraiswamy J, Kondreddi RR, Niyomrattanakit P, Lakshminarayana SB, Goh A, Xu HY, Liu W, Liu B, Lim JY, Ng CY, Qing M, Lim CC, Yip A, Wang G, Chan WL, Tan HP, Lin K, Zhang B, Zou G, Bernard KA, Garrett C, Beltz K, Dong M, Weaver M, He H, Pichota A, Dartois V, Keller TH, Shi PY. 2009. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A 106:20435–20439. doi: 10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen NM, Tran CN, Phung LK, Duong KT, Huynh Hle A, Farrar J, Nguyen QT, Tran HT, Nguyen CV, Merson L, Hoang LT, Hibberd ML, Aw PP, Wilm A, Nagarajan N, Nguyen DT, Pham MP, Nguyen TT, Javanbakht H, Klumpp K, Hammond J, Petric R, Wolbers M, Nguyen CT, Simmons CP. 2013. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis 207:1442–1450. doi: 10.1093/infdis/jis470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen YL, Abdul Ghafar N, Karuna R, Fu Y, Lim SP, Schul W, Gu F, Herve M, Yokohama F, Wang G, Cerny D, Fink K, Blasco F, Shi PY. 2014. Activation of peripheral blood mononuclear cells by dengue virus infection depotentiates balapiravir. J Virol 88:1740–1747. doi: 10.1128/JVI.02841-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Julander JG, Bantia S, Taubenheim BR, Minning DM, Kotian P, Morrey JD, Smee DF, Sheridan WP, Babu YS. 2014. BCX4430, a novel nucleoside analog, effectively treats yellow fever in a hamster model. Antimicrob Agents Chemother 58:6607–6614. doi: 10.1128/AAC.03368-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Julander JG, Furuta Y, Shafer K, Sidwell RW. 2007. Activity of T-1106 in a hamster model of yellow fever virus infection. Antimicrob Agents Chemother 51:1962–1966. doi: 10.1128/AAC.01494-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Julander JG, Shafer K, Smee DF, Morrey JD, Furuta Y. 2009. Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106. Antimicrob Agents Chemother 53:202–209. doi: 10.1128/AAC.01074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Julander JG, Jha AK, Choi JA, Jung KH, Smee DF, Morrey JD, Chu CK. 2010. Efficacy of 2′-C-methylcytidine against yellow fever virus in cell culture and in a hamster model. Antiviral Res 86:261–267. doi: 10.1016/j.antiviral.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Julander JG, Morrey JD, Blatt LM, Shafer K, Sidwell RW. 2007. Comparison of the inhibitory effects of interferon alfacon-1 and ribavirin on yellow fever virus infection in a hamster model. Antiviral Res 73:140–146. doi: 10.1016/j.antiviral.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patkar CG, Larsen M, Owston M, Smith JL, Kuhn RJ. 2009. Identification of inhibitors of yellow fever virus replication using a replicon-based high-throughput assay. Antimicrob Agents Chemother 53:4103–4114. doi: 10.1128/AAC.00074-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice CM, Lenches EM, Eddy SR, Shin SJ, Sheets RL, Strauss JH. 1985. Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science 229:726–733. doi: 10.1126/science.4023707. [DOI] [PubMed] [Google Scholar]
- 43.Bredenbeek PJ, Kooi EA, Lindenbach B, Huijkman N, Rice CM, Spaan WJ. 2003. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J Gen Virol 84:1261–1268. doi: 10.1099/vir.0.18860-0. [DOI] [PubMed] [Google Scholar]
- 44.Qu X, Pan X, Weidner J, Yu W, Alonzi D, Xu X, Butters T, Block T, Guo JT, Chang J. 2011. Inhibitors of endoplasmic reticulum α-glucosidases potently suppress hepatitis C virus virion assembly and release. Antimicrob Agents Chemother 55:1036–1044. doi: 10.1128/AAC.01319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo F, Zhao X, Gill T, Zhou Y, Campagna M, Wang L, Liu F, Zhang P, DiPaolo L, Du Y, Xu X, Jiang D, Wei L, Cuconati A, Block TM, Guo JT, Chang J. 2014. An interferon-beta promoter reporter assay for high throughput identification of compounds against multiple RNA viruses. Antiviral Res 107:56–65. doi: 10.1016/j.antiviral.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang D, Weidner JM, Qing M, Pan XB, Guo H, Xu C, Zhang X, Birk A, Chang J, Shi PY, Block TM, Guo JT. 2010. Identification of five interferon-induced cellular proteins that inhibit West Nile virus and dengue virus infections. J Virol 84:8332–8341. doi: 10.1128/JVI.02199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang J, Warren TK, Zhao X, Gill T, Guo F, Wang L, Comunale MA, Du Y, Alonzi DS, Yu W, Ye H, Liu F, Guo JT, Mehta A, Cuconati A, Butters TD, Bavari S, Xu X, Block TM. 2013. Small molecule inhibitors of ER alpha-glucosidases are active against multiple hemorrhagic fever viruses. Antiviral Res 98:432–440. doi: 10.1016/j.antiviral.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le Fur G, Vaucher N, Perrier ML, Flamier A, Benavides J, Renault C, Dubroeucq MC, Gueremy C, Uzan A. 1983. Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]RO5-4864 and [3H]PK 11195, by thermodynamic studies. Life Sci 33:449–457. doi: 10.1016/0024-3205(83)90794-4. [DOI] [PubMed] [Google Scholar]
- 49.Speth RC, Wastek GJ, Yamamura HI. 1979. Benzodiazepine receptors: temperature dependence of [3H]flunitrazepam binding. Life Sci 24:351–357. doi: 10.1016/0024-3205(79)90331-X. [DOI] [PubMed] [Google Scholar]
- 50.Taliani S, Da Settimo F, Da Pozzo E, Chelli B, Martini C. 2009. Translocator protein ligands as promising therapeutic tools for anxiety disorders. Curr Med Chem 16:3359–3380. doi: 10.2174/092986709789057653. [DOI] [PubMed] [Google Scholar]
- 51.Sieghart W, Ramerstorfer J, Sarto-Jackson I, Varagic Z, Ernst M. 2012. A novel GABA(A) receptor pharmacology: drugs interacting with the alpha(+) beta(-) interface. Br J Pharmacol 166:476–485. doi: 10.1111/j.1476-5381.2011.01779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zisterer DM, Williams DC. 1997. Peripheral-type benzodiazepine receptors. Gen Pharmacol 29:305–314. doi: 10.1016/S0306-3623(96)00473-9. [DOI] [PubMed] [Google Scholar]
- 53.Miller S, Sparacio S, Bartenschlager R. 2006. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J Biol Chem 281:8854–8863. doi: 10.1074/jbc.M512697200. [DOI] [PubMed] [Google Scholar]
- 54.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Green AM, Beatty PR, Hadjilaou A, Harris E. 2014. Innate immunity to dengue virus infection and subversion of antiviral responses. J Mol Biol 426:1148–1160. doi: 10.1016/j.jmb.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henderson EA, Alber DG, Baxter RC, Bithell SK, Budworth J, Carter MC, Chubb A, Cockerill GS, Dowdell VC, Fraser IJ, Harris RA, Keegan SJ, Kelsey RD, Lumley JA, Stables JN, Weerasekera N, Wilson LJ, Powell KL. 2007. 1,4-Benzodiazepines as inhibitors of respiratory syncytial virus. The identification of a clinical candidate. J Med Chem 50:1685–1692. [DOI] [PubMed] [Google Scholar]
- 57.Zhang P, Zhang N, Korba BE, Hosmane RS. 2005. Synthesis and in vitro anti-hepatitis B and C virus activities of ring-expanded (‘fat') nucleobase analogues containing the imidazo[4,5-e][1,3]diazepine-4,8-dione ring system. Bioorg Med Chem Lett 15:5397–5401. doi: 10.1016/j.bmcl.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 58.Basu A, Li B, Mills DM, Panchal RG, Cardinale SC, Butler MM, Peet NP, Majgier-Baranowska H, Williams JD, Patel I, Moir DT, Bavari S, Ray R, Farzan MR, Rong L, Bowlin TL. 2011. Identification of a small-molecule entry inhibitor for filoviruses. J Virol 85:3106–3119. doi: 10.1128/JVI.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schimer J, Cigler P, Vesely J, Grantz Saskova K, Lepsik M, Brynda J, Rezacova P, Kozisek M, Cisarova I, Oberwinkler H, Kraeusslich HG, Konvalinka J. 2012. Structure-aided design of novel inhibitors of HIV protease based on a benzodiazepine scaffold. J Med Chem 55:10130–10135. doi: 10.1021/jm301249q. [DOI] [PubMed] [Google Scholar]
- 60.Zmurko J, Neyts J, Dallmeier K. 2015. Flaviviral NS4b, chameleon and jack-in-the-box roles in viral replication and pathogenesis, and a molecular target for antiviral intervention. Rev Med Virol 25:205–223. doi: 10.1002/rmv.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zou J, Xie X, Lee le T, Chandrasekaran R, Reynaud A, Yap L, Wang QY, Dong H, Kang C, Yuan Z, Lescar J, Shi PY. 2014. Dimerization of flavivirus NS4B protein. J Virol 88:3379–3391. doi: 10.1128/JVI.02782-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bartenschlager R, Miller S. 2008. Molecular aspects of dengue virus replication. Future Microbiol 3:155–165. doi: 10.2217/17460913.3.2.155. [DOI] [PubMed] [Google Scholar]
- 63.Wang QY, Dong H, Zou B, Karuna R, Wan KF, Zou J, Susila A, Yip A, Shan C, Yeo KL, Xu H, Ding M, Chan WL, Gu F, Seah PG, Liu W, Lakshminarayana SB, Kang C, Lescar J, Blasco F, Smith PW, Shi PY. 2015. Discovery of dengue virus NS4B inhibitors. J Virol 89:8233–8244. doi: 10.1128/JVI.00855-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Wispelaere M, LaCroix AJ, Yang PL. 2013. The small molecules AZD0530 and dasatinib inhibit dengue virus RNA replication via Fyn kinase. J Virol 87:7367–7381. doi: 10.1128/JVI.00632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Cleef KW, Overheul GJ, Thomassen MC, Kaptein SJ, Davidson AD, Jacobs M, Neyts J, van Kuppeveld FJ, van Rij RP. 2013. Identification of a new dengue virus inhibitor that targets the viral NS4B protein and restricts genomic RNA replication. Antiviral Res 99:165–171. doi: 10.1016/j.antiviral.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 66.Xie X, Wang QY, Xu HY, Qing M, Kramer L, Yuan Z, Shi PY. 2011. Inhibition of dengue virus by targeting viral NS4B protein. J Virol 85:11183–11195. doi: 10.1128/JVI.05468-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Cleef KW, Overheul GJ, Thomassen MC, Marjakangas JM, van Rij RP. 2016. Escape mutations in NS4B render dengue virus insensitive to the antiviral activity of the paracetamol metabolite AM404. Antimicrob Agents Chemother 60:2554–2557. doi: 10.1128/AAC.02462-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cannalire R, Barreca ML, Manfroni G, Cecchetti V. 2016. A journey around the medicinal chemistry of hepatitis C virus inhibitors targeting NS4B: from target to preclinical drug candidates. J Med Chem 59:16–41. doi: 10.1021/acs.jmedchem.5b00825. [DOI] [PubMed] [Google Scholar]
- 69.Chang J, Block TM, Guo JT. 2015. Viral resistance of MOGS-CDG patients implies a broad-spectrum strategy against acute virus infections. Antivir Ther 20:257–259. doi: 10.3851/IMP2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. 2013. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol 87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. 2015. Core protein: a pleiotropic keystone in the HBV life cycle. Antiviral Res 121:82–93. doi: 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]