

ABSTRACT
Spring viremia of carp virus (SVCV) is an efficient pathogen causing high mortality in the common carp. Fish interferon (IFN) is a powerful cytokine enabling host cells to establish an antiviral response; therefore, the strategies that SVCV uses to avoid the cellular IFN response were investigated. Here, we report that the SVCV P protein is phosphorylated by cellular TANK-binding kinase 1 (TBK1), which decreases IFN regulatory factor 3 (IRF3) phosphorylation and suppresses IFN production. First, overexpression of P protein inhibited the IFN promoter activation induced by SVCV and the IFN activity activated by the mitochondrial antiviral signaling protein (MAVS) although TBK1 activity was not blocked by P protein. Second, P protein colocalized and interacted with TBK1. Dominant negative experiments suggested that the TBK1 N-terminal kinase domain interacted with P protein and was essential for P protein and IRF3 phosphorylation. Finally, P protein overexpression reduced the IRF3 phosphorylation activated by TBK1 and reduced host cellular ifn transcription. Collectively, our data demonstrated that the SVCV P protein is a decoy substrate for the host phosphokinase TBK1, preventing IFN production and facilitating SVCV replication.
IMPORTANCE TBK1 is a pivotal phosphokinase that activates host IFN production to defend against viral infection; thus, it is a potential target for viruses to negatively regulate IFN response and facilitate viral evasion. We report that the SVCV P protein functions as a decoy substrate for cellular TBK1, leading to the reduction of IRF3 phosphorylation and suppression of IFN expression. These findings reveal a novel immune evasion mechanism of SVCV.
INTRODUCTION
In aquatic viruses, spring viremia of carp virus (SVCV) belongs to the genus Vesiculovirus of the family Rhabdoviridae and causes significant mortality in the common carp (Cyprinus carpio) (1). Regarding its genome structure, SVCV encodes an ∼11-kb negative-sense, single-stranded RNA that encodes five proteins in the following order (3′ to 5′): nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), and viral RNA-dependent RNA polymerase (L) (2–4). The roles of these proteins in the replication and proliferation of rhabdovirus have been explored. N protein associates with viral RNA to form the nucleocapsid during viral assembly. M protein participates in the assembly and budding of virus, and G protein is involved in viral endocytosis. Moreover, as a phosphoprotein, the P protein is involved in several viral replication and assembly processes. As an example, the functional phosphorylated P protein interacts with the L protein to form a viral polymerase complex that interacts with the RNA template. In addition, the P protein also maintains the N protein in a soluble, encapsidation-competent form (1, 5).
In host cells, viral RNAs can be recognized by the cellular immune system, triggering an antiviral response (6). In the cytoplasm, RIG-I-like receptors (RLRs), including RIG-I and MDA5, are the major pattern-recognition receptors (PRRs) sensing exogenous viral RNAs (7, 8). Upon sensing the viral RNAs, the mitochondrial adaptor protein MAVS (also known as VISA, IPS-1, or Cardif) is recruited to and activated by RIG-I and MDA5, transducing the signal to the downstream molecules MITA (also known as STING, ERIS, or MYPS) and TANK-binding kinase 1 (TBK1) (9, 10). Then, activated TBK1 phosphorylates interferon (IFN) regulatory factor 3 (IRF3) and IRF7, which are translocated into the nucleus to trigger the transcription of IFN genes (11). IFN production initiates the transcription of more than 300 IFN-stimulated genes (ISGs), leading the infected cells and nearby uninfected cells to generate an antiviral state (12, 13). The RLR molecules play a pivotal role in IFN activation and are conserved in mammals and fish (14, 15). For example, MAVS and MITA in zebrafish (Danio rerio) are crucial to defend against both DNA and RNA viruses, and TBK1 in the crucian carp (Carassius carassius) is indispensable for IRF3 phosphorylation and IFN expression (16–18).
TBK1 is a crucial kinase that activates host IFN production; therefore, it is targeted by viruses as a major negative regulatory target to decrease the IFN response and facilitate viral replication (19, 20). Viruses have evolved multiple strategies to escape the TBK1-mediated antiviral system (21). In general, there are two essential mechanisms used by viruses to inhibit TBK1 activity: (i) physical interaction, such as that of the NS3 and NS2 proteins from the hepatitis C virus (HCV) that bind to TBK1 and disrupt the interaction between TBK1 and IRF3, reducing IRF3 activation, and (ii) chemical modification, such as that of the shorter form of the leader proteinase (Lbpro) from the foot-and-mouth disease virus (FMDV) and the papain-like protease domain 2 (PLP2) from mouse hepatitis virus A59 (MHV-A59) that mediate TBK1 deubiquitination and inactivate its kinase activity (22, 23).
As an efficient pathogen that causes high mortality in fish, SVCV likely has strategies to negatively regulate or evade the host immune response (24). In our previous study, the SVCV N protein degraded MAVS through the ubiquitin-proteasome pathway, reducing IFN transcription and facilitating viral genome replication and viral particle proliferation (25). To further investigate the immune evasion strategies of SVCV, we report here that the SVCV P protein functions as a TBK1 substrate to decrease IRF3 phosphorylation, reducing IFN transcription and facilitating viral replication.
MATERIALS AND METHODS
Cells and viruses.
Epithelioma papulosum cyprini (EPC) cells were maintained at 28°C in 5% CO2 in medium 199 (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen). HEK 293T cells were grown at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% FBS. SVCV was propagated in EPC cells until cytopathic effect (CPE) was observed; then the cultured medium with cells was stored at −80°C and prepared for use.
Gene cloning and plasmid construction.
The cDNA fragment encoding the P protein (DQ916053.1) was amplified by reverse transcription-PCR (RT-PCR) from the RNA of SVCV-infected cells and then cloned into pcDNA3.1(+) (Invitrogen) or pCMV-Myc vector (where CMV is cytomegalovirus) (Clontech). The open reading frames (ORFs) of zebrafish TBK1 (NM_001044748.2), MITA (NM_001278837.1), IRF3 (NM_001143904), and IRF7 (NM_200677.2) and of the truncated mutants of TBK1 were also subcloned into pCMV-HA, pCMV-Myc, and pCMV-Tag 2C vectors, respectively. C-terminally enhanced green fluorescent protein (EGFP)-tagged P protein (P-EGFP) was generated by inserting the ORF of the P protein into pEGFP-N3 vector (Clontech). To generate the expression plasmids DsRed-MAVS, DsRed-MITA, and DsRed-TBK1, the cDNA fragments encoding zebrafish MAVS (NM_001080584.2), MITA, and TBK1 were cloned into pDsRed-Monomer-C1 vector (Clontech). IFN-ϕ1pro and IFN-ϕ3pro luciferase reporter plasmids (IFNϕ1pro-Luc or IFNϕ3pro-Luc) and the expression plasmids for Flag-tagged MAVS, MITA, and TBK1 were described previously (25–27). All constructs were confirmed by DNA sequencing. The primers used for plasmid construction are listed in Table 1.
TABLE 1.
Primers used in this study
| Primer function and namea | Sequence (5′→3′) |
|---|---|
| Eukaryotic expression | |
| pcDNA3.1-P-F | CGCGGATCCATGTCTCTACATTCGAAATTG |
| pcDNA3.1-P-R | CCGCTCGAGTTACAACCTATATTTTTGATAC |
| pCMV-Myc-P-F | CCGGAATTCCGATGTCTCTACATTCGAAATTG |
| pCMV-Myc-P-R | CCGCTCGAGTTACAACCTATATTTTTGATAC |
| pCMV-HA/Myc-TBK1-F | CCGGAATTCCGATGCAGAGTACGGCCAAT |
| pCMV-Tag2c-TBK1-F | CGCGGATCCCGATGCAGAGTACGGCCAAT |
| pCMV-HA/Myc/Tag2C-TBK1-R | AACTCGAGTCACATCCGCTCCACTG |
| pCMV-HA/Myc/Tag2C-TBK1-ΔN-F | CCGGAATTCCGATGCAGAGTACGGCCAATTACCTGTGGATGATGTCCGACCTGACAGCGAACCTCTTC |
| pCMV-HA/Myc/Tag2C-TBK1-ΔN-R | AACTCGAGTCACATCCGCTCCACTG |
| pCMV-HA/Myc/Tag2C-TBK1-N-F | CCGGAATTCCGCTGGGTCAGGGAGCCACAGC |
| pCMV-HA/Myc/Tag2C-TBK1-N-R | AACTCGAGTTAGTTGTAAGTGTGTATGTAG |
| pCMV-HA/Myc/Tag2C-MITA-F | CCGGAATTCCGATGTCTGTGATGGGAGAA |
| pCMV-HA/Myc/Tag2C-MITA-R | CCGCTCGAG TTAGTTTTGTTTCATTGC |
| pCMV-HA/Myc/Tag2C-IRF3-F | CCGGAATTCCGATGACTCAAGCAAAACCG |
| pCMV-HA/Myc/Tag2C-IRF3-R | AACTCGAGTTAGCAGAGCTCCATCA |
| pCMV-HA/Myc/Tag2C-IRF7-F | CCGGAATTCCGATGCAGAGCACAAATGC |
| pCMV-HA/Myc/Tag2C-IRF7-R | AACTCGAGTTATTCCACTGAAGGCA |
| pEGFP-N3-P-F | CTAGCTAGCATGTCTCTACATTCGAAATTG |
| pEGFP-N3-P-R | CGCGGATCCCAACCTATATTTTTGATAC |
| pDsRed-TBK1-F | CGCGGATCCGCCACCATGCAGAGTACGGCCAAT |
| pDsRed-TBK1-R | CCGGAATTCCGCATCCGCTCCACTG |
| Real-time PCR | |
| IFN-EPC-F | ATGAAAACTCAAATGTGGACGTA |
| IFN-EPC-R | GATAGTTTCCACCCATTTCCTTAA |
| RIG-I-EPC-F | TGCTGGACCGGATGTGTTATCT |
| RIG-I-EPC-R | TGGTGATCGATGGTTCGATTCT |
| ISG15-1-EPC-F | CAGCCTTGAGGATGATTCCAG |
| ISG15-1-EPC-R | TGCCGTTGTAAATCAGTCG |
| ISG15-2-EPC-F | GCCTGGTATCACAGACAG |
| ISG15-2-EPC-R | ACATCTTGCACTGACATA |
| MAVS-EPC-F | GAATGTCCCTGTCCGAGAAA |
| MAVS-EPC-R | TCTGAACATGCTCGTTTGCAG |
| qP-F | TTGGACCTGGGATAGTGA |
| qP-R | CTTGCTTGGTTTGTGGG |
| qG-F | CGACCTGGATTAGACTTG |
| qG-R | AATGTTCCGTTTCTCACT |
| qM-F | TACTCCTCCCACTTACGA |
| qM-R | CAAGAGTCCGAGAAGGTC |
| qβ-actin-F | CACTGTGCCCATCTACGAG |
| qβ-actin-R | CCATCTCCTGCTCGAAGTC |
F, forward; R, reverse.
Luciferase activity assay.
EPC cells were seeded in 24-well plates and 24 h later cotransfected with 250 ng of luciferase reporter plasmid (IFNϕ1pro-Luc or IFNϕ3pro-Luc) and 25 ng of a Renilla luciferase internal control vector (pRL-TK; Promega). An empty vector pcDNA3.1(+) was used to maintain equivalent amounts of DNA in each well. At 48 h posttransfection, the cells were washed with phosphate-buffered saline (PBS) and lysed for measuring luciferase activity by a dual-luciferase reporter assay system, according to the manufacturer's instructions (Promega). The results are representative of more than three independent experiments, each performed in triplicate.
RNA extraction, reverse transcription, and qPCR.
Total RNAs were extracted by TRIzol reagent (Invitrogen). cDNA was synthesized using a GoScript reverse transcription system (Promega) according to the manufacturer's instructions. Quantitative real-time PCR (qPCR) was performed with Fast SYBR green PCR Master Mix (Bio-Rad) on a CFX96 Real-Time System (Bio-Rad). PCR conditions were as follows: 95°C for 5 min and then 40 cycles of 95°C for 20 s, 60°C for 20 s, and 72°C for 20 s. All primers used for qPCR are shown in Table 1, and the β-actin gene was used as an internal control. The relative fold changes were calculated by comparison to the corresponding controls using the 2−ΔΔCT (where CT is threshold cycle) method. Three independent experiments were conducted for statistical analysis.
Plaque formation assay.
EPC cells were seeded in 24-well plates. When the cells grew to 100% confluence, they were infected with SVCV in M199 culture medium with routine rocking. At 1 h postinfection, infection medium was removed, and infected EPC cells were then overlaid 1:1 with M199 culture medium containing 1% agarose. At 72 h postinfection, cells were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet.
Coimmunoprecipitation (co-IP) assay.
The HEK 293T cells seeded in 10-cm2 dishes overnight were transfected with a total of 10 μg of the plasmids indicated on the figures. At 24 h posttransfection, the medium was removed carefully, and the cell monolayer was washed twice with 10 ml of ice-cold PBS. Then the cells were lysed in 1 ml of radioimmunoprecipitation assay (RIPA) lysis buffer (1% NP-40, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM sodium orthovanadate [Na3VO4], 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.25% sodium deoxycholate) containing protease inhibitor cocktail (Sigma-Aldrich) at 4°C for 60 min on a rocker platform. The cellular debris was removed by centrifugation at 12,000 × g for 15 min at 4°C. The supernatant was transferred to a fresh tube and incubated with 30 μl of anti-hemagglutinin (HA)-agarose beads or anti-Flag affinity gel (Sigma-Aldrich) overnight at 4°C with constant agitation. These samples were further analyzed by immunoblotting. Immunoprecipitated proteins were collected by centrifugation at 5,000 × g for 1 min at 4°C, washed three times with lysis buffer, and resuspended in 50 μl of 2×SDS sample buffer. The immunoprecipitates and whole-cell lysates were analyzed by immunoblotting with the antibodies (Abs) indicated on the figures.
Immunoblot analysis.
Immunoprecipitates or whole-cell extracts were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad). The membranes were blocked for 1 h at room temperature in TBST buffer (25 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20, pH 7.5) containing 5% nonfat dry milk, probed with the primary Abs indicated on the figures at an appropriate dilution overnight at 4°C, washed three times with TBST, and then incubated with secondary Abs for 1 h at room temperature. After three additional washes with TBST, the membranes were stained with Immobilon Western chemiluminescent horseradish peroxidase (HRP) substrate (Millipore) and detected using an ImageQuant LAS 4000 system (GE Healthcare). Abs were used at the following dilutions: anti-β-actin (Cell Signaling Technology) at 1:1,000, anti-Flag/HA (Sigma-Aldrich) at 1:3,000, anti-Myc (Santa Cruz Biotechnology) at 1:2,000, and HRP-conjugated anti-rabbit IgG or anti-mouse IgG (Thermo Scientific) at 1:5,000. Results are representative of three independent experiments.
In vitro protein dephosphorylation assay.
Transfected HEK 293T cells were lysed as described above, except that the phosphatase inhibitors (Na3VO4 and EDTA) were omitted from the lysis buffer. Protein dephosphorylation was carried out in 100-μl reaction mixtures consisting of 100 μg of cell protein and 10 units (U) of calf intestinal phosphatase (CIP; Sigma-Aldrich). The reaction mixtures were incubated at 37°C for 40 min, followed by immunoblot analysis.
Fluorescence microscopy.
EPC cells were plated onto coverslips in six-well plates and transfected with the plasmids indicated on the figures for 24 h. Then the cells were washed twice with PBS and fixed with 4% paraformaldehyde (PFA) for 1 h. After three washes with PBS, the cells were stained with 1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI; Beyotime) for 15 min in the dark at room temperature. Finally, the coverslips were washed and observed with a Leica confocal microscope under a 63× oil immersion objective (LSM710; Zeiss).
Statistics analysis.
The statistical P values were calculated by one-way analysis of variance (ANOVA) with Dunnett's post hoc test (SPSS Statistics, version 19; IBM). A P value of <0.01 was considered statistically significant. Data are expressed as means ± standard deviations (SDs) of at least three independent experiments (n ≥ 3).
RESULTS
SVCV infection triggers the host cellular IFN response.
To investigate whether SVCV infection triggers the cellular IFN response in vitro, the expression patterns of several host and viral genes were examined. First, to measure the viral infectivity, SVCV at a multiplicity of infection (MOI) of 0.1 was used to infect EPC cells, and infectivity was measured by plaque assay. As shown in Fig. 1A, EPC cells were infected by SVCV efficiently. Then, after infection with SVCV at an MOI of 10 for 24 h, EPC cells were lysed, and the total RNAs from both the cells and the replicated viruses were extracted and monitored by qPCR. As shown in Fig. 1B, the p, n, m, and g transcripts from SVCV were significantly expressed in the infected cells, indicating that the cells were successfully infected with SVCV. Subsequently, the mRNA levels of ifn and the antiviral gene vig1 were also assessed, and they were remarkably upregulated after poly I·C stimulation and SVCV infection (Fig. 1C). These data demonstrated that SVCV infection induces the host cellular IFN response.
FIG 1.
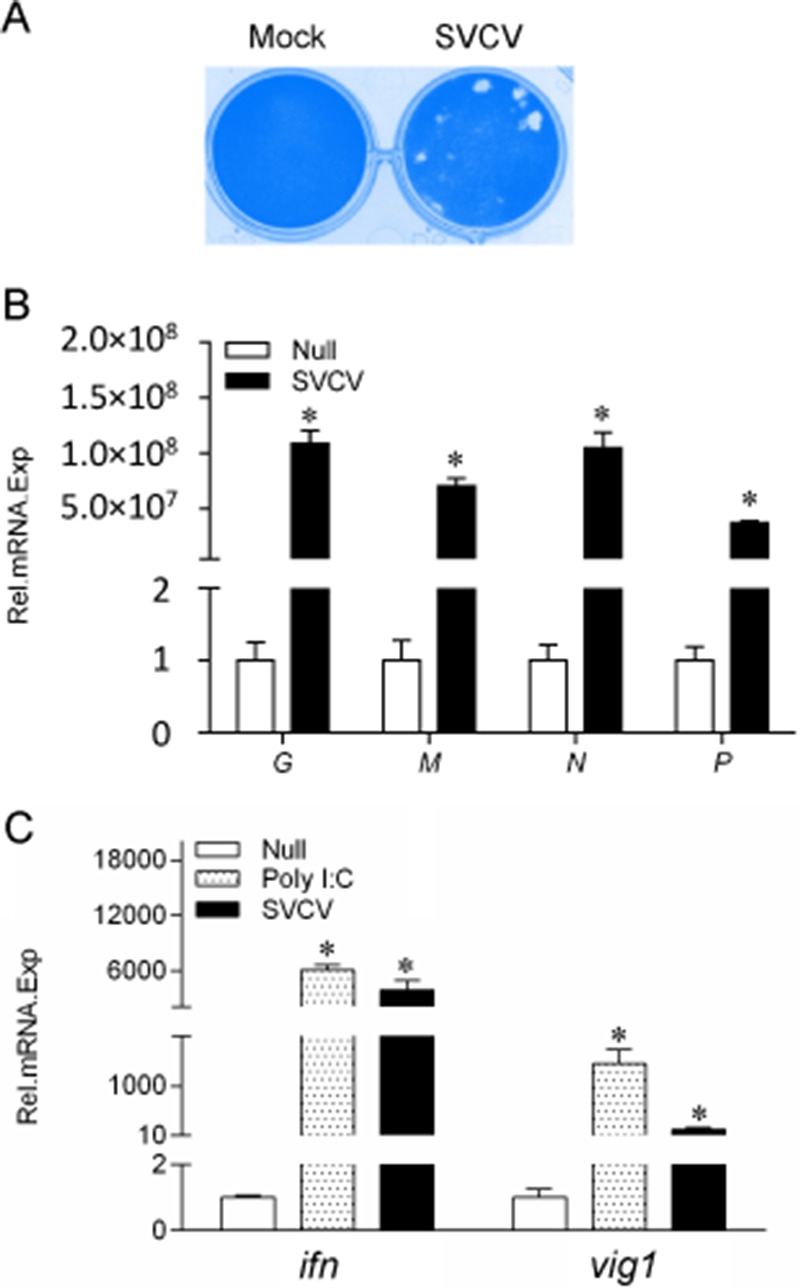
SVCV-induced expression of the genes of SVCV and host cells. (A) EPC cells were seeded in 24-well plates overnight and infected with SVCV (MOI of 0.1) for 72 h; the cells were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. (B and C) EPC cells were seeded in six-well plates overnight and infected with SVCV (MOI of 10) for 24 h. Then total RNAs were extracted to examine the mRNA levels of the g, m, n, and p genes of SVCV and the ifn and vig1 genes of host cells, as indicated, by qPCR. The β-actin gene was used as an internal control for normalization, and the relative expression is represented as fold induction relative to the expression level in control cells (set to 1). Error bars represent SDs obtained by measuring each sample in triplicate. Asterisks indicate significant differences from control values (*, P < 0.05).
P protein blocks RLR-mediated activation of the IFN-ϕ1 promoter.
Previous studies demonstrated that only IFN-ϕ1 and IFN-ϕ3 of the four type I IFNs (IFN-ϕ1 to IFN-ϕ4) in zebrafish were activated by poly I·C (a mimic of viral RNA), indicating that IFN-ϕ1 and IFN-ϕ3 should be involved in the antiviral process (18, 26). As shown in Fig. 2A, SVCV infection induced the activation of IFN-ϕ1pro; however, the IFN-ϕ1pro induction stimulated by SVCV was significantly impeded by overexpression of the P protein. Similarly, the P protein also suppressed SVCV-induced IFN-ϕ3pro activity (Fig. 2B), suggesting that the P protein inhibits the activation of IFN-ϕ1pro and IFN-ϕ3pro upon infection with SVCV. Furthermore, fish RLR factors are efficient for triggering IFN production (28); consequently, these constructs were used in a luciferase reporter gene assay. As shown in Fig. 2C, overexpression of MAVS and TBK1 upregulated the activation of IFN-ϕ1pro, and the activation of IFN-ϕ1pro induced by MAVS was inhibited by cotransfection with the P protein. However, ectopic expression of the P protein did not affect TBK1-stimulated IFN-ϕ1pro activity. Similarly, the upregulation of IFN-ϕ3pro activity activated by MAVS but not TBK1 was reduced by the P protein. Given that TBK1 is downstream of MAVS, these results indicated that the P protein likely decreased IFN-ϕ1 and IFN-ϕ3 production via the negative regulation of MAVS or TBK1.
FIG 2.

Inhibition of IFN-ϕ1pro and IFN-ϕ3pro activation by overexpression of P protein. (A and B) EPC cells were seeded in 24-well plates overnight and cotransfected with 250 ng of IFNϕ1pro-Luc or IFNϕ3pro-Luc, as indicated, and 25 ng of pRL-TK, plus 250 ng of empty vector or pcDNA3.1-P. At 24 h posttransfection, cells were infected with SVCV (MOI of 10). The luciferase activities were monitored at 24 h after stimulation. (C and D) EPC cells were seeded in 24-well plates and cotransfected with a MAVS- or TBK1-expressing plasmid and empty vector or pcDNA3.1-P, plus IFNϕ1pro-Luc or IFNϕ3pro-Luc, as indicate, at the ratio of 1:1:1. pRL-TK was used as a control. At 48 h posttransfection, cells were lysed for luciferase activity detection. Error bars are the SDs obtained by measuring each sample in triplicate. Asterisks indicate significant differences from control values (*, P < 0.05).
P protein colocalizes and interacts with MAVS and TBK1 in the cytosol.
To further investigate the function of the P protein, its subcellular location was monitored in EPC cells. Confocal microscopy revealed that the P-EGFP signal was mainly distributed in the cytoplasm (Fig. 3A). We cotransfected DsRed-MAVS, DsRed-MITA, or DsRed-TBK1 with P-EGFP. Red signal from MAVS, MITA, and TBK1 was observed in the cytosol. The P protein (green) overlapped with MAVS and TBK1 but not MITA (Fig. 3B, C, and D). These data suggested that the SVCV P protein might colocalize with MAVS and TBK1 in the cytosol. To investigate whether the P protein associates with MAVS and TBK1, 293T cells were cotransfected with P-myc and Flag-tagged RLR factors, including MAVS, MITA, TBK1, IRF3, and IRF7. Protein interactions were assessed by coimmunoprecipitation (co-IP) assays. Anti-Flag Ab-immunoprecipitated protein complexes containing MAVS, MITA, and TBK1 were also recognized with the anti-myc Ab, whereas IRF3 and IRF7 were not recognized by the anti-myc Ab (Fig. 3E), suggesting that the P protein interacts with MAVS, MITA, and TBK1 but not IRF3 and IRF7.
FIG 3.

P protein colocalizes and interacts with MAVS and TBK1. EPC cells seeded on microscopy coverglass in six-well plates were transfected with 2 μg of P-EGFP and 2 μg of empty vector (A) or DsRed-MAVS (B), DsRed-MITA (C), and DsRed-TBK1 (D). After 24 h, the cells were fixed and subjected to confocal microscopy analysis. Green signals represent overexpressed P protein, red signals represent overexpressed MAVS, MITA, or TBK1, and blue staining indicates the nucleus region. The yellow staining in the merged images indicates colocalization of P protein and MAVS, MITA, or TBK1 (original magnification, 63× oil immersion objective). Scale bar, 10 μm. All experiments were repeated at least three times with similar results. (E) 293T cells seeded in 10-cm2 dishes were transfected with the indicated plasmids (5 μg each). After 24 h, cell lysates were immunoprecipitated (IP) with an anti-Flag affinity gel. Then the immunoprecipitates and cell lysates were analyzed by immunoblotting (IB) with the anti-myc and anti-Flag Abs, respectively.
P protein is phosphorylated by the TBK1 N terminus.
Given that the P protein interacted with MAVS and TBK1, we sought to determine the interaction pattern between the P protein and MAVS or TBK1, which is a crucial phosphokinase in mammals. As shown in Fig. 4A and B, there was no difference when the overexpressed P protein was cotransfected with MAVS; however, a band shift was observed when the P protein was overexpressed with TBK1 in a dose-dependent manner. To identify whether the band shift was caused by phosphorylated P protein, cell lysate was treated with CIP, leading to the disappearance of the shifted bands (Fig. 4C). Given that the N-terminal domain is the functional kinase domain for TBK1, dominant negative mutants of TBK1 were generated to identify the functional domain in the P protein. As shown in Fig. 4D, compared to the robust P protein phosphorylation observed in the wild-type TBK1 group, TBK1-ΔN (lacking the N terminus) failed to phosphorylate the P protein. Finally, the TBK1 fragment interacting with the P protein was also examined using co-IP experiments. As shown in Fig. 4E, wild-type TBK1 bound to the P protein although this interaction was abolished when the N-terminal kinase domain was deleted. The N terminus of TBK1 alone also interacted with the P protein. These data suggested that the P protein can interact with and be phosphorylated by TBK1, which requires the TBK1 N-terminal kinase domain.
FIG 4.

P protein is phosphorylated by TBK1. (A and B) 293T cells were seeded in six-well plates overnight and cotransfected with 2 μg of P-myc and 2 μg of empty vector, MAVS-Flag, or TBK1-Flag (1 and 2 μg, respectively) for 24 h. The cell lysates were subjected to immunoblotting (IB) with the anti-myc, anti-Flag, and anti-β-actin Abs. (C) TBK1-phosphorylated P protein (pP) is reduced by CIP treatment. 293T cells were seeded in six-well plates overnight and transfected with the indicated plasmids (2 μg each) for 24 h. The cell lysates (100 μg) were left untreated or treated with CIP (10 U) for 40 min at 37°C. Then the lysates were detected by immunoblotting with anti-myc, anti-Flag, and anti-β-actin Abs. (D) P protein is phosphorylated by the N terminus of TBK1. 293T cells were seeded in six-well plates overnight and transfected with the indicated plasmids (2 μg each) for 24 h. The cell lysates were subjected to immunoblotting with the anti-myc, anti-Flag, and anti-β-actin Abs. All experiments were repeated at least three times with similar results. (E) TBK1 interacts with P protein via the N-terminal kinase domain. 293T cells seeded in 10-cm2 dishes were transfected with the indicated plasmids (5 μg each). After 24 h, cell lysates were immunoprecipitated with anti-HA-agarose beads. Then the immunoprecipitates and cell lysates were analyzed by immunoblotting with the anti-myc and anti-HA Abs, respectively. All experiments were repeated at least three times with similar results.
The TBK1 N terminus is essential for the phosphorylation of IRF3 and MITA.
To understand the biological effect of the interaction between the P protein and TBK1, the function of fish TBK1 was investigated. As shown in Fig. 5A and B, both IRF3 and MITA caused a band shift, exhibiting higher mobilities when they were cotransfected with TBK1-Flag in 293T cells. Subsequently, the cell lysates were incubated with CIP. As expected, the band shifts disappeared, demonstrating that IRF3 and MITA are phosphorylated by TBK1 in fish. Compared with TBK1, MAVS did not phosphorylate IRF3 or MITA (Fig. 5C). Furthermore, dominant negative TBK1 mutants were used to characterize the functional kinase domain of TBK1. Compared with the kinase activity of wild-type TBK1, TBK1-ΔN was unable to phosphorylate IRF3 (Fig. 5D) or MITA (Fig. 5E). These data indicated that the N-terminal kinase domain of TBK1 is also indispensable for IRF3 and MITA phosphorylation.
FIG 5.

The N-terminal kinase domain is essential for TBK1 to phosphorylate MITA and IRF3. (A and B) 293T cells were seeded in six-well plates overnight and transfected with the indicated plasmids (2 μg each) for 24 h. The cell lysates (100 μg) were left untreated or treated with CIP (10 U) for 40 min at 37°C. Then the lysates were detected by immunoblotting with the anti-myc, anti-Flag, and anti-β-actin Abs. (C) 293T cells were seeded in six-well plates overnight and transfected with 2 μg of MITA-myc or IRF3-myc and 2 μg of empty vector or MAVS-Flag for 24 h. The cell lysates were subjected to immunoblotting with the anti-myc, anti-Flag, and anti-β-actin Abs. (D and E) 293T cells were seeded in six-well plates overnight and transfected with the indicated plasmids (2 μg each) for 24 h. The cell lysates were subjected to immunoblotting with the anti-myc, anti-Flag, and anti-β-actin Abs. All experiments were repeated at least three times with similar results.
P protein decreases TBK1-mediated phosphorylation of IRF3.
Given that both SVCV P protein and cellular IRF3 can be phosphorylated by TBK1 and that the phosphorylation of IRF3 is indispensable for IFN expression, it is possible that SVCV uses the P protein to compete with cellular IRF3 for phosphorylation by TBK1. Figure 6A shows that increasing amounts of the P protein were phosphorylated by TBK1, and phosphorylated IRF3 was gradually reduced in a dose-dependent manner. Similarly, the TBK1-mediated phosphorylation of IRF7 (Fig. 6B) and MITA (Fig. 6C) was also decreased by exogenous expression of SVCV P protein. In conclusion, these data suggest that the SVCV P protein is an inhibitor of TBK1 kinase activity, resulting in the reduced phosphorylation of IRF3, IRF7, and MITA.
FIG 6.

P protein decreases TBK1-mediated phosphorylation of MITA, IRF3, and IRF7. 293T cells were seeded in six-well plates overnight and transfected with 1.5 μg of TBK1-myc and 1.5 μg of empty vector or P-myc (0.75 and 1.5 μg, respectively), together with 1.5 μg of IRF3-HA (A), IRF7-HA (B), or MITA-HA (C) for 24 h. Then the lysates were detected by immunoblotting with the anti-myc, anti-HA, and anti-β-actin Abs. All experiments were repeated at least three times with similar results.
P protein dampens the cellular IFN response and facilitates SVCV gene replication.
To investigate whether SVCV P protein interferes with the cellular IFN response and enhances viral replication, EPC cells were transfected with P protein and stimulated with SVCV. Total RNAs were extracted and monitored by qPCR. As shown in Fig. 7A, the expression of the ifn transcript in cells overexpressing P protein was reduced compared to the levels in control cells, and the reduced expression of several other host ISGs, such as isg15, rig-i, and mavs, was also observed. The expression of SVCV genes was also monitored. After infection with SVCV, the viral m, n, and p genes were upregulated in cells expressing the P protein (Fig. 7B). Then the viral titers in the supernatants of the infected EPC cells were confirmed by plaque assay, which showed that the titer in P protein-overexpressed cells was about 19 times that of the control cells (Fig. 7C). These data indicated that the P protein suppresses the cellular ifn response and facilitates SVCV proliferation.
FIG 7.
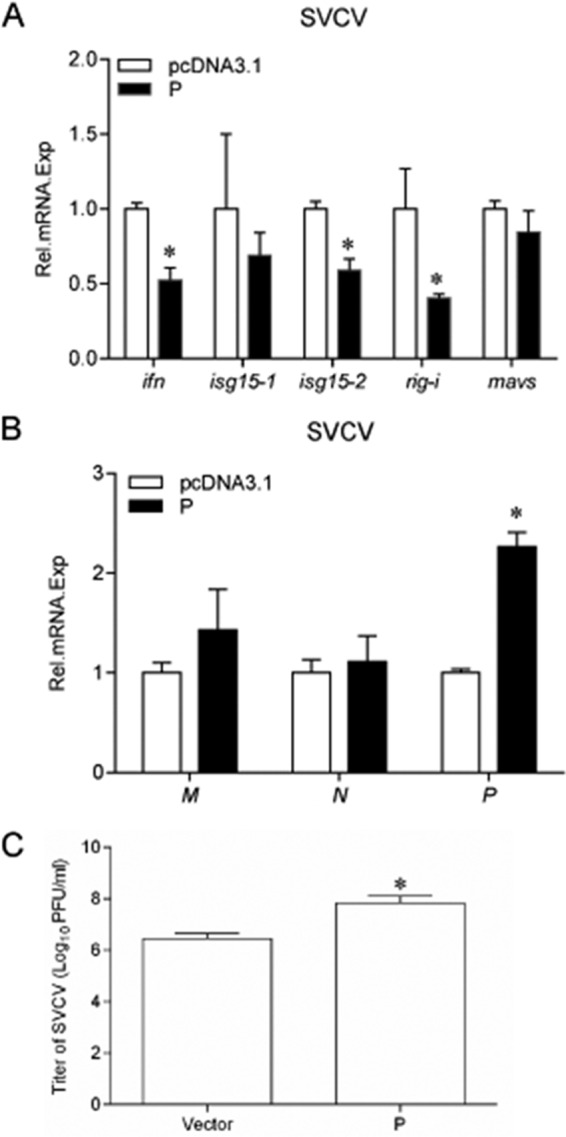
Overexpression of P protein increases virus replication in SVCV-infected EPC cells and inhibits the expression of ISGs. EPC cells seeded in six-well plates overnight were transfected with 2 μg of pcDNA3.1-P or empty vector and infected with SVCV (MOI of 10) at 24 h posttransfection. At 24 h after infection, total RNAs were extracted to examine the mRNA levels of cellular ifn, isg15-1, isg15-2, rig-i, and mavs (A) and the g, m, and p transcripts of SVCV (B) by qPCR. The relative transcription levels were normalized to the transcription level of the β-actin gene and are represented as fold induction relative to the transcription level in the control cells, which was set to 1. Error bars represent SDs obtained by measuring each sample in triplicate. Asterisks indicate significant differences from control values (*, P < 0.05). (C) P protein was overexpressed in EPC cells, cells were then infected with SVCV (MOI of 1) for 72 h, and the supernatants were harvested and used for standard plaque assays.
DISCUSSION
In a water-based environment, aquatic viruses can cause high mortality in fish because viral spread is enhanced (29). To date, significant research on aquatic viruses and fish immune responses against the pathogens has been performed; however, little is known about the evasion mechanisms of aquatic viruses from the fish immune system (30). Here, we report that the SVCV P protein, as a substrate of fish TBK1, competes with IRF3 for phosphorylation, leading to the reduction of IRF3 phosphorylation and IFN transcription.
The SVCV genome encodes only five proteins. These proteins not only participate in the process of viral particle assembly and replication but also should regulate the host immune response for viral evasion (31). Therefore, some of the five proteins must play multiple roles during the viral life cycle. For example, SVCV G protein forms the trimeric spikes on the viral outer surface to mediate endocytosis. In addition, G protein also induces autophagy in infected cells, which is beneficial for SVCV proliferation (32). The N protein associates with viral RNA, providing a helical symmetry to the viral nucleocapsid. In addition, the N protein mediates the degradation of host MAVS in a ubiquitin-proteasome-dependent manner for viral evasion (25). Previous studies have demonstrated that the M protein generates the bullet-shaped structure of SVCV and binds to the G protein embedded in the viral envelope (33). Several of our experiments indicated that the M protein reduces the global transcriptional level of host cells in a nonspecific manner (data not shown). These studies presume that SVCV proteins possessed additional functions beyond expectation.
Multiple strategies are employed by viruses to evade the host immune defense. In addition to escaping from direct detection by cellular sensors, interfering with or directly attacking the key molecules in host cells is also crucial for viral replication (33). IFN expression initiates the transcriptions of at least 300 ISGs which participate in the antiviral process. Therefore, upstream activating factors (such as those in the RLR system) for IFN transcription are frequently targeted and negatively regulated by viruses. In the RLR system, RIG-I and MDA5 sense viral double-stranded RNA (dsRNA) and interact with the adaptor MAVS to transfer the signals to TBK1, which recruits MITA and phosphorylates IRF3 to ultimately trigger IFN expression. Hence, each factor in the RLR axis could be a viral target. In mammals, for example, the cytoplasmic RIG-I level is decreased by human immunodeficiency virus (HIV) protease (34); MDA5 is inhibited by the N protein from human respiratory syncytial virus (RSV), and MAVS can associate with and be inhibited by the hepatitis B virus (HBV) protein HBx (35, 36). MITA can be cleaved into inactive fragments by the protease complex NS2B2 from the dengue virus (37). Several viruses negatively modulate the activity of TBK1, a crucial phosphokinase that defends against viral infections, leading to a reduction in the IFN response and promoting viral replication (21, 38). Similar to the SVCV P protein, the P protein from the Borna disease virus (BDV) also interacts with and is phosphorylated by TBK1, reducing IRF3 phosphorylation (39). Both BDV and SVCV are negative-sense RNA viruses (Mononegavirales) and encode N, P, M, L, and G proteins. In these viruses, the P proteins act as a TBK1 decoy and compete with IRF3 for phosphorylation. These observations indicate that although the natural reservoirs of SVCV and BDV are fish and mammals, respectively, the evasion mechanisms of these negative-sense RNA viruses might be conserved.
In addition to activating IFN production, the roles of TBK1 as an indispensable kinase also exist in host autophagic maturation, antibacterial response, cellular transformation, and oncogenesis (40–43). In the process of infection, viruses do not only combat the host's immune system but also utilize host cells for viral replication and proliferation. Thus, TBK1 mediates cell physiology. Therefore, the mechanism by which SVCV P protein impedes TBK1 kinase activity will be clearer with subsequent studies examining the biological function of TBK1.
The modification (i.e., phosphorylation) of viral protein is indispensable for the viral life cycle in addition to inhibiting the host immune response. Studies have revealed that several viral proteins are not active until modified by cellular protein kinases (44). There are three primary functions of the modified viral proteins. These include regulating viral genome replication and transcription. For example, when the NS1 protein from Periplaneta fuliginosa densovirus (PfDNV) was treated with a phosphatase, genome replication and transcription were reduced. A second function is changing the cellular location of viral proteins. IE63 from vesicular stomatitis virus (VSV), for example, is translocated from the nucleus to the cytoplasm after phosphorylation by the host cellular proteins CDK1 and CDK2. A third function is promoting viral assembly and release, as when Gag and Vpr proteins from human immunodeficiency virus 1 (HIV-1) interact to enhance their assembly into viral particles after phosphorylation by atypical protein kinase C (aPKC). In previous studies, recombinant P protein from VSV, which was isolated from Escherichia coli and was not phosphorylated, failed to support viral transcription in vitro, indicating that phosphorylation of the P protein is required for viral RNA synthesis (45, 46). However, the mechanism by which P protein function is controlled by phosphorylation is unclear. In this study, the SVCV P protein was phosphorylated by TBK1, leading to a reduction of IRF3 phosphorylation and viral evasion from the host immune system. Although the function of the phosphorylated P protein in viral proliferation has not yet been identified, further studies should identify the roles of activated SVCV P protein in the viral life cycle.
ACKNOWLEDGMENTS
We thank Feng Xiong (Institute of Hydrobiology, Chinese Academy of Sciences) for cellular RNA exaction and Fang Zhou (Institute of Hydrobiology, Chinese Academy of Sciences) for assistance with confocal microscopy analysis.
The National Basic Research Program of China provided funding to Y.-A.Z. under grant number 2014CB138601. The National Natural Science Foundation of China provided funding to S.L. under grant number 31502200. The Knowledge Innovation Program of the Chinese Academy of Sciences provided support to S.L. under grant number Y65E01-1-501.
We declare that we have no financial or commercial conflicts of interest.
REFERENCES
- 1.Ahne W, Bjorklund HV, Essbauer S, Fijan N, Kurath G, Winton JR. 2002. Spring viremia of carp (SVC). Dis Aquat Organ 52:261–272. doi: 10.3354/dao052261. [DOI] [PubMed] [Google Scholar]
- 2.Teng Y, Liu H, Lv JQ, Fan WH, Zhang QY, Qin QW. 2007. Characterization of complete genome sequence of the spring viremia of carp virus isolated from common carp (Cyprinus carpio) in China. Arch Virol 152:1457–1465. doi: 10.1007/s00705-007-0971-8. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann B, Schutze H, Mettenleiter TC. 2002. Determination of the complete genomic sequence and analysis of the gene products of the virus of Spring Viremia of Carp, a fish rhabdovirus. Virus Res 84:89–100. doi: 10.1016/S0168-1702(01)00441-5. [DOI] [PubMed] [Google Scholar]
- 4.Zhang QY, Gui JF. 2015. Virus genomes and virus-host interactions in aquaculture animals. Sci China Life Sci 58:156–169. doi: 10.1007/s11427-015-4802-y. [DOI] [PubMed] [Google Scholar]
- 5.Walker PJ, Dietzgen RG, Joubert DA, Blasdell KR. 2011. Rhabdovirus accessory genes. Virus Res 162:110–125. doi: 10.1016/j.virusres.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stark GR. 2007. How cells respond to interferons revisited: From early history to current complexity. Cytokine Growth Factor Rev 18:419–423. doi: 10.1016/j.cytogfr.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat Immunol 7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 8.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M Jr. 2008. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol 82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 10.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 12.Yoneyama M, Fujita T. 2009. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 13.Stetson DB, Medzhitov R. 2006. Antiviral defense: interferons and beyond. J Exp Med 203:1837–1841. doi: 10.1084/jem.20061377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang YB, Gui JF. 2012. Molecular regulation of interferon antiviral response in fish. Dev Comp Immunol 38:193–202. doi: 10.1016/j.dci.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Chang M, Collet B, Nie P, Lester K, Campbell S, Secombes CJ, Zou J. 2011. Expression and functional characterization of the RIG-I-like receptors MDA5 and LGP2 in rainbow trout (Oncorhynchus mykiss). J Virol 85:8403–8412. doi: 10.1128/JVI.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biacchesi S, LeBerre M, Lamoureux A, Louise Y, Lauret E, Boudinot P, Bremont M. 2009. Mitochondrial antiviral signaling protein plays a major role in induction of the fish innate immune response against RNA and DNA viruses. J Virol 83:7815–7827. doi: 10.1128/JVI.00404-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biacchesi S, Merour E, Lamoureux A, Bernard J, Bremont M. 2012. Both STING and MAVS fish orthologs contribute to the induction of interferon mediated by RIG-I. PLoS One 7:e47737. doi: 10.1371/journal.pone.0047737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun F, Zhang YB, Liu TK, Shi J, Wang B, Gui JF. 2011. Fish MITA serves as a mediator for distinct fish IFN gene activation dependent on IRF3 or IRF7. J Immunol 187:2531–2539. doi: 10.4049/jimmunol.1100642. [DOI] [PubMed] [Google Scholar]
- 19.Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. 2004. Differential requirement for TANK-binding kinase-1 in type I interferon responses to Toll-like receptor activation and viral infection. J Exp Med 199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weidberg H, Elazar Z. 2011. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci Signal 4:pe39. doi: 10.1126/scisignal.2002355. [DOI] [PubMed] [Google Scholar]
- 21.Zhao W. 2013. Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett 587:542–548. doi: 10.1016/j.febslet.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D, Fang LR, Li P, Sun L, Fan JX, Zhang QY, Luo R, Liu XT, Li K, Chen HC, Chen ZB, Xiao SB. 2011. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J Virol 85:3758–3766. doi: 10.1128/JVI.02589-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G, Chen G, Zheng DH, Cheng GH, Tang H. 2011. PLP2 of mouse hepatitis virus A59 (MHV-A59) targets TBK1 to negatively regulate cellular type I interferon signaling pathway. PLoS One 6:e17192. doi: 10.1371/journal.pone.0017192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanders GE, Batts WN, Winton JR. 2003. Susceptibility of zebrafish (Danio rerio) to a model pathogen, spring viremia of carp virus. Comp Med 53:514–521. [PubMed] [Google Scholar]
- 25.Lu LF, Li S, Lu XB, LaPatra SE, Zhang N, Zhang XJ, Chen DD, Nie P, Zhang YA. 2016. Spring viremia of carp virus n protein suppresses fish IFNϕ1 production by targeting the mitochondrial antiviral signaling protein. J Immunol 196:3744–3753. doi: 10.4049/jimmunol.1502038. [DOI] [PubMed] [Google Scholar]
- 26.Li S, Lu LF, Feng H, Wu N, Chen DD, Zhang YB, Gui JF, Nie P, Zhang YA. 2014. IFN regulatory factor 10 is a negative regulator of the IFN responses in fish. J Immunol 193:1100–1109. doi: 10.4049/jimmunol.1400253. [DOI] [PubMed] [Google Scholar]
- 27.Lu LF, Li S, Lu XB, Zhang YA. 2015. Functions of the two zebrafish MAVS variants are opposite in the induction of IFN1 by targeting IRF7. Fish Shellfish Immunol 45:574–582. doi: 10.1016/j.fsi.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 28.Sun F, Zhang YB, Liu TK, Gan L, Yu FF, Liu Y, Gui JF. 2010. Characterization of fish IRF3 as an IFN-inducible protein reveals evolving regulation of IFN response in vertebrates. J Immunol 185:7573–7582. doi: 10.4049/jimmunol.1002401. [DOI] [PubMed] [Google Scholar]
- 29.Workenhe ST, Rise ML, Kibenge MJT, Kibenge FSB. 2010. The fight between the teleost fish immune response and aquatic viruses. Mol Immunol 47:2525–2536. doi: 10.1016/j.molimm.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Reshi L, Wu JL, Wang HV, Hong JR. 2016. Aquatic viruses induce host cell death pathways and its application. Virus Res 211:133–144. doi: 10.1016/j.virusres.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 31.Xiao Y, Shao L, Zhang CW, An W. 2014. Genomic evidence of homologous recombination in spring viremia of carp virus: a negatively single-stranded RNA virus. Virus Res 189:271–279. doi: 10.1016/j.virusres.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Liu L, Zhu B, Wu S, Lin L, Liu G, Zhou Y, Wang W, Asim M, Yuan J, Li L, Wang M, Lu Y, Wang H, Cao J, Liu X. 2015. Spring viraemia of carp virus induces autophagy for necessary viral replication. Cell Microbiol 17:595–605. doi: 10.1111/cmi.12387. [DOI] [PubMed] [Google Scholar]
- 33.Kiuchi A, Roy P. 1984. Comparison of the primary sequence of spring viremia of carp virus M protein with that of vesicular stomatitis virus. Virology 134:238–243. doi: 10.1016/0042-6822(84)90290-3. [DOI] [PubMed] [Google Scholar]
- 34.Solis M, Nakhaei P, Jalalirad M, Lacoste J, Douville R, Arguello M, Zhao TJ, Laughrea M, Wainberg MA, Hiscott J. 2011. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol 85:1224–1236. doi: 10.1128/JVI.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE, Santangelo PJ. 2012. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol 86:8245–8258. doi: 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. 2011. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol 85:987–995. doi: 10.1128/JVI.01825-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, Lin YL. 2012. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog 8:e1002780. doi: 10.1371/journal.ppat.1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyahira AK, Shahangian A, Hwang SM, Sun R, Cheng GH. 2009. TANK-binding kinase-1 plays an important role during in vitro and in vivo type I IFN responses to DNA virus infections. J Immunol 182:2248–2257. doi: 10.4049/jimmunol.0802466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unterstab G, Ludwig S, Anton A, Planz O, Dauber B, Krappmann D, Heins G, Ehrhardt C, Wolff T. 2005. Viral targeting of the interferon-β-inducing Traf family member-associated NF-κB activator (TANK)-binding kinase-1. Proc Natl Acad Sci U S A 102:13640–13645. doi: 10.1073/pnas.0502883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang SY, Bradfute SB, Bruun JA, Hansen TE, Johansen T, Deretic V. 2012. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delhase M, Kim SY, Lee H, Naiki-Ito A, Chen Y, Ahn ER, Murata K, Kim SJ, Lautsch N, Kobayashi KS, Shirai T, Karin M, Nakanishi M. 2012. TANK-binding kinase 1 (TBK1) controls cell survival through PAI-2/serpinB2 and transglutaminase 2. Proc Natl Acad Sci U S A 109:E177–E186. doi: 10.1073/pnas.1119296109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clement JF, Meloche S, Servant MJ. 2008. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res 18:889–899. doi: 10.1038/cr.2008.273. [DOI] [PubMed] [Google Scholar]
- 43.Watson RO, Manzanillo PS, Cox JS. 2012. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor KE, Mossman KL. 2013. Recent advances in understanding viral evasion of type I interferon. Immunology 138:190–197. doi: 10.1111/imm.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barik S, Banerjee AK. 1991. Cloning and expression of the vesicular stomatitis-virus phosphoprotein gene in Escherichia coli: analysis of phosphorylation status versus transcriptional activity. J Virol 65:1719–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barik S, Banerjee AK. 1992. Phosphorylation by cellular casein kinase II is essential for transcriptional activity of vesicular stomatitis-virus phosphoprotein P. Proc Natl Acad Sci U S A 89:6570–6574. doi: 10.1073/pnas.89.14.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]