

Abstract
Purpose
To investigate the effect of ABCA4 mutation status on lipofuscin-related quantitative autofluorescence (qAF) in humans and on bisretinoid accumulation in mice.
Methods
Genotyped parents (n = 26; age 37–64 years) of patients with biallelic ABCA4-related retinopathy underwent in-depth retinal phenotyping including qAF imaging as a surrogate measure for RPE lipofuscin accumulation. In addition, bisretinoids as the main components of autofluorescent lipofuscin at the ocular fundus were quantified in Abca4−/−, Abca4+/−, and wild-type mice.
Results
Index patients showed a retinal phenotype characteristic for ABCA4-related retinopathy, including increased qAF levels. In contrast, qAF measures in carriers of only one ABCA4 mutation were not different from age-matched controls in this sample, and there was no difference between truncating and missense mutations. Also, none of these carriers presented an abnormal phenotype on conventional imaging. One parent with ABCA4-related retinopathy and increased qAF carried an additional ABCA4 mutation, explaining the phenotype under a recessive disease model (pseudodominance). Biochemical analysis in the mouse model revealed direct downstream products (A2PE-H2, at-RALdimer-PE) of the ABCA4 substrate N-Ret-PE to be similar in wild-type and Abca4+/− mice. Both bisretinoids were 12- to 18-fold increased in Abca4−/− mice. Levels of A2E and A2PE in Abca4+/− mice were in between those measured in wild-type and Abca4−/− mice.
Conclusions
This study indicates that carriers of monoallelic ABCA4 mutations are phenotypically normal. However, biochemical analysis in the Abca4-deficient mouse model suggests detectable effects of one mutation in ABCA4 on the molecular level. The findings may have implications for therapeutic approaches such as gene replacement therapy.
Keywords: quantitative fundus autofluorescence, monoallelic, Abca4, carrier, phenotype
Biallelic mutations in the adenosine triphosphate (ATP)-binding cassette (ABC) transporter ABCA4 are among the most common causes for inherited retinal disease and, thus, for loss of vision early in life. ABCA4-related retinopathy may clinically present as Stargardt disease, cone–rod dystrophy, or bull's-eye maculopathy.1–5 Despite this phenotypic heterogeneity, a common feature among most patients is an increased lipofuscin-related fundus autofluorescence (AF) upon excitation with blue light,6–9 which occurs before functional decline or the development of pattern-like fundus changes.7 Fundus AF intensity largely depends upon the concentration of lipofuscin in the retinal pigment epithelium (RPE),9 which considerably accumulates in patients with Stargardt disease.10,11 Similar observations have been reported in the Abca4−/− mouse, an established animal model for ABCA4-related retinopathy.12,13
There has been an ongoing debate on whether or not monoallelic ABCA4 mutations might also cause disease at the benign end of the spectrum of ABCA4-related retinopathy. For instance, single variations in ABCA4 have been implicated to be associated with age-related macular degeneration (AMD),14–16 but this finding could not be replicated by others.17–19 Moreover, late-onset Stargardt disease and a similar-appearing AMD subtype were suggested to be associated with monoallelic ABCA4 mutations.20,21 Some inconsistency between studies may be due to clinical categorization based on the retinal phenotype. Interpretation may then be biased by possible clinical misclassification of disease and/or uncertainty in comprehensively identifying the complete mutational profile in a diseased individual. If monoallelic ABCA4 mutations were indeed disease causing, either dominant-negative effects of the abnormal protein product or haploinsufficiency would be the pathophysiological mechanism. Both mechanisms would need to be considered when developing therapeutic gene delivery strategies for ABCA4-related retinopathy.
To further explore potential effects of monoallelic ABCA4 mutations, we investigated genotyped parents of patients with retinal disease due to biallelic mutations in ABCA4. To compare these genotypically defined probands with controls, quantitative fundus AF (qAF) imaging, which was recently used by Burke et al.8 to characterize patients with Stargardt disease, was used as a sensitive in vivo surrogate measure for RPE lipofuscin accumulation. Findings are to be discussed in the context of a biochemical analysis of bisretinoids, the major fluorophores and main molecular substrates of RPE lipofuscin, in eyes of a mouse model carrying a single mutant Abca4 allele.
Methods
Patients
In a monocenter cross-sectional study, patients and controls were examined between December 2013 and December 2014 at the Department of Ophthalmology of the University of Bonn, Germany. The study was in adherence with the Declaration of Helsinki. Institutional Review Board approval (Ethikkommission, Medizinischen Fakultät, Rheinische Friedrich-Wilhelms-Universität Bonn) and written patient consent were obtained.
The study included index patients with ABCA4-related retinopathy and their parents. ABCA4-related retinopathy was diagnosed based on characteristic findings on indirect ophthalmoscopy, optical coherence tomography, and conventional fundus AF imaging. The clinical diagnosis was confirmed by the presence of biallelic mutations in ABCA4, and segregation analysis was carried out with samples of the parents. All index patients and their parents underwent a complete ophthalmologic examination including best-corrected visual acuity (BCVA), slit-lamp examination, and indirect ophthalmoscopy with dilated pupils. Axial length and corneal curvature were measured using the IOL-Master 500 (Carl Zeiss Meditec, Jena, Germany). Healthy subjects without ocular disease served as controls.
To minimize an influence of factors known to alter qAF measurements,22 exclusion criteria were age ≥ 65 years, ethnicity other than Caucasian, significant lens opacities, dilated pupil diameter below 7 mm, unstable fixation, refractive error > ±6 diopters (spherical equivalent), and any other additional known ocular pathology or prior intraocular surgery.
Genetic Testing
Genomic DNA was extracted from blood lymphocytes by standard protocol. ABCA4 sequence analysis of all coding exons and flanking splice junctions was performed either by Sanger chain-terminating dideoxynucleotide sequencing after PCR amplification, by multiplex ligation-dependent probe amplification (MLPA) analysis (performed when only a single mutation was detected), or by next-generation sequencing (NGS) including a quantitative readout to detect large structural rearrangements using a gene panel covering 120 genes associated with inherited retinal disease as described previously.23 Validation of identified putatively pathogenic variants and segregation analysis were carried out by conventional sequencing. Bioinformatic analyses were performed using MutationTaster,24 SIFT,25 and Polyphen-226 programs to assess pathogenicity of identified mutations (Supplementary Table S1). Nomenclature of mutations followed standards of the Human Genome Variation Society (HGVS).
Image Acquisition and Analysis
Pupils were dilated by instillation of 0.5% tropicamide and 2.5% phenylephrine. All probands underwent a standardized imaging protocol consisting of fundus photography (Visucam; Carl Zeiss Meditec), spectral-domain optical coherence tomography (SD-OCT), and fundus AF imaging using a cSLO (488-nm excitation, Spectralis HRA+OCT; Heidelberg Engineering, Heidelberg, Germany).27
Quantitative AF was performed as originally described by Delori et al.22 Briefly, a Spectralis HRA (Heidelberg Engineering) was equipped with an internal fluorescence reference to account for fluctuations in laser power and detector sensitivity. The reference material was identical to that used by Delori et al. (Supplementary Fig. S1).22 The reference was mounted in the intermediate retinal plane of the camera such that it could be imaged simultaneously with the fundus. The system was calibrated with a master reference provided by Heidelberg Engineering to adjust the qAF scale to imaging devices used in previous studies.8,22 Calibration of the system was verified in 3 to 6 monthly intervals.
For image acquisition, the camera was positioned centered to the fovea using the near-infrared reflectance mode and the internal fixation light. After switching to the qAF mode (488- nm excitation and 500- to 680-nm detection), focus and alignment were adjusted to obtain a maximum and uniform signal. The retina was illuminated for 20 to 30 seconds to bleach visual pigment, and the detector sensitivity was adjusted to avoid overexposure. With optimal camera position a series of 12 successive images was recorded in the high-speed mode, a 30° field of view, and 768 × 768-pixel resolution.
For image analysis, each image of a series was checked for optimal image quality. Images were excluded in cases of inhomogeneous illumination, sectorial opacities (e.g., eyelashes, floater), or unstable fixation. Images were then averaged and saved without normalization. The right eye was used for analysis. If not applicable (e.g., due to poor image quality or anatomical abnormalities), the left eye was used instead. This was the case for six index patients and eight parents.
For further analysis, images were exported from the HEYEX software to a custom-made image analysis program (IGOR; WaveMetrics, Inc., Lake Oswego, OR, USA). The mean gray value of the fluorescent reference and of a circular region consisting of eight subsegments with an eccentricity of approximately 7° to 9° centered to the fovea was measured. Retinal vessels and areas of atrophy were excluded based on histogram analysis. Individual segments were excluded from further analysis if atrophy covered more than 50% of the segment. The qAF value was then calculated from the gray values of the segment and the reference, the offset of the laser, the magnification, the lens opacity (based on normative data),28 and a device-specific calibration factor.22 The overall qAF value was computed as the mean of the qAF values of the eight segments (qAF8).
Establishment of a Normative Database
Normative data were acquired from 110 healthy controls of Caucasian ethnicity between 8 and 64 years of age. Quantitative AF values increased with age and were comparable to previously published data (Supplementary Fig. S2).29 Repeatability was 6.4% within one session and 13.1% between sessions.
Quantification of Bisretinoids in Mouse Eyes
Three-month-old wild-type (strain 129/Sv), heterozygous (Abca4+/−), and knockout Abca4−/−(129S4/SvJae-Abca4tm1Ght)12 mice were raised under 12-hour cyclic light (40–50 lux) and fed a standard rodent diet (NIH-31, 7013; Harlan Teklad, Madison, WI, USA), all carrying the Rpe65:Leu450 allele but free of the known Rd8 mutation.30 Mouse studies were done in adherence to guidelines established by the University of California-Los Angeles Animal Research Committee and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. After euthanasia, eyes were removed and flash frozen in liquid nitrogen and stored at −80°C for further processing.
Bisretinoids such as N-retinylidene-N-retinylethanolamine (A2E), N-retinylidene-N-retinylphosphatidylethanolamine (A2PE), dihydro-A2PE (A2PE-H2), and all-trans-RAL-dimer PE-conjugates (at-RAL dimer-PE) were extracted from mouse eyes. Briefly, after removing the cornea and lens, a single mouse eyecup was homogenized in 1 mL PBS. Four milliliters chloroform/methanol (2:1, vol/vol) was added, and the samples were extracted with the addition of 4 mL chloroform and 3 mL dH2O, followed by centrifugation at 1000g for 10 minutes. Extraction was repeated with the addition of 4 mL chloroform. Organic phases were pooled, filtered, dried under a stream of argon, and redissolved in 100 μL 2-propanol. Bisretinoid extracts were analyzed by normal-phase HPLC with a silica column (Zorbax-Sil 5 μm, 250 × 4.6 mm; Agilent Technologies, Wilmington, DE, USA) as previously described.31 Absorption units at 435 nm were converted to picomoles using a calibration curve with an authentic A2E standard and the published molar extinction coefficient for A2E32; the identity of each bisretinoid peak was confirmed by online spectral analysis. Data are presented as picomoles and milli-absorbance (mAU) per eye for A2E and other bisretinoids, respectively.
Statistical Analysis
Data were firstly analyzed descriptively. For human participants, comparisons between groups were conducted using age-adjusted linear regression, modeled with log transformations of qAF and age. The 95% prediction intervals for each retinal segment for the control group were calculated using standard error of the forecast. The average qAF of each retinal segment was standardized by the mean qAF8 value for each group to calculate the relative qAF. One-way ANOVA with Bonferroni post hoc testing adjusting for multiple comparisons of differences between groups was performed to compare bisretinoid levels in the mouse model of Stargardt disease. All analyses were conducted using Stata 12.1 (StataCorp, College Station, TX, USA).
Results
Twenty-six parents of 17 unrelated index patients with ABCA4-related retinopathy were included in the study (Table). In each parent, segregation analysis revealed one of the two pathogenic ABCA4 mutations identified in the affected offspring.
Table.
Summary of Age, Quantitative Autofluorescence Levels (qAF8), and ABCA4 Mutations of All Subjects Investigated in This Study
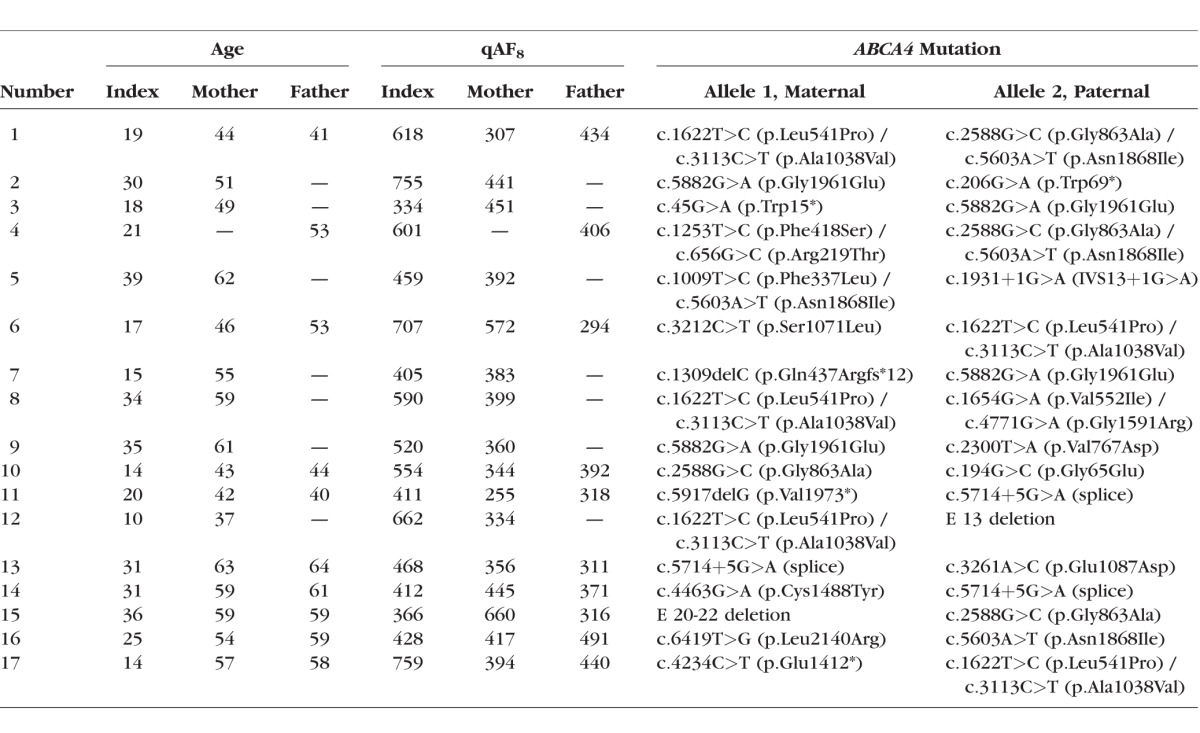
Index Patients With Biallelic ABCA4 Mutations
All index patients (mean age ± SD, 24.1 ± 9.1 years; range, 14–39 years) exhibited phenotypic findings characteristic for ABCA4-related retinopathy (Supplementary Fig. S3). Lipofuscin-related qAF8 levels were above the age-related 95% prediction interval of the control group in 16 out of the 17 patients, suggesting extensive disease-related lipofuscin accumulation (red triangles in Fig. 1; Table). One index patient had a qAF8 level within the normal range, most likely due to widespread dark flecks within the analyzed area (Supplementary Fig. S3, index of family 15).
Figure 1.
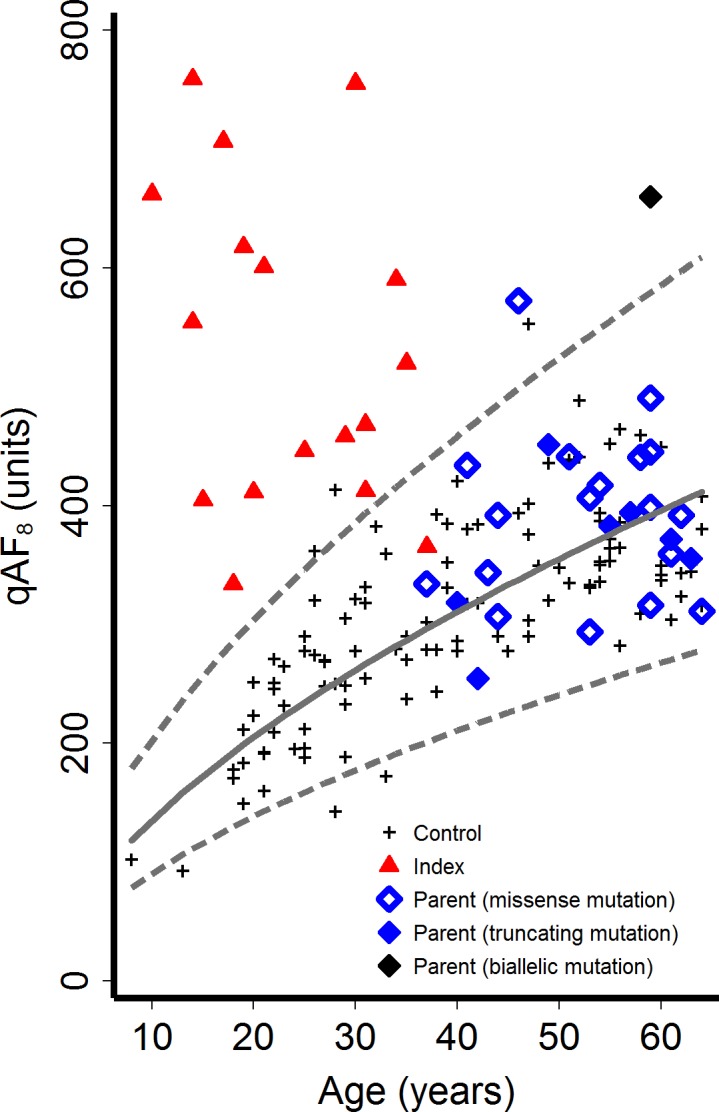
Quantitative autofluorescence values (qAF8) as a function of age. Dashed lines show 95% prediction interval of controls (black crosses, n = 110). Index patients (red triangles, n = 17) revealed elevated qAF8 values compared to controls. Parents with monoallelic ABCA4 mutations (blue diamonds, n = 25) were not different compared to controls in this sample. There was no difference between parents with truncating (solid blue diamonds) or missense (open blue diamonds) monoallelic ABCA4 mutations. One parent with elevated qAF8 values was found to carry a second independent ABCA4 mutation (black diamond).
Carriers of a Monoallelic ABCA4 Mutation
All parents (mean age ± SD 52.7 ± 8.1 years; range, 37–64 years) with one ABCA4 mutation had a best-corrected visual acuity (BCVA) of ≥20/20 except one (mother, family 5), who had mild refractive amblyopia (BCVA 20/32) in the right eye resulting from anisometropia. All probands except one (mother, family 15; see next paragraph) showed normal findings on ophthalmoscopy, SD-OCT, and conventional fundus AF imaging (Supplementary Fig. S3).
The overall qAF8 level and its variability in the parents with monoallelic ABCA4 mutations were not significantly different from controls (age-adjusted linear regression: t(135) = 1.045 P = 0.30; blue diamonds in Fig. 1) in this sample. Individual qAF8 values were within the age-adjusted 95% prediction interval of controls in 24 out of the 26 parents (Fig. 1). One subject with a monoallelic ABCA4 missense mutation (p.Ser1071Leu; mother, family 6) showed a slightly increased qAF8 at a level similar to an outlier of controls. Another parent (mother, family 15) with a deletion of the ABCA4 exons 20 to 22 revealed a substantially increased qAF8 compared to controls. Her visual acuity was 20/20 in both eyes, and she was asymptomatic and not aware of having a retinal disease. There were flecks of increased and decreased AF at the posterior pole in both eyes and a small paracentral geographic atrophy in the left eye. Sequencing of ABCA4 revealed an additional mild ABCA4 mutation, explaining the phenotype under a recessive disease model.
Despite the physiological age-dependent increase of lipofuscin-related qAF8 levels, parents with monoallelic ABCA4 mutations had lower qAF8 levels compared to their children with ABCA4-related retinopathy (Fig. 2, Supplementary Fig. S3). Levels of qAF8 were not significantly different between parents with truncating mutations (stop, frameshift, or splice site mutations) compared to those with missense mutations (t(135) = 0.961 P = 0.34, adjusted for the natural log of age; Figs. 1, 2) in this sample.
The spatial distribution of qAF levels was comparable between controls, index patients with two ABCA4 mutations, and their parents carrying one ABCA4 mutation. Highest values were located in the superotemporal and temporal segments, whereas lowest values were found in the nasal and inferior segments (Figs. 3).
Figure 3.

Spatial distribution of quantitative autofluorescence (qAF). The relative spatial distribution of qAF values (a) along the qAF8 circle (b) was comparable between controls, index patients, and parents with monoallelic ABCA4 mutations. The values represent the qAF of each segment relative to the mean of all segments (qAF8). Error bars show the 95% confidence interval. S, superior; ST, superiortemporal; T, temporal; IT, inferiortemporal; I, inferior; IN, inferiornasal; N, nasal; SN, superonasal.
Figure 2.

Quantitative autofluorescence (qAF) in exemplary families with either truncating (a) or missense (b) ABCA4 mutations. Left: Fundus autofluorescence images and corresponding color-coded qAF images in two index patients and their parents ([a] family 10, [b] family 11). Although gray levels appear similar, color-coded images reveal elevated qAF levels in the index patients. Right: Age-dependent qAF8 of the index patients (red triangles) and their parents (blue squares and dots) with monoallelic ABCA4 mutations. Dashed lines: 95% prediction interval of normal controls (black crosses).
Changes of Bisretinoid Levels in Abca4+/− Mice
To analyze the biochemical effects of monoallelic Abca4 mutations, bisretinoid levels from the whole eye of 3-month-old Abca4+/− mice were compared to those in wild-type (Abca4+/+) and knockout (Abca4−/−) mice at the same age. Levels of N-retinylidene-phosphatidylethanolamine (N-Ret-PE), the precursor of all tested bisretinoids, were not different between the groups (Supplementary Fig. S4). Compared to wild-type (Abca4+/+) mice, levels of at-RAL dimer-PE and A2PE-H2 were only slightly increased in Abca4+/− mice, but revealed a 12- and 18-fold increase, respectively, in Abca4−/− mice (Figs. 4a, 4b). In contrast, the levels of A2PE and total A2E were increased 3- and 6-fold in Abca4+/− and Abca4−/− mice, respectively, compared to wild-type (Abca4+/+) controls (Figs. 4c, 4d).
Figure 4.

Bisretinoid levels ([a]: at-RAL dimer, [b]: A2PE-H2, [c]: A2-PE, [d]: total A2E) from eye cups of 3-month-old wild-type mice (black, n = 6, Abca4+/+), mice with monoallelic Abca4 mutation (blue, n = 5, Abca4+/−), and Abca4 knockout mice (red, n = 6, Abca4−/−). mAU/eye, milli absorbance units per eye. Error bars show standard deviation. **P < 0.01; ***P < 0.001.
Discussion
The results of this study indicate that one ABCA4 mutation is not sufficient to cause changes typical for ABCA4-related retinopathy in humans, confirming the recessive nature of ABCA4-related retinopathy. Carriers of monoallelic mutations were indistinguishable from controls when investigated with a multimodal imaging approach including quantitative assessment of lipofuscin-related fundus AF (qAF). The latter method can be considered highly sensitive for detecting very early manifestations of ABCA4-related retinal disease,7,13 even though a minority of patients with specific ABCA4 mutations and/or specific phenotypic changes may reveal qAF levels in the normal range.8
In general, genotype/phenotype correlation in ABCA4-related retinopathy is a complex issue that is still under debate. Historically, a dose effect of the number and severity of ABCA4 mutations on retinal disease manifestation has been suggested.33,34 Accordingly, individuals with two severe ABCA4 mutations may manifest an early widespread structural and functional decline,35 while individuals with a combination of one severe and one mild or of two mild mutations present with the Stargardt disease phenotype confined to the posterior pole. Monoallelic ABCA4 mutations have been hypothesized to cause late-onset Stargardt disease or enhanced susceptibility to AMD. In these studies patients were defined by their retinal phenotype and genetic testing that was mostly limited to mutational analysis of ABCA4.14–16,20,21
The approach of this study differs from that in previous reports in that individuals were included based on their carrier state of monoallelic mutations in ABCA4 instead of their phenotype. A second mutation in ABCA4 or in any other gene conferring risk of retinal disease would be present only according to its (mostly low) frequency in the general population. Our cohort involved subjects similar in age and with monoallelic ABCA4 mutations similar to those in patients reported in recent studies that suggested a potential association between monoallelic ABCA4 mutations and atrophic subforms of AMD or late-onset Stargardt disease (Supplementary Table S2). Provided that ABCA4 indeed had a causative role in patients of these previous studies, it is possible that a second ABCA4 mutation was missed. In line with this assumption, the only parent in our study with a phenotype compatible with late-onset Stargardt disease (mother, family 15) turned out to carry a mild ABCA4 mutation in trans to the deletion she had passed on to the index patient. Alternatively, retinal disease in some patients in the previous studies may have been due to mutation(s) in a gene other than ABCA4.
In accordance with the human imaging data, direct downstream products of the ABCA4 substrate N-Ret-PE,36 A2PE-H2 and at-RAL dimer-PE were distinctly increased in Abca4−/− mice while levels were similar in Abca4+/− and wild-type controls. However, Abca4+/− mice showed levels of A2E and A2PE in between those measured in wild-type and Abca4−/− mice. This suggests a more complex situation on the molecular level (at least in mice), where the product of the normal allele does not fully compensate for a loss of functional product from the mutant allele. Different dynamics of formation and degradation of retinal bisretinoids might underlie these findings, including, for instance, saturation of specific biochemical processes when only one allele is functional.
These results have to be interpreted with reference to recent findings that revealed differences in the composition of mouse and human lipofuscin,37 which cannot be assessed using quantitative measures of lipofuscin-related AF. Besides A2E and A2PE, multiple other bisretinoid species and fluorophores have been detected in the RPE,38–40 which might be more important for the development of degenerative changes. However, their relative contributions to the overall lipofuscin-related AF signal intensity are currently unknown.
Accordingly, one cannot exclude subclinical effects of monoallelic ABCA4 mutations that remained undetected by means of all imaging modalities used herein. For instance, there may be an overall increased bisretinoid load that differs from normal in its composition, cellular toxicity, and spectral AF properties. Future studies using more precise spectral molecular imaging tools might reveal more details of the underlying molecular pathways. However, if such hypothetical and subclinical processes would affect retinal health later in life, a gradual onset of morphologic and/or functional changes might be expected. The lack of retinal alterations in carriers of monoallelic ABCA4 mutations up to an age of 64 years in our cohort suggests absence of such processes or a threshold damage that needs to be overcome before overt manifestation of retinal disease.
Although it would be interesting to study carriers of monoallelic ABCA4 mutations over 65 years of age, there is currently no established method to correct for the increasing variability of lens opacities with increasing age when recording qAF measures. Moreover, if carriers with monoallelic ABCA4 mutations presented with macular disease in the seventh or eighth decade, it might be difficult to differentiate between a monogenic effect and a genetically unrelated susceptibility to multifactorial AMD, which is highly prevalent in the older population. Thus, if rare variants in ABCA4 would indeed have a (modest) effect on AMD, very large cohorts and high-quality phenotyping would be needed to confirm such a hypothesis. In any case, one may not expect a risk for developing severe visual symptoms due to monoallelic ABCA4 mutations even at older age because supposedly early fundus changes (such as increased lipofuscin-related qAF) should precede functional decline.
Apart from the inability to differentiate specific fluorophores within the RPE, limitations of this study include the low number of study participants and the relatively old age of some participants (>60 years). The latter is accompanied by increasing variability of lens opacities, resulting in more variable and less reliable qAF measures. However, the uniformity of the observation and the strict criteria for image quality, which is essential for qAF imaging, suggest acceptable validity of our results.
The finding that carriers of monoallelic ABCA4 mutations were phenotypically normal in our setting has implications for future therapeutic approaches. Gene replacement therapy is currently pursued,41 but challenges include the large size of the ABCA4 coding region, the relatively inefficient viral transduction of photoreceptors, and a possibly low disease-specific efficiency of gene delivery.42 However, the lack of haploinsufficiency in the pathogenesis of ABCA4-mediated retinal and macular degenerations shown here suggests that gene therapy may be effective despite these challenges.
Supplementary Material
Acknowledgments
We thank Markus Preising for providing the genotype of family 17. We thank Francois Delori for providing the IGOR software that was developed in conjunction with the Department of Ophthalmology at Columbia University, New York, New York, United States.
Supported by the ProRetina Deutschland, Aachen, Germany, and the BONFOR research program of the University of Bonn, Bonn, Germany (Grant O-137.0018). The Department of Ophthalmology, University of Bonn, receives research support from Heidelberg Engineering. The sponsor or funding organization had no role in the design or conduct of this research. No sponsor or funding agency had any involvement in the design, collection, analysis, and interpretation of the data; manuscript writing; or the decision to submit the manuscript for publication. HJB and CB are employees of Bioscientia, which is part of a publicly traded diagnostic company.
Disclosure: P.L. Müller, None; M. Gliem, None; E. Mangold, None; H.J. Bolz, Bioscientia (E); R.P. Finger, None; M. McGuinness, None; C. Betz, Bioscientia (E); Z. Jiang, None; B.H.F. Weber, None; R.E. MacLaren, None; F.G. Holz, Heidelberg Engineering (C); R.A. Radu, None; P. Charbel Issa, None
References
- 1. Allikmets R,, Singh N,, Sun H,, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997; 15: 236–246. [DOI] [PubMed] [Google Scholar]
- 2. Cremers FP,, van de Pol DJ,, van Driel M,, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet. 1998; 7: 355–362. [DOI] [PubMed] [Google Scholar]
- 3. Maugeri A,, Klevering BJ,, Rohrschneider K,, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000; 67: 960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klevering BJ,, Deutman AF,, Maugeri A,, Cremers FP,, Hoyng CB. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005; 243: 90–100. [DOI] [PubMed] [Google Scholar]
- 5. Michaelides M,, Chen LL,, Brantley MA,, Jr, et al. ABCA4 mutations and discordant ABCA4 alleles in patients and siblings with bull's-eye maculopathy. Br J Ophthalmol. 2007; 91: 1650–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Delori FC,, Staurenghi G,, Arend O,, Dorey CK,, Goger DG,, Weiter JJ. In vivo measurement of lipofuscin in Stargardt's disease--fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1995; 36: 2327–2331. [PubMed] [Google Scholar]
- 7. Cideciyan AV,, Aleman TS,, Swider M,, et al. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet. 2004; 13: 525–534. [DOI] [PubMed] [Google Scholar]
- 8. Burke TR,, Duncker T,, Woods RL,, et al. Quantitative fundus autofluorescence in recessive Stargardt disease. Invest Ophthalmol Vis Sci. 2014; 55: 2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Delori FC,, Dorey CK,, Staurenghi G,, Arend O,, Goger DG,, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci. 1995; 36: 718–729. [PubMed] [Google Scholar]
- 10. Eagle RC,, Jr, Lucier AC,, Bernardino VB,, Jr, Yanoff M. Retinal pigment epithelial abnormalities in fundus flavimaculatus: a light and electron microscopic study. Ophthalmology. 1980; 87: 1189–1200. [DOI] [PubMed] [Google Scholar]
- 11. Birnbach CD,, Jarvelainen M,, Possin DE,, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994; 101: 1211–1219. [DOI] [PubMed] [Google Scholar]
- 12. Weng J,, Mata NL,, Azarian SM,, Tzekov RT,, Birch DG,, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell. 1999; 98: 13–23. [DOI] [PubMed] [Google Scholar]
- 13. Charbel Issa P,, Barnard AR,, Singh MS,, et al. Fundus autofluorescence in the Abca4−/− mouse model of Stargardt disease--correlation with accumulation of A2E, retinal function and histology. Invest Ophthalmol Vis Sci. 2013; 54: 5602–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Allikmets R,, Shroyer NF,, Singh N,, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997; 277: 1805–1807. [DOI] [PubMed] [Google Scholar]
- 15. Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet. 2000; 67: 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Souied EH,, Ducroq D,, Gerber S,, et al. Age-related macular degeneration in grandparents of patients with Stargardt disease: genetic study. Am J Ophthalmol. 1999; 128: 173–178. [DOI] [PubMed] [Google Scholar]
- 17. Stone EM,, Webster AR,, Vandenburgh K,, et al. Allelic variation in ABCR associated with Stargardt disease but not age-related macular degeneration. Nat Genet. 1998; 20: 328–329. [DOI] [PubMed] [Google Scholar]
- 18. Guymer RH,, Heon E,, Lotery AJ,, et al. Variation of codons 1961 and 2177 of the Stargardt disease gene is not associated with age-related macular degeneration. Arch Ophthalmol. 2001; 119: 745–751. [DOI] [PubMed] [Google Scholar]
- 19. De La Paz MA,, Guy VK,, Abou-Donia S,, et al. Analysis of the Stargardt disease gene (ABCR) in age-related macular degeneration. Ophthalmology. 1999; 106: 1531–1536. [DOI] [PubMed] [Google Scholar]
- 20. Fritsche LG,, Fleckenstein M,, Fiebig BS,, et al. A subgroup of age-related macular degeneration is associated with mono-allelic sequence variants in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2012; 53: 2112–2118. [DOI] [PubMed] [Google Scholar]
- 21. Westeneng-van Haaften SC,, Boon CJ,, Cremers FP,, Hoefsloot LH,, den Hollander AI,, Hoyng CB. Clinical and genetic characteristics of late-onset Stargardt's disease. Ophthalmology. 2012; 119: 1199–1210. [DOI] [PubMed] [Google Scholar]
- 22. Delori F,, Greenberg JP,, Fischer J,, et al. Quantitative measurements of autofluorescence with the scanning laser ophthalmoscope. Invest Ophthalmol Vis Sci. 2011; 52: 9379–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eisenberger T,, Neuhaus C,, Khan AO,, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One. 2013; 8: e78496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwarz JM,, Rödelsperger C,, Schuelke M,, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010; 7: 575–576. [DOI] [PubMed] [Google Scholar]
- 25. Ng PC,, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003; 31: 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adzhubei I,, Jordan DM,, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013; Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed]
- 27. Helb HM,, Charbel Issa P,, Fleckenstein M,, et al. Clinical evaluation of simultaneous confocal scanning laser ophthalmoscopy imaging combined with high-resolution spectral-domain optical coherence tomoggraphy. Acta Ophthalmol. 2010; 88: 842–849. [DOI] [PubMed] [Google Scholar]
- 28. van de Kraats J,, van Norren D. Optical density of the aging human ocular media in the visible and the UV. J Opt Soc Am A Opt Image Sci Vis. 2007; 24: 1842–1857. [DOI] [PubMed] [Google Scholar]
- 29. Greenberg JP,, Duncker T,, Woods RL,, Smith RT,, Sparrow JR,, Delori FC. Quantitative fundus autofluorescence in healthy eyes. Invest Ophthalmol Vis Sci. 2013; 54: 5684–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mattapallil MJ,, Wawrousek EF,, Chan CC,, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012; 53: 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Radu RA,, Hu J,, Yuan Q,, et al. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 2011; 286: 18593–18601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parish CA,, Hashimoto M,, Nakanishi K,, Dillon J,, Sparrow J. Isolation and one-step preparation of A2E and iso-A2E fluorophores from human retinal pigment epithelium. Proc Natl Acad Sci U S A. 1998; 95: 14609–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rozet JM,, Gerber S,, Souied E,, et al. Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur J Hum Genet. 1998; 6: 291–295. [DOI] [PubMed] [Google Scholar]
- 34. Shroyer NF,, Lewis RA,, Allikmets R,, et al. The rod photoreceptor ATP-binding cassette transporter gene, ABCR, and retinal disease: from monogenic to multifactorial. Vision Res. 1999; 39: 2537–2544. [DOI] [PubMed] [Google Scholar]
- 35. Fujinami K,, Zernant J,, Chana RK,, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2014; 122: 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Quazi F,, Lenevich S,, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012; 3: 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ablonczy Z,, Higbee D,, Grey AC,, Koutalos Y,, Schey KL,, Crouch RK. Similar molecules spatially correlate with lipofuscin and N-retinylidene-N-retinylethanolamine in the mouse but not in the human retinal pigment epithelium. Arch Biochem Biophys. 2013; 539: 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mata NL,, Tzekov RT,, Liu X,, Weng J,, Birch DG,, Travis GH. Delayed dark-adaptation and lipofuscin accumulation in abcr+/- mice: implications for involvement of ABCR in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2001; 42: 1685–1690. [PubMed] [Google Scholar]
- 39. Kim SR,, Fishkin N,, Kong J,, Nakanishi K,, Allikmets R,, Sparrow JR. Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc Natl Acad Sci U S A. 2004; 101: 11668–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mata NL,, Weng J,, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A. 2000; 97: 7154–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Auricchio A,, Trapani I,, Allikmets R. Gene therapy of ABCA4-associated diseases. Cold Spring Harb Perspect Med. 2015; 5: a017301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Charbel Issa P,, De Silva SR,, Lipinski DM,, et al. Assessment of tropism and effectiveness of new primate-derived hybrid recombinant AAV serotypes in the mouse and primate retina. PLoS One. 2013; 8: e60361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.