

Abstract
Considerable evidence suggests that rare leukemia cells with stem cell features, including self-renewal capacity and drug resistance, are primarily responsible for both disease maintenance and relapses. Traditionally, these so-called leukemia stem cells (LSCs) have been identified in the laboratory by their ability to engraft acute myeloid leukemia (AML) into immunocompromised mice. For many years, only those rare AML cells characterized by a hematopoietic stem cell (HSC) CD34+CD38− phenotype were believed capable of generating leukemia in immunocompromised mice. However more recently, significant heterogeneity in the phenotypes of those AML cells that can engraft immunocompromised mice has been demonstrated. AML cells that engraft immunocompromised mice have also been shown to not necessarily represent either the founder clone or those cells responsible for relapse. A recent study found that the most immature phenotype present in an AML correlated with genetically-defined risk groups and outcomes, but was heterogeneous. The patients with AML cells expressing a primitive HSC phenotype (CD34+CD38− with high aldehyde dehydrogenase activity) manifested significantly lower complete remission rates, as well as poorer event-free and overall survivals. Leukemias whose most primitive cells displayed more mature phenotypes demonstrated better outcomes. The strong clinical correlations suggest that the most immature phenotype detectable within a patient’s AML might serve as a biomarker for “clinically-relevant” LSCs.
Keywords: leukemia stem cells, acute myeloid leukemia, hematopoietic stem cells
Introduction
Most acute myeloid leukemia (AML) patients achieve complete remissions (CRs) with standard induction chemotherapy, but the majority subsequently relapse and succumb to the disease. It is becoming clear that tumor heterogeneity is one of the important factors in the dissociation between response and survival in AML. Although some of this tumor heterogeneity can be explained by subclonal progression within the malignant cells, the presence of leukemia cells at various stages of differentiation is also a major contributor. [1] Most data suggest that AML retains some semblance of the normal hematopoietic hierarchical structure: i.e., rare leukemia cells with stem cell features, including self-renewal capacity, give rise to partially differentiated progeny that comprise the bulk of the leukemia but possess only limited proliferative potential. These rare leukemia-initiating cells, or so-called leukemia stem cells (LSCs), are postulated to be responsible for relapse by resisting traditional cytotoxic chemotherapies that are usually highly active against the leukemia bulk. [2–15] LSCs appear to exhibit inherent drug resistance at least in part by co-opting normal stem cells’ intrinsic defense mechanisms such as quiescence, efflux pumps, and detoxifying enzymes. Moreover, LSCs, like normal hematopoietic stem cells (HSCs), upregulate the ‘don’t eat me signal’ CD47, presumably as a mechanism to avoid immune-mediated killing. [16] Recent data also suggest a prominent role for the stem cell microenvironment or niche in protecting LSCs from both cytotoxic and immunologic therapies. Importantly, cross talk between LSCs and their niche has now been clearly shown to be critical to the growth and maintenance of the leukemic clone. [17, 18]
The LSC Concept: Historical Perspective
As initially postulated by Ashley, cancer-initiating cells must survive long enough to accumulate the 3–7 genetic mutations necessary to generate cancer. [19, 20] Nowell hypothesized that the inherent longevity and extensive proliferative capacity of stem cells made them ideal candidates for cancer initiating cells. [21] However, longevity and extensive proliferative capacity are not traits restricted to classical stem cells. To some degree, myeloid progenitors beyond the level of HSCs also retain these properties.
The first clear evidence supporting the LSC concept was published more than 40 years ago, when Fialkow et al demonstrated clonal hematopoiesis involving both the erythroid and myeloid lineages in patients with chronic myeloid leukemia (CML). [22] In 1994, Lapidot and colleagues [8] established the capability to recapitulate leukemia after transplantation into immunocompromised mice as the gold standard for identifying LSCs. In these early mouse experiments LSCs were located strictly within the 34+38− cell compartment, suggesting a homogenous HSC phenotype. [3, 8] Moreover, in most AML patients the leukemic CD34+CD38− cells that engrafted immunocompromised mice could be separated from normal HSCs by their expression of the stem cell marker aldehyde dehydrogenase 1 (ALDH). Normal HSCs exhibited high ALDH expression (CD34+CD38−ALDHhigh), while the putative LSCs expressed intermediate levels (CD34+CD38−ALDHint). [5, 7, 12] However, in a significant fraction of AML patients no leukemia cell subset will engraft immunocompromised mice, even using the newer, more permissive mouse models. [2, 10, 14]
LSC Heterogeneity
Many studies have now suggested that the phenotype of putative LSCs is heterogeneous. AML cells of various differentiation phenotypes, including CD34+CD38+ and CD34−, have been shown capable of engrafting immunocompromised mice. [4, 10, 11, 13] Still other groups have suggested that putative LSCs can exhibit heterogeneous expression of ALDH. [5, 7, 23, 24] Sarry et al found that the engrafting AML cells can be heterogeneous even within the same patient. [11]
Our group found that the majority of core-binding factor (CBF) AML cells present in minimal residual disease (MRD) exhibited a CD34+CD38−ALDHint phenotype [5], even though such cells represented only about 1–10% of the total leukemia burden at diagnosis. [6] Moreover, their presence after therapy was highly associated with subsequent clinical relapse. [5] Thus, we hypothesized that the most primitive hematopoietic phenotype present in the AML may serve as a clinical biomarker for LSCs. [6] However, several patients had no detectable CD34+ AML cells, as others have also described [4, 10, 11, 13], and others had leukemia cells that were CD34+CD38−ALDHhigh. [5] To better understand the heterogeneity and clinical significance of the most immature phenotype present in a leukemia, patients with newly-diagnosed AML prospectively entered on a large multi-institutional clinical trial were studied. [6] As our earlier work predicted, the most immature hematopoietic cellular phenotype present within a specific leukemia was found to be heterogeneous, ranging from CD34− to that of primitive HSCs (i.e., CD34+CD38−ALDHhigh). [6] In most patients, the most primitive AML phenotype found was CD34+CD38−. The CD34+CD38− leukemia cells from about 60% of these patients displayed intermediate ALDH expression as previously described [5, 7, 12], while normal CD34+CD38− HSCs expressed high levels of ALDH. In the other 40% of patients harboring CD34+CD38− leukemia cells, the primitive AML cells exhibited high ALDH activity. No CD34+ leukemia cells could be detected in about a quarter of patients. [6]
Clinical significance of LSCs
Despite abundant research around the LSC concept, there has been limited data that LSCs are indeed responsible for disease resistance or relapse. Several groups have reported that the frequency of CD34+CD38− leukemia cells correlated with prognosis [14, 25], but as just described, some leukemias do not have a CD34+CD38− population to assess. [4, 10, 11, 13] Engraftability of AML cells in immunocompromised mice has also been shown to be associated with a poor clinical outcome. [2, 9, 10] However, the mouse engraftment assay may more accurately reflect the proliferative potential of the leukemic cells [26] and/or their interactions with the mouse microenvironment [27], than it does their role in disease maintenance and relapse. Accordingly, a recent study showed that AML cells that engrafted into immunocompromised mice may not represent either the founder clone or those responsible for relapse. [1] Thus, these data together with the fact that no AML subset in many patients will engraft immunocompromised mice, suggest that other means for LSC identification are needed to allow their study clinically.
Regardless of their phenotype or tumorigenic potential in immunocompromised mice, leukemic cells that persist after therapy (i.e., MRD) are arguably the most clinically important. Our group studied the clinical significance of an AML’s most primitive hematopoietic phenotype, since we found it to be enriched during MRD. [5] Despite demonstrating substantial heterogeneity overall within AML patients, the most immature phenotypes were much more consistent within individual genetically-defined risk groups. [6] The majority of AML patients whose most immature phenotype was CD34+CD38−ALDHhigh harbored poor-risk cytogenetics or FLT3 internal tandem duplications. The most immature phenotype found in all CBF and most intermediate-risk AMLs was CD34+CD38−ALDHint, and the most immature phenotype in the most favorable AMLs, NPM1 as a single mutation and APLs, was usually CD34+CD38+ or CD34−. [6]
Not surprisingly given the strong association with poor-risk genetics, patients harboring AML cells with a primitive HSC phenotype (CD34+CD38−ALDHhigh) display significantly lower event-free and overall survivals (Figure 1). [5–7, 23, 24] Patients whose most immature AML cells were CD34− displayed the best event-free and overall survivals [6], as others have also described. [28] Patients whose most immature AML cells had a CD34+CD38−ALDHint phenotype showed an intermediate prognosis (Figure 1). [6]
Figure 1. Clinical outcomes of AML patients based on the most immature leukemic cell phenotype.
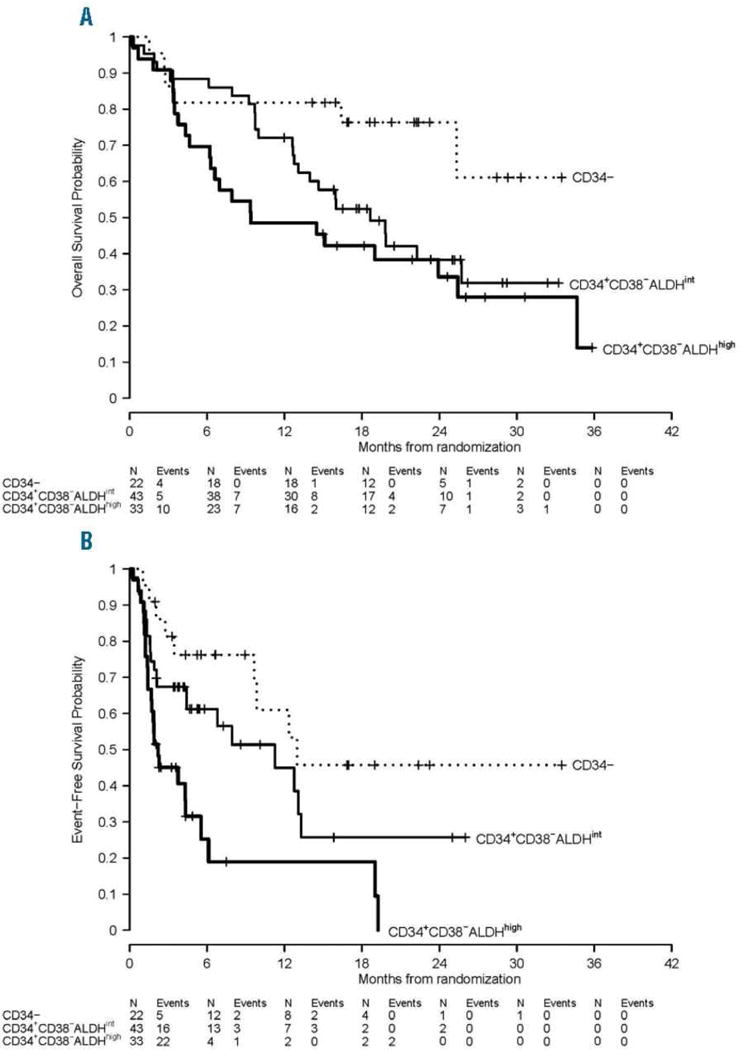
(A) Overall survival and (B) event-free survival by the most immature phenotype detectable in leukemia cells. (Adapted from Gerber et al, 2016 [6])
These data raise the possibility that the correlation of the AML’s most immature phenotype with outcome may be a function of the stage of hematopoietic differentiation at which the leukemogenic mutation develops. As normal CD34+CD38−ALDHhigh HSCs differentiate into more committed progenitors, both CD34 and ALDH expression decrease while CD38 expression increases (Figure 2A). [29–33] In addition, expression of resistance mechanisms (e.g., quiescence, efflux pumps, and detoxifying enzymes) also decreases with differentiation. [34] The most favorable AMLs appear to arise from more differentiated progenitors (CD34−) and the least favorable from primitive HSCs (CD34+CD38−ALDHhigh) (Figure 2B – 2D). The differentiation state of the AML’s cell of origin also appears prognostic within genetically-defined risk groups. Some NPM1-mutated AMLs and APLs appear to arise from CD34+ progenitors, and they appear to do worse than the more common CD34− varieties of these AMLs. [5, 35–38]
Figure 2. Leukemia stem cell heterogeneity as a function of the stage of hematopoietic differentiation at which the leukemogenic mutation develops.
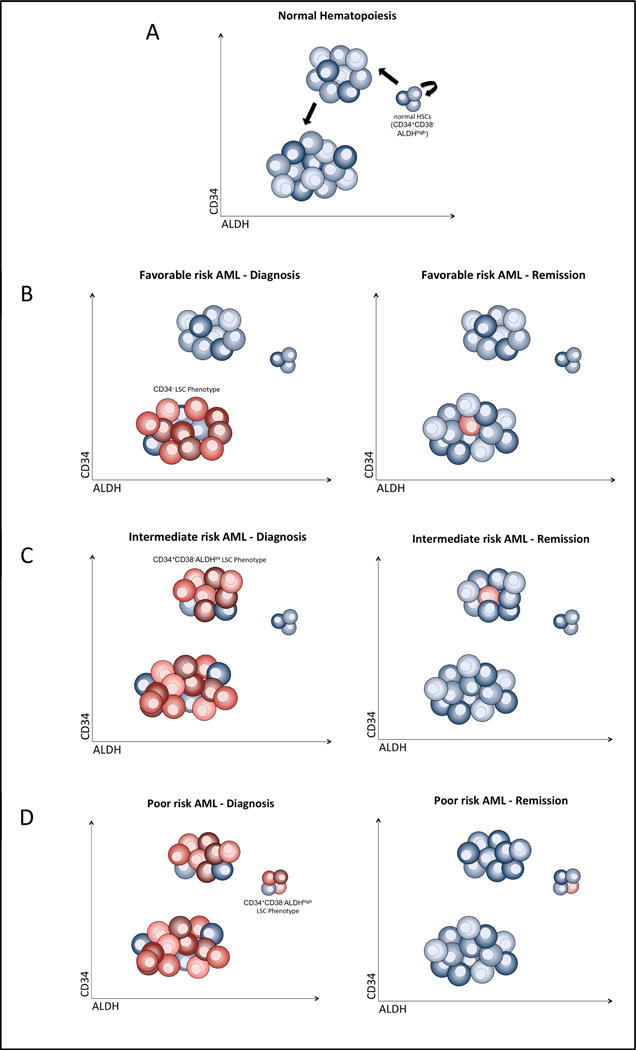
(A) As normal CD34+CD38−ALDHhigh HSCs (blue) differentiate into more committed progenitors, both CD34 and ALDH expression decreases. LSCs (red) are phenotypically heterogeneous with (B) the most favorable AMLs arising from more differentiated progenitors (CD34−), (C) intermediate-risk AMLs from less differentiated CD34+CD38−ALDHint, and (D) the least favorable AMLs from primitive HSCs (CD34+CD38−ALDHhigh). At remission, MRD is enriched for the most immature phenotype present in the leukemia (i.e., LSCs).
The LSC Microenvironment
It is now clear that the pathophysiology of AML involves a complex interplay between the leukemic cells and their surrounding bone marrow microenvironment. [17] In fact, creating a more humanized bone marrow microenvironment allowed for engraftment of traditionally “hard to engraft leukemias,” such as APL and CBF AML, further highlighting the importance of crosstalk between the human bone marrow microenvironment and the leukemia cells in disease pathology. [39] Like it does for normal hematopoietic progenitors, the bone marrow niche provides critical signals for the growth and maintenance of AML cells. However, the bone marrow microenvironment is highly complex, and thus the critical interactions between the niche and leukemia cells can also be expected to be multifaceted.
Increasing evidence suggests an important role for LSC interactions with the bone marrow microenvironment through the chemokine receptor CXCR4, which is important in the homing and support of HSCs. Normally, HSCs use CXCR4 on their surface to interact with CXCL12 secreted from various cells of the bone marrow to create HSC niches. Like normal HSCs, LSCs home to CXCL12+ areas of the bone marrow and increased expression of CXCR4 on leukemic cells predicts poor outcomes in leukemia patients. [40, 41] Intriguingly, in vitro and in vivo models have shown that homing of LSCs to the marrow can be disrupted by treatments targeting the CXCR4/CXCL12 axis, and chemotherapy resistance can be overcome. [42–46]
Combination therapies using LSC/microenvironment-targeted treatments have shown promise. The CXCR4 inhibitor, plerixafor, in combination with anti-TGFbeta and cytarabine was capable of prolonging survival in a mouse model of AML. [47] Interestingly, studies have shown an increase in CXCR4 expression on AML cells in response to chemotherapy, which further highlights the important role of microenvironment and leukemia cell interactions in AML pathogenesis. [48] Anti-VLA-4 antibodies used to disrupt the binding of VLA-4 on the surface of LSCs to fibronectin in the bone marrow microenvironment in combination with cytarabine significantly prolonged survival in a mouse model of AML. [49] Overall, these data demonstrate the potential of combinations targeting both LSCs and their interactions with the bone marrow microenvironment.
The bone marrow microenvironment’s expression of CYP3A4 and cytidine deaminase was recently shown to be at least partially responsible for the bone marrow stroma’s ability to protect leukemia cells from etoposide and cytarabine, respectively, both in vitro and in vivo. [50] Importantly, inhibiting CYP3A4 was able to restore the activity of etoposide against AML cells in the presence of bone marrow stroma. [50] Bone marrow stromal expression of CYP26, the major means of retinoid inactivation, also appears to protect LSCs from retinoids, a possible reason why these drugs have demonstrated little clinical activity in non-APL AML despite substantial in vitro activity. [51] Thus, expression of drug-metabolizing enzymes appears to be a novel mechanism of microenvironment-mediated drug resistance, allowing the bone marrow niche to create a sanctuary site from drugs. [50]
Clinically targeting LSCs
Despite the increasing evidence that relatively treatment-resistant LSCs are in part responsible for relapses following successful induction of complete remissions [5, 6, 10, 16, 25], there remains no clinical proof of the LSC concept: i.e., targeting these cells will result in improved outcomes. In order for an LSC-based target to have clinical utility, it must not only be expressed on LSCs, but if co-expressed by any normal cells, it must also have an acceptable toxicity profile. Given the heterogeneity of LSCs across different AMLs, it is also likely that one target will not be effective for all AML patients.
Several cell surface markers have been proposed as potential LSC-associated targets (Table 1). [29, 52–56] These phenotypic LSC markers have largely been identified based on their presence on AML cells that will engraft immunocompromised mouse models or from RNA sequencing of sorted cell populations. In some cases, including CD25, CD47, CD123, and CLL-1, the markers have been shown to correlate with AML patient outcomes. [55–58] CLL-1 expression on residual CD34+CD38− leukemic cells after induction was a better predictor of outcome than traditional MRD monitoring in one small series. [56] In another report, the high expression of a combination of CD123 with CLL-1 on CD34+ cells was also a strong prognostic marker for relapse in AML patients who had achieved remission. [59]
Table 1. Proposed phenotypic LSC markers.
A compilation of proposed phenotypic LSC-specific markers in AML, their alternate name(s), known biological function(s), and some of the cell type(s) they are known to be positive on.
| Alternate name(s) | Function(s) | Cell Type(s) | |
|---|---|---|---|
| CD25 | IL-2Rα | T cell proliferation, inflammatory responses | T cells, B cells |
| CD26 | Dipeptidyl peptidase-4 (DPP4) | Immune regulation, T cell activation, apoptosis, and glucose metabolism | T cells, B cells, NK cells, macrophages |
| CD33 | Sialic acid binding Ig-like lectin 3 (Siglec-3) | Sialic acid dependent binding, apoptosis in AML | Myeloid cells, few lymphoid cells |
| CD47 | Integrin associated protein (IAP) | Apoptosis, proliferation, adhesion, migration, and immune recognition; phagocytosis block | T cells, B cells, NK cells, macrophages, progenitor cells, and many others |
| CD96 | T cell activation, increased late expression (Tactile) | Immune adhesion, antigen presentation | T cells, NK cells, some B cells |
| CD123 | IL-3R | Proliferation and differentiation during hematopoiesis | Hematopoietic progenitor cells |
| CLL-1 | C-type lectin-like molecule 1, C-type lectin domain family 12 member A (CLEC12A) | Cell adhesion, cell-cell signaling, inflammation, and immune responses | Monocytes, macrophages, dendritic cells, granulocytes |
| TIM3 | T cell immunoglobulin mucin-3 | Autoimmunity and allergy | Th1 cells |
Several of these targets have already been, or are actively being, studied clinically. Monoclonal antibody therapy targeting CD33+ cells did show efficacy in both relapsed and elderly AML patients. [60–62] However, lack of overall survival despite higher remission rates suggested CD33 was probably expressed primarily by differentiated leukemia cells and not LSCs. [65] Moreover, given the ubiquitous expression of CD33 on many blood cell types, cytopenias were a common side effect. [63, 64] Interestingly, the one subgroup that appeared to have an overall survival improvement was the favorable cytogenetic AMLs [65, 66]; these data may represent additional evidence that favorable AMLs arise from more differentiated, CD33+, hematopoietic progenitors, while the LSCs from other subtypes arise from CD33− progenitors.
Anti-CLL-1 antibodies were capable of inducing in vitro complement-dependent cytotoxicity (CDC) in CLL-1 expressing AML cell lines and primary blasts in both in vitro and in vivo mouse models. [65] It does not appear that clinical trials targeting CLL-1 in AML have been undertaken. Anti-CD123 as an LSC-targeted therapy in AML has also shown promise. Though early experiments suggested a nonconventional role for upregulated CD123 on LSCs, more recent ex vivo studies have shown that anti-CD123 is capable of reducing IL-3-mediated proliferation in primary AML cells. [54, 66, 67] In addition, cytokine-induced killer cells transduced with a chimeric antigen receptor (CAR) targeting CD123 have shown potent killing of CD123 positive AML cell lines in vitro as well as primary AML blasts while sparing normal HSCs ex vivo. [68] Trials targeting CD123 are in progress, but thus far have generally involved patients that were refractory to standard therapy so its role in preventing relapses remains unclear. [66] The ‘don’t eat me signal,’ CD47, was shown to be expressed on most primary AML specimens, bulk tumor as well as LSCs, compared with normal bone marrow HSCs which expressed lower levels. [26] Clinical trials targeting CD47 are just beginning.
In addition to targeting LSCs directly, clinical trials targeting the LSC microenvironment are also in progress. Inhibition of the CXCR4/CXCL12 axis is being studied to mobilize leukemia from its protective environment as a means to increase its sensitivity to chemotherapy. A phase I/II clinical trial of plerixafor to inhibit CXCR4 in relapsed, refractory AML showed it to be safe and capable of mobilizing AML blasts [69], but definitive clinical activity awaits the results of ongoing trials. Targeting other adhesion molecules such as CD44 [70] or V-CAM [71] could also overcome microenvironment-mediated drug resistance. However, it is possible that the microenvironment-mediated drug resistant phenotype is maintained even after malignant cells are displaced from their bone marrow niche. [72] Based on the detoxifying effects of CYPs in the microenvironment [50, 51], our group has developed several clinical trials aimed at overcoming this potential mechanism for LSC resistance.
Conclusion
The failure of complete remissions to reliably translate into cures in AML can be explained by the LSC paradigm. Unfortunately, the definitive proof for the clinical importance of LSCs – that targeting them improves outcome – is currently lacking. Thus, the true clinical relevance of LSCs has remained the focus of considerable debate. The long-standing definition of LSCs focusing on immunocompromised mouse models of engraftment has led to potentially contradictory results that have proven difficult to translate into the clinic and across AML subtypes. Not only are such assays cumbersome and non-quantitative, but they also reveal that varying cell phenotypes are capable of engrafting leukemia in mice; further, these assays may have little correlation to ultimate disease outcomes. [1, 4, 10, 11, 13] Beyond that, a significant fraction of AML patients has no leukemia cell subset that will engraft. [2, 10, 14]
Most studies have found that both phenotypic and genetic heterogeneity is less evident in MRD present during first CR than at disease diagnosis (Figure 2). [5, 73, 74] As a “more homogeneous” leukemia cell population, first CR may present an optimal time to target these cells with novel approaches. Moving forward, focusing on the most primitive cell phenotype present within the patients’ AML cells may provide a broadly applicable means of studying clinically relevant LSCs as well as appropriate therapies to target these cells. Moreover, about 30–40% of AML patients lack any usual cytogenetic or genetic prognostic factors, and even when present such prognostic factors may not be available for days or weeks. The most immature phenotype present within a patient’s AML can be readily determined in essentially all patients by flow cytometry within hours of diagnosis. Rapid risk-stratification may be particularly useful for patients harboring CD34+CD38−ALDHhigh leukemia cells, which appear to identify high-risk patients often refractory to induction chemotherapy. A CD34+CD38−ALDHhigh leukemic phenotype could also be used to guide patients toward allogeneic transplantation when no prognostic cytogenetic or genetic abnormalities are present.
Highlights.
Relapses occur in acute myeloid leukemia because current treatments do not clinically target and eliminate leukemia stem cells.
Several lines of evidence now question the clinical relevance of leukemia cells that engraft in immunocompromised mice, the “gold standard” for identifying LSCs.
The leukemia clone’s most immature phenotype is heterogeneous for CD34, CD38, and aldehyde dehydrogenase expression, but correlates with genetically-defined risk groups and outcomes.
Identifying the most immature phenotype within a patient’s leukemia and studying this population for actionable targets may bypass the reliance on mouse models to identify LSCs.
Acknowledgments
This work was supported in part by the National Institutes of Health [grants P01 CA015396 and P30 CA006973].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klco JM, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25(3):379–92. doi: 10.1016/j.ccr.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ailles LE, et al. Growth characteristics of acute myelogenous leukemia progenitors that initiate malignant hematopoiesis in nonobese diabetic/severe combined immunodeficient mice. Blood. 1999;94(5):1761–72. [PubMed] [Google Scholar]
- 3.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 4.Cheung AM, et al. FLT3/internal tandem duplication subclones in acute myeloid leukemia differ in their engraftment potential in NOD/SCID mice. Leuk Res. 2010;34(1):119–22. doi: 10.1016/j.leukres.2009.07.035. [DOI] [PubMed] [Google Scholar]
- 5.Gerber JM, et al. A clinically relevant population of leukemic CD34(+)CD38(−) cells in acute myeloid leukemia. Blood. 2012;119(15):3571–7. doi: 10.1182/blood-2011-06-364182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerber JM, et al. Association of acute myeloid leukemia’s most immature phenotype with risk groups and outcomes. Haematologica. 2016;101(5):607–16. doi: 10.3324/haematol.2015.135194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoang VT, et al. The rarity of ALDH(+) cells is the key to separation of normal versus leukemia stem cells by ALDH activity in AML patients. Int J Cancer. 2015;137(3):525–36. doi: 10.1002/ijc.29410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapidot T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 9.Monaco G, et al. Engraftment of acute myeloid leukemia in NOD/SCID mice is independent of CXCR4 and predicts poor patient survival. Stem Cells. 2004;22(2):188–201. doi: 10.1634/stemcells.22-2-188. [DOI] [PubMed] [Google Scholar]
- 10.Pearce DJ, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood. 2006;107(3):1166–73. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarry JE, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J Clin Invest. 2011;121(1):384–95. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuurhuis GJ, et al. Normal hematopoietic stem cells within the AML bone marrow have a distinct and higher ALDH activity level than co-existing leukemic stem cells. PLoS One. 2013;8(11):e78897. doi: 10.1371/journal.pone.0078897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taussig DC, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115(10):1976–84. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Terwijn M, et al. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS One. 2014;9(9):e107587. doi: 10.1371/journal.pone.0107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martelli MP, et al. CD34+ cells from AML with mutated NPM1 harbor cytoplasmic mutated nucleophosmin and generate leukemia in immunocompromised mice. Blood. 2010;116(19):3907–22. doi: 10.1182/blood-2009-08-238899. [DOI] [PubMed] [Google Scholar]
- 16.Jaiswal S, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–85. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghiaur G, Wroblewski M, Loges S. Acute Myelogenous Leukemia and its Microenvironment: A Molecular Conversation. Semin Hematol. 2015;52(3):200–6. doi: 10.1053/j.seminhematol.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Kim JA, et al. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015;75(11):2222–31. doi: 10.1158/0008-5472.CAN-14-3379. [DOI] [PubMed] [Google Scholar]
- 19.Ashley DJ. The two “hit” and multiple “hit” theories of carcinogenesis. Br J Cancer. 1969;23(2):313–28. doi: 10.1038/bjc.1969.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogelstein B, Kinzler KW. The Path to Cancer –Three Strikes and You’re Out. N Engl J Med. 2015;373(20):1895–8. doi: 10.1056/NEJMp1508811. [DOI] [PubMed] [Google Scholar]
- 21.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 22.Fialkow PJ, Gartler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci U S A. 1967;58(4):1468–71. doi: 10.1073/pnas.58.4.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearce DJ, et al. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells. 2005;23(6):752–60. doi: 10.1634/stemcells.2004-0292. [DOI] [PubMed] [Google Scholar]
- 24.Ran D, et al. Heterogeneity of leukemia stem cell candidates at diagnosis of acute myeloid leukemia and their clinical significance. Exp Hematol. 2012;40(2):155–65.e1. doi: 10.1016/j.exphem.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Vergez F, et al. High levels of CD34+CD38low/−CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica. 2011;96(12):1792–8. doi: 10.3324/haematol.2011.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rombouts WJ, Martens AC, Ploemacher RE. Identification of variables determining the engraftment potential of human acute myeloid leukemia in the immunodeficient NOD/SCID human chimera model. Leukemia. 2000;14(5):889–97. doi: 10.1038/sj.leu.2401777. [DOI] [PubMed] [Google Scholar]
- 27.Risueno RM, et al. Identification of T-lymphocytic leukemia-initiating stem cells residing in a small subset of patients with acute myeloid leukemic disease. Blood. 2011;117(26):7112–20. doi: 10.1182/blood-2011-01-329078. [DOI] [PubMed] [Google Scholar]
- 28.Zeijlemaker W, et al. Absence of leukaemic CD34+ cells in acute myeloid leukaemia is of high prognostic value: a longstanding controversy deciphered. Br J Haematol. 2015 doi: 10.1111/bjh.13572. [DOI] [PubMed] [Google Scholar]
- 29.Gerber JM, et al. Genome-wide comparison of the transcriptomes of highly enriched normal and chronic myeloid leukemia stem and progenitor cell populations. Oncotarget. 2013;4(5):715–28. doi: 10.18632/oncotarget.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghiaur G, et al. Regulation of human hematopoietic stem cell self-renewal by the microenvironment’s control of retinoic acid signaling. Proc Natl Acad Sci U S A. 2013;110(40):16121–6. doi: 10.1073/pnas.1305937110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones RJ, et al. Assessment of aldehyde dehydrogenase in viable cells. Blood. 1995;85(10):2742–6. [PubMed] [Google Scholar]
- 32.Jones RJ, et al. Characterization of mouse lymphohematopoietic stem cells lacking spleen colony-forming activity. Blood. 1996;88(2):487–91. [PubMed] [Google Scholar]
- 33.Krause DS, et al. CD34: structure, biology, and clinical utility. Blood. 1996;87(1):1–13. [PubMed] [Google Scholar]
- 34.Raaijmakers MH, et al. Quantitative assessment of gene expression in highly purified hematopoietic cells using real-time reverse transcriptase polymerase chain reaction. Exp Hematol. 2002;30(5):481–7. doi: 10.1016/s0301-472x(02)00787-7. [DOI] [PubMed] [Google Scholar]
- 35.Ahmad EI, et al. The biological characteristics of adult CD34+ acute promyelocytic leukemia. Med Oncol. 2012;29(2):1119–26. doi: 10.1007/s12032-011-9895-y. [DOI] [PubMed] [Google Scholar]
- 36.Breccia M, et al. Negative prognostic value of CD34 antigen also if expressed on a small population of acute promyelocytic leukemia cells. Ann Hematol. 2014;93(11):1819–23. doi: 10.1007/s00277-014-2130-0. [DOI] [PubMed] [Google Scholar]
- 37.Dang H, et al. CD34 expression predicts an adverse outcome in patients with NPM1-positive acute myeloid leukemia. Hum Pathol. 2013;44(10):2038–46. doi: 10.1016/j.humpath.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Lee JJ, et al. CD34 expression is associated with poor clinical outcome in patients with acute promyelocytic leukemia. Am J Hematol. 2003;73(3):149–53. doi: 10.1002/ajh.10337. [DOI] [PubMed] [Google Scholar]
- 39.Reinisch A, et al. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med. 2016;22(7):812–21. doi: 10.1038/nm.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rombouts EJ, et al. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104(2):550–7. doi: 10.1182/blood-2004-02-0566. [DOI] [PubMed] [Google Scholar]
- 41.Colmone A, et al. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861–5. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- 42.Kuhne MR, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19(2):357–66. doi: 10.1158/1078-0432.CCR-12-2333. [DOI] [PubMed] [Google Scholar]
- 43.Nervi B, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206–14. doi: 10.1182/blood-2008-06-162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tavor S, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64(8):2817–24. doi: 10.1158/0008-5472.can-03-3693. [DOI] [PubMed] [Google Scholar]
- 45.Zeng Z, et al. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther. 2006;5(12):3113–21. doi: 10.1158/1535-7163.MCT-06-0228. [DOI] [PubMed] [Google Scholar]
- 46.Zeng Z, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113(24):6215–24. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tabe Y, et al. TGF-beta-Neutralizing Antibody 1D11 Enhances Cytarabine-Induced Apoptosis in AML Cells in the Bone Marrow Microenvironment. PLoS One. 2013;8(6):e62785. doi: 10.1371/journal.pone.0062785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sison EA, et al. Dynamic chemotherapy-induced upregulation of CXCR4 expression: a mechanism of therapeutic resistance in pediatric AML. Mol Cancer Res. 2013;11(9):1004–16. doi: 10.1158/1541-7786.MCR-13-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsunaga T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9(9):1158–65. doi: 10.1038/nm909. [DOI] [PubMed] [Google Scholar]
- 50.Alonso S, et al. Human bone marrow niche chemoprotection mediated by cytochrome P450 enzymes. Oncotarget. 2015;6(17):14905–12. doi: 10.18632/oncotarget.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su M, et al. All-Trans Retinoic Acid Activity in Acute Myeloid Leukemia: Role of Cytochrome P450 Enzyme Expression by the Microenvironment. PLoS One. 2015;10(6):e0127790. doi: 10.1371/journal.pone.0127790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hosen N, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104(26):11008–13. doi: 10.1073/pnas.0704271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jan M, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci U S A. 2011;108(12):5009–14. doi: 10.1073/pnas.1100551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jordan CT, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–84. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 55.Majeti R, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–99. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Rhenen A, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110(7):2659–66. doi: 10.1182/blood-2007-03-083048. [DOI] [PubMed] [Google Scholar]
- 57.Ikegawa S, et al. CD25 expression on residual leukemic blasts at the time of allogeneic hematopoietic stem cell transplant predicts relapse in patients with acute myeloid leukemia without complete remission. Leuk Lymphoma. 2016;57(6):1375–81. doi: 10.3109/10428194.2015.1099644. [DOI] [PubMed] [Google Scholar]
- 58.Nakase K, et al. Prognostic Relevance of Cytokine Receptor Expression in Acute Myeloid Leukemia: Interleukin-2 Receptor alpha-Chain (CD25) Expression Predicts a Poor Prognosis. PLoS One. 2015;10(9):e0128998. doi: 10.1371/journal.pone.0128998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roug AS, et al. hMICL and CD123 in combination with a CD45/CD34/CD117 backbone - a universal marker combination for the detection of minimal residual disease in acute myeloid leukaemia. Br J Haematol. 2014;164(2):212–22. doi: 10.1111/bjh.12614. [DOI] [PubMed] [Google Scholar]
- 60.Burnett AK, Mohite U. Treatment of older patients with acute myeloid leukemia–new agents. Semin Hematol. 2006;43(2):96–106. doi: 10.1053/j.seminhematol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 61.Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011;30(9):1009–19. doi: 10.1038/onc.2010.511. [DOI] [PubMed] [Google Scholar]
- 62.Tsimberidou AM, et al. Mylotarg, fludarabine, cytarabine (ara-C), and cyclosporine (MFAC) regimen as post-remission therapy in acute myelogenous leukemia. Cancer Chemother Pharmacol. 2003;52(6):449–52. doi: 10.1007/s00280-003-0671-3. [DOI] [PubMed] [Google Scholar]
- 63.Giles F, Estey E, O’Brien S. Gemtuzumab ozogamicin in the treatment of acute myeloid leukemia. Cancer. 2003;98(10):2095–104. doi: 10.1002/cncr.11791. [DOI] [PubMed] [Google Scholar]
- 64.Taussig DC, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–92. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao X, et al. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica. 2010;95(1):71–8. doi: 10.3324/haematol.2009.009811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He SZ, et al. A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymphoma. 2015;56(5):1406–15. doi: 10.3109/10428194.2014.956316. [DOI] [PubMed] [Google Scholar]
- 67.Jin L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 68.Tettamanti S, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 69.Uy GL, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917–24. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gul-Uludag H, et al. Polymeric nanoparticle-mediated silencing of CD44 receptor in CD34+ acute myeloid leukemia cells. Leuk Res. 2014;38(11):1299–308. doi: 10.1016/j.leukres.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 71.Matsunaga T, et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia. 2008;22(2):353–60. doi: 10.1038/sj.leu.2405017. [DOI] [PubMed] [Google Scholar]
- 72.Alonso S, et al. Bone Marrow Niche - Multiple Myeloma Cross-Talk Generates Bortezomib Resistance. Blood. 2015;126(23):914–914. [Google Scholar]
- 73.Mardis ER. Genome sequencing and cancer. Curr Opin Genet Dev. 2012;22(3):245–50. doi: 10.1016/j.gde.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zeijlemaker W, Gratama JW, Schuurhuis GJ. Tumor heterogeneity makes AML a “moving target” for detection of residual disease. Cytometry B Clin Cytom. 2014;86(1):3–14. doi: 10.1002/cyto.b.21134. [DOI] [PubMed] [Google Scholar]