

Abstract
Regenerating the diseased tissue is one of the foremost concerns for the millions of patients who suffer from tissue damage each year. Local delivery of cell-laden hydrogels offers an attractive approach for tissue repair. However, due to the typical macroscopic size of these cell constructs, the encapsulated cells often suffer from poor nutrient exchange. These issues can be mitigated by incorporating cells into microscopic hydrogels, or microgels, whose large surface-to-volume ratio promotes efficient mass transport and enhanced cell-matrix interactions. Using microfluidic technology, monodisperse cell-laden microgels with tunable sizes can be generated in a high-throughput manner, making them useful building blocks that can be assembled into tissue constructs with spatially controlled physicochemical properties. In this review, we examine microfluidics-generated cell-laden microgels for tissue regeneration applications. We provide a brief overview of the common biomaterials, gelation mechanisms, and microfluidic device designs that are used to generate these microgels, and summarize the most recent works on how they are applied to tissue regeneration. Finally, we discuss future applications of microfluidic cell-laden microgels as well as existing challenges that should be resolved to stimulate their clinical application.
Textual Abstract
This review provides an overview of how cell-laden microfluidic microgels are generated, summarizes their most recent applications in tissue regeneration, and discusses future applications as well as existing challenges.
1. Introduction
Millions of patients suffer from diseased or damaged tissue each year. Although tissue transplantation can be used to treat these patients, its application is limited by a severe shortage of donor tissue. Tissue engineering offers a solution by engineering tissues to replace the lost functions.1 The extracellular matrix, a critical component of natural tissue that is composed of a variety of proteins as well as both soluble and insoluble macromolecules, regulates tissue dynamics by influencing cellular processes such as proliferation, differentiation, migration, and apoptosis through bi-directional molecular interactions with encapsulated cells.2 Currently, a popular tissue engineering approach is to isolate and incorporate a patient’s autologous cells into three-dimensional scaffolds that mimic the functions of the extracellular matrix. These cell-laden scaffolds, which provide an environment for new tissue generation, can be inserted into the diseased area of a patient’s body to guide the structure and function of the new tissue.
The material comprising the scaffold determines its physical, biological, and mass transport properties, which are crucial design variables to consider depending on the target tissue. Many synthetic polymers, including poly(glycolic acid), poly(lactic acid), and poly(lactic-co-glycolic acid), have been used as scaffold materials, but they require surgical incisions for placement into the patient’s body.3 In contrast, hydrogels, which are three-dimensional polymer matrices formed by crosslinking hydrophilic homopolymers, copolymers, or macromers, can be delivered into the body in a minimally invasive manner. Moreover, hydrogels are not only biocompatible but also structurally and compositionally similar to the extracellular matrix.4 Despite these favorable properties, encapsulation of cells within macroscopic hydrogels often leads to limited cell-cell contact and communication as well as poor nutrient exchange due to a low rate of diffusion and suboptimal distance between extracellular molecules.5
This issue can be resolved by forming hydrogel microspheres, or microgels, whose large surface area-to-volume ratio promotes effective nutrient and water transfer as well as improve cell-matrix interactions, thereby maintaining long-term viability of the encapsulated cells.6,7 Cell-laden microgels have been used in tissue engineering applications as building blocks for complex tissue mimics,8 co-culture systems for developing three-dimensional organ models,9 and controlled microenvironments for directing stem cell differentiation.10 In all of these applications, control over the size and size distribution of the microgels is important as they can influence the phenotypes of the encapsulated cells.11,12 Although many microfabrication techniques can entrap cells, they suffer from low throughput inherent in batch processing. A promising alternative is microfluidic techniques, which can be used to rapidly generate monodisperse microgels with tunable sizes simply by manipulating and controlling the flow of multiple immiscible liquids.
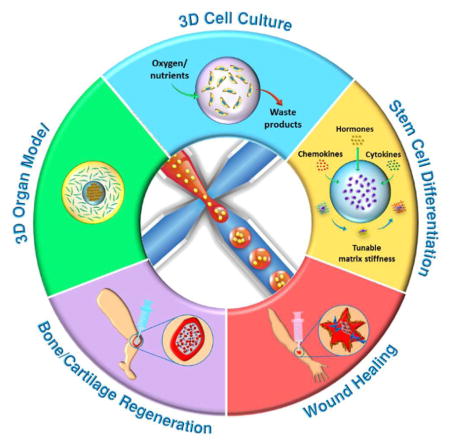
Here, we review microfluidics-generated cell-laden microgels for tissue regeneration applications. We first briefly cover the methods for gelation and materials used to produce microgels. We next describe typical microfluidic device designs used to generate microgels and strategies for incorporating cells into these microgels. This will be followed by highlighting the advantages of microfluidics-generated microgels over conventional hydrogels. After summarizing the most recent works on cell-laden microgels for tissue regeneration (Fig. 1), we will speculate on the potential applications of these microgels in other tissue engineering applications.
Fig. 1.

Schematic overview of the applications of cell-laden microfluidic microgels for tissue regeneration.
2. Principle of gelation
Aqueous hydrogel precursor solutions generally undergo either physical or chemical gelation to form solidified microgels. The gelation method is usually chosen based on a variety of factors, such as the type of polymer used for crosslinking, strategy for cell encapsulation, and specific tissue regeneration application. Comprehensive overviews of common gelation mechanisms can be found in recent reviews.13,14 Here, we provide a brief overview of the gelation principles for common physical and chemical gelation strategies, as well as outline their respective advantages and disadvantages when applied to the preparation of microfluidic microgels (Table 1).
Table 1.
The advantages and disadvantages of common physical and chemical gelation strategies for fabricating microfluidic microgels.
| Gelation Strategy | Advantage | Disadvantage | Ref. |
|---|---|---|---|
| Physical Gelation | |||
| Electrostatic interaction |
|
|
15 |
| Thermally-mediated gelation |
|
|
26 |
| Hydrogen bond |
|
|
15, 30 |
|
| |||
| Chemical Gelation | |||
| Photopolymerization |
|
|
31, 32 |
| Michael addition |
|
|
5, 33, 34 |
| Enzymatic reaction |
|
|
35–39 |
2.1 Physical gelation
Physical gelation can be initiated by an environmental trigger, such as changes in pH or temperature, upon which macromolecules crosslink via noncovalent, secondary interactions such as hydrophobic, electrostatic, and hydrogen bonding.15,16 Although the gelation process can be carried out under mild conditions, the physical hydrogel network typically has weak mechanical strength and shows poor stability in tissues.17 Here, we briefly overview several common physical gelation methods, including electrostatic interaction (ionic crosslinking), thermally-mediated gelation, and hydrogen bonding.
Being one of the most frequently used physical methods, crosslinking based on electrostatic interactions can be carried out between either a polymer and a small molecule or two polymers of opposite charges. For the first case, a classic example would be the crosslinking of alginate with Ca2+ ions, which attach to the carboxylic acid groups of alginate molecules.18–20 An example of the latter case would be ionic complementary peptides with alternating positively and negatively charged units that can self-assemble into crosslinked networks.15 The crosslinking process can be triggered by pH changes, which ionize the functional groups that induce gelation. For instance, acetic acid has been used to trigger the release of Ca2+ from Ca-EDTA complexes mixed in alginate, leading to gelation upon the ions’ contact with alginate molecules.21 However, changes in pH should be gradual and kept within a certain limit so that cell viability is not significantly reduced. An advantage of this strategy is that ionic species in the extracellular fluid can bind competitively with components in the ionically crosslinked network, thereby gradually degrading the hydrogel.
Another popular physical strategy is thermally-mediated solgel transition.22–25 For instance, gelatin solutions are emulsified at 40 °C, and the droplets are subsequently cooled down to 5 °C to induce gelation.26 The advantage of this method lies in its biocompatibility because it doesn’t require any toxic reagents. However, the precursor solution must be maintained at a temperature that is higher than the gelation point when operating the emulsification step in a microfluidic device. Otherwise, temperatures below the gelling point could lead to fluctuations in the viscosity of the polymer solution, making it difficult to produce monodisperse droplets.
The last physical gelation strategy is hydrogen bond-mediated crosslinking, which requires a mixture of two or more natural polymers with compatible geometries that contain chemical groups capable of forming hydrogen bonds.27–29 The viscoelastic properties of the resulting polymer mixture are more gel-like than those of the individual polymers, making the mixture suitable for injection.30 The main advantage of this method is its excellent biocompatibility because no chemical crosslinking agent is required and the constituent polymers are usually found naturally in the ECM. However, hydrogels assembled using this strategy may degrade rapidly, as an influx of water will quickly dilute the hydrogen-bonded network.15
2.2 Chemical gelation
Compared to physical hydrogels, hydrogels produced by chemical gelation via covalent crosslinking show greater stability, longer durability, and better mechanical properties.17 However, the use of chemical or enzymatic crosslinkers may cause cytotoxicity, and the low specificity of some chemical reactions may lead to undesired interactions with proteins and cells. Several frequently implemented chemical crosslinking strategies are presented here, including photopolymerization, Michael addition, and enzymatic reaction.
During photopolymerization, UV light homolytically splits photoinitiator molecules into free radicals, which initiate the polymerization of a liquid monomer or macromer into a covalently crosslinked hydrogel that is mechanically strong and stable.31 The gelation process not only can be carried out rapidly at room or physiological temperature, usually taking less than a second to a few minutes, but can also be controlled temporally and spatially.32 Due to a high degree of control, photopolymerization can effect in situ gelation after solutions of photosensitive polymers conform to irregularly-shaped tissue defects. The disadvantage of this crosslinking strategy is that the released radicals can react with and damage cellular components. Therefore, parameters such as the type of photoinitiator, photoinitiator concentration, UV light intensity, and total UV exposure time must be finely tuned to ensure high cell viability.
Another chemical gelation method is the milder and most common type of Michael addition, thiol-Michael addition, in which a nucleophilic thiolate reacts with an electrophile, such as an unsaturated ester, to form a thioester linkage.5 Thiol-Michael addition only requires a small amount of catalysts, occurs under mild reaction conditions without the use of heat or light, and does not produce any degradation products.33 However, one particular issue to note is that insufficient mixing of reactants will result in heterogeneous gelation and lead to inconsistent cellular responses to the biochemical and mechanical cues of the hydrogel.34 Therefore, gelation kinetics must be fine-tuned to achieve a balance between the mixing and gelation rates. Since gelation usually occurs on the order of minutes, droplets encapsulating the reactants can first be flowed through serpentine microfluidic channels, which induce passive mixing, before undergoing complete gelation.
Finally, enzymatic reaction provides a more physiologically relevant chemical crosslinking strategy, as most enzymes used for hydrogel crosslinking are responsible for catalyzing reactions that occur naturally in the human body.35–38 For example, one of the most commonly employed enzymes is transglutaminase, which is responsible for forming fibrin clots during wound healing. It can catalyze the formation of covalent bonds between the free amine groups and the γ-carboxamide groups of protein, resulting in highly stable bonds that are resistant to proteolytic degradation.39 In addition to occurring at neutral pH and moderate temperatures, enzymatic reactions also avoid any undesired side reactions or toxicity due to the high substrate specificity of the enzymes. Moreover, cell-responsive enzymes can be designed so that the degradation rate of the hydrogel can be coupled with cellular signals secreted during tissue regeneration.
3. Gel materials
One of the most important factors to consider when designing microgels is the constituent material. Since cells are suspended in an aqueous precursor solution prior to microgel formation inside of the microfluidic device, the gel material must be biocompatible and promote cell survival. Ideally, it should also provide biochemical and mechanical cues to enhance tissue regeneration, and possess a degradation rate that is closely coupled with the rate of tissue growth. Currently, there is no single type of material that meets all of these demands; but by carefully considering their respective merits and drawbacks (Table 2), we may judiciously apply these materials to specific tissue regeneration applications. Here we briefly overview the properties of several gel materials, and readers may refer to these comprehensive reviews for their detailed chemical functionalization and principle of gelation.40, 41
Table 2.
The advantages and disadvantages of typical natural polysaccharides, proteins, and synthetic polymers that are crosslinked to form microgels.
| Biomaterial | Advantage | Disadvantage | Reference |
|---|---|---|---|
| Natural Polysaccharides | |||
| Alginate |
|
|
42–44, 64, 71 |
| Hyaluronic acid |
|
|
55–57, 64, 71 |
| Agarose |
|
|
62,64, 71 |
| Dextran |
|
|
64 |
| Chitosan |
|
|
55, 64–67, 69 |
|
| |||
| Proteins | |||
| Collagen |
|
|
71, 72, 76, 78, 79 |
| Fibrin |
|
|
55, 80–82 |
| Gelatin |
|
|
71, 83–88 |
|
| |||
| Synthetic Polymers | |||
| PEG |
|
|
5, 64, 89, 90 |
| Pluronic |
|
|
92–95 |
3.1 Natural polysaccharides
Natural polysaccharides are frequently used as materials for cell encapsulation because they are biocompatible and capable of gelling at mild physiological conditions.18 However, since natural polysaccharides are isolated from living organisms, their physicochemical properties depend on the specific species from which they are derived and may also face immunogenicity issues. Here, we introduce several common polysaccharides used for gel formation, including alginate, hyaluronic acid, agarose, dextran, and chitosan, whose chemical structures are presented in Fig. 2A.
Fig. 2.

Chemical structures of common gelation materials. (A) Structures of natural polysaccharides. (B) Structures of synthetic polymers.
Alginate, an inexpensive and cytocompatible polymer, is an unbranched polysaccharide comprising varying ratios and arrangements of β-D-mannuronic acid and α-L-guluronic acid residues.42 The interaction of multivalent ions, such as Ca2+, Co2+, and Ba2+, with alginate leads to the formation of ionic bridges between the ions and the carboxylic acid groups of α-L-guluronic acid, and subsequently results in a three-dimensional network.43 The concentration of alginate can be modulated to produce microgels with varying stiffness. However, achieving stable microfluidic emulsification at moderately high alginate concentrations is relatively difficult due to a significant increase in viscosity that is induced by the rapid gelation of alginate.44 To mitigate this issue, the droplet formation and gelation processes must be separated in time, which can be accomplished using one of three general strategies: internal gelation, external gelation, and coalescence-induced gelation.45 The most representative works have been reported as well as summarized by Kumacheva and coworkers.26,46–49
In the internal gelation approach, a precursor polymer solution is mixed with an initiator or crosslinking agent, which is subsequently activated upon contact with a compound in the continuous phase, thereby triggering gelation.48,50 In one study by Tan et al., droplets were formed using a dispersed phase composed of an alginate solution containing cells and CaCO3 nanoparticles as well as a continuous phase comprising oil with acetic acid.44 As acetic acid diffused into the droplet, the acidity of the alginate solution increased, thereby releasing Ca2+ ions from the nanoparticles and inducing gelation. However, prolonged direct contact of the cells with CaCO3 nanoparticles in the alginate solution can lead to physical damage to the cells and decrease their viability. In order to reduce the amount of damage, the cells and CaCO3 nanoparticles can be flowed through the microchannels in two different alginate solutions before emulsification.51 Also, instead of mixing acetic acid with the continuous oil phase, it can be directly added after droplet formation, thereby reducing the exposure time of cells to acidic conditions.
For external gelation, the crosslinking agent and the alginate solution are initially physically separated. After droplets encapsulating the alginate solution are formed, they come into contact with another liquid that contains the crosslinking agent, which diffuses into the droplets and initiates gelation. Compared to internal gelation, this strategy enables control over the morphology of the resulting microgel through modulation of the amount of Ca2+ ions that diffuse into the droplets.26 Kim et al. co-flowed an alginate solution containing cells and another aqueous alginate solution through a microfluidic flow-focusing device, and generated droplets with a continuous phase comprising an oleic acid solution containing CaCl2.52 The alginate solution gelled as CaCl2 diffused into the droplets.
In coalescence-induced gelation, droplets containing a precursor polymer solution and droplets encapsulating a crosslinking agent coalesce with each other one-by-one to form crosslinked microgels.53 Therefore, the productivity of microgel formation depends on the probability of collisions between two separate droplets.46 Using this technique, Liu et al. generated sodium alginate droplets and CaCl2 droplets in two separate flow-focusing channels, which subsequently led to two circular expansion chambers where the two types of droplets collided and merged with one another, triggering gelation.54
Hyaluronic acid (HA), a linear polysaccharide composed of alternating units of the disaccharide β-1,4-D-glucuronic acid-β-1,3-N-acetyl-D-glucosamine, is a non-sulfated glycosaminoglycan that is found throughout the body in connective, epithelial, and neural tissues.55 It plays an active role in wound repair, cellular signaling, morphogenesis, and organization of the ECM.56 Additionally, HA interacts with cell-surface receptors to influence tissue organization, stimulates the production of collagen II and aggrecan, and promotes cell proliferation.55 With excellent biocompatibility and non-immunogenicity, the enzymatically degradable HA is a biologically-relevant material for fabricating hydrogels.57 HA-based hydrogels can be generated using a variety of gelation strategies, including photopolymerization, Michael addition, Schiff-base reaction, thermally-induced crosslinking, and covalent crosslinking.57,58 Out of these methods, photopolymerization and Michael addition are the most frequently used strategies for producing HA hydrogels. For instance, the carboxylic acids on the HA backbone can easily be modified with methacrylic anhydride to form methacrylated HA, whose network properties can be controlled by varying the length of UV exposure, HA molecular weight, concentration, and number of reactive groups.56,59 Another study by Segura’s group reported the assembly of hyaluronic acid-based building blocks, which were crosslinked via pseudo Michael addition, into an injectable microporous scaffold that can be loaded with cells during scaffold formation.60
Agarose is a neutral, bioinert polysaccharide comprising the polymers β-1,3-linked D-galactose and α-1,4-linked 3,6-anhydro-L-galactose in alternating order.18 The gelation of agarose is thermally reversible, as it maintains a liquid form at physiological 37 °C and gels upon cooling below 20 °C, which is a result of the aggregation of double helices produced by the entanglement of anhydro bridges on each agarose molecule. Therefore, emulsification of agarose solutions is usually performed at temperatures above 37 °C, and microgels are subsequently formed by cooling the droplets to below the gelation temperature. The mechanical properties of the resulting agarose microgels can be tuned by adjusting the agarose concentration.61 Although agarose does not have active adsorption sites for proteins or adhesion sites for cells, cell adhesion proteins or collagen can be incorporated to promote attachment of anchorage-dependent cells.62,63
Dextran, homopolysaccharides consisting mainly of α-1,6-linked D-glucopyranose residues, are synthesized by lactic acid bacteria.64 Although they are highly hydrophilic and chemically similar to glycosaminoglycans, they are resistant to cell adhesion and protein adsorption. To ameliorate this issue, the abundant hydroxyl groups of dextran can be chemically modified to incorporate specific cell-binding sites. Dextran-based hydrogels can be generated by either physical or chemical crosslinking, but the more common approach is by photopolymerization of dextran that has been functionalized with polymerizable groups such as methacrylates.
Chitosan, a linear polysaccharide consisting of β-1,4-linked D-glucosamine and N-acetyl-D-glucosamine units, is derived from the naturally occurring chitin and has a similar structure to glycosaminoglycans.55 It has excellent biocompatibility, antibacterial properties, as well as low toxicity and immunostimulatory activities.65 Furthermore, its hydrophilic nature supports cell adhesion, proliferation, and differentiation.64 Chitosan hydrogels can be formed through thermally-induced or pH-triggered gelation and enzymatically degraded by lysozymes in vivo,66 releasing harmless natural metabolites as degradation products.67 It is often mixed with other polymers to improve its mechanical properties, and its amine groups enable the conjugation of biomolecules to control its biochemical properties.67 Fan et al. used metal-free click chemistry to generate an oxanorbornadiene-functionalized chitosan and azido-functionalized hyaluronan composite hydrogel, whose crosslinking and resulting microstructure determine its characteristics such as equilibrium swelling, mechanical properties and degradation kinetics.68 However, one particular disadvantage is that it is hard to obtain ultra-pure, medical-grade chitosan, so impurities such as allergans may affect its rate of de-polymerization and biological activity.69
3.2 Proteins
Proteins are attractive materials for generating cell-laden microgels because their chemical properties and fibrous structure closely mimic those of the extracellular matrix, thereby providing a confined microenvironment that promotes the adherence, growth, migration, and differentiation of the encapsulated cells.70 Here, we provide a brief overview of several frequently used proteins, including collagen, fibrin, and gelatin.
Collagen, the most abundant protein in the human body that helps maintain structural integrity, consists of repeating (Gly-X-Y)n units that form left-handed helical α-chains, which pack together to form triple helices that are stabilized by specific covalent crosslinks between neighboring collagen molecules.64 Of the numerous forms of collagen, Type I collagen is the most commonly used due to its abundance, good biodegradability, low antigenicity after removal of telopeptides, and cell-binding ability.71,72 Collagen has frequently been used as an ECM substrate in applications such as bioartificial liver studies.73–75 However, extended application of collagen as a robust material for tissue regeneration has been hampered by its rapid degradation rate and hence weak mechanical strength. Increasing the collagen concentration will result in a modest increase in strength, but will also restrict cell migration and nutrient diffusion.76 Glutaraldehyde crosslinking has been explored to enhance the mechanical properties of collagen-based gels, but it can cause cytotoxicity and biocompatibility issues.64 Another strategy is to generate collagen-containing composite hydrogels, often accomplished by incorporating photocrosslinkable hyaluronic acid into collagen gels, to increase the elastic modulus and mechanical strength.77 Another issue with collagen is that its polymerization mechanism is thermally driven and thus requires exposure to elevated temperatures for at least 30 minutes to ensure complete gelation.78 Thus, an additional incubation step is often required after microfluidic synthesis of collagen-containing emulsion droplets.79
Fibrin, an important ECM component that regulates wound healing, is polymerized from a precursor plasma protein called fibrinogen. Upon vascular injury, the enzyme thrombin is released and cleaves peptide fragments from soluble fibrinogen to generate insoluble fibrin peptides that form fibrils, which eventually assemble into a fibrin network that is crosslinked upon addition of the transglutaminase Factor XIII.80 The clotting time is the main factor in determining characteristics of the resulting microstructure such as fiber size and porosity.80 For instance, higher thrombin concentration leads to more rapid gelation, resulting in a tighter network structure with smaller pore sizes. Since thrombin has a Na+ binding site that controls its activation kinetics,81 addition of sodium chloride can increase the gelation time to several minutes.82 In addition to fibrin’s excellent biocompatibility, cell-adhesiveness, and biodegradability, the mechanical properties of fibrin hydrogels can be tuned by varying the initial concentrations of fibrinogen and thrombin. One of the main advantages of fibrin is that it forms a provisional matrix, which serves as a temporary scaffold that promotes the invasion of leukocytes and endothelial cells until the diseased tissue is completely regenerated.82 This means that fibrin clots are naturally designed with appropriate cell-mediated degradation kinetics to balance the rate of tissue regeneration and matrix remodeling, which is hard to achieve with synthetic materials or even other more permanent natural polymers in the ECM.
Gelatin, the denatured form of collagen, comprises a variety of amino acids such as glycine and proline.18 Bioactive and cell-adhesive motifs in gelatin promote cell attachment and proliferation, while matrix metalloprotease (MMP)-sensitive degradation sites enable gelatin-containing microgels to be easily degraded to release the encapsulated cells.7,83 Furthermore, gelatin is cytocompatible and shows lower antigenicity compared to collagen.84 At room temperature, gelatin solidifies due to its extensive physical bonds. However, it liquefies upon heating to above the physiological temperature of 37 °C, making it impractical to use gelatin directly as an injectable in situ curable gel. Although chemical crosslinking agents such as glutaraldehyde can be used to strengthen the mechanical properties of gelatin, they can lead to cytotoxicity.85 As a more cell-friendly alternative, methacrylate groups can be incorporated into the amine-containing side groups of gelatin, rendering the gel photocrosslinkable via activation of a photoinitiator.86 An additional advantage of this strategy is that the mechanical and chemical properties of the gel can be tuned by adjusting parameters in the methacrylation and photocrosslinking processes.83,87,88
3.2 Synthetic polymers
Synthetic polymers have emerged as attractive alternative scaffold materials to naturally derived ECM proteins, which are limited by their immunogenicity and poor mechanical properties.89 Compared to natural materials, pure synthetic polymers have lower risks of toxicity, immunogenicity, and infection.64 Additional advantages of synthetic polymers include the ability to be photopolymerized, tunable mechanical properties, and facile manipulation of scaffold structure, chemical composition, and degradability.90 However, synthetic polymers lack critical biological functions that bioactive natural materials possess, including cell adhesion and biodegradation, but this can easily be compensated for by functionalizing them with bioactive molecules such as cell-adhesive peptides and enzyme-sensitive peptides. Here, we introduce two common synthetic polymers, poly(ethylene glycol) (PEG) and Pluronic, whose chemical structures are shown in Fig. 2B.
PEG is an FDA-approved hydrophilic polymer comprising a PEG diol with two hydroxyl end groups, which can be converted into other functional groups to generate hydrogels with different chemical properties.89 Although PEG is biocompatible and non-immunogenic, it has poor degradability and is resistant to cell adhesion and protein adsorption.5 In order to improve its degradability, enzyme-sensitive peptide sequences can be incorporated into PEG hydrogels to make the degradation process responsive to cellular signals and cell-secreted enzymes.89 Likewise, addition of adhesive proteins such as fibronectin introduces proteolytic and cell adhesion sites into the resulting gel. There are three general crosslinking strategies for fabricating PEG hydrogels: radiation of PEG polymers, free radical polymerization of PEG acrylates, and chemical reactions such as Michael addition and enzymatic reaction.89 Out of these strategies, the most commonly used is photopolymerization, which has the advantage of inducing in situ gelation of a PEG macromer solution at physiological temperature and pH.89 Moreover, with a careful choice of photoinitiator chemistry and concentration as well as light intensity, cell exposure to radicals can be minimized, making the gelation process more cell-friendly.
Pluronic, a commercial surfactant comprising the triblock copolymer poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) (PEO-PPO-PEO), has been used as an injectable thermosensitive hydrogel.91 Above the critical micellization concentration, the amphiphilic copolymer blocks can form micelles in water, which become entangled with one another upon heating, thereby leading to gelation.91,92 For instance, an aqueous solution of Pluronic F127 copolymer (>20 w/v%) can be gelled in situ when heated to body temperature. Although Pluronic has demonstrated excellent biocompatibility both in vitro and in vivo, Pluronic hydrogels suffer from rapid erosion at the injection site, non-biodegradability, and poor mechanical properties.92–95 Nevertheless, its biodegradability can be improved by linking ester, urea, or carbonate bonds to the copolymers.96,97 Furthermore, other crosslinking strategies, such as photopolymerization and Michael addition, can be used to improve the stability and mechanical properties of Pluronic gels.91
4. Design of microfluidic device for microgel generation
In order to generate microgels with the desired size, geometry, or biochemical and mechanical properties, microfluidic devices must be carefully designed with appropriate dimensions as well as features for manipulating the spatial organization of the constituent cells. Here, we briefly explain how general microfluidic devices can be used to fabricate microgels, and provide schematics of typical designs.
4.1 Microfluidic devices
4.1.1 PDMS-based microfluidic devices
Poly(dimethylsiloxane) (PDMS) is one of the most widely used materials for fabricating microfluidic devices.98 Several features make it an attractive material for prototyping, including its low cost, optical transparency, and biocompatibility.99 Additionally, with the development of soft lithography for PDMS, not only has the prototyping process been greatly simplified such that new concepts can easily be tested in PDMS-based devices, but also more complicated devices could be designed with components such as pneumatically activated valves. These valves can restrict fluid movement through pressurization and deflection of an adjacent channel, which is only made possible by the elastomeric nature of PDMS and cannot be accomplished using rigid materials such as silicon or glass.100
A common PDMS device design for microgel generation is based on the flow-focusing geometry (Fig. 3A), in which a dispersed liquid phase flows through the middle channel and a second immiscible continuous liquid phase flows through the two outer channels.101 Typically in this design, the dispersed phase contains a gel solution mixed with cells while the continuous phase comprises a stream of immiscible oil that exerts a shear force on and focuses the dispersed phase through a narrow cross-junction, resulting in a periodic break-up of the dispersed phase and formation of gel-encapsulated emulsion droplets. The main advantage of this design is that the number of encapsulated cells per droplet and the droplet diameter can be controlled simply by tuning the gel and oil flow rates. Since a droplet production rate of 1 kHz per device can easily be achieved,10 parallelization of a large number of devices may facilitate the scale-up of microfluidic fabrication of microgels.
Fig. 3.

Typical microfluidic device designs. (A) PDMS flow-focusing. (B) PDMS-based pneumatically-actuated microvalves. (C) Capillary co-flow. (D) Capillary flow-focusing.
Although the flow-focusing configuration is a reliable method for producing monodisperse spherical microgels, it is not amenable to producing gels with other geometries, which could serve as important tissue constructs that mimic the natural tissue architecture. For instance, in a hepatic lobule, which is the basic unit of liver tissue, hepatocytes are arranged in a cord-like structure surrounded by endothelial cells in a sinusoidal distribution.102 The complex spatial patterning of tissue architecture can be recreated using PDMS devices with pneumatically-actuated microvalves (Fig. 3B), which can be selectively manipulated to precisely control fluid movement within the microchannels. During device operation, flexible membranes are deflected outwards upon application of air pressure, and effectively serve as micro-molding barriers that keep the precursor polymer solution in a desired geometric configuration until the gelation process is completed. By sequentially actuating these microvalves, additional layers with complex patterns can be added successively, forming a biomimetic microgel. Despite the versatility that this technology affords, its fabrication process can be tedious as it requires multilayer soft lithography to build the valve system.
4.1.2 Capillary microfluidic devices
Aside from commonly used PDMS-based devices, capillary microfluidic devices offer an alternative method of generating highly monodisperse emulsion droplets with controllable sizes. Capillary microfluidic devices are fabricated by coaxially aligning circular and square glass capillaries on glass slides.103 First, a square capillary is securely attached to a glass slide. Next, a cylindrical capillary, usually with an outer diameter of 1–2 mm, is tapered to create an orifice, simply by heating and then pulling the capillary with a micropipette puller. The size of the resulting orifice represents the inner diameter of the circular capillary, which can be further adjusted with a microforge to precisely define the diameter of the injection nozzle for the inner disperse phase.7 To ensure accurate coaxial alignment of both capillaries, the outer diameter of the circular capillary must match the inner dimensions of the square capillary, which allows the tapered circular capillary to be slid into the square capillary to form the microfluidic device.
By adjusting the geometric configuration of this setup, two different methods for droplet generation can be achieved using capillary microfluidics. The first method utilizes a co-flow geometry (Fig. 3C), in which two fluids injected through the circular and square capillaries are flowing in the same direction to generate coaxial flow. In contrast to the co-flow configuration, the flow-focusing geometry injects the two fluids from opposite ends of the same square capillary (Fig. 3D), which allows the inner fluid to be hydrodynamically flow-focused as it is forced through the capillary orifice by the outer fluid. Both methods can produce monodisperse emulsion droplets upon extrusion of the inner fluid from the capillary orifice under low rates of fluid flow. However, if either flow rate exceeds a certain threshold, a jet will form in which the inner fluid travels in a long stream before droplets are generated downstream, leading to greater variation in droplet size. Compared to the co-flow configuration, the flow-focusing setup is better suited for generating small monodisperse droplets that contain particulate suspensions in the inner phase, which could otherwise accumulate and block the capillary orifice in the co-flow configuration.
The adoption of glass as a constituent material, as well as their geometric configuration, affords capillary microfluidic devices with several advantages. First, the compatibility of glass with common organic solvents extends the range of applications in which these devices can be used, such as chemical and material synthesis.98 Second, the wettability of capillary devices can be precisely controlled by treating the inner wall with a surface modifier. For instance, treatment with n-octadecyltrimethoxyl silane makes the capillary wall hydrophobic while treatment with 2-[methoxy(polyethyleneoxy)-propyl] trimethoxy silane makes the wall hydrophilic.104 Finally, the coaxial assembly of glass capillary allows these devices to generate truly three-dimensional fluid flow, which robustly confines sample flow to a streamline at the center of the capillaries, enabling precise control over the size and uniformity of the emulsion droplets.
4.2 Size control
Precise regulation of the size and size distribution of microgels is essential to the preparation of cell-laden microgels for various applications in tissue regeneration. The diameter of the microgel must be controlled within a certain range to maintain a microenvironment that promotes high cell viability and proliferation. For instance, if the diameter of the resulting microgel were to fall below 60 μm, there may not be a sufficient number of encapsulated cells to promote cell contact and proliferation.103 Conversely, the microgel should not exceed 200 μm in diameter so that oxygen can be readily exchanged across the gel to maintain long-term cell survival.105 Furthermore, a narrow size distribution of cell-laden microgels is desired in applications such as the co-culture of multiple cell types and the study of the role of cell confinement and intercellular distance in influencing cell fate, which require accurate control over the average number of cells in each microgel.18
Bulk emulsification techniques, which often utilize some form of mechanical agitation, suffer from a lack of control over the size distribution of the microdroplets.106,107 In contrast, microfluidic methods offer enhanced control over the droplet size and significantly reduce the size distribution of the microdroplets.108–110 As a result, microfluidics provides a facile method for modulating the gel size and controlling the size distribution within a narrow range. A common strategy for selectively tuning the droplet diameter is to vary the flow rate ratio between the dispersed and continuous phases. For a specific device geometry and combination of input liquids, increasing this ratio leads to larger droplet diameters. Stable and accurate droplet formation using this method requires the maintenance of a constant pressure difference between the aqueous liquid and oil channels throughout the entire microfluidic fabrication process, which can easily be achieved using syringe pumps.111 A more robust alternative strategy is to adjust the dimensions of the input microchannels. For instance, increasing the width of the channels enables the formation of droplets with larger diameters.
5. Strategy for cell deposition
Microfluidic microgels can be classified into two general categories based on the spatial deposition of the cells. Depending on the specific application, cells can be encapsulated within the microgel or grown on the surface of the gel (Fig. 4). Here, we explain why one strategy may be preferred over the other.
Fig. 4.

Schematic illustrating two different strategies for cell deposition: inside the microgel and on the surface of the microgel. Note: Zoomed-in area of schematic is not drawn to scale. The size of a typical cell (20 μm) is approximately three orders of magnitude greater than that of a typical polymer network mesh (1–10 nm).
5.1 In the microgel
True recapitulation of the native cellular microenvironment requires a 3D matrix with distinct physicochemical properties. Although macroscale 3D culture systems have been used as alternatives to conventional monolayer cultures, they are not feasible for long-term cell culture due to a maximum oxygen diffusion limit of 200 μm.112 Therefore, cell-laden microgels with diameters between 100 and 400 μm have been fabricated to ensure that the encapsulated cells stay safely within oxygen diffusion constraints. Additionally, important factors that influence the behavior of cells in the microenvironment, such as extracellular matrix components, soluble biomolecular signals, and biophysical cues, can be incorporated into the microgels to provide the cells with more physiologically relevant microenvironments.
Since cells are often pre-mixed with a precursor solution before emulsification, the fabrication process must be cytocompatible, which limits the number of appropriate materials and gelation methods that can be used. For instance, if the temperature required for gelation exceeds that of physiological conditions or if the crosslinking agent and droplet-forming oil contain cytotoxic components, then cell viability could be compromised. Nevertheless, this strategy is particularly useful for in vivo applications in which cell-laden microgels can provide a minimally invasive means of delivery to a localized wound area as well as protect the cells from the host immune response and external stress during injection.
5.2 On the microgel
Culturing cells on the microgel surface provides an alternative method of in vitro cell culture that possesses several advantages over conventional monolayer culture. First, due to the significantly higher surface-to-volume ratio of microgels, a greater number of cells can be cultured with smaller volumes of culture media.113 Second, compared to those of plastic or glass surfaces, the mechanical and chemical properties of microgels can be tailored to replicate physiological conditions more faithfully. Third, cell-seeded microgels can be directly used as injectable tissue constructs, whereas multiple extra steps are required to collect monolayer-cultured cells and attach them onto microgels for subsequent injection into the body.
As described in the previous section, cell encapsulation within microgels enables a more accurate 3D recapitulation of the native microenvironment compared to 2D strategies. Despite this, there are several reasons why cell culture on the 2D surface of microgels would be preferred instead. First, cells grown inside microgels are restricted within the inner space of the gel, thereby limiting the ability of the cells to proliferate.113 In contrast, cells grown on the surface are not tightly confined, enabling them to proliferate more quickly so that a greater number of cells can be collected within a shorter amount of time. Second, detachment and collection of cells growing on the microgel surface are much easier than extracting cells encapsulated within the microgel, which often requires enzymatic or thermally-mediated degradation. Finally, compared to cell-encapsulated microgels, these microgels can be generated prior to cell adherence to the surface, thereby offering more flexibility in the type of precursor solution and gelation conditions.
6. The advantages of microfluidic microgel compared with conventional hydrogel
Conventional hydrogels, characterized by their high water content and broad range of physical properties, can be engineered in shape, size, and form to mimic the extracellular environment of natural tissue. Due to their mechanical and chemical versatility, hydrogels have found applications in medicine as soft contact lenses,114 adhesives for wound closure,14 biosensors,115 etc. However, applications in stem cell research, cell therapy, and tissue engineering demand additional features from hydrogels such as compatibility with cells and biomimicry of the native extracellular matrix.11 To maintain high cell viability, there must be effective oxygen and nutrient transfer, which is largely dictated by the size of the hydrogel. Furthermore, since the properties and functions of cells are influenced by the distance between cells as well as their structural arrangement, the size and size distribution of the hydrogel must be strictly controlled.116
Whereas conventional suspension polymerization, spraying, and precipitation techniques do not provide sufficient control over the dimensions of the generated particles, microfluidic methods offer facile control over the size, size distribution, shape, and morphology of the resulting particles, making them particularly useful for the generation of microgels with polydispersities of under 2–3%.26,109,117,118 Since encapsulated cells require steady diffusion of nutrients and oxygen into the gel and waste products out of the gel, the large size of hydrogels, which typically range from 500 to 1000 μm, makes it difficult to support efficient transport for cells in the inner core, thereby leading to reduced cell viability. In contrast, the size of microfluidic microgels can be effectively controlled within the ideal range of 60 to 200 μm, which helps maintain high cell viability due to the microgels’ significantly greater surface-to-volume ratio that enhances nutrient and waste exchange.
In addition to the gel size, uniformity in size distribution is another important criterion to consider, especially for applications in directed-assembly tissue engineering, where building blocks composed of cell-laden gels are arranged in a desired geometry with specific shape and size to mimic functional tissue units.119 Since the size of stem cell aggregates can influence the differentiation of cells into specific lineages,120 cell-laden gels should ideally be monodisperse to produce uniform aggregates that differentiate into the same lineage. Otherwise, biomimetic tissue units assembled from building blocks with heterogeneous cell populations could result in poor functionality.
7. Cell-laden microfluidic microgels for tissue regeneration
Microfluidic technologies have been used to generate cell-laden microgels with varying properties and geometries for use in a range of tissue engineering applications. Here, we overview the latest representative works that exploit the advantages of cell-laden microfluidic microgels for tissue regeneration, and provide a brief summary in Table 3.
Table 3.
Microfluidic microgels for tissue regeneration applications
| Microfluidic Device | Polymerization Strategy | Biomaterial | Tissue Regeneration Application | Ref. |
|---|---|---|---|---|
| Capillary flow-focusing | Photopolymerization | Gel-MA | Injectable BMSC-laden microgels (160 μm) with prolonged release of BMP-2 growth factor as osteogenic tissue constructs | 7 |
| PDMS flow-focusing | Electrostatic interaction | Alginate | 3D core-shell microgel scaffold (169 μm) for co-culture of hepatocyte and fibroblasts as in vitro liver model for drug screening | 21 |
| PDMS flow-focusing | Michael addition | Heparin, PEG | Bioactive microgels (120 μm) containing growth factors FGF-2 and Nodal for directed endodermal differentiation of mESCs | 10 |
| PDMS flow-focusing | Electrostatic interaction | Alginate | Alginate-RGD microgels for formation of hMSC spheroids (36, 46, 62, 84 μm) and induction of osteogenic differentiation | 146 |
| PDMS devices with integrated pneumatic microvalves | Thermal | Collagen, gelatin, agarose | Cell-laden microgels (200 μm thick) of different shapes, with single and multiple microchannels of varying shapes, for constructing biomimetic liver lobule comprising a co-culture of hepatocytes and endothelial cells, as a 3D tissue model for in vitro drug toxicity screening | 9 |
| PDMS flow-focusing | Photopolymerization | Gel-MA, silica | Cell-seeded Gel-MA microgels (100 μm) with a protective silica hydrogel shell for cell culture and injectable tissue construct | 113 |
| PDMS flow-focusing | Michael addition | PEG-VS + RGD, K, and Q peptides | Injectable scaffold assembled from annealed microgel building blocks (30–150 μm) for accelerated wound healing | 176 |
| PDMS flow-focusing | Electrostatic interaction | Alginate, collagen | Co-culture of hepatocyte-EPC composite spheroids (~80 μm) in alginate-collagen microgels for enhancing hepatocellular functions | 162 |
7.1 Cell encapsulation for 3D cell culture
One of the most frequent application of microgels is the encapsulation of cells for in vitro cell culture and expansion.104,121–131 Since a 3D microenvironment more faithfully mimics the cell’s native niche, physicochemically well-defined 3D matrices are preferred for in vitro cell culture. Micron-sized hydrogels are attractive candidates because they not only promote the exchange of nutrients and metabolic wastes during cell growth and tissue development but also enable the tuning of physical and chemical properties of the gel. Furthermore, combined with microfluidic technologies, monodisperse cell-laden microgels with tailored properties can be generated to serve as 3D culturing units for studying how various microenvironmental properties and cues affect cell fate. Additionally, parallelization and automation of microfluidic systems enable high-throughput screening of microgel properties, which will facilitate the discovery of in vitro conditions that more closely mimic those of the in vivo microenvironment. Readers are referred to many excellent reviews on cell encapsulation within microfluidics-generated microgels for 3D cell culture.12,18,26,45,119,132–135 We will instead focus on other applications of cell-laden microfluidic microgels in tissue regeneration.
7.2 Controlled microenvironment for stem cell differentiation
Stem cells, which can renew themselves and differentiate into specialized cell types, play a major role in the regeneration of tissues and restoration of tissue functions.136–138 Due to the ability of murine embryonic stem (ES) cells to differentiate into any type of cell in the adult body, many differentiation protocols have been developed for differentiating ES cells into adult tissues for tissue engineering.139–141 However, it is often difficult to control the tightly regulated microenvironmental factors that induce specific differentiation pathways when large quantities of stem cell–derived tissue constructs are needed. Thus, a scalable strategy for tailoring the stem cell microenvironment with important growth factors is required. Microfluidics, a large-scale bioprocessing technique, shows great promise as it not only facilitates rapid nutrient and waste exchange, but also reduces hydrodynamic stresses from stirring during suspension culture of stem cells.142 Moreover, compared with monolayer cultures, the expansion and differentiation of encapsulated stem cells can be accomplished efficiently in a one-step process in microfluidic microgels.135 Furthermore, the monodispersity of microfluidic droplets is particularly important in microscale tissue engineering, in which the spheroid size influences stem cell behavior and differentiation potential.143,144
Microfluidic double emulsion (DE) droplets provide well-defined, uniform, and compartmentalized microenvironments for cell culture, with the oil shell serving as a selective barrier that enables an influx of nutrients, oxygen, and small molecules from the external environment while retaining biomacromolecules in the inner core.145 In our recent work, we used microfluidic DEs as miniature bioreactors to rapidly generate human mesenchymal stem cell (hMSC) spheroids, which were subsequently encapsulated in alginate-RGD microgels and demonstrated enhanced osteogenic differentiation (Fig. 5A).146 Compared to previous water-in-oil (W/O) single-emulsion cultures, this platform enhances the viability of encapsulated cells by introducing an additional outer aqueous phase to the external oil phase, which has been shown to be incompatible for long-term cell culture.147 Moreover, by varying the cell density during the encapsulation process, the size of the spheroid can be precisely controlled. This is important because small spheroids lead to more homogeneous chondrogenic differentiation while larger spheroids form more heterogeneous tissues.148 For instance, average spheroid sizes of 36, 46, 62, and 84 μm can be obtained with densities of 2, 5, 10, and 20 million cells/mL in 200-μm DE droplets (Fig. 5B). Additionally, one of the greatest advantages of this system is that it only takes 150 min for the encapsulated cells to aggregate into spheroids (Fig. 5C), whereas other technologies require approximately 1 to 4 days.149–152 For microgel fabrication, hMSCs suspended in an alginate-RGD solution were used as the inner phase during DE formation. Upon successful cell aggregation, the spheroids were released into a solution containing calcium ions, which induced rapid crosslinking of alginate molecules and subsequent formation of microgels. Assessment of the osteogenic differentiation of hMSC spheroids encapsulated in alginate-RGD microgels showed that they contained greater quantities of calcium deposits and higher alkaline phosphatase activity after seven days in culture (Fig. 5D), suggesting that spheroid behavior can be controlled by modulating its microenvironment using this system.
Fig. 5.

Microfluidic DE droplets serve as miniature bioreactors to rapidly generate human mesenchymal stem cell (hMSC) spheroids, which were subsequently encapsulated in alginate-RGD microgels and demonstrated enhanced osteogenic differentiation. (A) Schematic diagram of DE droplet generation and subsequent spheroid formation. Two PDMS flow-focusing devices are connected serially, with the first device producing W/O emulsions and the second device supplementing an outer aqueous phase to generate W/O/W DE emulsions. Cells encapsulated in DE droplets assemble into a single spheroid, which can then be released with or without microgel encapsulation. (B) Diameter of spheroids measured at different densities of encapsulated cells (n ≥ 50). (Data = mean ± SD) (C) Time course images showing the formation of spheroids in 150 min. (D) Control of microenvironment for spheroid culture using DE droplets. (a, b) Confocal images showing phalloidin staining and immunostaining for E-cadherin, integrin a5b1 for spheroids encapsulated in alginate or alginate-RGD gels and cultured in normal media for 3 days. (c, d) Images of alizarin red staining and alkaline phosphatase activity staining using BCIP/NBT as substrate for spheroids cultured in osteogenic medium for 7 days. Adapted from ref. 127 with permission from Nature Publishing Group.
Previously, there has not been any growth factor-containing microgel used for ES cell differentiation. To explore this possibility, Siltanen et al. generated bioactive microgels (120 μm) composed of heparin and PEG, and incorporated the growth factors FGF-2 and Nodal to enhance the differentiation of mouse embryonic stem cells (mESCs) toward a definitive endoderm.10 Heparin-methacrylate and PEG-diacrylate macromers were mixed with 8-arm PEG-thiol inside the microchannels and emulsion droplets were produced at rates of up to ~1 kHz, with gelation occurring via Michael addition (Fig. 6A–6C). This crosslinking strategy has the benefits of being specific, cytocompatible, and independent from free radicals. To test the ability of the microgels to enhance endodermal differentiation, undifferentiated mESCs were encapsulated in the microgels and supplemented with FGF-2 and Nodal/Activin by following a definitive endoderm differentiation protocol. The mESCs showed >95% viability two hours after encapsulation (Fig. 6D–a), and formed mESC spheroids that expanded in size and broke free from the microgels (Fig. 6D–b). After 5 days of culture, data from qRT-PCR indicated that mESCs encapsulated in the microgels and subjected to a one-time dose of FGF-2 and Nodal expressed a 33- and 65-fold increase in the endoderm markers FoxA2 and Sox17, respectively, compared to undifferentiated mESCs (Fig. 6E). In contrast, cells cultured in monolayer (control) on fibronectin with continuous FGF-2/Nodal supplementation, 3D spheroids cultured on matrigel, and mESCs encapsulated in microgels without FGF-2 or Nodal expressed 2- and 5-fold, 10- and 11-fold, and 3- and 7-fold increases in FoxA2 and Sox17, respectively (Fig. 6E). These results indicate that compared to standard 2D or 3D differentiation procedures with continuous growth factor supplementation, encapsulation of mESCs in heparin microgels provides a well-defined, definitive endoderm-inducing microenvironment where a one-time dose of growth factors was sufficient to induce significantly higher expression levels of endoderm markers.
Fig. 6.

Fabrication of heparin microgel with microfluidic flow-focusing device. (A, B) Heparin-methacrylate, PEG-diacrylate, and 8-arm PEG-thiol undergo Michael addition crosslinking to form heparin hydrogels. (C) Microfluidic flow-focusing device for mixing and emulsifying hydrogel precursors. Numbers represent inlets for (1) carrier oil, (2–3) hydrogel precursors, and (4) collection outlet. Scale bar = 300 μm. (D) Encapsulated mESC viability and EB formation. (a) Representative Live/Dead staining of mESCs 2 h after encapsulation. (b) Embryoid body formation. Scale bar = 200 μm. (E) Growth factor adsorption and mESC-to-DE differentiation in microgels. Definitive endoderm marker expression after 5 days of differentiation with or without Nodal and FGF-2. Error bars represent standard deviation for n = 3 samples. Significance indicated at the p < 0.05 level. Adapted from ref. 10 with permission from Elsevier.
7.3 Co-culture systems as 3D organ models
Tissues are three-dimensional structures composed of a multitude of cells that are surrounded and separated by extracellular matrices.153 The spatial organization of a cell determines how it interacts with other cells, which subsequently effects the transmission of molecular signals involved in cell movement and formation of boundaries in tissues. As a result, tissue function is affected by a variety of cues such as intercellular signaling and interaction between cells and their surrounding extracellular matrices.154,155 Therefore, in order to fabricate artificial tissues that more realistically mimic the in vivo microenvironment, methods are needed that can spatially pattern multiple types of cells embedded in biocompatible extracellular matrices.
Chen et al. used droplet-based microfluidics to controllably assemble 3D core-shell microgel scaffolds comprising a hepatocyte-filled aqueous core and a fibroblast-laden alginate shell.21 A PDMS flow-focusing device was used to generate water-water-oil (W/W/O) DEs with four phases: an inner phase consisting of hepatocytes (HepG2 cells) in cell culture medium, a middle phase composed of fibroblasts (NIH-3T3 cells) mixed with alginate solution and Ca-EDTA, an outer phase made up of oil, and an additional oil phase containing 0.15% acetic acid that triggers Ca2+ release from the Ca-EDTA complex to form a crosslinked alginate network (Fig. 7A, 7B). The mechanical strength and portability of the microgels enable them to be stored at −80 °C, thawed at 37 °C, and then cultured again without damaging the structure or significantly reducing the viability of the cells (Fig. 7C–a). Moreover, the alginate microgels (169 μm in diameter) are well-suited for long-term culture as their high permeability facilitates nutrient and metabolite exchange. An additional benefit of using unmodified alginate is that it deters cell attachment and prevents migration of the fibroblasts. Maintaining spatial separation between the hepatocytes and fibroblasts is of particular importance as random co-cultures of these two types of cells will result in lower liver-specific functions due to an imbalance between homotypic and heterotypic cell-cell interactions.156 To assess the enhancement of liver-specific functions of the core-shell spheroid, two microgel samples were cultured using the same amount of hepatocytes in the core, one with and the other without fibroblasts in the shell (Fig. 7C–b, c). Compared to its monotypic counterpart, the co-cultured spheroids demonstrated increased albumin secretion and urea synthesis (Fig. 7D), which are two major biomarkers of the liver used in drug screening.157 These results suggest that this 3D core-shell microgel scaffold is a good in vitro liver model and may be applied to rapid drug screening.
Fig. 7.

(A) Microfluidic construction of the 3D scaffold consisting of an aqueous core and a hydrogel shell. (a) Crosslinking of the alginate network by triggered release of Ca2+ from the Ca-EDTA complex. (b) Schematic diagram of the PDMS device. (c) Fabrication of core–shell droplets using W/W/O DEs as templates. (d) Monodisperse core–shell droplets generated using droplet-based microfluidics. The alginate shell is labeled with fluorescein, as shown in the inset. (B) Simultaneous assembly of hepatocytes in the core and fibroblasts in the shell, forming an artificial liver in a droplet. Cell viability is characterized by the calcein AM/EthD-1 staining kit. Scale bar = 100 μm. (C) Co-culture of hepatocytes and fibroblasts in the core–shell spheroids. (a) High viability of cells encapsulated in the spheroids after being frozen at −80 °C for two weeks and then thawed at 37 °C. (b) Co-culture of cells encapsulated in the spheroids for 10 days showing high cell viability. (c) The viability of cells cultured for 14 days decreases slightly. Cell viability is characterized by the calcein AM/EthD-1 staining kit. Scale bar = 100 μm. (D) Comprehensive assays of liver-specific functions of the hepatocyte/fibroblast co-culture and the hepatocyte only culture. (a) Albumin secretion and (b) urea synthesis of HepG2/NIH-3T3 co-culture and HepG2 culture measured over seven days. *p < 0.01. Adapted from ref. 18 with permission from the Royal Society of Chemistry.
Although shear-induced droplet formation, employed by microfluidic technologies such as flow-focusing and T-junction devices, can produce monodisperse spherical cell-laden microgels, it does not support the formation of other shapes, which could provide additional insight into the relationship between ECMs and cell fate and behavior.158 An alternative approach called pneumatic-aided micro-molding (PAM) makes it possible to not only generate cell-laden microgels with precisely controlled sizes and well-defined geometries, but also fabricate single and multiple microchannels in the microgels using various combinations of hydrogels and homogeneous or heterogeneous cell types.9 The central components of PAM are pre-designed pneumatic microvalves integrated into PDMS microfluidic devices using multilayer soft lithography, which enables precise manipulation of fluid and micron-sized objects. The basic operating principle is as follows (Fig. 8A): cells are first pre-mixed with a hydrogel precursor solution and loaded into the chamber of the device; then an array of microvalves are activated by external gas pressure, during which the PDMS membrane of the valves deflect outwards to form a 3D physical barrier that serves as a micro-mold to restrict the cell-hydrogel mixture; after gelation through polymerization, the microgels (200 μm thick) are recovered by deactivating the microvalves to release the barrier. Based on this principle, a two-step PAM configuration consisting of an inner ring-shaped and an outer U-shaped microvalve enables precise manipulation of heterogeneous cell types and ECM components in 3D (Fig. 8B), which is essential for fabricating functional tissue constructs that can mimic the complex organization of in vivo tissue architectures.159–161 As a proof of concept, two-step PAM was used to construct a 3D liver lobule-like microtissue by aligning endothelial cells (HUVEC-C) along radially patterned hepatocytes (HepG2) to mimic the natural architecture of the liver lobule in which cords of hepatocytes radiate from a central vein and are separated by sinusoid-like endothelial cells (Fig. 8C–a, b, c). Compared to 2D and 3D monolayer-cultured HepG2 cells, the liver lobule-like microtissues exhibited lower cell viability given the same treatment of acetaminophen (APAP), which causes hepatotoxicity upon overdose (Fig. 8C–d). This result indicates that the HepG2 cells cultured in the microtissues were more sensitive to APAP toxicity, likely due to the microtissues’ ability to better maintain heterotypic cell-cell and cell-ECM interactions, making the microtissues more reliable 3D tissue models for in vitro drug toxicity screening.
Fig. 8.

Flexible fabrication of cell-laden microgels using PAM. (A) Schematic illustrating the fabrication process of PAM. (a) 3D view of the PAM process. (b) Cross-sectional view of microvalve manipulation during PAM. (B) Generation of multi-compartmental cell-laden microgels. Diagram depicting a two-step PAM protocol. (C) Application of PAM for liver tissue engineering. (a, b) Biomimetic construction of a 3D liver lobule-like microtissue. (a) Illustration of the liver lobule, which consists of radially arranged hepatic cords lined by hepatic sinusoids. (b) Schematic delineating the experimental procedure for constructing the biomimetic microtissue. HepG2 cells were molded to form a hepatic cord-like network (green pattern), and then HUVEC-C cells were co-immobilized to generate a hepatic sinusoid-like network (red pattern), which together recreated the liver lobule-like morphology (merged pattern). (c) Morphological demonstration of the 3D liver lobule-like microtissue on-chip and after harvesting and deposition into media. (iii) The fabricated liver lobule-like microtissue consisted of (i) a hepatic cord-like network and (ii) a hepatic sinusoid-like network. Scale bars = 500 μm. (d) Analysis of APAP-induced hepatotoxicity via the biomimetic microtissue. The data are given as means ± SD and collected from three independent experiments. *p < 0.05; **p < 0.01. Adapted from ref. 9 with permission from the Royal Society of Chemistry.
In our latest work, we developed a microfluidic DE droplet-based micro-encapsulation technology for co-culturing hepatocytes and endothelial progenitor cells (EPCs) to form composite spheroids, which were subsequently encapsulated into alginate-collagen microgels and demonstrated enhanced hepatocellular functions.162 DE droplets were generated using two PDMS flow-focusing devices, with the inner phase comprising a 5:1 mixture of hepatocytes and EPCs (HepEPCs) suspended in an alginate solution with an optimized 1:1 mixture of EPC (EGM-2) and hepatocyte culture media. Following spheroid formation after only 4 hr in culture, the oil shell was removed and the inner phase was exposed to a calcium chloride solution, which induced polymerization and led to the formation of alginate microgels (Fig. 9A–a). The resulting microgels were smaller than 200 μm and contained encapsulated composite spheroids of ~80 μm (Fig. 9A–b, c), which is vital to maintaining cell viability as sizes greater than ~150 μm will induce necrosis in the spheroid core.163 Results showed that EPCs significantly improved hepatocyte functions, such as syntheses of albumin and urea, at a HepEPC co-culture ratio of 5:1 (Fig. 9B). Further increases in EPC fraction led to reduced hepatocyte performance, suggesting that this synergistic effect is only manifested at certain co-culture ratios. To examine whether conducive matrix cues would further enhance the synergistic effect, collagen I—a matrix substrate that helps maintain hepatocellular functions—was added to the gelation solution to form alginate-collagen (Alg-col) microgels. As a result, the amount of cumulative albumin synthesized was significantly higher in the HepEPC-in-Alg-col group than in the Hep-in-Alg-col and HepEPC-in-Alg groups (Fig. 9C), indicating that matrix cues provide additional support to enhance the functions of hepatocytes in co-culture. Overall, this DE micro-encapsulation system offers several advantages over existing technologies. First, production of spheroids of ~80 μm is difficult to achieve using existing methods, which usually generate microgels with sizes of around 500–1000 μm,164 presenting a diffusion barrier to the detoxification process of many bioartificial livers in clinical trials.165 Second, this method enables the production of microgels containing single spheroids and prevents the formation and fusion of multiple spheroids within the same gel, which could ultimately lead to spheroid sizes that are greater than the 150-μm limit for ensuring high cell viability. Finally, whereas other strategies require the generation of spheroids before micro-encapsulation, the DE technology enables facile one-step generation of microgel-encapsulated hepatocyte spheroids with tunable biochemical and mechanical properties.
Fig. 9.

(A) (a) Micro-encapsulated spheroid production (Left and middle: Bright field image taken at time 0 and 4 h. Right: Microgel formation after oil removal). Scale bar = 200 μm. (b) Size distribution of DE droplet. (c) Size distribution of spheroid and microgel. (B) (a) Tracking of cell organization in the composite spheroids at different co-culture ratios. Scale bar = 50 μm. (b) Cumulative albumin release (**p < 0.01 between 1:5 and 1:1/3:1/0:1). (c) Cumulative urea secretion (*p < 0.05 between 1:5 and 1:1/3:1/0:1). (d) Basal CYP3A4 activity measured by a luminogenic assay (**p < 0.01 between 1:5 and 0:1). The figure legends denote co-culture cell ratios (EPC:hepatocytes). (Data represent mean ± S.E.M., n = 3). (C) (a) Characterization (immunostaining against hepatocyte (albumin) and EPC (vWF) markers, Live/Dead staining and staining of bile canaliculi) of single or co-culture spheroids cultured in different conditions. Scale bar = 50 μm. (b) Cumulative albumin release. (c) Cumulative urea secretion. (d) Basal CYP3A4 activity measured by a luminogenic assay (*p < 0.05 between HepEPC in Alg-col and HepEPC in Alg). (e) Induction of CYP3A4 activity after treatment with 10×10−6 M Dex for 72 h. (Data represent mean ± S.E.M., n = 3). Adapted from ref. 143 with permission from John Wiley and Sons.
7.4 Bone/cartilage regeneration
Stem cell transplantation is a promising strategy for treating bone and cartilage injuries. Although direct injection provides a minimally invasive method for delivering cells to the repair site, it is limited by low cell retention and engraftment, which may be attributed to mechanical shear forces that damage the cell membrane during the injection process.166 To overcome this limitation, stem cells have been suspended in hydrogels that can be injected and solidified in situ. However, these hydrogels suffer from insufficient oxygen and nutrient exchange due to the large size of the bone defects, which impedes bone regeneration.167 Alternatively, microgels not only maintain cell viability by facilitating efficient nutrient and waste exchange but also protect the scaffold from shear stress during injection. Due to the controlled sizes, monodispersity, and high encapsulation efficiency of microfluidic microgels, they have been used to encapsulate cells for bone/cartilage regeneration applications.
Weitz’s group engineered methacrylated gelatin (Gel-MA) microgels encapsulating bone marrow-derived mesenchymal stem cells (BMSCs) and the osteogenic growth factor, bone morphogenic protein-2 (BMP-2), that may serve as injectable tissue constructs for enhancing bone formation and ossification.7 A capillary flow-focusing microfluidic device was used to generate the microgels, with the dispersed phase consisting of Gel-MA supplemented with the photoinitiator 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropriophenone, BMSCs, and BMP-2, and the continuous phase comprising the perfluorinated oil HFE-7500. The size of the W/O emulsions was controlled by adjusting the flow rate of each of the two phases. After collection, the W/O emulsions were exposed to UV light (365 nm) for 20 s and polymerized into microgels that were approximately 160 μm in diameter. Adherent cells readily spread inside the microgels, and there were a large number of BMSCs that migrated from the interior of the microgel to the surface after 4 weeks of culture, indicating that the BMSCs were actively involved in the regeneration process. Cells released from the microgels demonstrated similar viability and morphology as those directly grown on tissue culture plastic. There was an early reduction in alkaline phosphatase activity, which usually signals the differentiation of BMSCs into osteoblasts. Meanwhile, the percentage of calcium deposits within the microgels increased to almost 70% after 4 weeks of culture, demonstrating the in vivo osteogenic potential of the BMSC-laden microgels. Moreover, the Gel-MA microgels demonstrated a prolonged release of BMP-2 that lasted for 1 week, which is advantageous for repairing bone defects as it promotes bone formation and ossification longer than do most hydrogels. In a model of rabbit femoral defect, Gel-MA microgels delivering both BMSCs and BMP-2 induced the most extensive in vivo bone formation and hence the highest therapeutic efficacy out of the three groups: microgels loaded with BMSCs, with and without BMP-2, and the control group of microgels alone.
7.5 Microgel scaffold for wound healing
Injectable materials have been developed as a minimally invasive means of implanting stem cells into wound sites for regenerating healthy tissue.168 The efficacy of these materials in promoting tissue regeneration depends on how closely coupled are the rate of material degradation and tissue development, so that newly forming tissues can gradually replace the materials at a similar rate.55 For example, if the degradation rate of the material is too fast, an insufficient amount of scaffolding will hamper tissue ingrowth. In contrast, a rate too slow may promote fibrosis and lead to improper tissue development.150 Hydrogels, whose degradation rates can easily be tuned by modulating the density of the crosslinked network as well as the polymeric backbone, have been widely used as injectable scaffold materials.170–173 However, this approach relies on material degradation prior to tissue integration, which may not be suitably applied in different degradation environments. A more robust strategy—the building-block approach—uses microgels to form scaffolds by assembling them into interconnected lattices with micron-sized pores, thereby eliminating the need to tune the degradation rate of the material for different wound sites, which possess varying physical and chemical characteristics.
Khademhosseini’s group cultured cardiac side population (CSP) cells on the surface of Gel-MA microgels, and coated the cell-seeded microgels with an additional layer of silica hydrogel to protect the cells against external stress during injection procedures.113 The Gel-MA microgels were fabricated using a PDMS-based microfluidic flow-focusing device, with the dispersed aqueous phase consisting of Gel-MA and a photoinitiator, and the continuous oil phase consisting of mineral oil supplemented with a surfactant (Fig. 10A). As the droplets exited the outlet of the device and passed through a plastic connection tubing, they were polymerized in situ under 850-mW UV light for 5 min. The elastic modulus of the resulting microgels, as determined by AFM-assisted nano-indentation, was 1.87 kPa (±0.23), suggesting that they are capable of withstanding shear stress upon injection from a surgical syringe. CSP cells were cultured on Gel-MA microgels with a diameter of 100 μm, which provide sufficient surface area for multiple cell attachment while also being small enough in size to be injected through syringe needles (Fig. 10B–a, b). When the cell-laden microgels were placed in tissue culture plates, the cells migrated to the plastic surface and proliferated to cover the entire surface, demonstrating the ability of these microgel-cultured CSP cells to spread to the surrounding environment, which is a vital step following cell transplantation to target tissues in a wound site (Fig. 10B–c). Lastly, the Gel-MA microgels were coated with a sol-gel-generated silica hydrogel (Fig. 10C), which acts as a shell that protects the cells on the surface of the microgel from exposure to reactive oxidative species, mechanical stress during the injection process, and the host immune response, which are common external factors responsible for low post-transplantation cell viability.174,175 Upon exposure to an oxidative stress environment, the CSP cells protected by a silica hydrogel shell only showed an 8% decrease in viability, compared to a 50% decrease for CSP cells without the protective shell (Fig. 10D), suggesting that the cell-seeded microgels combined with a silica hydrogel shell are promising injectable tissue constructs for applications in wound healing.
Fig. 10.

(A) (a) Microfluidic fabrication of Gel-MA microgels. Aqueous droplets made of Gel-MA pre-gel solution, generated from a microfluidic flow-focusing device, were photopolymerized to form Gel-MA microgels. (b, c) Microscopic images of (b) microfluidic flow-focusing device generating Gel-MA droplets and (c) Gel-MA microgels fabricated by UV-initiated photopolymerization of Gel-MA droplets. (B) (a) Gel-MA microgels were used as an in vitro platform to culture cells on the surface. (b) Microscopic images of CSP cells cultured on Gel-MA microgels. The cells adhered on the surface of Gel-MA microgels and proliferated over time. Scale bar = 100 μm. (c) CSP cells on Gel-MA microgels were placed on a cell-adhesive surface, and their adhesion and proliferation over time were monitored. Scale bar = 50 μm. (C) (a) Schematic of the fabrication of a protective silica hydrogel shell on Gel-MA microgel. (b) Optical microscopic images of Gel-MA microgels and silica-coated Gel-MA microgels. (D) (a) Fluorescent images of CSP cells on Gel-MA microgels (left) and Gel-MA microgels coated with silica hydrogel (right), subjected to oxidative stress. The cells were stained with calcein-AM (green) and ethidium homodimer-1 (red) to visualize live and dead cells, respectively. Scale bar = 50 μm. (b) Viability of the CSP cells was quantified by the percentage of live cells (*p < 0.05). Adapted from ref. 94 with permission from the American Chemical Society. Copyright 2014.
Griffin et al. fabricated microporous annealed particle (MAP) gels by using the activated enzyme Factor XIII (FXIIIa) to anneal microfluidics-generated microgel building blocks into a scaffold containing interconnected microporous networks.176 The microgels were produced in a PDMS flow-focusing device, with the dispersed aqueous phase composed of a mixture of bioactive 4-arm PEG-vinyl sulphone (PEG-VS), cell-adhesive peptide (RGD), transglutaminase peptide substrates (K and Q), and MMP-sensitive peptide sequences, and the continuous oil phase consisting of mineral oil supplemented with a surfactant. Serving as building blocks, the microgels were annealed to each other via FXIIIa-mediated non-canonical amide linkage between the peptides K and Q (Fig. 11A). Prior to scaffold annealing, the FXIIIa-supplemented microgel mixture containing live cells can be injected into a wound site and solidify into the shape of the cavity (Fig. 11A–d). After annealing of the MAP scaffolds, the cells began to spread and form extensive networks in vitro. A murine skin wound-healing model was used to test the ability of the MAP scaffolds to support in vivo cell migration and bulk tissue integration. After five days post-injection, 60% of the wound area remained in the group with MAP scaffolds compared to 100% for the non-porous control (Fig. 11B), demonstrating that MAP scaffolds facilitate significantly more rapid wound closure. In a seven-day in vivo wound-healing experiment on BALB/c mice, the MAP scaffolds contributed to 39% wound closure, compared to only 19% for the no-treatment control, 7% for the physically matched non-porous control, and 10% for non-annealing microgels, suggesting that microgel annealing is essential to rapid wound closure. Also, unannealed microporous gels fabricated using a porogen-based casting method led to 27% wound closure, indicating that micron-sized pores greatly facilitate wound healing in vivo. Furthermore, the interconnected network of micropores in the MAP scaffolds promoted bulk tissue integration, as evidenced by the co-localization of endothelial cells and pericytes in the scaffolds, which signals the development of a vascular network. In summary, MAP scaffolds have the following advantages: 1) their injectability enables them to completely fill wounds of any shape and size; 2) their microporosity facilitates rapid cellular invasion and network formation; and 3) the modular nature of their assembly allows for the fabrication of microgel building blocks with a diverse range of chemical and physical properties that can be used for tissue regeneration in a wide range of physiological niches.
Fig. 11.

(A) Microfluidic generation of microgel building blocks for the creation of MAP scaffolds. (a) Schematic illustrating microgel formation using a microfluidic W/O emulsion system. A pre-gel and crosslinker solution are segmented into monodisperse droplets followed by in-droplet mixing and crosslinking via Michael addition. (b) Microgels are purified into an aqueous solution and annealed using FXIIIa to generate a microporous scaffold, either in the presence of cells or as a pure scaffold. (c) Fluorescent images showing purified microgel building blocks (left) and a subsequent cell-laden MAP scaffold (right). (d) MAP scaffolds are moldable to macroscale shapes, and can be injected to form complex shapes that are maintained after annealing. (e) This process can be performed in the presence of live cells. (B) MAP scaffolds promote fast wound closure in SKH1-Hrhr and Balb/c epidermal mouse models. (a) Quantification of wound closure over a five-day period shows statistically significant wound-closure rates for MAP scaffolds when compared with non-porous bilateral controls (ND6). Statistical significance performed using standard two-tailed t-test (*p<0.05). (b) Representative images of wound closure during a five-day in vivo wound-healing model in SKH1-Hrhr mice. (c) Representative images of wound closure during seven-day in vivo Balb/c experiments. (d) Quantification of wound-closure data from Balb/c in vivo wound healing. Adapted from ref. 157 with permission from Nature Publishing Group.
8. Potential applications of cell-laden microfluidic microgels in tissue regeneration
The advantages of microfluidics-generated cell-laden microgels, such as uniform size, could make it possible to overcome existing technical challenges in 3D bioprinting. Based on layer-by-layer deposition, 3D bioprinting offers great control over the spatial patterning of functional components. Using this technology, artificial tissues with functions that closely mimic those of native tissue can be generated by replicating the intricate interaction between cells and their microenvironment on the microscale. Tissue constructs can be assembled from building blocks comprised of either a cell-laden hydrogel precursor solution or cell-encapsulated microspheres.
Upon activation by thermal-, photo- or pH-mediated mechanisms, hydrogel solutions containing cells can be crosslinked in situ as they are injected through the orifice of the printing nozzle.175 Although the small inner diameter of the nozzle allows for x-y resolutions of 50 μm or less, it causes a pressure build-up that could reach 20 psi or greater,177,178 which leads to substantial mechanical stress that damages the cells and reduces their physiological functions. Furthermore, since cells are prepared and dispersed in an aqueous precursor solution, there is a loss of cell-cell and cell-substrate contact during and after the printing process. A long period of culture, often a week or more, is required for the cells to degrade the hydrogels and gradually reform cell-cell junctions. As a result, cells that require stable cell-cell and cell-substrate signaling may lose their functions and undergo cell de-differentiation or apoptosis.
The microsphere building-block approach, in which the basic units are composed of cell spheroids or microspheres fully coated with cells, enables the generation of microspheres containing tens of thousands of cells. The microspheres are extruded through a large nozzle with an inner diameter of hundreds of microns, which significantly mitigates the amount of shear stress and leads to increased cell viability.175 Moreover, cells on the microspheres have already formed stable cell-cell junctions and cell-substrate adhesion, so ECM proteins such as fibronectin, collagen, and laminin are deposited on the microspheres,162 leading to improved cellular activity, viability, and tissue function.
An existing problem with using microspheres as building blocks for 3D bioprinting is the inability to produce homogeneous microspheres and cell spheroids. Variation in microsphere size results in non-uniform cell dispersion and leads to unstable tissue structures, as microspheres with different sizes will form unstable stacks that are prone to move or shift, which reduces both the mechanical strength of the printed tissue and its spatial resolution. A promising alternative is microfluidic cell-laden microgels, whose monodispersity allows them to be closely packed into multiple layers, thereby forming a tissue construct that has good mechanical stability as well as homogeneous physicochemical properties. The ability of microfluidics to produce microgels with varying biochemical and mechanical properties, combined with 3D bioprinting’s control over the spatial assembly of functional components, may facilitate the generation of macroscale biomimetic tissue constructs for organ transplantation or for in vitro drug testing.
9. Concluding remarks and future perspectives
Cell-laden scaffolds for tissue regeneration should possess good biocompatibility, controlled degradability, mechanical stability, uniform size, a conducive cellular microenvironment with appropriate biochemical and biophysical cues, and appropriate mass transport properties. Combined with a judicious selection of biomaterials, microfluidic technologies can be used to generate monodisperse cell-laden microgels with easily tunable sizes, biophysical properties, and biochemical cues. These microgels may serve as building blocks that can be assembled into mesoscale tissue structures with functions that closely mimic those of native tissue. However, several challenges must be overcome before clinical implementation of microfluidic microgels can be realized.
One of the foremost concerns is the scale-up demand of bio-manufacturing macroscale tissue assemblies.179 For example, the average weight of an adult human liver is approximately 1.56 kg,180 with around 139 million cells per gram of liver,181 which means that nearly 220 billion cells in total would be required to regenerate an entire functional liver. Assuming that cell-laden microgels can be generated at a rate of ~1 kHz,10 and that the average encapsulation density is ~25 cells per microgel, then non-stop microgel production from 100 microfluidic devices running in parallel for an entire day would be required to achieve such a feat. In an early study by Nisisako et al., a mass-production module composing of 256 O/W emulsion-forming units was used to produce monodisperse acrylic microgels at a rate of ~0.3 kg per hour, demonstrating scale-up potential.110 However, for complex droplet-forming devices that demand high-resolution spatial patterning of wettability,182,183 more robust designs are needed to ensure controlled droplet formation during large-scale parallelization. A few groups have developed complex devices that obviate the need for localized surface modification of channel hydrophilicity, but they are limited either by low droplet production rates or by the types of emulsions that can be generated.184,185 Therefore, there is a need to develop devices that can support large-scale production of microgels with complex geometries, such as core-shell morphology.
Another issue impeding the application of cell-laden microgels in tissue implantation is the lack of proper vascularization. Biomimetic tissue constructs should be composed of precisely organized components in 3D so that different cell types, ECM, and vasculature can properly interact with each other to maintain normal tissue functions.186 Out of these components, a well-perfused microvasculature is perhaps most vital to the viability of the tissue construct, as cells need to be within 200 μm of a blood vessel, which facilitates efficient exchange of nutrients and wastes as well as transport of growth factors and cellular signals.187 Therefore, organized vascular networks must be incorporated into cell-laden microgels before they are injected into a defect site so that the regenerated tissue can readily integrate with the host vasculature. Proper vascularization requires precise spatial and temporal patterning of different cell types and chemical cues that control the vascular organization and remodeling processes.188 Various methods have been demonstrated for fabricating vascular networks in hydrogels, such as using gelatin as a sacrificial material to form hollow channels189 or using PDMS molds to form collagen-filled microchannels that are seeded with endothelial cells.190 In order to reliably produce microgels with branched vascular networks, strategies need to be developed for adapting these techniques to a miniaturized microfluidic device and automating the series of steps that are involved in the fabrication process.
The development of whole-organ decellularization techniques during the past decade may mitigate the vascularization dilemma and enable the fabrication of microfluidic microgels that can maximally recreate the complex architecture and functionality of native tissue. After removal of all cellular and immunogenic species, the remaining ECM of the decellularized organ not only retains relevant biological signals but also preserves the microarchitecture of the native tissue, including an intact vascular system that could easily integrate with the patient’s circulatory system.191,192 Since the specific composition and organization of the ECM depend on the tissue source from which the ECM is derived,193 the use of tissue-specific ECMs as scaffolds for tissue regeneration applications may result in biomimetic tissue constructs that better recapitulate the functions of the native tissue. Therefore, one can envision the incorporation of tissue-specific decellularized ECM into a hydrogel precursor solution to produce microfluidic microgels that not only contain cells encapsulated within a physiologically relevant microenvironment but also promote the bulk integration of the vascular system with surrounding tissue. In concert with advances in tissue engineering and microfluidic technology, the clinical application of cell-laden microfluidic microgels for tissue regeneration sees a bright future.
Acknowledgments
Funding support from NIH (HL109442, AI096305, GM110494), Guangdong Innovative and Entrepreneurial Research Team Program NO.2013S086, and Global Research Laboratory Program (Korean NSF GRL; 2015032163) is also acknowledged.
Biographies

Weiqian Jiang received dual BS degrees in Biomedical Engineering and Electrical and Computer Engineering from Duke University in 2013. He is currently a graduate student majoring in Biomedical Engineering at Columbia University. His current research focuses on microfluidics and tissue engineering.

Dr. Mingqiang Li received his BS degree from the University of Science and Technology of China in 2009, followed by a PhD degree from the Chinese Academy of Sciences in 2015. He is working as a postdoctoral research scientist with Prof. Kam W. Leong at Columbia University on microfluidics and biomaterials.

Dr. Zaozao Chen received his BS and MS degrees from Southeast University, followed by a PhD degree from the University of North Carolina at Chapel Hill. He is working as a postdoctoral research scientist with Prof. Kam W. Leong at Columbia University, focusing on development of human “Organs-on-a-Chip” systems as disease models and drug screening platforms.

Kam W. Leong is the Samuel Y. Sheng Professor of Biomedical Engineering at Columbia University, and holds a joint faculty position in the Department of Systems Biology. His lab focuses on non-viral gene delivery, direct cell reprogramming, tissue-chip system, and genome editing. He is the Editor-in-Chief of Biomaterials, and is a member of the National Academy of Engineering and National Academy of Inventors.
References
- 1.Langer R, Vacanti JP. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 2.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 3.Lee KY, Mooney DJ. Chem Rev. 2001;101:1869–1877. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 4.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Adv Mater. 2009;21:3307–3329. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicodemus GD, Bryant SJ. Tissue Eng, Part B. 2008;14:149–165. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen R, Curran SJ, Curran JM, Hunt JA. Biomaterials. 2006;27:4453–4460. doi: 10.1016/j.biomaterials.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Zhao X, Liu S, Yildirimer L, Zhao H, Ding R, Wang H, Cui W, Weitz D. Adv Funct Mater. 2016;26:2809–2819. [Google Scholar]
- 8.Yanagawa F, Kaji H, Jang YH, Bae H, Yanan D, Fukuda J, Qi H, Khademhosseini A. J Biomed Mater Res, Part A. 2011;97:93–102. doi: 10.1002/jbm.a.33034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma C, Tian C, Zhao L, Wang J. Lab Chip. 2016;16:2609–2617. doi: 10.1039/c6lc00540c. [DOI] [PubMed] [Google Scholar]
- 10.Siltanen C, Yaghoobi M, Haque A, You J, Lowen J, Soleimani M, Revzin A. Acta Biomater. 2016;34:125–132. doi: 10.1016/j.actbio.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seliktar D. Science. 2012;336:1124–1128. doi: 10.1126/science.1214804. [DOI] [PubMed] [Google Scholar]
- 12.Allazetta S, Lutolf MP. Curr Opin Biotechnol. 2015;35:86–93. doi: 10.1016/j.copbio.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Malda J, Visser J, Melchels FP, Jungst T, Hennink WE, Dhert WJ, Groll J, Hutmacher DW. Adv Mater. 2013;25:5011–5028. doi: 10.1002/adma.201302042. [DOI] [PubMed] [Google Scholar]
- 14.Ghobril C, Grinstaff MW. Chem Soc Rev. 2015;44:1820–1835. doi: 10.1039/c4cs00332b. [DOI] [PubMed] [Google Scholar]
- 15.Hoare TR, Kohane DS. Polymer. 2008;49:1993–2007. [Google Scholar]
- 16.Pape ACH, Dankers PYW. In: Supramolecular Polymer Networks and Gels. Seiffert S, editor. Springer International Publishing; 2015. pp. 253–279. [Google Scholar]
- 17.Jiang Y, Chen J, Deng C, Suuronen EJ, Zhong Z. Biomaterials. 2014;35:4969–4985. doi: 10.1016/j.biomaterials.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Velasco D, Tumarkin E, Kumacheva E. Small. 2012;8:1633–1642. doi: 10.1002/smll.201102464. [DOI] [PubMed] [Google Scholar]
- 19.Leslie SK, Nicolini AM, Sundaresan G, Zweit J, Boyan BD, Schwartz Z. J Mater Chem B. 2016;4:3515–3525. doi: 10.1039/c6tb00035e. [DOI] [PubMed] [Google Scholar]
- 20.Mazutis L, Vasiliauskas R, Weitz DA. Macromol Biosci. 2015;15:1641–1646. doi: 10.1002/mabi.201500226. [DOI] [PubMed] [Google Scholar]
- 21.Chen Q, Utech S, Chen D, Prodanovic R, Lin JM, Weitz DA. Lab Chip. 2016;16:1346–1349. doi: 10.1039/c6lc00231e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee H, Chung S, Kim MG, Lee LP, Lee JY. Adv Healthcare Mater. 2016;5:1638–1645. doi: 10.1002/adhm.201600048. [DOI] [PubMed] [Google Scholar]
- 23.Lee YB, Shin YM, Kim EM, Lim J, Lee J-Y, Shin H. Adv Healthcare Mater. 2016;5:2320–2324. doi: 10.1002/adhm.201600210. [DOI] [PubMed] [Google Scholar]
- 24.Xu QH, He CL, Ren KX, Xiao CS, Chen XS. Adv Healthcare Mater. 2016;5:1979–1990. doi: 10.1002/adhm.201600292. [DOI] [PubMed] [Google Scholar]
- 25.Cheng YL, He CL, Ding JX, Xiao CS, Zhuang XL, Chen XS. Biomaterials. 2013;34:10338–10347. doi: 10.1016/j.biomaterials.2013.09.064. [DOI] [PubMed] [Google Scholar]
- 26.Tumarkin E, Kumacheva E. Chem Soc Rev. 2009;38:2161–2168. doi: 10.1039/b809915b. [DOI] [PubMed] [Google Scholar]
- 27.Anthamatten M. In: Supramolecular Polymer Networks and Gels. Seiffert S, editor. Springer International Publishing; 2015. pp. 47–99. [Google Scholar]
- 28.Xu FM, Wang HB, Zhao J, Liu XS, Li DD, Chen CJ, Ji J. Macromolecules. 2013;46:4235–4246. [Google Scholar]
- 29.Chen Y, Ballard N, Gayet F, Bon SAF. Chem Commun. 2012;48:1117–1119. doi: 10.1039/c2cc16670d. [DOI] [PubMed] [Google Scholar]
- 30.Sionkowska A. Prog Polym Sci. 2011;36:1254–1276. [Google Scholar]
- 31.Nguyen KT, West JL. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 32.Fedorovich NE, Oudshoorn MH, van Geemen D, Hennink WE, Alblas J, Dhert WJ. Biomaterials. 2009;30:344–353. doi: 10.1016/j.biomaterials.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 33.Fu Y, Kao WJ. J Biomed Mater Res, Part A. 2011;98:201–211. doi: 10.1002/jbm.a.33106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darling NJ, Hung YS, Sharma S, Segura T. Biomaterials. 2016;101:199–206. doi: 10.1016/j.biomaterials.2016.05.053. [DOI] [PubMed] [Google Scholar]
- 35.Davis NE, Ding S, Forster RE, Pinkas DM, Barron AE. Biomaterials. 2010;31:7288–7297. doi: 10.1016/j.biomaterials.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin R, Moreira Teixeira LS, Dijkstra PJ, Zhong Z, van Blitterswijk CA, Karperien M, Feijen J. Tissue Eng, Part A. 2010;16:2429–2440. doi: 10.1089/ten.TEA.2009.0764. [DOI] [PubMed] [Google Scholar]
- 37.Jin R, Teixeira LS, Dijkstra PJ, van Blitterswijk CA, Karperien M, Feijen J. Biomaterials. 2010;31:3103–3113. doi: 10.1016/j.biomaterials.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 38.Yung CW, Bentley WE, Barbari TA. J Biomed Mater Res, Part A. 2010;95:25–32. doi: 10.1002/jbm.a.32740. [DOI] [PubMed] [Google Scholar]
- 39.Teixeira LS, Feijen J, van Blitterswijk CA, Dijkstra PJ, Karperien M. Biomaterials. 2012;33:1281–1290. doi: 10.1016/j.biomaterials.2011.10.067. [DOI] [PubMed] [Google Scholar]
- 40.Thiele J, Ma Y, Bruekers SMC, Ma S, Huck WTS. Adv Mater. 2014;26:125–148. doi: 10.1002/adma.201302958. [DOI] [PubMed] [Google Scholar]
- 41.Ghobril C, Grinstaff MW. Chem Soc Rev. 2015;44:1820–1835. doi: 10.1039/c4cs00332b. [DOI] [PubMed] [Google Scholar]
- 42.Topuz F, Henke A, Richtering W, Groll J. Soft Matter. 2012;8:4877–4881. [Google Scholar]
- 43.Morch YA, Donati I, Strand BL, Skjak-Braek G. Biomacromolecules. 2006;7:1471–1480. doi: 10.1021/bm060010d. [DOI] [PubMed] [Google Scholar]
- 44.Tan WH, Takeuchi S. Adv Mater. 2007;19:2696–2701. [Google Scholar]
- 45.Luo R-C, Chen C-H. Soft. 2012;01:1–23. [Google Scholar]
- 46.Zhang H, Tumarkin E, Sullan RMA, Walker GC, Kumacheva E. Macromol Rapid Commun. 2007;28:527–538. [Google Scholar]
- 47.Velasco D, Tumarkin E, Kumacheva E. Small. 2012;8:1633–1642. doi: 10.1002/smll.201102464. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, Tumarkin E, Peerani R, Nie Z, Sullan RMA, Walker GC, Kumacheva E. J Am Chem Soc. 2006;128:12205–12210. doi: 10.1021/ja0635682. [DOI] [PubMed] [Google Scholar]
- 49.Raz N, Li JK, Fiddes LK, Tumarkin E, Walker GC, Kumacheva E. Macromolecules. 2010;43:7277–7281. [Google Scholar]
- 50.Sabhachandani P, Motwani V, Cohen N, Sarkar S, Torchilin V, Konry T. Lab Chip. 2016;16:497–505. doi: 10.1039/c5lc01139f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akbari S, Pirbodaghi T. Microfluid Nanofluid. 2013;16:773–777. [Google Scholar]
- 52.Kim C, Chung S, Kim YE, Lee KS, Lee SH, Oh KW, Kang JY. Lab Chip. 2011;11:246–252. doi: 10.1039/c0lc00036a. [DOI] [PubMed] [Google Scholar]
- 53.Shah RK, Kim J-W, Agresti JJ, Weitz DA, Chu L-Y. Soft Matter. 2008;4:2303–2309. [Google Scholar]
- 54.Liu K, Ding HJ, Liu J, Chen Y, Zhao XZ. Langmuir. 2006;22:9453–9457. doi: 10.1021/la061729+. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Rodrigues J, Tomas H. Chem Soc Rev. 2012;41:2193–2221. doi: 10.1039/c1cs15203c. [DOI] [PubMed] [Google Scholar]
- 56.Burdick JA, Prestwich GD. Adv Mater. 2011;23:H41–56. doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan H, Ramirez CM, Miljkovic N, Li H, Rubin JP, Marra KG. Biomaterials. 2009;30:6844–6853. doi: 10.1016/j.biomaterials.2009.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burdick JA, Prestwich GD. Adv Mater. 2011;23:H41–H56. doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bian L, Hou C, Tous E, Rai R, Mauck RL, Burdick JA. Biomaterials. 2013;34:413–421. doi: 10.1016/j.biomaterials.2012.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sideris E, Griffin DR, Ding Y, Li S, Weaver WM, Di Carlo D, Hsiai T, Segura T. ACS Biomater Sci Eng. 2016 doi: 10.1021/acsbiomaterials.6b00444. [DOI] [PubMed] [Google Scholar]
- 61.Kumachev A, Greener J, Tumarkin E, Eiser E, Zandstra PW, Kumacheva E. Biomaterials. 2011;32:1477–1483. doi: 10.1016/j.biomaterials.2010.10.033. [DOI] [PubMed] [Google Scholar]
- 62.Ling Y, Rubin J, Deng Y, Huang C, Demirci U, Karp JM, Khademhosseini A. Lab Chip. 2007;7:756–762. doi: 10.1039/b615486g. [DOI] [PubMed] [Google Scholar]
- 63.Tumarkin E, Tzadu L, Csaszar E, Seo M, Zhang H, Lee A, Peerani R, Purpura K, Zandstra PW, Kumacheva E. Integr Biol. 2011;3:653–662. doi: 10.1039/c1ib00002k. [DOI] [PubMed] [Google Scholar]
- 64.Puppi D, Chiellini F, Piras AM, Chiellini E. Prog Polym Sci. 2010;35:403–440. [Google Scholar]
- 65.De Souza R, Zahedi P, Allen CJ, Piquette-Miller M. Biomaterials. 2009;30:3818–3824. doi: 10.1016/j.biomaterials.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 66.Bhattarai N, Gunn J, Zhang M. Adv Drug Delivery Rev. 2010;62:83–99. doi: 10.1016/j.addr.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 67.Koev ST, Dykstra PH, Luo X, Rubloff GW, Bentley WE, Payne GF, Ghodssi R. Lab Chip. 2010;10:3026–3042. doi: 10.1039/c0lc00047g. [DOI] [PubMed] [Google Scholar]
- 68.Fan M, Ma Y, Mao J, Zhang Z, Tan H. Acta Biomater. 2015;20:60–68. doi: 10.1016/j.actbio.2015.03.033. [DOI] [PubMed] [Google Scholar]
- 69.Holme HK, Davidsen L, Kristiansen A, Smidsrod O. Carbohydr Polym. 2008;73:656–664. doi: 10.1016/j.carbpol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 70.Griffith LG, Swartz MA. Nat Rev Mol Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 71.Kim B-S, Park I-K, Hoshiba T, Jiang H-L, Choi Y-J, Akaike T, Cho C-S. Prog Polym Sci. 2011;36:238–268. [Google Scholar]
- 72.Glowacki J, Mizuno S. Biopolymers. 2008;89:338–344. doi: 10.1002/bip.20871. [DOI] [PubMed] [Google Scholar]
- 73.Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Lab Chip. 2010;10:51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ho CT, Lin RZ, Chen RJ, Chin CK, Gong SE, Chang HY, Peng HL, Hsu L, Yew TR, Chang SF, Liu CH. Lab Chip. 2013;13:3578–3587. doi: 10.1039/c3lc50402f. [DOI] [PubMed] [Google Scholar]
- 75.Lee SA, No da Y, Kang E, Ju J, Kim DS, Lee SH. Lab Chip. 2013;13:3529–3537. doi: 10.1039/c3lc50197c. [DOI] [PubMed] [Google Scholar]
- 76.Wallace D. Adv Drug Delivery Rev. 2003;55:1631–1649. doi: 10.1016/j.addr.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 77.Brigham MD, Bick A, Lo E, Bendali A, Burdick JA, Khademhosseini A. Tissue Eng, Part A. 2009;15:1645–1653. doi: 10.1089/ten.tea.2008.0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moraes C, Simon AB, Putnam AJ, Takayama S. Biomaterials. 2013;34:9623–9631. doi: 10.1016/j.biomaterials.2013.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hong S, Hsu HJ, Kaunas R, Kameoka J. Lab Chip. 2012;12:3277–3280. doi: 10.1039/c2lc40558j. [DOI] [PubMed] [Google Scholar]
- 80.Rowe SL, Lee S, Stegemann JP. Acta Biomater. 2007;3:59–67. doi: 10.1016/j.actbio.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lechtenberg BC, Johnson DJ, Freund SM, Huntington JA. Proc Natl Acad Sci U S A. 2010;107:14087–14092. doi: 10.1073/pnas.1005255107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davis HE, Miller SL, Case EM, Leach JK. Acta Biomater. 2011;7:691–699. doi: 10.1016/j.actbio.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 83.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A. Biomaterials. 2010;31:5536–5544. doi: 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Y, Meng H, Liu Y, Narkar A, Lee BP. ACS Appl Mater Interfaces. 2016;8:11980–11989. doi: 10.1021/acsami.6b01364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sisson K, Zhang C, Farach-Carson MC, Chase DB, Rabolt JF. Biomacromolecules. 2009;10:1675–1680. doi: 10.1021/bm900036s. [DOI] [PubMed] [Google Scholar]
- 86.Chen YC, Lin RZ, Qi H, Yang Y, Bae H, Melero-Martin JM, Khademhosseini A. Adv Funct Mater. 2012;22:2027–2039. doi: 10.1002/adfm.201101662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aubin H, Nichol JW, Hutson CB, Bae H, Sieminski AL, Cropek DM, Akhyari P, Khademhosseini A. Biomaterials. 2010;31:6941–6951. doi: 10.1016/j.biomaterials.2010.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qi H, Du Y, Wang L, Kaji H, Bae H, Khademhosseini A. Adv Mater. 2010;22:5276–5281. doi: 10.1002/adma.201002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu J. Biomaterials. 2010;31:4639–4656. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Drury JL, Mooney DJ. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 91.Lin C, Zhao P, Li F, Guo F, Li Z, Wen X. Mater Sci Eng, C. 2010;30:1236–1244. [Google Scholar]
- 92.Yu L, Ding J. Chem Soc Rev. 2008;37:1473–1481. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 93.Lee SY, Tae G. J Controlled Release. 2007;119:313–319. doi: 10.1016/j.jconrel.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 94.Sosnik A, Cohn D. Biomaterials. 2004;25:2851–2858. doi: 10.1016/j.biomaterials.2003.09.057. [DOI] [PubMed] [Google Scholar]
- 95.Ricci EJ, Lunardi LO, Nanclares DM, Marchetti JM. Int J Pharm. 2005;288:235–244. doi: 10.1016/j.ijpharm.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 96.Cohn D, Sosnik A, Levy A. Biomaterials. 2003;24:3707–3714. doi: 10.1016/s0142-9612(03)00245-x. [DOI] [PubMed] [Google Scholar]
- 97.Cohn D, Lando G, Sosnik A, Garty S, Levi A. Biomaterials. 2006;27:1718–1727. doi: 10.1016/j.biomaterials.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 98.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 99.Sia SK, Whitesides GM. Electrophoresis. 2003;24:3563–3576. doi: 10.1002/elps.200305584. [DOI] [PubMed] [Google Scholar]
- 100.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 101.Anna SL, Bontoux N, Stone HA. Appl Phys Lett. 2003;82:364–366. [Google Scholar]
- 102.Liu Y, Li H, Yan S, Wei J, Li X. Biomacromolecules. 2014;15:1044–1054. doi: 10.1021/bm401926k. [DOI] [PubMed] [Google Scholar]
- 103.Shah RK, Shum HC, Rowat AC, Lee D, Agresti JJ, Utada AS, Chu L-Y, Kim J-W, Fernandez-Nieves A, Martinez CJ, Weitz DA. Mater Today. 2008;11:18–27. [Google Scholar]
- 104.Choi CH, Wang H, Lee H, Kim JH, Zhang L, Mao A, Mooney DJ, Weitz DA. Lab Chip. 2016;16:1549–1555. doi: 10.1039/c6lc00261g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Herrero EP, Valle EMMD, Galán MA. Biotechnol Prog. 2007;23:940–945. doi: 10.1021/bp070047p. [DOI] [PubMed] [Google Scholar]
- 106.Tawfik DS, Griffiths AD. Nat Biotechnol. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 107.Huebner A, Sharma S, Srisa-Art M, Hollfelder F, Edel JB, Demello AJ. Lab Chip. 2008;8:1244–1254. doi: 10.1039/b806405a. [DOI] [PubMed] [Google Scholar]
- 108.Utada AS, Lorenceau E, Link DR, Kaplan PD, Stone HA, Weitz DA. Science. 2005;308:537–541. doi: 10.1126/science.1109164. [DOI] [PubMed] [Google Scholar]
- 109.Xu S, Nie Z, Seo M, Lewis P, Kumacheva E, Stone HA, Garstecki P, Weibel DB, Gitlin I, Whitesides GM. Angew Chem. 2005;117:734–738. doi: 10.1002/anie.200462226. [DOI] [PubMed] [Google Scholar]
- 110.Nisisako T, Torii T. Lab Chip. 2008;8:287–293. doi: 10.1039/b713141k. [DOI] [PubMed] [Google Scholar]
- 111.Deveza L, Ashoken J, Castaneda G, Tong X, Keeney M, Han L-H, Yang F. ACS Biomater Sci Eng. 2015;1:157–165. doi: 10.1021/ab500051v. [DOI] [PubMed] [Google Scholar]
- 112.Colton CK. Adv Drug Delivery Rev. 2014;67–68:93–110. doi: 10.1016/j.addr.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 113.Cha C, Oh J, Kim K, Qiu Y, Joh M, Shin SR, Wang X, Camci-Unal G, Wan KT, Liao R, Khademhosseini A. Biomacromolecules. 2014;15:283–290. doi: 10.1021/bm401533y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nicolson PC, Vogt J. Biomaterials. 2001;22:3273–3283. doi: 10.1016/s0142-9612(01)00165-x. [DOI] [PubMed] [Google Scholar]
- 115.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv Mater. 2006;18:1345–1360. [Google Scholar]
- 116.Bhatia SN, Balis UJ, Yarmush ML, Toner M. FASEB J. 1999;13:1883–1900. doi: 10.1096/fasebj.13.14.1883. [DOI] [PubMed] [Google Scholar]
- 117.Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K. Prog Polym Sci. 2008;33:448–477. doi: 10.1016/j.progpolymsci.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nie ZH, Xu SQ, Seo M, Lewis PC, Kumacheva E. J Am Chem Soc. 2005;127:8058–8063. doi: 10.1021/ja042494w. [DOI] [PubMed] [Google Scholar]
- 119.Kachouie NN, Du Y, Bae H, Khabiry M, Ahari AF, Zamanian B, Fukuda J, Khademhosseini A. Organogenesis. 2010;6:234–244. doi: 10.4161/org.6.4.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Khademhosseini A, Peppas NA. Adv Healthcare Mater. 2013;2:10–12. doi: 10.1002/adhm.201200444. [DOI] [PubMed] [Google Scholar]
- 121.Tsuda Y, Morimoto Y, Takeuchi S. Langmuir. 2010;26:2645–2649. doi: 10.1021/la902827y. [DOI] [PubMed] [Google Scholar]
- 122.Rossow T, Heyman JA, Ehrlicher AJ, Langhoff A, Weitz DA, Haag R, Seiffert S. J Am Chem Soc. 2012;134:4983–4989. doi: 10.1021/ja300460p. [DOI] [PubMed] [Google Scholar]
- 123.Rossow T, Bayer S, Albrecht R, Tzschucke CC, Seiffert S. Macromol Rapid Commun. 2013;34:1401–1407. doi: 10.1002/marc.201300353. [DOI] [PubMed] [Google Scholar]
- 124.Shi Y, Gao X, Chen L, Zhang M, Ma J, Zhang X, Qin J. Microfluid Nanofluid. 2013;15:467–474. [Google Scholar]
- 125.Steinhilber D, Rossow T, Wedepohl S, Paulus F, Seiffert S, Haag R. Angew Chem, Int Ed Engl. 2013;52:13538–13543. doi: 10.1002/anie.201308005. [DOI] [PubMed] [Google Scholar]
- 126.Headen DM, Aubry G, Lu H, Garcia AJ. Adv Mater. 2014;26:3003–3008. doi: 10.1002/adma.201304880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Allazetta S, Kolb L, Zerbib S, Bardy J, Lutolf MP. Small. 2015;11:5647–5656. doi: 10.1002/smll.201501001. [DOI] [PubMed] [Google Scholar]
- 128.Hackelbusch S, Rossow T, Steinhilber D, Weitz DA, Seiffert S. Adv Healthcare Mater. 2015;4:1841–1848. doi: 10.1002/adhm.201500359. [DOI] [PubMed] [Google Scholar]
- 129.Utech S, Prodanovic R, Mao AS, Ostafe R, Mooney DJ, Weitz DA. Adv Healthcare Mater. 2015;4:1628–1633. doi: 10.1002/adhm.201500021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang Y, Zhao L, Tian C, Ma C, Wang J. Anal Methods. 2015;7:10040–10051. [Google Scholar]
- 131.Wieduwild R, Krishnan S, Chwalek K, Boden A, Nowak M, Drechsel D, Werner C, Zhang Y. Angew Chem, Int Ed Engl. 2015;54:3962–3966. doi: 10.1002/anie.201411400. [DOI] [PubMed] [Google Scholar]
- 132.Chung BG, Lee KH, Khademhosseini A, Lee SH. Lab Chip. 2012;12:45–59. doi: 10.1039/c1lc20859d. [DOI] [PubMed] [Google Scholar]
- 133.Seiffert S. Chemphyschem. 2013;14:295–304. doi: 10.1002/cphc.201200749. [DOI] [PubMed] [Google Scholar]
- 134.Seiffert S. Angew Chem, Int Ed Engl. 2013;52:11462–11468. doi: 10.1002/anie.201303055. [DOI] [PubMed] [Google Scholar]
- 135.Leong W, Wang DA. Trends Biotechnol. 2015;33:653–666. doi: 10.1016/j.tibtech.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 136.Bianco P, Robey PG. Nature. 2001;414:118–121. doi: 10.1038/35102181. [DOI] [PubMed] [Google Scholar]
- 137.Daley GQ, Scadden DT. Cell. 2008;132:544–548. doi: 10.1016/j.cell.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 138.Passier R, van Laake LW, Mummery CL. Nature. 2008;453:322–329. doi: 10.1038/nature07040. [DOI] [PubMed] [Google Scholar]
- 139.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 140.Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. Proc Natl Acad Sci U S A. 2002;99:4391–4396. doi: 10.1073/pnas.032074999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jukes JM, Both SK, Leusink A, Sterk LM, van Blitterswijk CA, de Boer J. Proc Natl Acad Sci U S A. 2008;105:6840–6845. doi: 10.1073/pnas.0711662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wilson JL, McDevitt TC. Biotechnol Bioeng. 2013;110:667–682. doi: 10.1002/bit.24802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Agarwal P, Zhao S, Bielecki P, Rao W, Choi JK, Zhao Y, Yu J, Zhang W, He X. Lab Chip. 2013;13:4525–4533. doi: 10.1039/c3lc50678a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schukur L, Zorlutuna P, Cha JM, Bae H, Khademhosseini A. Adv Healthcare Mater. 2013;2:195–205. doi: 10.1002/adhm.201200194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang Y, Ho YP, Chiu YL, Chan HF, Chlebina B, Schuhmann T, You L, Leong KW. Biomaterials. 2013;34:4564–4572. doi: 10.1016/j.biomaterials.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chan HF, Zhang Y, Ho YP, Chiu YL, Jung Y, Leong KW. Sci Rep. 2013;3:3462. doi: 10.1038/srep03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen F, Zhan Y, Geng T, Lian H, Xu P, Lu C. Anal Chem. 2011;83:8816–8820. doi: 10.1021/ac2022794. [DOI] [PubMed] [Google Scholar]
- 148.Kitagawa F, Takei S, Imaizumi T, Tabata Y. Tissue Eng, Part C. 2013;19:438–448. doi: 10.1089/ten.TEC.2012.0166. [DOI] [PubMed] [Google Scholar]
- 149.Sakai S, Ito S, Inagaki H, Hirose K, Matsuyama T, Taya M, Kawakami K. Biomicrofluidics. 2011;5:13402. doi: 10.1063/1.3516657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tung YC, Hsiao AY, Allen SG, Torisawa YS, Ho M, Takayama S. Analyst. 2011;136:473–478. doi: 10.1039/c0an00609b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jeong GS, Song JH, Kang AR, Jun Y, Kim JH, Chang JY, Lee SH. Adv Healthcare Mater. 2013;2:119–125. doi: 10.1002/adhm.201200070. [DOI] [PubMed] [Google Scholar]
- 152.Yoon S, Kim JA, Lee SH, Kim M, Park TH. Lab Chip. 2013;13:1522–1528. doi: 10.1039/c3lc41322e. [DOI] [PubMed] [Google Scholar]
- 153.Place ES, Evans ND, Stevens MM. Nat Mater. 2009;8:457–470. doi: 10.1038/nmat2441. [DOI] [PubMed] [Google Scholar]
- 154.Vogel V, Sheetz M. Nat Rev Mol Cell Biol. 2006;7:265–275. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 155.Kraehenbuehl TP, Langer R, Ferreira LS. Nat Methods. 2011;8:731–736. doi: 10.1038/nmeth.1671. [DOI] [PubMed] [Google Scholar]
- 156.Khetani SR, Bhatia SN. Nat Biotechnol. 2008;26:120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- 157.Uygun BE, Soto-Gutierrez A, Yagi H, Izamis ML, Guzzardi MA, Shulman C, Milwid J, Kobayashi N, Tilles A, Berthiaume F, Hertl M, Nahmias Y, Yarmush ML, Uygun K. Nat Med. 2010;16:814–820. doi: 10.1038/nm.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mitragotri S, Lahann J. Nat Mater. 2009;8:15–23. doi: 10.1038/nmat2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cheung YK, Gillette BM, Zhong M, Ramcharan S, Sia SK. Lab Chip. 2007;7:574–579. doi: 10.1039/b700869d. [DOI] [PubMed] [Google Scholar]
- 160.Rivron NC, Rouwkema J, Truckenmuller R, Karperien M, De Boer J, Van Blitterswijk CA. Biomaterials. 2009;30:4851–4858. doi: 10.1016/j.biomaterials.2009.06.037. [DOI] [PubMed] [Google Scholar]
- 161.Cheng Y, Zheng F, Lu J, Shang L, Xie Z, Zhao Y, Chen Y, Gu Z. Adv Mater. 2014;26:5184–5190. doi: 10.1002/adma.201400798. [DOI] [PubMed] [Google Scholar]
- 162.Chan HF, Zhang Y, Leong KW. Small. 2016;12:2720–2730. doi: 10.1002/smll.201502932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lin RZ, Chang HY. Biotechnol J. 2008;3:1172–1184. doi: 10.1002/biot.200700228. [DOI] [PubMed] [Google Scholar]
- 164.Tomei AA, Manzoli V, Fraker CA, Giraldo J, Velluto D, Najjar M, Pileggi A, Molano RD, Ricordi C, Stabler CL, Hubbell JA. Proc Natl Acad Sci U S A. 2014:111. doi: 10.1073/pnas.1402216111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Struecker B, Raschzok N, Sauer IM. Nat Rev Gastroenterol Hepatol. 2014;11:166–176. doi: 10.1038/nrgastro.2013.204. [DOI] [PubMed] [Google Scholar]
- 166.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, O’Sullivan C, Collins L, Chen Y, Minami E, Gill EA, Ueno S, Yuan C, Gold J, Murry CE. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 167.Kaully T, Kaufman-Francis K, Lesman A, Levenberg S. Tissue Eng, Part B. 2009;15:159–169. doi: 10.1089/ten.teb.2008.0193. [DOI] [PubMed] [Google Scholar]
- 168.Discher DE, Mooney DJ, Zandstra PW. Science. 2009;324:1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Alijotas-Reig J, Fernandez-Figueras MT, Puig L. Clin Rev Allergy Immunol. 2013;45:97–108. doi: 10.1007/s12016-012-8348-5. [DOI] [PubMed] [Google Scholar]
- 170.Patterson J, Siew R, Herring SW, Lin AS, Guldberg R, Stayton PS. Biomaterials. 2010;31:6772–6781. doi: 10.1016/j.biomaterials.2010.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hennink WE, van Nostrum CF. Adv Drug Delivery Rev. 2012;64:223–236. [Google Scholar]
- 172.Holloway JL, Ma H, Rai R, Burdick JA. J Controlled Release. 2014;191:63–70. doi: 10.1016/j.jconrel.2014.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Purcell BP, Lobb D, Charati MB, Dorsey SM, Wade RJ, Zellars KN, Doviak H, Pettaway S, Logdon CB, Shuman JA, Freels PD, Gorman JH, 3rd, Gorman RC, Spinale FG, Burdick JA. Nat Mater. 2014;13:653–661. doi: 10.1038/nmat3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Laflamme MA, Murry CE. Nat Biotechnol. 2005;23:845–856. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 175.Murphy SV, Atala A. Nat Biotechnol. 2014;32:773–785. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- 176.Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T. Nat Mater. 2015;14:737–744. doi: 10.1038/nmat4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Smith CM, Christian JJ, Warren WL, Williams SK. Tissue Eng. 2007;13:373–383. doi: 10.1089/ten.2006.0101. [DOI] [PubMed] [Google Scholar]
- 178.Chang CC, Boland ED, Williams SK, Hoying JB. J Biomed Mater Res, Part B. 2011;98:160–170. doi: 10.1002/jbm.b.31831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chan HF, Ma S, Leong KW. Regener Biomater. 2016;3:87–98. doi: 10.1093/rb/rbw009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Molina DK, DiMaio VJ. Am J Forensic Med Pathol. 2012;33:368–372. doi: 10.1097/PAF.0b013e31823d29ad. [DOI] [PubMed] [Google Scholar]
- 181.Sohlenius-Sternbeck AK. Toxicol In Vitro. 2006;20:1582–1586. doi: 10.1016/j.tiv.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 182.Abate AR, Krummel AT, Lee D, Marquez M, Holtze C, Weitz DA. Lab Chip. 2008;8:2157–2160. doi: 10.1039/b813405g. [DOI] [PubMed] [Google Scholar]
- 183.Abate AR, Thiele J, Weinhart M, Weitz DA. Lab Chip. 2010;10:1774–1776. doi: 10.1039/c004124f. [DOI] [PubMed] [Google Scholar]
- 184.Nisisako T, Ando T, Hatsuzawa T. Lab Chip. 2012;12:3426–3435. doi: 10.1039/c2lc40245a. [DOI] [PubMed] [Google Scholar]
- 185.Romanowsky MB, Abate AR, Rotem A, Holtze C, Weitz DA. Lab Chip. 2012;12:802–807. doi: 10.1039/c2lc21033a. [DOI] [PubMed] [Google Scholar]
- 186.Zheng Y, Roberts MA. Nat Mater. 2016;15:597–599. doi: 10.1038/nmat4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jain RK, Au P, Tam J, Duda DG, Fukumura D. Nat Biotechnol. 2005;23:821–823. doi: 10.1038/nbt0705-821. [DOI] [PubMed] [Google Scholar]
- 188.Rouwkema J, Khademhosseini A. Trends Biotechnol. 2016;34:733–745. doi: 10.1016/j.tibtech.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 189.Golden AP, Tien J. Lab Chip. 2007;7:720–725. doi: 10.1039/b618409j. [DOI] [PubMed] [Google Scholar]
- 190.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA, Stroock AD. Proc Natl Acad Sci U S A. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Arenas-Herrera JE, Ko IK, Atala A, Yoo JJ. Biomed Mater. 2013;8:014106. doi: 10.1088/1748-6041/8/1/014106. [DOI] [PubMed] [Google Scholar]
- 192.Khademhosseini A, Langer R. Nat Protoc. 2016;11:1775–1781. doi: 10.1038/nprot.2016.123. [DOI] [PubMed] [Google Scholar]
- 193.Badylak SF, Taylor D, Uygun K. Annu Rev Biomed Eng. 2011;13:27–53. doi: 10.1146/annurev-bioeng-071910-124743. [DOI] [PMC free article] [PubMed] [Google Scholar]