

Abstract
Mutations of the mitochondrial citrate carrier (CIC) SLC25A1 cause combined d-2- and l-2-hydroxyglutaric aciduria (dl-2HGA; OMIM #615182), a neurometabolic disorder characterized by developmental delay, hypotonia, and seizures. Here, we describe the female child of consanguineous parents who presented neonatally with lactic acidosis, periventricular frontal lobe cysts, facial dysmorphism, recurrent apneic episodes, and deficient complex IV (cytochrome c oxidase) activity in skeletal muscle. Exome sequencing revealed a homozygous SLC25A1 missense mutation [NM_005984.4: c.593G>A; p.(Arg198His)] of a ubiquitously conserved arginine residue putatively situated within the substrate-binding site I of CIC. Retrospective review of the patient’s organic acids confirmed the d- and l-2-hydroxyglutaric aciduria typical of dl-2HGA to be present, although this was not appreciated on initial presentation. Cultured patient skin fibroblasts showed reduced survival in culture, diminished mitochondrial spare respiratory capacity, increased glycolytic flux, and normal mitochondrial bulk, inner membrane potential, and network morphology. Neither cell survival nor cellular respiratory parameters were improved by citrate supplementation, although oral citrate supplementation did coincide with amelioration of lactic acidosis and apneic attacks in the patient. This is the fifth clinical report of CIC deficiency to date. The clinical features in our patient suggest that this disorder, which can potentially be recognized either by molecular means or based on its characteristic organic aciduria, should be considered in the differential diagnosis of pyruvate dehydrogenase deficiency and respiratory chain disorders.
One-Sentence Summary A novel homozygous missense substitution in SLC25A1 was identified in a neonate presenting with lactic acidosis, intracerebral cysts, and an apparent mitochondrial complex IV defect in muscle.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2016_536) contains supplementary material, which is available to authorized users.
Keywords: Citrate transporter, Cytochrome c oxidase, Exome sequencing, Lactic acidosis, Mitochondrial disease, SLC25A1
Introduction
The mitochondrial citrate carrier (CIC) SLC25A1 is an inner membrane antiporter that mediates the exchange of a tricarboxylate dianion (e.g., citrate2−, isocitrate2−) for another tricarboxylate dianion, a dicarboxylate (e.g., malate2−), or phosphoenolpyruvate (Palmieri 2004). The major described function of the CIC is to shuttle mitochondrially synthesized citrate to the cytoplasm, where it (1) furnishes (via citrate lyase) acetyl-coA to support fatty acid and sterol synthesis and (2) exerts feedback control over glycolysis by allosterically inhibiting phosphofructokinase. Recently, recessive mutations of SLC25A1 in 20 persons with combined d,l-2-hydroxyglutaric aciduria (dl-2HGA; OMIM #615182), a disorder characterized clinically by severe developmental delay, hypotonia, and seizures, have been described (Edvardson et al. 2013; Nota et al. 2013). With subsequent reports, the clinical phenotype of dl-2HGA has since been expanded to include secondary microcephaly, hypoplasia or agenesis of the corpus callosum, optic nerve hypoplasia, dysmorphic features, lactic acidosis, and recurrent apneic crises (Chaouch et al. 2014; Mühlhausen et al. 2014; Prasun et al. 2015). Apart from a proposed defect of mitochondrial citrate efflux, little is known about the downstream pathophysiology of this condition, and the cellular bioenergetic implications of this basic transport defect remain poorly understood. Here, we report the clinical, biochemical, and molecular findings in a female neonate with a homozygous SLC25A1 mutation and a novel presentation with congenital lactic acidosis, leukoencephalopathy with cystic frontal lobe lesions, and dysmorphic features reminiscent of pyruvate dehydrogenase deficiency.
Materials and Methods
Patient DNA and Cellular Analyses
All procedures were in accord with the declaration of Helsinki. Informed consent was obtained from all study participants prior to enrollment. The research was approved by the Children’s Hospital Research Ethics Board. DNA extraction, sequencing, and exome analysis were performed as previously described (McDonell et al. 2013). Primary patient fibroblast cultures were established based on a 2 mm sterile skin biopsy according to standard clinical protocols. All other experiments conducted in control and patient fibroblasts were performed as previously described by our group (Antoun et al. 2015).
Results
Clinical Description
The patient, a female, was the first child to first cousins of mixed Caucasian background. Routine antenatal ultrasound imaging was normal. The pregnancy was complicated by gestational diabetes and preeclampsia, for which labor was induced; delivery was by emergency Caesarian section at 35 weeks, 6 days, due to fetal distress. Birth weight was 2.16 kg (third–tenth centile). The baby briefly required bag-mask ventilation, followed by a further 15 min of continuous positive pressure, following which breathing was satisfactory and the patient was maintained in room air. Dysmorphic features were noted, including midface hypoplasia, a thin, smooth philtrum, micrognathia, and numerous small raised capillary hemangiomata in a general distribution. At 20 h of age, the patient developed apneic spells and stridor, in conjunction with abnormal jerking movements; a lactic acidosis was noted (lactate = 8.6 mmol/L; pH 7.36; bicarbonate = 17 mmol/L). With aggressive fluid and respiratory support, over the following 24 h, her serum lactate decreased to 2.7 mmol/L, but failed to normalize entirely, and has remained persistently elevated since birth (range, 2.5–8.6 mmol/L; typical resting values, 5–6 mmol/L). Echocardiography was normal. Cranial imaging, initially by ultrasound and subsequently by MRI, revealed diffuse white matter deficiency with cerebral volume loss, bilateral periventricular frontal lobe cysts, and severe hypoplasia of the corpus callosum (Fig. 1). Funduscopy revealed bilateral optic nerve hypoplasia; visual evoked potentials were absent. Her early clinical course was characterized by hypotonia, poor feeding, temperature instability, and progressively frequent episodes of central apnea. Tracheostomy was performed at age 3 months. Repeat MRI at that time showed progressive volume loss, hypomyelination, and presence of a lactate doublet on spectroscopy (Fig. 1). At 3 years of age, the patient remained ventilator dependent, with little psychomotor development, a paucity of spontaneous movement, frequent apneic crises, and periods of generalized extensor posturing.
Fig. 1.
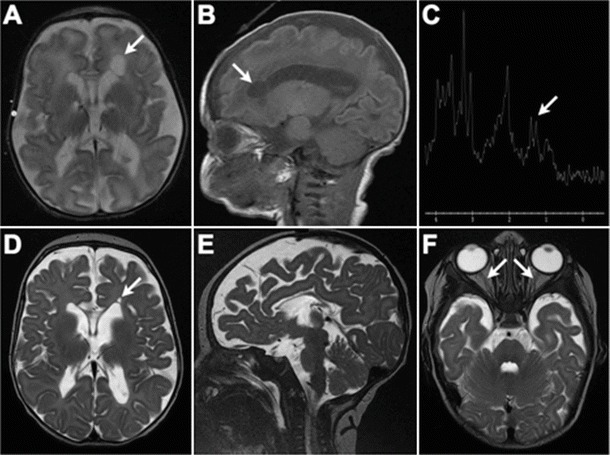
Cranial MRI appearance at 5 days (a, b) and 3 months of age (c–f). (a) and (b) Diffuse deficiency of cerebral white matter with enlarged fluid spaces and marked hypoplasia of the corpus callosum. Arrow indicates connatal (frontal lobe) cyst; a similar (smaller) right-sided cyst was present but is not well shown on this axial slice. (c) Lactate doublet is present on MR spectroscopy. (d) Evolution of changes in panels a and b. Myelination is markedly reduced for age. Basal ganglia, brainstem, and cerebellum have a relatively normal appearance. (f) Small optic nerves and optic chiasm (not shown)
Selected biochemical investigations in this patient included the following: plasma amino acids and acylcarnitine profile were normal. CSF lactate was 3.9 mmol/L (vs. 3.1 mmol/L in serum). CSF amino acids were within normal limits. Urine organic acids showed increased excretion of 2-ketoglutaric, fumaric, glutaric, 2-hydroxyglutaric, and (inconsistently) lactic and succinic acids. Muscle biopsy (age, 4 months) showed subtle nonspecific fiber size variation, normal Gomori trichrome staining, and normal NADH, SDH, and cytochrome oxidase histochemical stains. The semi-thin toluidine blue staining revealed subsarcolemmal clearing, with accumulation of non-membrane-bound glycogen; this was confirmed at the ultrastructural level. Note was also made of occasional small aggregates of mitochondria with nonspecific pleomorphism (Supplemental Fig. 1). Respiratory chain testing revealed an isolated, marked decrease in complex IV in skeletal muscle activity, but not in cultured skin fibroblasts (Table 1).
Table 1.
Selected activities in skeletal muscle and cultured fibroblasts
| Frozen skeletal muscle | ||
|---|---|---|
| Enzyme | Patient muscle activity (μmol/min/g wet weight) | Reference interval (μmol/min/g wet weight) |
| I+III (NADH-cytochrome C reductase) | 0.71 | 0.53–2.72 |
| II+III (succinate-cytochrome C reductase) | 1.24 | 0.55–3.46 |
| IV (cytochrome oxidase) | 0.09 (↓) | 0.8–6.03 |
| Citrate synthase | 4.81 | 3.48–8.03 |
| Cultured skin fibroblasts | ||
| Enzyme | Patient fibroblasts (nmol/min/mg protein) | Reference interval (nmol/min/mg protein) |
| Pyruvate dehydrogenase (native) | 0.67 ± 0.05 (n = 3) | 0.7–2.5 |
| Pyruvate dehydrogenase (dichloroacetate stimulated) | 0.89 ± 0.05 (n = 3) | 0.9–2.5 |
| II+III (succinate-cytochrome C reductase) | 5.85 ± 0.40 (n = 8) | 4–12 |
| IV (cytochrome c oxidase) | 4.11 ± 0.57 (n = 8) | 4–12 |
| Lactate: pyruvate ratio | 25.79 ± −4.38a(n = 4) | 10–25a |
anmol/h/mg protein
Molecular Diagnosis
In order to identify the causative genetic lesion in this patient, we performed whole-exome sequencing of the proband. Among the resulting list of rare (allele frequency <1%) homozygous coding variants in our proband was a novel missense substitution in SLC25A1 (NM_005984.3:c.593G>A; p.Arg198His; confirmed by Sanger sequencing) (Supplemental Fig. 2). This substitution is predicted to be damaging by multiple algorithms (Adzhubei et al. 2010; Kumar et al. 2009; Schwarz et al. 2014) and has not been observed in any of the 3513 exomes previously sequenced at our facility. It is not represented in the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/), although a different substitution of this same position, p.Arg198Cys, is represented at extremely low frequency (2/121148 alleles, 0.002%). With arginine 198 having previously been established to be essential for CIC’s transporter function (see “Discussion”), dl-2HGA was suspected as the most likely diagnosis. Enantiomeric separation of 2-hydroxyglutaric acids in the patient’s urine (VUmc Amsterdam; patient age 4 years, 2 months) showed hitherto unappreciated increases in the excretion of both d-2-hydroxyglutaric acid (248.6 mmol/mol creatinine, reference 2.8–17.0 mmol/mol creatinine) and l-2-hydroxyglutaric acid (50.0 mmol/mol creatinine, reference 1.3–18.9 mmol/mol creatinine), confirming the diagnosis. On retrospective review of the patient’s previous (semiquantitative) urine organic acids analyses, citrate was detected repeatedly, albeit in relatively small amounts.
The lactic acidosis, connatal white matter cysts, and reduced muscle complex IV activity in our patient were collectively indicative of a defect of mitochondrial energy metabolism. To produce a more detailed assessment of the patient’s mitochondrial dysfunction, we assessed several parameters (basal and maximal respiration, mitochondrial bulk, inner membrane potential, and mitochondrial network structure) in primary patient fibroblasts (Fig. 2). Cells were exceedingly fragile in culture on standard media, displaying exaggerated senescence when passaged below ~60 % confluence. Micro-oximetry of the fibroblasts showed a dramatic decrease in spare respiratory capacity (Fig. 2a, b); this was accompanied by an increase in glucose-supported extracellular acidification (Fig. 2c), a proxy measure of overall glycolytic flux. The oxygen consumption rate (OCR) to extracellular acidification rate (ECAR) ratio, a comparison of the cell’s reliance on oxidative phosphorylation versus glycolysis, was dramatically lower in patient cells than in controls (Fig. 2d). High-throughput confocal microscopy of fixed cells stained for the outer membrane transporter TOMM-20 (Fig. 3a, b) revealed a normal-appearing mitochondrial network. Lastly, flow cytometric assays for (1) average cellular mitochondrial bulk and (2) mitochondrial inner membrane potential showed no statistical difference in either of these parameters versus controls (Fig. 3c, d).
Fig. 2.
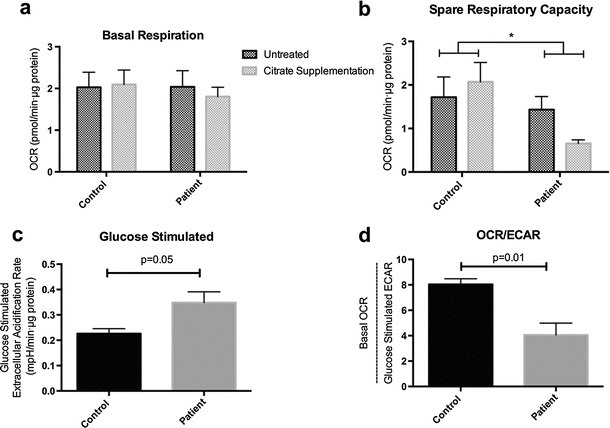
SLC25A1-deficient patient cells exhibit lessened spare respiratory capacity and increased glycolytic flux. Micro-oximetry was performed, as per Antoun et al. (2015), on cells grown in standard medium and on cells grown in medium supplemented with 4 mM citrate. Oxygen consumption rate (OCR) was corrected for non-mitochondrial oxygen consumption and normalized to protein content. (a) Basal OCR of patient fibroblasts does not differ significantly from that of control fibroblasts. (b) Spare respiratory capacity is reduced in the patient fibroblasts; this was not ameliorated by citrate supplementation. (c) Glucose-stimulated extracellular acidification rate (ECAR), reflecting glycolytic flux, is markedly increased in the patient cells (n = 3). (d) OCR/ECAR ratio (basal OCR divided by glucose-driven ECAR) is markedly reduced in the patient, i.e., the patient cells are relatively reliant on glycolysis (n = 3). Values are presented as mean ± SEM, analyzed by unpaired, two-tailed student’s t-test
Fig. 3.
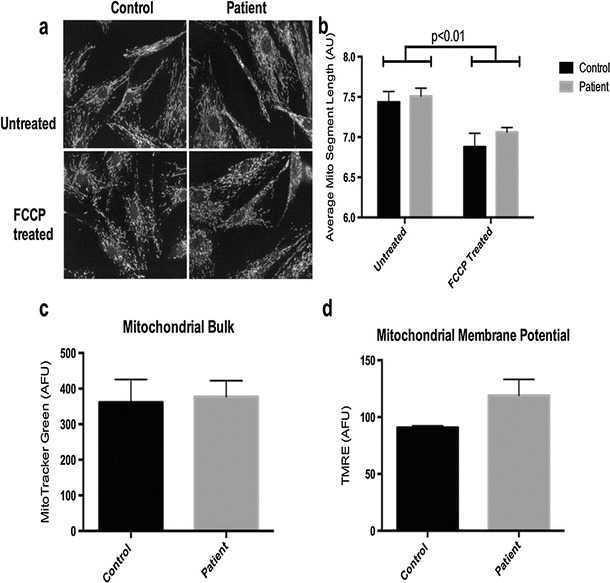
Normal patient mitochondrial networking, drug-induced fragmentation, total mitochondrial bulk, and inner membrane charge in skin-derived fibroblasts. (a, b) High-throughput confocal imaging of fixed control (left) and patient (right) fibroblasts ± acute 60 μM FCCP treatment; immunostained for the outer membrane transporter TOMM-20 (n = 4). (c) Mitochondrial content (MitoTracker Green) (n = 3) and (d) inner membrane potential (TMRE) (n = 3) measured by flow cytometry. Values are presented as mean ± SEM; analyzed by (b) two-way ANOVA or (c) nonparametric Mann-Whitney test for unequal variance and (d) unpaired, two-tailed student’s t-test
The patient was prescribed an oral citrate supplement (3 mg/kg/day) at 33 months of age, and this coincided with an improvement in the frequency of her apneic attacks, increased spontaneous respiratory drive (i.e., lessened reliance on her mandatory “backup” ventilator rate and an increase in patient-triggered breaths). Blood lactate concentration decreased significantly (8.9 mmol/L to 3.8 mmol/L) during the first week of therapy, although lactate remained persistently elevated thereafter (last measurement, 5.2 mmol/L), and urinary 2-hydroxyglutarate excretion has also remained markedly increased. The patient’s clinic performance status remains poor.
Discussion
This is the fifth report of dl-2HGA, our proband being the twentieth affected individual reported to date (Nota et al. 2013; Chaouch et al. 2014; Prasun et al. 2015; Edvardson et al. 2013). As far as we are aware, no other patient with dl-2HGA has been described to have a specific respiratory chain defect (complex IV deficiency) on muscle biopsy. In general terms, the findings in the patient suggest impaired oxidative phosphorylation, increased reliance on glycolysis, and preservation of mitochondrial bulk, inner membrane charge, and overall network structure. Contributing factors in the patient’s lactic acidosis could include (1) NADH accumulation due to respiratory chain dysfunction and/or (2) increased glycolytic flux, by allosteric disinhibition of phosphofructokinase (Newsholme et al. 1977).
The arginine residue (Arg198) altered by our proband’s substitution is likely to be particularly crucial for CIC’s carrier function. In the orthologous S. cerevisiae citrate transport protein CTP, the equivalent position (Arg189) is proposed to form direct physical contacts with citrate at one of the protein’s two putative citrate-binding sites (Ma et al. 2007). Substitution of Arg189 with a cysteine drastically compromises both the kinetics and the selectivity of CTP, with a >300-fold increase in K m, ~1,000-fold decrease in V max, and a greatly increased affinity for dicarboxylic acids (e.g., malate) rather than citrate (Ma et al. 2007; Aluvila et al. 2010). An in silico structural model based on the homologous mitochondrial carrier ANT1 (PDB: 10KC) (Pebay-Peyroula et al. 2003) situates the side chain of Arg198 within CIC’s positively charged site 1 pocket (Supplemental Fig. 3), and substitution of this residue with a significantly less basic histidine residue is likely to change the steric and electrostatic configuration of site 1.
Our patient’s clinical presentation was initially considered to be suggestive of pyruvate dehydrogenase deficiency. As proved to be the case here, PDH-like facial dysmorphism, periventricular frontal lobe cysts, and hyperlactatemia are nonspecific signs that can be seen in many disorders of cellular energy metabolism. Because dl-2HGA is rare and its characteristic organic aciduria may be overlooked, panel- or exome-based molecular testing may be the most practical route to a diagnosis in some cases. The apparent clinical improvement in apneic episodes on oral citrate supplementation has been described in one other reported individual, although in both instances, treatment was started well into the disease course (Mühlhausen et al. 2014). The effect, if any, of earlier initiation of therapy on the remainder of the dl-2HGA phenotype (developmental delay, microcephaly, structural brain malformations, seizures, etc.) remains to be assessed prospectively in suitable cases.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors gratefully acknowledge the contribution of the proband and her family, without whose participation this work could not be undertaken.
Compliance with Ethics Guidelines
Authors’ Contributions
All authors contributed to the conception and design of the study. JLM, JeM, OA, PC, and ML acquired the clinical, biochemical, and pathological data. SM produced primary experimental data. Data were analyzed and interpreted by AS, SM, JeM, OA, JS, JaM, DB, and ML. The manuscript was drafted by AS, SM, and ML. Critical revisions were performed by all authors.
Guarantor
Dr. Matthew A. Lines
Competing Interest Statement
Amanda Smith declares she has no conflict of interest.
Skye McBride declares she has no conflict of interest.
Julien Marcadier declares he has no conflict of interest.
Jean Michaud declares he has no conflict of interest.
Osama Y. Al-Dirbashi declares he has no conflict of interest.
Jeremy Schwartzentruber declares he has no conflict of interest.
Chandree Beaulieu declares she has no conflict of interest.
Sherri L. Katz declares she has no conflict of interest.
Jacek Majewski declares he has no conflict of interest.
Dennis E. Bulman declares he has no conflict of interest.
Michael T. Geraghty declares he has no conflict of interest.
Mary-Ellen Harper declares she has no conflict of interest.
Pranesh Chakraborty declares he has no conflict of interest.
Matthew A. Lines declares he has no conflict of interest.
Funding
This work was supported by Genome Canada, the Canadian Institutes of Health Research (CIHR), and the Ontario Genomics Institute (OGI-049), with additional funding from Genome Québec and Genome British Columbia. SM and ML are supported by MitoCanada Foundation
Ethics Approval and Informed Consent
All procedures followed were in accordance with the Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans (2010) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients participating in the study. The research protocol was approved by the Children’s Hospital of Eastern Ontario Research Ethics Board.
Footnotes
Competing interests: None declared
Contributor Information
Matthew A. Lines, Email: mlines@cheo.on.ca
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;4:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluvila S, Kotaria R, Sun J, et al. The yeast mitochondrial citrate transport protein: molecular determinants of its substrate specificity. J Biol Chem. 2010;285:27314–27326. doi: 10.1074/jbc.M110.137364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoun G, McBride S, Vanstone JR et al (2015) Detailed biochemical and bioenergetic characterization of FBXL4-related encephalomyopathic mitochondrial DNA depletion. JIMD Rep (epub ahead of print) [DOI] [PMC free article] [PubMed]
- Chaouch A, Porcelli V, Cox D, et al. Mutations in the mitochondrial citrate carrier SLC25A1 are associated with impaired neuromuscular transmission. J Neuromusclular Disord. 2014;1:75–90. doi: 10.3233/JND-140021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Porcelli V, Jalas C, et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J Med Genet. 2013;50:240–245. doi: 10.1136/jmedgenet-2012-101485. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;7:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Ma C, Remani S, Sun J, et al. Identification of the substrate binding sites within the yeast mitochondrial citrate transport protein. J Biol Chem. 2007;282:17210–17220. doi: 10.1074/jbc.M611268200. [DOI] [PubMed] [Google Scholar]
- McDonell LM, Mirzaa GM, Alcantara D, et al. Mutations in STAMBP, encoding a deubiquitinating enzyme, cause microcephaly-capillary malformation syndrome. Nat Genet. 2013;45:556–562. doi: 10.1038/ng.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlhausen C, Salomons GS, Lukacs Z, et al. Combined D2-/L2-hydroxyglutaric aciduria (SLC25A1 deficiency): clinical course and effects of citrate treatment. J Inherit Metab Dis. 2014;37:775–781. doi: 10.1007/s10545-014-9702-y. [DOI] [PubMed] [Google Scholar]
- Newsholme EA, Sugden PH, Williams T. Effect of citrate on the activities of 6-phosphofructokinase from nervous and muscle tissues from different animals and its relationship to the regulation of glycolysis. Biochem J. 1977;166:123–129. doi: 10.1042/bj1660123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nota B, Struys EA, Pop A. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am J Hum Genet. 2013;92:627–631. doi: 10.1016/j.ajhg.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, et al. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Prasun P, Young S, Salomons G, et al. Expanding the clinical spectrum of mitochondrial citrate carrier (SLC25A1) deficiency: facial dysmorphism in siblings with epileptic encephalopathy and combined D, L-2-hydroxyglutaric aciduria. JIMD Rep. 2015;19:111–115. doi: 10.1007/8904_2014_378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.