

Abstract
Little is known about the heritability of chemotherapy activity or the identity of genes that may enable the individualization of cancer chemotherapy. Although numerous genes are likely to influence chemotherapy response, current candidate gene-based pharmacogenetics approaches require a priori knowledge and the selection of a small number of candidate genes for hypothesis testing. In this study, an ex vivo familial genetics strategy using lymphoblastoid cells derived from Centre d'Etude du Polymorphisme Humain reference pedigrees was used to discover genetic determinants of chemotherapy cytotoxicity. Cytotoxicity to the mechanistically distinct chemotherapy agents 5-fluorouracil and docetaxel were shown to be heritable traits, with heritability values ranging from 0.26 to 0.65 for 5-fluorouracil and 0.21 to 0.70 for docetaxel, varying with dose. Genome-wide linkage analysis was also used to map a quantitative trait locus influencing the cellular effects of 5-fluorouracil to chromosome 9q13-q22 [logarithm of odds (LOD) = 3.44], and two quantitative trait loci influencing the cellular effects of docetaxel to chromosomes 5q11–21 (LOD = 2.21) and 9q13-q22 (LOD = 2.73). Finally, 5-fluorouracil and docetaxel were shown to cause apoptotic cell death involving caspase-3 cleavage in Centre d'Etude du Polymorphisme Humain lymphoblastoid cells. This study identifies genomic regions likely to harbor genes important for chemotherapy cytotoxicity using genome-wide linkage analysis in human pedigrees and provides a widely applicable strategy for pharmacogenomic discovery without the requirement for a priori candidate gene selection.
Significant interpatient variability in response to chemotherapy is consistently observed across patient populations (1, 2). Initial candidate gene evaluations of severe toxicity to chemotherapeutic agents have revealed specific examples of pharmacogenetically relevant single-nucleotide polymorphisms (3, 4), and previous studies of twins detailed a substantial influence of inheritance on general measures of hepatic drug metabolism (5, 6). However, little is known about the heritability of chemotherapy activity and current candidate gene strategies require the a priori selection of individual candidates from among the potentially numerous genes that may regulate the action of a drug (2). Unbiased genome-wide approaches are needed, but traditional methods for assessing genetic contribution (e.g., family studies of patients or volunteers) are obstructed for chemotherapy outcomes due to the rarity of simultaneous occurrence of a specific tumor type among family members and the unsuitability of these agents for use in normal volunteer subjects. Whole-genome association studies in clinical populations have a theoretical basis as a strategy for the discovery of markers influencing drug response (7, 8); however, such studies are currently limited by sample size, the availability of relevant populations, and the expense of genotyping (9, 10).
Therefore, an ex vivo familial genetics strategy involving lymphoblastoid cells derived from Centre d'Etude du Polymorphisme Humain (CEPH) reference pedigrees was used to quantify the impact of genetic variation on chemotherapy cytotoxicity and to provide a model for the discovery of genes influencing the activity of this important class of medications. The CEPH resource is an easily accessible collection of multigeneration families, on which extensive microsatellite and single-nucleotide polymorphism genotype data are freely available (11–13). This experimental platform provides a unique opportunity for the application of familial genetic strategies to cancer pharmacogenomic discovery, allowing for the rigorous testing of samples from multiple families under identical conditions. Thus, heritability estimates and gene discovery efforts become feasible and are not complicated by differences between familial environments or related confounding variables. In addition, candidate genes or polymorphisms identified using the CEPH resource can be used to provide specific hypotheses for testing in human pharmacogenetic studies. This system was used to demonstrate that cytotoxicity to the mechanistically distinct chemotherapy agents 5-fluorouracil or docetaxel are heritable traits in CEPH pedigrees and to identify multiple quantitative trait loci (QTL) associated with the activity of these drugs. In addition, 5-fluorouracil and docetaxel were shown to cause apoptotic cell death involving caspase-3 cleavage in CEPH lymphoblastoid cells. These results provide a framework for the discovery of genes that may lead to the individualization of cancer chemotherapy.
Materials and Methods
Cell Lines. Epstein–Barr virus-immortalized lymphoblastoid cells derived from CEPH reference pedigrees were obtained from Coriell Cell Repositories (http://locus.umdnj.edu/ccr). Lymphoblastoid cell lines from the following CEPH pedigrees were used in this study: 66, 1362, 1420, 1421, 1424, 1345, 1346, 1347, 1356, 1408, 1416, 1418, 1447, 1451, 37, 23, 2, 12, 21, 28, 35, 45, 1328, 13281, 13291, 13292, 13293, 13294, 1330, 1331, 1332, 1333, 1334, 1340, 1341, 1344, 1346, 1349, 1350, and 1353. Cells were cultured in RPMI medium 1640 containing 2 mmol/liter l-glutamine, 100 units/ml penicillin, 100 μg/liter streptomycin, 0.25 μg/liter amphotericin B, and 15% heat-inactivated FBS (Invitrogen).
Drugs. 5-Fluorouracil (Sigma) was prepared at working concentrations in complete media before addition to cells. Docetaxel (Aventis Pharma, Bridgewater, NJ) was prepared as a 10 mM stock in 25% polysorbate 80/10% ethanol, and diluted to working concentrations in complete media before addition to cells. The final concentration of polysorbate 80/ethanol was <0.001% in all experimental wells.
Cytotoxicity Measurements. Cells used for experiments were derived from fresh cultures sent from Coriell Cell Repositories. Cells were seeded in 96-well plates at a density of 1 × 105/ml and incubated for 18 h at 37°C, 5% CO2. Drugs were then added in the following concentrations: docetaxel (vehicle only), 0.1, 0.5, 1.0, 5, 10, 50, and 100 nM; and 5-fluorouracil (vehicle only), 0.76, 1.92, 3.84, 5.77, 7.68, 19.2, 38.4, and 76.8 μM. Cells were
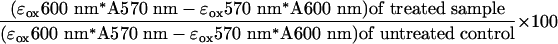 |
[1] |
incubated for 96 h. Alamar blue reagent (BioSource International, Camarillo, CA) was added, and cells were incubated an additional 18 h. Absorbance at 570 and 600 nM was measured, and viability relative to untreated control was calculated according to the manufacturer's instructions by using the formula shown in Eq. 1.
Where εox600 nm = 117,216 (molar extinction coefficient of oxidized Alamar blue reagent at 600 nm), εox570 nm = 80,586 (molar extinction coefficient of oxidized Alamar blue reagent at 570 nm), A570 nm = absorbance of sample at 570 nm, and A600 = absorbance of sample at 600 nm. Three replicates were performed for each data point. IC50 values were calculated by four-parameter logistic regression by using prism 3.0 (GraphPad, San Diego).
Rate of Cell Growth. Rate of cell growth was measured by calculating the percent reduction of Alamar blue after drug treatment as described above. Percent reduction was calculated according to the manufacturer's instructions by using the formula shown in Eq. 2.
Where εred570 nm = 155,677 (molar extinction coefficient of reduced Alamar blue at 570 nm), εred600 nm = 14,652 (molar extinction coefficient of reduced Alamar blue at 600 nm), A′600 nm = absorbance of negative control wells which contained media plus Alamar blue but no cells at 600 nm, and A′570 = absorbance of negative control wells that contained media plus Alamar blue but no cells at 570 nm.
Random Coefficient Regression (RCR) Model. The RCR model was used to find a random slope derived from the dose–response curve for each subject. It is assumed that specific coefficients for each subject are a random sample from a population of possible coefficients. RCR was used to assess the relationship between drug dosage and cellular viability, by incorporating all available data. The model applied was the following: yijk = β0 + si + (β1 + di)Xij + eij, where yijk represents the response of the ith subject at the jth dose at kth replication, β0 and β1 are respectively the fixed intercept and the slope, si is the random deviation of the ith subject's intercept from β0, di is the random deviation of the ith subject's slope from β1, and eij is the random error. The β1 coefficient represents the population mean rate of dose–response changes in cellular viability, and di measures the average slope deviations from the population mean slope β1. In the linkage analysis for finding putative QTLs for drug cytotoxicity, the di random coefficients were used for the average dose–response change in the viability response modeling. The procedure MIXED of sas Ver. 8.2 (SAS Institute, Cary, NJ) for Linux OS was used to extract the si and di random coefficients.
Heritability/Linkage Analysis. Microsatellite marker genotypes were selected from the CEPH database, Ver. 9.0 (www.cephb.fr/cephdb). After careful quality assurance analysis, 983 nonredundant markers with a high degree of heterogeneity, ranging from 75 markers on chromosome 1 to 20 markers on chromosome 22, were used. The viability responses were verified to be approximately normally distributed (data not shown). Genetic marker distances from the Weber map (http://research.marshfieldclinic.org/genetics) were used in all analyses. Narrow sense heritability
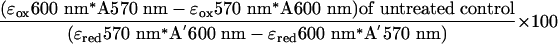 |
[2] |
estimates (h2) and linkage analysis were performed by using the variance components approach as implemented in segpath (14). This model partitions the correlation structure among relatives into the additive effect of a QTL, a pseudopolygenic component, and a residual nonfamilial variance component. A parameter to allow for additional resemblance between siblings was included, which can account for the net effect of differences in exposures between generations (e.g., cohort effects, secular trends, etc.). The null hypothesis was set by restricting the amount of the narrow sense heritability (h2) due to the putative locus of the QTL to 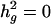 (but estimating all other parameters,
(but estimating all other parameters, 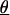 ). Likelihood ratio tests of null against the alternative hypothesis that a putative QTL effect is present have 50:50 mixture of a χ2 with 1° of freedom and a point mass at 0, were calculated as χ2 = 2*logeL(H1) –2* logeL(H0) and the LOD = χ2/(2*loge (10)), where L(H1) is the likelihood of the data at the joint maximum likelihood estimates of the parameters
). Likelihood ratio tests of null against the alternative hypothesis that a putative QTL effect is present have 50:50 mixture of a χ2 with 1° of freedom and a point mass at 0, were calculated as χ2 = 2*logeL(H1) –2* logeL(H0) and the LOD = χ2/(2*loge (10)), where L(H1) is the likelihood of the data at the joint maximum likelihood estimates of the parameters 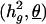 and L(H0) is the likelihood at the restricted maximum likelihood subspace where
and L(H0) is the likelihood at the restricted maximum likelihood subspace where 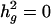 .
.
Immunoblot Analysis. Cells were seeded in T25 flasks at a density of 1 × 105/ml and incubated for 18 h at 37°C, 5% CO2. Cells were then treated for 48 h with 10 nM docetaxel, 19.2 μM 5-fluorouracil, or vehicle only. Cells were collected by centrifugation, washed once in PBS, and resuspended in SDS sample buffer: 10 mM Tris, pH 8.0/1.5 mM MgCl2/15 mM NaCl/0.5% (octylphenoxy)polyethoxyethanol (Igepal) CA-630, supplemented with 1 mM phenylmethylsufonyl fluoride (Sigma), and complete protease inhibitor mixture (Sigma). Protein concentrations were quantified by using the Bradford reagent (Bio-Rad), and 75 μg of protein per lane was loaded onto 4–12% precast gels (Bio-Rad) for SDS/PAGE electrophoresis. After electrophoresis, proteins were transferred to nitrocellulose membranes (Hybond) by electroblotting, blocked for 2 h in 5% nonfat milk, and incubated overnight at 4°C with the following dilutions of primary antibody in 5% nonfat milk: α-cleaved caspase-3 (Cell Signaling Technology, Beverly, MA, 1:2,000) or α-beta actin (Novus Biologicals, Littleton, CO, 1:10,000). Membranes were incubated in secondary antibody, and proteins were visualized by chemiluminescence detection (Amersham Pharmacia).
Results
Phenotypic Variation and Heritability of 5-Fluorouracil Cytotoxicity. To assess the utility of the CEPH pedigrees as a discovery tool for genes influencing chemotherapy activity, the extent of variation in sensitivity of CEPH lymphoblastoid cells to death caused by incubation with 5-fluorouracil, a uracil analog widely used to treat colorectal and breast tumors, was analyzed. Four hundred twenty-seven lymphoblastoid cell lines derived from members of 38 CEPH reference families were exposed to 5-fluorouracil (0.768–76.8 μM), and viability relative to untreated controls was determined by using the Alamar blue vital dye indicator assay. Significant variation in 5-fluorouracil cytotoxicity was observed for each dose examined (Table 1), indicating that genetic background is likely to be an important factor in determining 5-fluorouracil sensitivity. The overall population mean IC50 (drug concentration inhibiting cell growth by 50% relative to untreated control) was 19.9 μM. This value is similar to 5-fluorouracil IC50 values observed across the National Cancer Institute NCI60 panel of human tumor cell lines, which exhibit a mean IC50 of 17.6 μM, with a range of 1–501 μM (http://dtp.nci.nih.gov).
Table 1. Variability in drug cytotoxicity.
| Drug | Dose, M | Mean viability* | Range† |
|---|---|---|---|
| 5-Fluorouracil | 7.68 × 10-7 | 0.917 | 0.57-1.0 |
| 1.92 × 10-6 | 0.789 | 0.47-1.0 | |
| 3.84 × 10-6 | 0.674 | 0.35-1.0 | |
| 5.77 × 10-6 | 0.603 | 0.27-1.0 | |
| 7.68 × 10-6 | 0.579 | 0.22-1.0 | |
| 1.92 × 10-5 | 0.514 | 0.07-1.0 | |
| 3.84 × 10-5 | 0.481 | 0.04-1.0 | |
| 7.68 × 10-5 | 0.382 | 0.02-1.0 | |
| Docetaxel | 1 × 10-10 | 0.978 | 0.63-1.0 |
| 5 × 10-10 | 0.959 | 0.43-1.0 | |
| 1 × 10-9 | 0.939 | 0.26-1.0 | |
| 5 × 10-9 | 0.459 | 0.06-1.0 | |
| 1 × 10-8 | 0.366 | 0.02-1.0 | |
| 5 × 10-8 | 0.253 | 0.01-1.0 | |
| 1 × 10-7 | 0.255 | 0.001-0.91 |
Overall population mean (n = 427) for cell viability at each dose.
Minimum and maximum cell viability values in the population at each dose.
A heritability estimation was then performed to quantify the impact of inherited factors on cytotoxicity to 5-fluorouracil. High heritability values for 5-fluorouracil cytotoxicity were observed at each dose, ranging from 0.26 to 0.65 (Fig. 1A). The heritability of cytotoxicity in this system is similar to or greater than the heritabilities of most common human phenotypes studied to date, including plasma triglyceride levels (0.19–0.55), body mass index (0.32–0.59), and measures of lung function (0.06–0.52) (15). Rate of cell growth was not a heritable trait (heritability <0.05), indicating that variation in cellular response to 5-fluorouracil is not simply due to differential growth characteristics among cell lines. These results provide an objective demonstration that genetic inheritance is a key determinant of 5-fluorouracil cytotoxicity.
Fig. 1.

Heritability and linkage analysis of 5-fluorouracil cytotoxicity. (A) Dose–response curve for 5-fluorouracil. Data points represent the overall population mean (n = 427) for viability relative to untreated controls at each dose. Numbers represent the proportion of phenotypic variance that can be explained by genetic factors (heritability). Vertical bars represent the standard deviation for cell viability across the population. (B) Linkage results for chromosome 9 using percent viability at individual doses of 5-fluorouracil as separate phenotypes. The highest overall LOD score (3.44) was observed at marker D9S253, using viability at 19.2 μM5-fluorouracil as the phenotype. The positions of selected genetic markers are shown, and the approximate 1 LOD interval is underlined along the x axis. Linkage results in this region for other 5-fluorouracil doses are also shown.
Genome-Wide Linkage Analysis of 5-Fluorouracil Cytotoxicity. To identify loci influencing 5-fluorouracil cytotoxicity, an initial genome-wide linkage analysis was performed by using a sensitivity parameter derived from all collected data points of an individual dose–response curve as the phenotype. This parameter, generated by using a RCR model (ref. 16; see Materials and Methods), estimates the subject-specific rate of decrease in cellular viability as drug dose increases (rate of dose–response). Nine hundred eighty-three highly informative microsatellite markers were used in this genome-wide linkage scan. Using a variance-components linkage analysis based on identity-by-descent allele sharing (14), chromosomes 9 and 16 showed preliminary evidence for linkage (Table 2).
Table 2. Regions showing preliminary evidence for linkage using the RCR-derived rate of dose response as the phenotype.
| Drug | Maximum LOD* | Chromosome† |
|---|---|---|
| 5-Fluorouracil | 1.55 | 9 (99.4 cM) |
| 1.95 | 16 (73.98 cM) | |
| Docetaxel | 1.24 | 5 (109.63 cM) |
| 1.66 | 6 (100.91 cM) | |
| 1.5 | 9 (100.74 cM) |
LOD score >1.0 required for preliminary evidence of linkage.
Genetic map location of maximum LOD score within QTL peak.
To further these preliminary linkage results and analyze drug dose-related changes in gene effects, linkage analysis was repeated by using cell viability at each dose as separate phenotypes. One chromosomal region with supportive evidence of linkage was identified on chromosome 9q13-q22. The maximum logarithm of odds (LOD) score observed in this region was 3.44, at marker D9S253 (Fig. 1B and Table 3). The influence of genetic factors in this region on 5-fluorouracil cytotoxicity varies in relation to drug dose, and the highest LOD score was observed at 19.2 μM 5-fluorouracil. Thus, by using relative viability at each dose as separate phenotypes, dose-specific gene effects can be revealed. Interestingly, an overlapping region of chromosome 9 has previously been implicated in 5-f luorouracil cytotoxicity by comparative genomic hybridization of 5-fluorouracil-sensitive and -resistant human colorectal cancer cell lines (17), providing support for the influence of this region on 5-fluorouracil response.
Table 3. Regions showing supportive evidence for linkage using individual doses of drug as separate phenotypes.
| Drug | Maximum LOD* | Chromosome† | Approximate 1 LOD interval |
|---|---|---|---|
| 5-Fluorouracil | 3.44 | 9 (94.85 cM) | D9S175-D9S1162 |
| Docetaxel | 2.21 | 5 (97.21 cM) | D5S502-D5S1965 |
| 2.73 | 9 (94.85 cM) | D9S175-D9S1162 |
LOD score >2.0 required for supportive evidence of linkage.
Genetic map location of maximum LOD score within QTL peak.
Phenotypic Variation and Heritability of Docetaxel Cytotoxicity. The utility of this approach as a discovery tool for genetic modifiers of cytotoxicity was then explored by using the mechanistically distinct chemotherapy agent docetaxel, a microtubule-stabilizing drug commonly used to treat breast and lung tumors. Cells from the same 38 CEPH pedigrees were exposed to docetaxel (0.1–100 nM), and viability relative to untreated controls was determined. Large intersample variation in docetaxel sensitivity was observed at each dose (Table 1), suggesting that genetic background is also likely to be an important factor in determining docetaxel sensitivity. The overall population mean IC50 for docetaxel treatment was 4.67 nM. This value is again similar to IC50 values observed across the NCI60 panel of human tumor cell lines, which exhibit a mean docetaxel IC50 of 23.4 nM, with a range of 0.31–100 nM (http://dtp.nci.nih.gov). A heritability estimation was then performed to quantify the impact of inherited factors on docetaxel sensitivity. High heritability values for docetaxel cytotoxicity were observed at each dose, ranging from 0.21 to 0.70 (Fig. 2A).
Fig. 2.

Heritability and linkage analysis of docetaxel cytotoxicity. (A) Dose–response curve for docetaxel. Data points represent the overall population mean (n = 427) for viability relative to untreated controls at each dose. Numbers represent the heritability of cytotoxicity at each dose. Vertical bars represent the standard deviation for cell viability across the population. (B) Linkage results for chromosome 5 using percent viability at individual doses of docetaxel as separate phenotypes. The highest overall LOD score (2.21) was observed at marker D5S459, using viability at 1 nM docetaxel as the phenotype. (C) Linkage results for chromosome 9 using percent viability at individual doses of docetaxel as phenotypes. The highest overall LOD score (2.73) was observed at marker D9S1690, using viability at 100 nM docetaxel as the phenotype. The positions of selected genetic markers are shown and the ≈1 LOD intervals are underlined along the x axes. Linkage results in these regions for other docetaxel doses are also shown.
Genome-Wide Linkage Analysis of Docetaxel Cytotoxicity. To search for loci influencing docetaxel cytotoxicity, a genome-wide linkage analysis was then performed by using the RCR-derived rate of dose–response for docetaxel as the phenotype, by using the same 983 microsatellite markers and methodology that were used in the analysis of 5-fluorouracil cytotoxicity. Chromosomes 5, 6, and 9 were identified as showing preliminary evidence for linkage (Table 2). To further these results, linkage analysis was repeated by using cell viability at each dose as separate phenotypes. Two regions showing supportive dose-dependent evidence of linkage were identified: chromosome 5q11-q21, maximum LOD = 2.21 at marker D5S459 (Fig. 2B and Table 3), and chromosome 9q13-q22, maximum LOD = 2.73 at marker D9S1690 (Fig. 2C and Table 3). These data demonstrate that the CEPH resource can be used to map loci influencing the activity of mechanistically distinct chemotherapy agents and provide a general framework for pharmacogenomic discovery.
Chemotherapy Treatment Induces Apoptotic Cell Death Involving Caspase-3. The mechanism of cell death caused by 5-fluorouracil or docetaxel treatment in CEPH lymphoblastoid cells was then explored. Because both docetaxel and 5-fluorouracil are known to induce apoptosis involving caspase-3 activation in tumor cells (18, 19), caspase-3 cleavage was assessed in two randomly selected CEPH lymphoblastoid cell lines after drug treatment. Cells were treated for 48 h with docetaxel (10 nM), 5-fluorouracil (19.2 μM), or vehicle only, and the presence of activated caspase-3 was analyzed by Western blot. Both docetaxel and 5-fluorouracil treatment induced apoptosis in CEPH cells, as evidenced by the appearance of apoptotic caspase-3 cleavage fragments (Fig. 3). These data indicate that the mechanism of cell death caused by docetaxel or 5-fluorouracil treatment in CEPH lymphoblastoid cells is similar to that observed in tumor cells.
Fig. 3.

Treatment with 5-fluorouracil or docetaxel induces apoptosis. Two CEPH lymphoblastoid cell lines (GM10857 and GM12005) were treated for 48 h with 10 nM docetaxel, 19.2 μM 5-fluorouracil, or vehicle only. Cells were harvested and analyzed for the presence of the p17 and p19 proteolytic fragments of caspase-3 by immunoblot analysis. Both docetaxel and 5-fluorouracil induced apoptotic cell death involving caspase-3 cleavage.
Discussion
There is a pressing need for the development of genome-wide strategies to identify genes influencing chemotherapy response. CEPH lymphoblastoid cells have been used to identify genomic loci and candidate genes influencing sodium–lithium countertransport (13), natural variation in gene expression (11), transcriptional response to ionizing radiation (12), and allelic variation in gene expression (20), demonstrating the applicability of this model system to a broad range of biological phenotypes. Therefore, a familial genetics strategy using the CEPH resource was applied to demonstrate the heritability of chemotherapy toxicity and to identify genomic loci influencing drug activity.
This study provides an objective demonstration that chemotherapy cytotoxicity is a heritable trait in humans. The heritability of chemotherapy cytotoxicity in this system was quite high for each dose of both 5-fluorouracil and docetaxel. The heritability of drug response is lowest at low drug concentrations, when these agents have a relatively small effect on cell viability. However, heritability values are high at increased drug concentrations, when these agents have a major impact on cell survival. This is likely due to the fact that large interindividual differences in drug sensitivity become more apparent at higher drug doses. This high degree of heritability suggests that genetic inheritance is a key component regulating chemotherapy sensitivity and provides evidence that inherited genetic variation may be an important determinant of cytotoxicity to a broad range of chemotherapeutic agents.
Also presented here is the discovery of QTLs influencing chemotherapy cytotoxicity using genome-wide linkage analysis in human pedigrees. Under the guidelines proposed by Lander and Kruglyak (21) for interpreting LOD scores (LOD score thresholds of 2.2 as suggestive and 3.6 as significant), the linkage results in this study are classified as “suggestive.” However, such categorization is only approximate. For example, Rao and Gu (22) have recently proposed significance at a more relaxed LOD score threshold of 2.21 or greater. It is of interest to note that the same region of chromosome 9 was identified as the strongest QTL for cytotoxicity to both 5-fluorouracil and docetaxel in our study. Further analysis will be required to determine whether there is a common pharmacodynamic variable present in this region influencing sensitivity to both drugs, or whether there are distinct but closely linked genes contributing to the cytotoxicity phenotype for each drug. Because this region of chromosome 9 has previously been identified as being important for 5-fluorouracil sensitivity in colorectal tumor cell lines (17), it is possible that this region of the genome harbors a gene that is of general importance for chemotherapy cytotoxicity. The availability of high-resolution single-nucleotide polymorphism genotype data for a subset of CEPH individuals used in this analysis [produced by the International Haplotype Map Project (www.hapmap.org)] will provide a useful resource for the fine-mapping of this region.
Multiple lines of evidence suggest that cell death induced by 5-fluorouracil or docetaxel treatment is similar in CEPH lymphoblastoid cells and tumor cells. First, the drug sensitivities observed in our study mimicked those observed in the NCI60 panel of tumor cell lines. The population mean IC50 values for both 5-fluorouracil and docetaxel were similar to the mean IC50 values observed in the NCI60 panel and well within the observed ranges. Indeed, the population mean IC50 for 5-fluorouracil in our study, 19.9 μM, is nearly identical to the NCI60 mean IC50 of 17.6 μM. Second, a similar region of chromosome 9 correlating with 5-fluorouracil sensitivity was identified both in our study and by comparative genomic hybridization of 5-fluorouracil-resistant and -sensitive colorectal tumor cell lines in a previous study (17). Finally, docetaxel and 5-fluorouracil induce apoptotic cell death involving caspase-3 cleavage in both CEPH lymphoblastoid cells and tumor cells (18, 19). These data suggest that loci influencing chemotherapy cytotoxicity identified in CEPH lymphoblastoid cells are likely to be relevant in human cancers.
These results demonstrate that CEPH lymphoblastoid cells are a tractable model system for the discovery of genetic loci influencing chemotherapy cytotoxicity in a way not possible in human patients. Alternatives to this approach are case-control studies performed using a priori-selected candidate genes thought to effect response to the drug under investigation. Although such approaches have revealed clinically relevant associations, disadvantages include the low likelihood of selecting the correct gene from the myriad known and unknown genes that may influence drug response. The data presented here will improve the chance of success of such association studies by identifying regions likely to contain genetic polymorphisms with substantial influence on drug effects. In addition, the high degree of heritability of chemotherapy cytotoxicity found with two mechanistically distinct drugs (antimetabolite and tubulin inhibitor) suggests that this system may be broadly applicable and can be expanded to other therapeutic areas where pharmacogenomic discovery is difficult, including ion channel inhibition, receptor signaling, and cellular transport. These data provide an objective basis for optimism that the development of genetic tools for individualized chemotherapy is an achievable goal.
Acknowledgments
We thank Drs. Mark Johnston, Rick Wilson, Wayne Yokoyama, and members of the McLeod Laboratory for critical review of the manuscript. This work was supported by the National Institutes of Health Pharmacogenetics Research Network (GM63340).
Abbreviations: QTL, quantitative trait locus; LOD, logarithm of odds; CEPH, Centre d'Etude du Polymorphisme Humain; RCR, random coefficient regression.
References
- 1.Evans, W. E. & McLeod, H. L. (2003) N. Engl. J. Med. 348, 538–549. [DOI] [PubMed] [Google Scholar]
- 2.Watters, J. W. & McLeod, H. L. (2003) Biochim. Biophys. Acta 1603, 99–111. [DOI] [PubMed] [Google Scholar]
- 3.McLeod, H. L., Relling, M. V., Liu, Q., Pui, C. H. & Evans, W. E. (1995) Blood 85, 1897–1902. [PubMed] [Google Scholar]
- 4.Johnston, P. G., Lenz, H. J., Leichman, C. G., Danenberg, K. D., Allegra, C. J., Danenberg, P. V. & Leichman, L. (1995) Cancer Res. 55, 1407–1412. [PubMed] [Google Scholar]
- 5.Vesell, E. S. & Page, J. G. (1968) Science 161, 72–73. [DOI] [PubMed] [Google Scholar]
- 6.Vesell, E. S. & Page, J. G. (1968) Science 159, 1479–1480. [DOI] [PubMed] [Google Scholar]
- 7.Risch, N. & Merikangas, K. (1996) Science 273, 1516–1517. [DOI] [PubMed] [Google Scholar]
- 8.Lander, E. S. (1996) Science 274, 536–539. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein, D. B., Tate, S. K. & Sisodiya, S. M. (2003) Nat. Rev. Genet. 4, 937–947. [DOI] [PubMed] [Google Scholar]
- 10.Kwok, P. Y. & Gu, Z. (1999) Mol. Med. Today 5, 538–543. [DOI] [PubMed] [Google Scholar]
- 11.Cheung, V. G., Conlin, L. K., Weber, T. M., Arcaro, M., Jen, K. Y., Morley, M. & Spielman, R. S. (2003) Nat. Genet. 33, 422–425. [DOI] [PubMed] [Google Scholar]
- 12.Jen, K. Y. & Cheung, V. G. (2003) Genome Res. 13, 2092–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schork, N. J., Gardner, J. P., Zhang, L., Fallin, D., Thiel, B., Jakubowski, H. & Aviv, A. (2002) Hypertension 40, 619–628. [DOI] [PubMed] [Google Scholar]
- 14.Province, M. A., Rice, T. K., Borecki, I. B., Gu, C., Kraja, A. & Rao, D. C. (2003) Genet. Epidemiol. 24, 128–138. [DOI] [PubMed] [Google Scholar]
- 15.Ober, C., Abney, M. & McPeek, M. S. (2001) Am. J. Hum. Genet. 69, 1068–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corbett, J., Kraja, A., Borecki, I. B. & Province, M. A. (2003) BMC Genet. 4 Suppl. 1, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rooney, P. H., Stevenson, D. A., Marsh, S., Johnston, P. G., Haites, N. E., Cassidy, J. & McLeod, H. L. (1998) Cancer Res. 58, 5042–5045. [PubMed] [Google Scholar]
- 18.Kolfschoten, G. M., Hulscher, T. M., Duyndam, M. C., Pinedo, H. M. & Boven, E. (2002) Biochem. Pharmacol. 63, 733–743. [DOI] [PubMed] [Google Scholar]
- 19.Wu, X. X., Kakehi, Y., Mizutani, Y., Lu, J., Terachi, T. & Ogawa, O. (2001) Int. J. Oncol. 19, 19–24. [DOI] [PubMed] [Google Scholar]
- 20.Yan, H., Yuan, W., Velculescu, V. E., Vogelstein, B. & Kinzler, K. W. (2002) Science 297, 1143. [DOI] [PubMed] [Google Scholar]
- 21.Lander, E. & Kruglyak, L. (1995) Nat. Genet. 11, 241–247. [DOI] [PubMed] [Google Scholar]
- 22.Rao, D. C. & Gu, C. (2001) Adv. Genet. 42, 487–498. [DOI] [PubMed] [Google Scholar]