

Abstract
Integrin activation leads to an increased affinity for the extracellular matrix and regulates many cellular processes such as cell adhesion and migration. To capture the process of integrin inside-out activation in a purified system, we describe here methods to isolate platelet αIIbβ3 integrins in the inactive state and to incorporate them into phospholipid nanodiscs each bearing a single lipid-embedded αIIbβ3 integrin. We delineate a simple enzyme-linked immunosorbent assay that can be used in conjunction with binding of an activation specific monoclonal antibody, PAC1, to monitor the affinity of the integrin before and after the addition of activators such as the talin head domain. The system has been used to show that binding of the talin head domain to both the integrin and the phospholipid bilayer is necessary and sufficient to activate and promote molecular extension of unclustered integrins in the absence of force. This method can be used to test various integrin-binding proteins, membrane phospholipid compositions and different types of integrins. It can also serve as the nidus for synthetic assembly of adhesion plaque like complexes from purified proteins.
Keywords: Integrin, Talin, Nanodiscs, Activation, Platelets, Membrane scaffold protein (MSP)
1. Introduction
Increased ligand binding affinity of integrins, or activation, is central to many cellular processes such as cell migration, extracellular matrix assembly, hemostasis and immune response (1). Integrins are noncovalent heterodimers of transmembrane α- and β-subunits, each with a single transmembrane and cytoplasmic domain. Their activation involves a series of transitions from bent low-affinity states to extended high-affinity states with an open head piece (2). This extensive conformational change is initiated through interactions at the integrin cytoplasmic domain to regulate their affinity for extracellular ligands, a process referred to as “inside-out” signaling (3–5).
Integrin activation has been analyzed by forward and reverse genetics; however, the many unknowns present in cells and the possible complex effects of over-expression or deletion make it mandatory to complement these cell-based studies with analyses in purified systems. Previous in vitro methods analyzed the interaction of cytoplasmic proteins with the α and β integrin tails and measured activation induced by non-physiological agents such as divalent cations or monoclonal antibodies. Although these methodologies reveal valuable information about integrin activation, they fail to capture the entire process of physiological inside-out signaling. Moreover, it is now clear that physiological activation is regulated through interactions of the α- and β-transmembrane and cytoplasmic domains and some of these interactions require embedding in a phospholipid bilayer.
Here we describe methods to purify the inactive αIIbβ3 integrin from platelets, to synthesize phospholipid bilayer nanodiscs, to generate integrin nanodiscs each bearing a single lipid-embedded integrin, and how to use these to study physiologically-relevant activation of unclustered integrins.
2. Materials
2.1. KYGRGDS column preparation
Coupling buffer: 0.1 M NaHCO3, 0.5 M NaCl, pH 8.3.
Cyanogen bromide (CNBr) Sepharose: 15 mL of resin (GE Healthcare).
KYGRGDS peptide: 50 mg.
2.2. Integrin αIIbβ3 preparation
Calpeptin: 2.76 mM solubilized in DMSO.
Leupeptin: 11.7 mM solubilized in dH2O.
PMSF: 200 mM solubilized in ethanol.
Protease inhibitor E64 (Sigma Aldrich) [10 mM].
Wash buffer: 20 mM Tris, 150 mM NaCl, pH 7.4.
Lysis buffer: 200 mL of 20 mM Tris, 150 mM NaCl, pH 7.4, 1% Triton X-100, 0.5 mM CaCl2, 5 mM PMSF, 10 μM Leupeptin, 10 μM protease inhibitor E64, 2.76 μM Calpeptin.
Platelet-rich plasma, 100 units (~5 liter total) (see Note 1).
Centrifuge pots, ideally 500 mL disposable.
Integrin buffer: 20 mM Tris, 150 mM NaCl, pH 7.4, 1 mM CaCl2, 1 mM MgCl2, 0.1% Triton X-100.
Con A elution buffer: 20 mM Tris, 150 mM NaCl, pH 7.4, 1 mM CaCl2, 1 mM MgCl2, 0.1% Triton X-100, 5 mM Leupeptin, 200 mM methyl α-D mannopyranoside.
Concanavalin A Sepharose (Con A) column containing 25 mL of resin (GE Healthcare).
Heparin Sepharose: 10 mL of resin (GE Healthcare).
A 15 mL KYGRGDS column.
Aquacide II (Calbiochem).
Dialysis bag of MWCO 25,000 (Spectra/Por).
Sephacryl S-300 column 16/60 (GE Healthcare).
High pH buffer: 20 mM Tris, 0.5 M NaCl, pH 8.5.
Low pH buffer: 20 mM Acetate, 0.5 M NaCl, pH 4.5.
Competitive small molecule inhibitor eptifibatide.
2.3. Integrin activity ELISA (enzyme-linked immunosorbent assay)
This ELISA is used to assay the: i) purified integrin activity (Section 3.2), ii) integrin nanodiscs activity (Section 3.6), and iii) the integrin nanodisc activation (Section 3.7). The only difference between the assays is the presence of 0.1% Triton X-100 in the buffer with purified integrins (Section 3.2), which is not present in the buffer for the nanodisc assays (Sections 3.6 and 3.7) (see Note 2).
Anti-β3 mouse monoclonal AP3 antibody (ATCC).
Anti-β3 polyclonal antibody Rb8053, rabbit sera (Ginsberg lab and many others).
Purified Mouse IgM anti-human αIIbβ3 integrin PAC1 antibody (for active integrin) (Becton Dickinson).
Anti-LIBS mouse monoclonal antibody, anti-LIBS2 or anti-LIBS6 (integrin activating antibodies) (Millipore).
Competitive small molecule inhibitor eptifibatide.
HRP-anti-mouse IgM.
HRP-anti-rabbit IgG.
Plate reader for chemi-luminescence.
Microfluor2 plates.
Purified integrins as prepared in Section 3.1.
HBS-T buffer: 20 mM HEPES, 150 mM NaCl, pH 7.4, 0.5 mM CaCl2 and 0.5mM MgCl2, 0.1% Triton X-100.
Blocking buffer: HBS-T with 10% BSA.
ECL Western Blotting Substrate (Pierce).
2.4. Purification of recombinant MSP1
Luria Bertani medium (LB).
100 mM Isopropyl-beta-d-thiogalactopyranoside (IPTG), dioxane free solubilized in ddH2O.
MSP1 lysis buffer: 40 mM Tris, 0.3 M NaCl, pH 8.0, 1% Triton X-100, 1 mM PMSF.
Nickel column containing 5 mL of resin charged with Ni2+ according to manufacturer’s instructions.
His-MSP1 cloned into your vector of choice, we used pET-28a (Novagen).
E. coli BL21 (DE3) strain.
MSP1 wash buffer 1: 40 mM Tris, 0.3 M NaCl, pH 8.0, 1% Triton X-100.
MSP1 wash buffer 2: 40 mM Tris, 0.3 M NaCl, pH 8.0, 50 mM cholate.
MSP1 wash buffer 3: 40 mM Tris, 0.3 M NaCl, pH 8.0.
MSP1 wash buffer 4: 40 mM Tris, 0.3 M NaCl, 50 mM imidazole, pH 8.0.
MSP1 elution buffer: 40 mM Tris, 0.3 M NaCl, 0.3 M imidazole, pH 8.0.
MSP1 buffer: 10 mM Tris, 0.1 M NaCl, pH 7.4, 1 mM EDTA.
Sonicator or French pressure cell press (French press).
2.5. Purification of recombinant Talin head domain (THD)
Luria Bertani medium (LB).
100 mM Isopropyl-beta-d-thiogalactopyranoside (IPTG), dioxane free solubilized in ddH2O.
Nickel column containing 5 mL of resin charged with Ni2+ according to manufacturer’s instructions.
Sonicator or French pressure cell press (French press).
Recombinant human talin head. We used residues 1–433 cloned in pET-30a (Novagen) with a C-terminus hexahistidine tag (see Note 3).
E. coli BL21 (DE3) pLysS.
Nickel buffer A: 20 mM Tris, 0.5 M NaCl, 20 mM imidazole, pH 8.0.
Nickel buffer B: 20 mM Tris, 0.5 M NaCl, 0.5 M imidazole, pH 8.0.
Tris-buffered saline (TBS): 20 mM Tris, 150 mM NaCl, pH 7.4.
2.6. Integrin nanodiscs
Purified integrin αIIbβ3 in integrin buffer (at least 10 μM). Use a fresh tube from the −80°C.
Purified membrane scaffold protein (MSP) MSP1D1 as prepared in Section 3.3.
Synthetic lipids: 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DMPG) from Avanti Polar Lipids. They are solubilized in 2.5 mL Chloroform (DMPC) or Chloroform/methanol mixture (2:1 volume ratio) (DMPG) at a concentration of 10 mg/mL. All lipids should be freshly made.
SM-2 Biobeads from Bio-rad.
Tris-buffered saline (TBS): 20 mM Tris, 150 mM NaCl, pH 7.4.
Lipid resuspension buffer: 10 mM Tris, 100mM NaCl, pH 7.4, 100 mM cholate.
Superdex 200 (16/300) size exclusion column (GE Healthcare).
TBS + 0.5 mM CaCl2.
2.7. Integrin nanodisc activation ELISA
Same material as described in Section 2.3. with the exception of the HBS-T buffer which is now replaced with HBS buffer.
HBS buffer: 20 mM HEPES, 150 mM NaCl, pH 7.4, 0.5 mM CaCl2 and 0.5 mM MgCl2.
Integrin nanodiscs as prepared in Section 3.5.
Blocking buffer: HBS with 10% BSA.
2.8. Integrin nanodiscs activation by THD
Same material as described in Section 2.7.
Recombinant Talin Head domain (THD) as prepared in Section 3.4.
3. Methods
Here we describe how to purify the inactive αIIbβ3 integrin from platelets, to synthesize phospholipid bilayer nanodiscs, to generate integrin nanodiscs each bearing a single lipid-embedded integrin, and how to use them to study inside-out regulation of unclustered integrins leading to molecular extension in the absence of force or other membrane proteins. The overall procedure is outlined in Fig. 1.
Fig. 1.

Diagram of the major steps for isolation of the integrin αIIbβ3 from platelets, followed by an integrin activity ELISA, purification of the MSP1 protein, purification of the talin head domain (THD), preparation of the integrin nanodiscs, testing for the integrin nanodiscs activity, and measurement of the integrin activation within a nanodisc by THD.
3.1. Purification of integrin αIIbβ3 (GP IIb–IIIa) from platelets
Many methods have been described for purification of the platelet fibrinogen receptor (6) integrin αIIbβ3 or Glycoprotein IIb-III3. However, for the experiment described here, it is critical to isolate the inactive integrin fraction. The procedure involves lectin affinity chromatography on concanavalin-A Sepharose followed by heparin Sepharose. The inactive integrin is further purified by separating it from the active integrin by the use of a KYGRGDS affinity purification column, which binds the active integrin fraction (7). The flow through containing the inactive integrin is further purified by gel filtration chromatography on a Sephacryl S-300 column.
3.1.1. Preparation of a KYGRGDS column
Dissolve 50 mg of KYGRGDS peptide in 1 ml of ddH2O and dilute in 15 mL coupling buffer. Determine the peptide concentration by A280 (we use 1.637 as extinction coefficient).
Prepare 15 mL CNBr sepharose as per the manufacturer’s instructions and combine the sucked dry slurry with the peptide solution. Incubate at 4°C overnight with rocking.
Determine the A280 of the supernatant in order to determine if the coupling was efficient (see Note 4).
Pack the KYGRGDS resin in a column.
If the column is to be used later, store in 20% ethanol.
3.1.2. Platelet washing and lysis
Collect the platelet enriched plasma in 500 mL centrifuge pots (see Note 5) and centrifuge once at 300 × g for 10 min at room temperature (RT) to remove red blood cells (see Note 6).
Transfer the supernatants to new centrifuge pots taking extra care not to disturb the loose pellet of red blood cells and centrifuge at 1,800 × g for 30 min at RT to pellet the platelets (see Note 6).
Discard the supernatants and gently resuspend each pellet in 200 mL wash buffer and centrifuge at 1,800 × g for 60 min at RT. Repeat to remove the majority of plasma proteins (see Note 6).
Solubilize all the platelet pellets in 200 mL lysis buffer and leave rocking at 4°C overnight.
Centrifuge the lysate at 30,000 × g for 60 min at 4°C.
Collect the supernatant and discard the Triton X-100 insoluble pellet (see Note 7).
3.1.3. Con A affinity chromatography
The concanavalin A binds specifically to mannosyl and glucosyl residues of polysaccharides and glycoproteins. Here it is used to concentrate the platelet glycoproteins such as thrombospondin, the αIIbβ3 integrin, and fibrinogen.
Equilibrate the Con A Sepharose column with integrin buffer at 4°C.
Load the platelet lysate over a 25 mL Con A Sepharose column using a flow rate of 1 mL/min and recirculate overnight at 4°C (see Note 8).
Wash at 4°C the Con A Sepharose column with integrin buffer until the A280 goes down to 0.02.
Elute the bound material with 200 mL of Con A elution buffer and collect 4 mL fractions at 4°C.
Run the eluted fractions on SDS-PAGE for analysis (see Note 9). Pool the fractions containing the integrins and keep them at 4°C(see Note 10).
3.1.4. Heparin Sepharose chromatography
Heparin is a highly sulphated glycosaminoglycan which serves as an anticoagulant by selectively binding antithrombin III. This column here is primarily used to remove the thrombospondin which is a major contaminant of the Con A eluate.
Prepare 10 mL of Heparin Sepharose resin (~20 mL slurry) in a 50 mL plastic tube. Fill the tube with ddH2O and centrifuge at 3,500 × g for 30 min at 4°C.
Discard the supernatant and fill the tube with integrin buffer to equilibrate the resin. Resuspend the resin and centrifuge at 3,500 × g for 30 min at 4°C. Repeat this step to wash the resin properly.
Add the Heparin Sepharose to the pooled fractions eluted from the Con A column and leave rocking overnight at 4°C.
Spin the Heparin beads at 3,500 × g for 30 min at 4°C and collect the supernatant. Repeat this step if necessary until the supernatant is clear.
3.1.5. KYGRGDS affinity chromatography
Active integrins recognize the RGD sequence, therefore the active integrin will bind to the immobilized KYGRGDS peptide on the column and get separated from the inactive integrins in the flow through.
Wash the KYGRGDS column with 3 changes of high and low pH buffer and then equilibrate with two column volumes of integrin buffer.
Apply the Heparin Sepharose supernatant to the KYGRGDS using a flow rate of 1 mL/min and recirculate overnight at 4°C.
Collect the flow through containing the inactive integrins. Generally, >90% of the integrins are inactive and found in the flow through.
To recover the active integrin bound to the KYGRGDS column, wash the column with integrin buffer and eluted the active integrin with 20 μM eptifibatide compound in integrin buffer. This active integrin fraction can be used for other types of experiments.
3.1.6. Concentration of the inactive integrins
Load the KYGRGDS flow through, inactive integrin, into a dialysis bag of MWCO 25,000.
Cover a flat container with Aquacide II, place the dialysis bag into the bed of aquacade II and cover with more Aquacide II (see Note 11). Place at 4°C.
Constantly monitor the remaining volume until it is <10 mL. Approximately every hour, peel off the Aquacide II from the dialysis bag and cover again. According to the manufacture’s instructions, 1 g of aquacide will absorb 5 mL of water in 1 hour.
3.1.7. Sephacryl S-300 gel filtration chromatography
Fibrinogen is the major contaminant left from the preparation (MW under non-reducing conditions = 340,000 kDa) and can be separated from the αIIbβ3 integrin using gel filtration.
Pre-equilibrate the Sephacryl S-300 16/60 column with integrin buffer.
Load the concentrated inactive integrin onto the Sephacryl S-300 at RT using a flow rate of 1 mL/min. Collect 4 mL fractions.
Run the fractions on SDS-PAGE for analysis (see Note 9).
Keep the integrin fractions, taking extra care to discard the fractions containing fibrinogen.
Determine the integrin concentration using a BCA assay.
Freeze the samples at −80 in small aliquots.
3.2. Integrin activity ELISA
Here we describe an ELISA method to measure integrin activation in vitro. Our ELISA assay relies on the selective binding of integrin ligands and ligand-mimetic antibodies to activated integrins as shown in Fig. 2. The basis of the integrin αIIbβ3 activation assay is the mouse monoclonal antibody PAC1, as it is a ligand-mimetic antibody that binds to the ligand-binding site in the integrin. The PAC1 antibody binds only the active form of the integrin, so here it is used to measure the fraction of active integrins in our preparation as shown in Fig. 2A.
Fig. 2.

Schematic of assays for measuring activation of detergent solubilized αIIbβ3.
(A). The integrins are immobilized using the AP3 antibody and the PAC1 antibody is used to measure the integrins in their active conformation.
(B). To measure the background signal, eptifibatide is used to block the PAC1 antibody binding site.
(C). To measure the maximum PAC1 binding to the integrins, the anti-LIBS antibody is used to activate the integrins. Anti-LIBS2 or -LIBS6 are IgG and PAC1 is an IgM, therefore the secondary antibody which is an anti-IgM will only detect PAC1 binding.
(D). Measurement of the fraction of active integrins in the detergent solubilized preparation.
To accurately measure the background of the assay, we use eptifibatide to block PAC1 binding to the integrins as shown in Fig. 2B. Eptifibatide, also known as integrilin, is a cyclic heptapeptide that behaves as an RGD-mimetic and therefore competes for binding with PAC1.
To measure the maximum integrin activation, we measure PAC1 binding in the presence of an activating antibody (anti-LIBS) that binds the extracellular domain of β3 (8) as shown in Fig. 2C. As a supplementary control, one can use the rabbit anti-αIIbβ3 (Rb8053) to measure the total integrin loading.
Coat each well of the plate with 100 μL of 5 μg/mL of AP3 antibody. Cover the plate and incubate at 4°C overnight. Make sure that the solution covers the entire well and that there are no air bubbles.
Remove the coating solutions and block with 150 μL of blocking buffer at 37°C for 1 hour.
Remove the blocking buffer and wash 3 times with 150 μL HBS-T buffer.
Add 75 μL of purified integrin solution (6 μg/mL) according to the layout in Table 1. Incubate at RT for 3 hours.
Remove the binding mixture and wash 3 times with 150 μL HBS-T buffer.
-
PAC1 detection: Add 75 μL of the antibody PAC1 (1:4,000), with the appropriate controls as described in Table 1: i) 20 μM eptifibatide as negative control, and ii) anit-LIBS as positive control (1:100).
Rb8053 detection: Add 75 μL of the detection antibody Rb8053 to measure the amount of integrin capturing (1:1,000).
Incubate at RT for 1.5 hours.
Wash 3 times with 150 μL HBS-T buffer.
Add 75 μL of secondary antibody: i) HRP-anti-mouse IgM for PAC1 (1:1,000), or ii) HRP-anti-rabbit IgG for Rb8053 (1:1,000). Incubate at RT for 1.5 hours.
Wash 3 times with 150 μL HBS-T buffer.
Add 75 μL of ECL to each well. Incubate 5 to 10 min at RT.
Read the luminescence using a plate reader.
Analyse the data as shown in Fig 2. The activation indices are calculated as 100 × (L−L0)/(Lmax − L0), where L = luminescence in the presence of 20 μM eptifibatide, and Lmax = luminescence in presence of anti-LIBS antibody.
Table 1.
Plate layout for integrin activity assay (see Note 12).

|
In this experiment, 3% of the integrins are active so the major portion of our integrin preparation (97%) is in the inactive conformation.
3.3. Membrane scaffold protein (MSP) preparation
Nanoparticulate phospholipid bilayer disks, termed nanodiscs, can be assembled using a class of amphipathic helical proteins termed membrane scaffold proteins (MSP). Here we used MSP1 that forms nanodiscs of approximately 10 nm in diameter and 5.5 nm thickness (9).
3.3.1. MSP1 Expression
Inoculate 100 mL of LB containing the appropriate antibiotic with E. coli BL21 (DE3) culture containing His-MSP1 fusion protein expression construct. Grow overnight at 37°C in a rotary shaker incubator with agitation at 225 rpm.
Use 1 mL of the overnight culture to inoculate 1 L of LB with the appropriate antibiotic and grow the culture to an approximate OD600 of 0.6.
Induce the protein expression by adding 1 mM IPTG to the culture and grow for 3 hours.
Harvest the bacteria by centrifugation at 3,600 × g for 20 min and store the pellet at −20°C until use.
3.3.2. Purification of MSP1
Resuspend the cell pellet in 50 mL of ice cold MSP1 lysis buffer.
Lyse the cells using sonication or a French press while keeping the sample on ice.
Clear the lysate by centrifugation at 30,000 × g for 30 min at 4°C.
Load the supernatant onto the nickel column.
Wash the column with 4 bed volumes of each of the following: (1) MSP1 wash buffer 1, (2) MSP1 wash buffer 2, (3) MSP1 wash buffer 3, (4) MSP1 wash buffer 4.
Elute the MSP1 with the MSP1 elution buffer and collect 4 mL fractions.
Check for purity by SDS-PAGE (MW = 24.66 kDa).
Pool the fractions containing MSP1 and dialyze against MSP1 buffer at 4°C.
Determine the protein concentration using a BCA assay or using the absorbance at 280 nM (extinction coefficient of 0.869).
Freeze the protein in small aliquots and store at −80°C.
3.4. Talin head domain (THD) preparation
Here we describe how to purify the recombinant talin head (THD) as a hexahistidine tagged protein for easy purification.
3.4.1. Talin head domain expression
Inoculate 100 mL of LB containing the appropriate antibiotic with E. coli BL21 (DE3) pLysS culture containing THD-His fusion protein expression construct. Grow overnight at 37°C in a rotary shaker incubator with agitation at 225 rpm.
Use 1 mL of the overnight culture to inoculate 1 L of LB with appropriate antibiotic and grow the culture to an approximate OD600 of 0.5.
Transfer the culture to a 20 °C shaker for 1 hour.
Induce the protein expression by adding 0.5 mM IPTG to the culture and grow for 10 hours or overnight at 20 °C.
Harvest the bacteria by centrifugation at 3,600 × g for 20 min and store the pellet at −20°C until use.
3.4.2. Purification of Talin head domain
Resuspend the cell pellet in 50 mL of ice cold Nickel buffer A.
Lyse the cells using sonication or a French press while keeping the sample on ice.
Clear the lysate by centrifugation at 30,000 × g for 30 min at 4°C.
Load the supernatant onto the nickel column.
Wash the column with 5 bed volumes of Nickel buffer A.
Elute the THD with a gradient of Nickel buffer B and collect 3 mL fractions.
Check for purity by SDS-PAGE (MW = 53 kDa).
Pool the fractions containing THD and dialyze twice for 1 hour against 1 L TBS containing 2 mM EDTA at 4°C (see Note 13).
Dialyze twice for 1 hour against 1 L TBS at 4°C.
Determine the protein concentration using a BCA assay.
Freeze the protein in small aliquots and store at −80°C.
3.5. Integrin nanodiscs preparation
Many membrane proteins have been successfully embedded into nanodiscs (10–12), including integrins (13). The self-assembly process begins with a mixture of the purified integrin αIIbβ3 and MSP1 in the presence of a detergent, the phospholipids are then added and the detergent removed (see Note 14).
Mix 339 μL of DMPC (3.39 mg) with 344 μL of DMPG (3.44 mg) (1:1 ratio) into a glass tube and mix thoroughly.
Dry the lipids into a glass tube with a steady flow of nitrogen air and leave in the fume hood overnight.
Solubilize the MSP1 in ddH2O such that the final concentration is 200 μM.
Solubilize the dried lipids in 200 μL of lipid resuspension buffer, giving a lipid concentration of 50 mM. Vortex until the lipids are fully solubilized. This may take several hours.
Mix 72 μL of lipid solution with 200 μL of 200 μM MSP1 in dH2O and 10 μM purified inactive integrin. The final ratio of lipids:MSP:protein is 90:1:0.05 in a total volume of 472 μL.
Add some CaCl2 to the mixture to reach a 0.5 mM final concentration as the lipid and MSP1 solutions do not have calcium in them (see Note 15).
Prewash 1 mL of SM-2 Biobeads with methanol (3 times), water (7 times) and TBS (twice) by centrifugation at 3,500 × g for 5 min at RT.
Add 1 mL of SM-2 Biobeads to the nanodiscs tube to remove the detergent (see Note 16). Incubate overnight with gentle agitation at room temperature.
Remove the supernatant containing the integrin nanodiscs from the bio-beads.
Equilibrate a Superdex 200 (16/300) size exclusion column with TBS + 0.5 mM CaCl2.
Load 0.25 mL of the integrin nanodiscs solution onto the Superdex 200 size exclusion column. Use a flow rate of 0.5 mL/min and collect fractions of 0.3 mL. Two runs are necessary to purify ~0.5 mL of integrin nanodiscs mixture.
Run an SDS-PAGE to identify the fractions containing both integrins and nanodiscs.
Pool the fractions containing the integrin nanodiscs as shown in Fig 3. The integrin nanodiscs and empty nanodiscs should be separated.
Fig. 3.

Profile of the Superdex 200 (16/300) gel filtration chromatography. The fractions which contain the integrin nanodiscs and empty nanodiscs are shown. The column was run in TBS + 0.5 mM CaCl2 at a flow rate of 0.5 mL/min and fractions of 0.3 mL were collected.
3.6. Integrin nanodisc activity
This section describes how to test the activity of the integrins within the nanodiscs using a method almost identical to that described in Section 3.2. It is critical to know which portion of the integrin nanodiscs is active: the less, the better. Measuring integrin activation with talin for example with a preparation that is largely active could be difficult.
Follow the method outlined for the integrin activity ELISA, see Section 3.2, but throughout use HBS buffer instead of HBS-T buffer.
Instead of adding 75 μL of purified integrin, add 75 μL of purified integrin nanodiscs.
Analyse the data as shown in Fig 4.
Fig. 4.
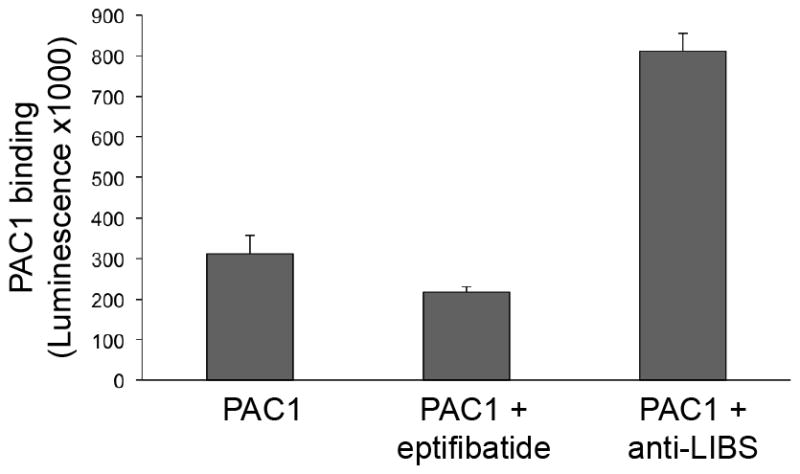
Measurement of the fraction of active integrin within the integrin nanodiscs preparation.
In our experiment, 16% of the integrin nanodiscs were active as determined by PAC1 binding. Therefore, the major portion of the integrin nanodiscs (84%) can, in theory, be activated by adding other components to the experimental setup as described in Section 3.7.
3.7. Integrin nanodisc activation
This method provides a powerful tool in screening for players in integrin signaling and discovering new regulators of talin mediated integrin activation. Obviously this method does not only apply to talin, other regulators, such as kindlins, can be added to the system (14,15).
Again, it is the same experiment as for the integrin nanodiscs activity ELISA, see Section 3.6, except that the plate layout is slightly more complex because here we will add THD to activate the integrins.
-
PAC1 detection: Add 75 μL of the antibody PAC1 (1:4,000), with the appropriate controls as described in Table 2: i) 2.5 μM of THD for activation, ii) 20 μM eptifibatide as negative control, and iii) anti-LIBS as positive control (1:100).
Rb8053 detection: Add 75 μL of the detection antibody Rb8053 to measure the amount of integrin capturing (1:1,000).
Analyse the data as shown in Fig 5A. Calculate the activation indices as described in Section 3.2 and as show in Fig 5B. In our experiment 21% of the integrin nanodiscs alone are active and 62% in the presence of 2.5 μM THD.
Calculate the increase in activation using AIwith-THD − AIintegrin-alone, where AI stands for activation indices. In our experiment, adding 2.5 μM of THD produced a 40% increase in integrin nanodiscs activation.
Table 2.
Plate layout for integrin nanodisc activation assay (see Note 12).

|
Fig. 5.

Measurement of the integrin activation by talin head domain (THD).
(A). Measurement of the active integrin nanodiscs in the absence and presence of THD.
(B). Integrin nanodiscs activation index in the absence and presence of THD. There is a 40% increase in active integrins in the presence of 2.5 μM THD.
Acknowledgments
This work was supported by the American Heart Association 12SDG11610043 (A.R. Gingras) and NIH (M.H. Ginsberg).
Footnotes
It is important to have the platelet-rich plasma delivered at room temperature. Cold can lead to platelet activation and aggregation.
The Triton X-100 in the buffer is used to keep the purified integrin soluble in solution. Within a nanodisc, the integrins are inserted into a phospholipid bilayer and adding detergent in those experiments would disrupt that phospholipid bilayer and therefore affect the integrins.
The talin head domain has been extensively studied and smaller sub-domains, such as the F2–F3 region could work just as well for integrin activation (16–19).
In our experiment more than 60% of the peptide was coupled to the resin, but we did not perform any test to verify if the efficiency can be improved by using more resin or less peptide.
The handling and disposal of human blood products should be performed with biohazard precautions.
It is important to keep the centrifuge temperature around 20°C to prevent platelet aggregation and activation.
If necessary, the supernatant can be frozen at −80°C until further use. This crude lysate can then be thawed rapidly and centrifuged at 30,000 × g for 60 min at 4°C to remove residual cytoskeleton.
This can be performed using a peristaltic pump or an FPLC.
Under reducing conditions, αIIb has a MW of ~120 kDa and β3 has a MW of ~95 kDa. Thrombospondin has a MW of 160 kDa and fibrinogen alpha, beta and gamma chains of ~95, ~55 and ~51.5 kDa respectively.
If necessary, the eluate can be frozen at −80°C until further use.
There are several simple and relatively inexpensive methods for concentrating protein solutions. Dialysis against Aquacide II (Calbiochem), which removes water through the dialysis tubing, is what we use. Alternatively, centrifugal concentrators or ultrafiltration membranes may be used.
For the ELISA each experiment has to be performed in triplicate.
It is important to dialyze all traces of nickel from the THD preparation because residual Ni2+ on the hexahistidine tag can cause lipid precipitation problems.
The preparation has been described by Ye et. al., 2010 (13) and it was shown that each nanodisc contained a single αIIbβ3 integrin.
It is important to keep a divalent cation present with integrins as the α and β subunits could separate.
Use a 1.5 mL Eppendorf as the integrin nanodiscs mixture is about 0.5 mL and filling the tube with SM-2 Biobeads will result in approximately 1 mL of beads added.
References
- 1.Calderwood DA. Integrin activation. J Cell Sci. 2004;117:657–666. doi: 10.1242/jcs.01014. [DOI] [PubMed] [Google Scholar]
- 2.Shattil SJ, Hoxie JA, Cunningham M, et al. Changes in the platelet membrane glycoprotein IIb. IIIa complex during platelet activation. J Biol Chem. 1985;260:11107–11114. [PubMed] [Google Scholar]
- 3.Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol. 2011;27:321–345. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- 4.Anthis NJ, Wegener KL, Ye F, et al. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 2009;28:3623–3632. doi: 10.1038/emboj.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Travis MA, Humphries JD, Humphries MJ. An unraveling tale of how integrins are activated from within. Trends Pharmacol Sci. 2003;24:192–197. doi: 10.1016/S0165-6147(03)00069-5. [DOI] [PubMed] [Google Scholar]
- 6.Fitzgerald LA, Leung B, Phillips DR. A method for purifying the platelet membrane glycoprotein IIb–IIIa complex. Anal Biochem. 1985;151:169–177. doi: 10.1016/0003-2697(85)90067-3. [DOI] [PubMed] [Google Scholar]
- 7.Kouns WC, Hadvary P, Haering P, et al. Conformational modulation of purified glycoprotein (GP) IIb-IIIa allows proteolytic generation of active fragments from either active or inactive GPIIb-IIIa. J Biol Chem. 1992;267:18844–18851. [PubMed] [Google Scholar]
- 8.Frelinger AL, 3rd, Du XP, Plow EF, et al. Monoclonal antibodies to ligand-occupied conformers of integrin alpha IIb beta 3 (glycoprotein IIb–IIIa) alter receptor affinity, specificity, and function. J Biol Chem. 1991;266:17106–17111. [PubMed] [Google Scholar]
- 9.Denisov IG, Grinkova YV, Lazarides AA, et al. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 10.Katayama H, Wang J, Tama F, et al. Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc Natl Acad Sci U S A. 2010;107:3453–3457. doi: 10.1073/pnas.1000100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denisov IG, Sligar SG. Cytochromes P450 in nanodiscs. Biochim Biophys Acta. 2011;1814:223–229. doi: 10.1016/j.bbapap.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raschle T, Hiller S, Yu TY, et al. Structural and functional characterization of the integral membrane protein VDAC-1 in lipid bilayer nanodiscs. J Am Chem Soc. 2009;131:17777–17779. doi: 10.1021/ja907918r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye F, Hu G, Taylor D, et al. Recreation of the terminal events in physiological integrin activation. J Cell Biol. 2010;188:157–173. doi: 10.1083/jcb.200908045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moser M, Legate KR, Zent R, et al. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 15.Ye F, Kim C, Ginsberg MH. Molecular mechanism of inside-out integrin regulation. J Thromb Haemost. 2011;9(Suppl 1):20–25. doi: 10.1111/j.1538-7836.2011.04355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouaouina M, Harburger DS, Calderwood DA. Talin and signaling through integrins. Methods Mol Biol. 2012;757:325–347. doi: 10.1007/978-1-61779-166-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elliott PR, Goult BT, Kopp PM, et al. The Structure of the talin head reveals a novel extended conformation of the FERM domain. Structure. 2010;18:1289–1299. doi: 10.1016/j.str.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Alvarez B, de Pereda JM, Calderwood DA, et al. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 19.Anthis NJ, Wegener KL, Critchley DR, et al. Structural diversity in integrin/talin interactions. Structure. 2010;18:1654–1666. doi: 10.1016/j.str.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]