

In this study, Dodgson et al. used a genome-wide screen for gene deletions that impair the fitness of aneuploid yeast and identified the deubiquitinase Ubp3 as a key regulator of aneuploid cell homeostasis. They found that Ubp3 is a guardian of aneuploid cell fitness conserved across species.
Keywords: aneuploidy, Ubp3, deubiquitinase, proteasome
Abstract
Aneuploidy—or an unbalanced karyotype in which whole chromosomes are gained or lost—causes reduced fitness at both the cellular and organismal levels but is also a hallmark of human cancers. Aneuploidy causes a variety of cellular stresses, including genomic instability, proteotoxic and oxidative stresses, and impaired protein trafficking. The deubiquitinase Ubp3, which was identified by a genome-wide screen for gene deletions that impair the fitness of aneuploid yeast, is a key regulator of aneuploid cell homeostasis. We show that deletion of UBP3 exacerbates both karyotype-specific phenotypes and global stresses of aneuploid cells, including oxidative and proteotoxic stress. Indeed, Ubp3 is essential for proper proteasome function in euploid cells, and deletion of this deubiquitinase leads to further proteasome-mediated proteotoxicity in aneuploid yeast. Notably, the importance of UBP3 in aneuploid cells is conserved. Depletion of the human homolog of UBP3, USP10, is detrimental to the fitness of human cells upon chromosome missegregation, and this fitness defect is accompanied by autophagy inhibition. We thus used a genome-wide screen in yeast to identify a guardian of aneuploid cell fitness conserved across species. We propose that interfering with Ubp3/USP10 function could be a productive avenue in the development of novel cancer therapeutics.
Aneuploidy—or an unbalanced karyotype in which whole chromosomes are gained or lost—results from chromosome missegregation during mitotic or meiotic cell divisions. Such genomic imbalance causes a variety of phenotypes on both the cellular and organismal levels and is the leading cause of spontaneous abortion in humans. Whole organisms with constitutional aneuploidy are generally less fit than their euploid counterparts and exhibit developmental abnormalities as well as reduced viability or longevity. Interestingly, however, the majority of human cancer cells is also aneuploid, with >90% of solid tumors exhibiting some degree of genomic imbalance (Weaver and Cleveland 2006; Nagaoka et al. 2012).
Aneuploid cells, regardless of origin, are known to express the vast majority of the genes encoded on their extra chromosomes at both the RNA and protein levels (Mao et al. 2003; Torres et al. 2007; Williams et al. 2008; Pavelka et al. 2010; Stingele et al. 2012; Dephoure et al. 2014). The expression of these extra proteins leads to a plethora of phenotypes in aneuploid yeast cells, including impaired proliferation, genomic instability, vesicle transport defects, and proteotoxic and oxidative stresses among others (Torres et al. 2007; Sheltzer et al. 2011; Oromendia et al. 2012; Dephoure et al. 2014; Dodgson et al. 2016). These phenotypes are thought to be caused primarily by the sum of many gene imbalances that, on their own, have little or no phenotype, as copy number changes of individual dosage-sensitive genes cannot explain the proliferation defects of aneuploid yeast (Torres et al. 2007; Pavelka et al. 2010; Oromendia et al. 2012; Bonney et al. 2015). However, particular aneuploidies have been shown to cause chromosome-specific phenotypes, indicating that the consequences of aneuploidy represent a combination of the overall genomic imbalance as well as the results of specific gene duplications or deletions (Torres et al. 2007; Pavelka et al. 2010; Chen et al. 2015; Dodgson et al. 2016).
Proteins must be properly folded and assembled into complexes with balanced stoichiometries. If this does not occur, proteins are degraded. The processes of protein folding, complex assembly, and degradation are mediated by chaperones and the ubiquitin–proteasome system (UPS). Aneuploid yeast cells are sensitive to compounds that interfere with protein folding and degradation, suggesting that aneuploidy places a burden on these pathways (Torres et al. 2007; Oromendia et al. 2012). Similarly, aneuploid mammalian cells are sensitive to chaperone and autophagy inhibitors, indicating that limited protein quality control pathway function is a universal feature of the aneuploid state (Tang et al. 2011; Donnelly et al. 2014).
We previously conducted a genome-wide screen for genes whose deletion specifically impairs the fitness of aneuploid Saccharomyces cerevisiae using haploid strains containing single extra chromosomes, known as disomes (Dodgson et al. 2016). The deletion of one gene, UBP3, encoding a deubiquitinating enzyme (DUB) was found to be detrimental to the growth of most disomic yeast strains analyzed. Here we investigate the mechanism of this synthetic fitness defect and identify both chromosome-specific and global weaknesses of aneuploid cells exacerbated by deletion of UBP3. The aneuploidy-wide effects of UBP3 deletion can be explained by the observation that Ubp3 is required for efficient UPS function, with deletion of this gene impairing protein degradation in both euploid and aneuploid cells. We further demonstrate that the anti-proliferative effect conferred by UBP3 deletion on aneuploid cells is conserved in higher eukaryotes. Depletion or inhibition of the human homolog USP10 reduces aneuploid human cell fitness. We conclude that pleiotropic hub proteins such as Ubp3 act as aneuploidy-tolerating proteins and that interference with such proteins affects aneuploid cell viability across species.
Results
Deletion of UBP3 broadly impairs aneuploid budding yeast cell fitness
Using synthetic genetic array (SGA) analysis, we previously generated a data set of synthetic fitness interactions between gene deletions and the condition of yeast aneuploidy (Dodgson et al. 2016). The deletion of UBP3 exhibited among the most severe synthetic negative fitness interactions with all disomes queried in high-throughput screens of the yeast knockout collection (overall rank = 11) (Fig. 1A). We confirmed this synthetic fitness defect by de novo deletion of UBP3 in the yeast disomes. Colony sizes of disomic yeast deleted for UBP3 upon dissection were substantially reduced relative to euploid counterparts (Fig. 1B). Additionally, the proliferation rates of the disomes lacking UBP3 are, in most cases, much slower than that of a euploid ubp3Δ strain (Fig. 1C). In fact, disomy for either chromosome XII or XIV is synthetic lethal with deletion of UBP3; such strains cannot be recovered without loss of large segments of the disomic chromosome or whole-genome-doubling events, which buffer against the adverse effects of aneuploidy (data not shown). The degree of this synthetic fitness defect correlates to some extent with the amount of extra genetic material present in aneuploid cells, with strains containing larger extra chromosomes tolerating UBP3 deletion more poorly (Fig. 1D). This is true for both the colony size and doubling time measurements, although we note that the two do not correspond perfectly, likely due to the impact of additional parameters such as germination and growth in the absence of a quorum influencing colony size. Importantly, the synthetic defect is not a function solely of the slow growth of disomic yeast, as deletion of UBP3 did not impair the proliferation of slow-growing temperature-sensitive cdc mutants (Supplemental Fig. S1).
Figure 1.
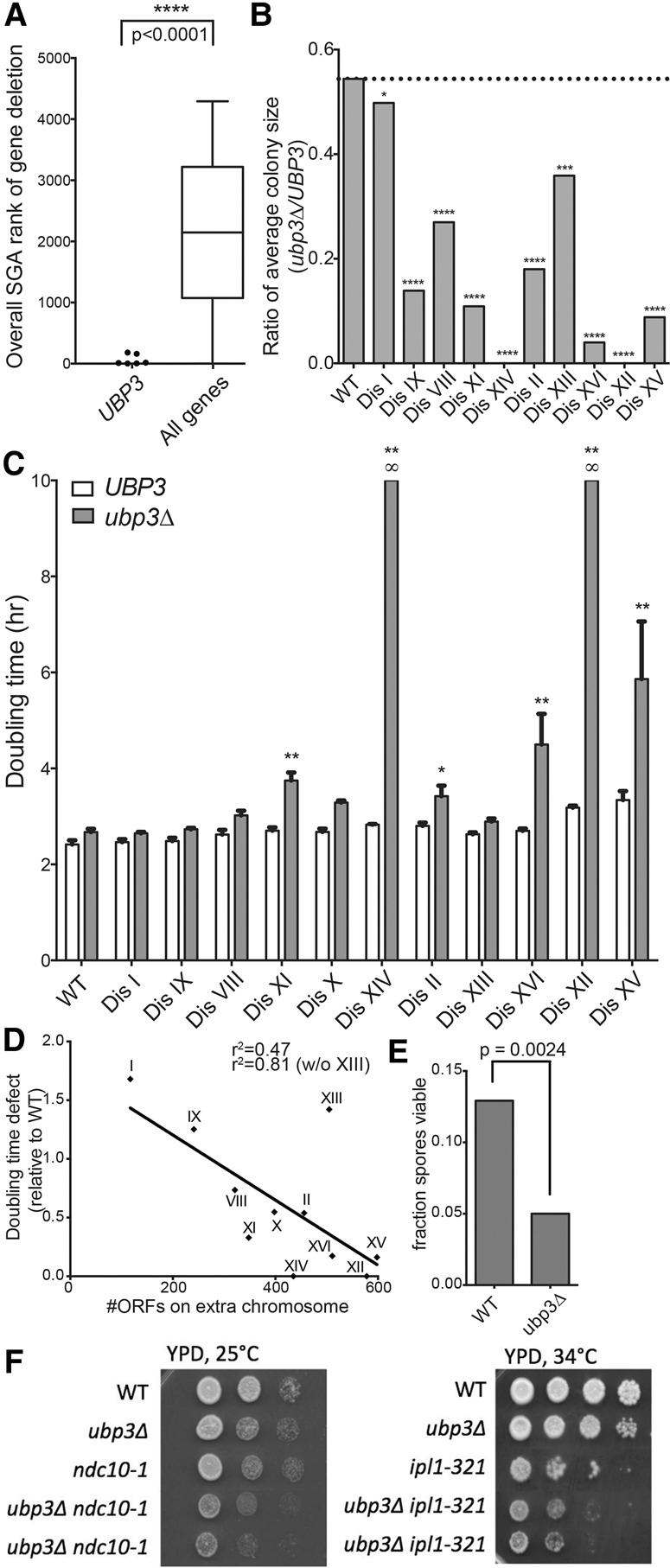
Deletion of UBP3 impairs the proliferation of aneuploid yeast cells. (A) The SGA ranks of the UBP3 deletions in terms of fitness defect conferred on each disome are plotted at the left. The distribution of all gene ranks across the seven disomic SGA screens is plotted at the right for comparison. The distributions are significantly different. P < 0.0001, Mann-Whitney test. Data from are from Dodgson et al. (2016). (B) Euploid ubp3Δ cells were crossed to each disome and dissected to generate ubp3Δ disomic strains. Resulting colony size ratios (ubp3Δ/UBP3) for each disome and the euploid control are shown. (*) P < 0.05; (***) P < 0.001; (****) P < 0.0001, Student's t-test. Colony size ratios equal to zero are indicative of synthetic lethality. (C) Doubling times of disomes either wild type (WT) or deleted for UBP3 were determined by growing cells in YPD at 25°C and taking OD600 measurements every 15 min over 24 h. SD is shown. (*) P < 0.05; (**) P < 0.001, Student's t-test. Disomes are ordered by increasing size of the extra chromosome. Doubling times marked as infinite (∞) represent synthetic lethality. (D) The proliferation defect (ubp3Δ/UBP3) relative to the euploid control is plotted as a function of the number of ORFs encoded on the disomic chromosome. Linear regression analysis gives an r2 value of 0.47 (r2 = 0.81 if disome XIII is excluded). (E) Triploid strains of S. cerevisiae containing either three (wild-type) or zero (ubp3Δ) copies of UBP3 were induced to sporulate. The resulting tetrads were dissected, and the number of spores that formed colonies was counted. n = 3 biological replicates of 80 dissected spores. χ2 test, df = 1; P = 0.0024. (F) Cultures of the indicated strains were grown overnight in YPD, and 10-fold serial dilutions were plated on YPD plates at the indicated temperatures.
Deletion of UBP3 not only impaired the fitness of disomic yeast strains, it was also detrimental in other yeast aneuploidy models. When triploid yeast strains are induced to undergo meiosis, the resultant spores harbor karyotypes ranging from euploid to highly aneuploid (Charles et al. 2010). For this reason, spore viability of triploid yeast is very low; however, deletion of all three copies of UBP3 in a triploid strain further reduced spore viability, demonstrating incompatibility with the aneuploid cell state (Fig. 1E). Furthermore, deletion of UBP3 exhibits a synthetic genetic defect with mutations that cause increased chromosome missegregation. Temperature-sensitive mutations in genes essential for chromosome segregation are lethal at the restrictive temperature (37°C), but, when cells with mutations in these genes are grown at intermediate (semipermissive) temperatures, these strains are viable but missegregate chromosomes at a high rate, leading to the accumulation of aneuploid cells (Oromendia et al. 2012). We found that the growth defects of strains harboring the temperature-sensitive mutations ipl1-321 or ndc10-1 were exacerbated by deletion of UBP3 at semipermissive temperatures (Fig. 1F). We conclude that deletion of UBP3 causes broad fitness impairment of aneuploid cells of various origins and karyotypes.
Deletion of UBP3 specifically impairs vesicle transport in disome XVI cells
UBP3 encodes a highly pleiotropic DUB that removes monoubiquitin from substrates (Baker et al. 1992; Brew and Huffaker 2002). Due to the diversity of its substrates, the deletion of UBP3 has been implicated in a plethora of cellular processes, including vesicle transport, proteostasis, stress granule formation, the response to DNA damage, and autophagy (Baxter and Craig 1998; Cohen et al. 2003a; Bilsland et al. 2007; Oling et al. 2014; Müller et al. 2015; Nostramo et al. 2016). It was therefore plausible that deletion of UBP3 had a variety of consequences on aneuploid cells either dependent on or independent of karyotype. That is, UBP3 deletion may impair disomic fitness through different pathways in each disomic strain, through a global mechanism common among aneuploid cells, or through a combination of both.
We showed previously that aneuploidy impairs vesicle transport in most disomic yeast strains (Dodgson et al. 2016). Given the role of Ubp3 in this process (Cohen et al. 2003a,b), we examined secretion of the glycoprotein Ccw14 in disomes lacking UBP3. In wild-type cells, Ccw14 becomes covalently attached to the yeast inner cell wall and is therefore insoluble and mostly undetectable in Western blots (Fig. 2A; Dodgson et al. 2016). Accumulation of Ccw14 in either its faster-migrating ER form or its slower-migrating post-Golgi form indicates the presence of secretory delays. Deletion of UBP3 resulted in clear secretory defects at multiple steps in the pathway across all ubp3Δ strains tested (Fig. 2A). However, these defects were exacerbated only in the disome XVI ubp3Δ strain. Accumulation of the ER form of Ccw14 was as extreme in disome XVI ubp3Δ cells as in a known secretory mutant, sec7-1 (Fig. 2A). Thus, deletion of UBP3 causes a chromosome-specific vesicle-trafficking defect in disome XVI cells.
Figure 2.
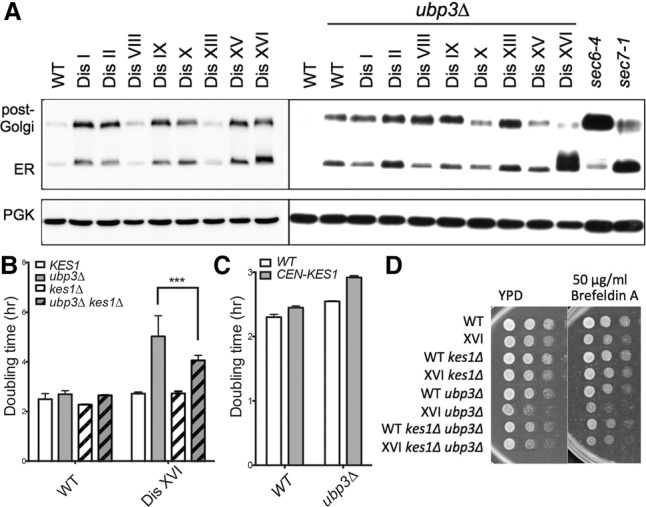
Deletion of UBP3 enhances the vesicle transport defect of disome XVI cells. (A) Cells were grown to mid-log phase in YPD at room temperature. Ccw14 mobility was analyzed by Western blot analysis. The ER and post-Golgi forms of the protein are indicated. Known secretory mutants sec6-4 and sec7-1 are shown for comparison. Pgk1 was used as a loading control. The lysates for the blot at the left (disomes wild type [WT] for UBP3) are from Dodgson et al. (2016). (B,C) Doubling times of the indicated strains were determined as described in Figure 1. (***) P < 0.0001, Student's t-test. (D) Cultures of the indicated strains were grown overnight in YPD, and 10-fold serial dilutions were plated on YPD plates with or without the indicated concentration of Brefeldin A at 30°C.
We next attempted to determine whether the trafficking defect of disome XVI ubp3Δ cells could be traced to duplication of a specific gene on chromosome XVI. A known chromosome XVI-encoded dosage-sensitive gene involved in Golgi function is KES1 (Fairn et al. 2007). Restoration of haploid KES1 gene dosage in disome XVI ubp3Δ partially suppressed the proliferation defect of these cells (Fig. 2B). Consistent with this observation, we found that euploid cells lacking UBP3 are sensitive to the duplication of KES1; introduction of a centromeric plasmid harboring the KES1 gene increased the doubling time of the ubp3Δ strain while having little effect on wild type (Fig. 2C). Restoration of euploid KES1 dosage also partially rescued the increased sensitivity of the disome XVI ubp3Δ strain to the vesicle transport-inhibiting drug Brefeldin A (Fig. 2D). However, although duplication of KES1 partially explains the fitness defect conferred by deletion of UBP3 on disome XVI, it does not explain the severe Ccw14 secretion defect of disome XVI ubp3Δ cells. Disome XVI ubp3Δ cells carrying only one copy of the KES1 gene behaved indistinguishably from disome XVI ubp3Δ cells with two copies of KES1 in the Ccw14 secretion assay (Supplemental Fig. S2). Our results indicate that duplication of KES1 contributes to the extreme protein-trafficking defect in these cells, albeit without affecting Ccw14 secretion. We conclude that chromosome-specific phenotypes explain at least part of the fitness defects conferred by deletion of UBP3 on the disomic strains. Thus, karyotype is an important determinant of the magnitude of the response of aneuploid cells to even broadly detrimental mutations.
Proteotoxic stress is increased in disomic yeast strains lacking UBP3
In addition to chromosome-specific phenotypes, the disomes share cellular stresses such as genomic instability, proteotoxic stress, and oxidative stress (Sheltzer et al. 2011; Oromendia et al. 2012; Dephoure et al. 2014). UBP3 was originally isolated as a multicopy suppressor of a heat-shock response-defective mutant and has since been shown to contribute to protein aggregate clearance in proteotoxically stressed cells (Baxter and Craig 1998; Oling et al. 2014). Indeed, disomes lacking UBP3 are particularly heat-sensitive, with ubp3Δ disomes VIII, IX, XI, XV, and XVI showing large doubling time increases at 37°C (Supplemental Fig. S3). Given the high levels of proteotoxicity exhibited by disomic yeast, we assessed the effect of UBP3 deletion on this phenotype using the previously described proteotoxicity reporter VHL-GFP (Kaganovich et al. 2008; Oromendia et al. 2012). The expression of human VHL in yeast in the absence of its binding partners, Elongin B and Elongin C, leads to its ubiquitination and degradation (McClellan et al. 2005; Kaganovich et al. 2008). In proteotoxically stressed cells, however, VHL is prone to form aggregates, which are visible as VHL-GFP foci. Using this assay, we observed a substantial increase in the number of disomic cells with discernible VHL-GFP foci under unstressed conditions (Fig. 3A). Remarkably, deletion of UBP3 caused such significant proteotoxic stress in the disomes that a notable fraction of ubp3Δ cells contained more than one VHL-GFP focus, an event rarely observed in wild-type cells (Fig. 3B). The extent of this severe proteotoxicity, defined as the number of cells containing multiple VHL-GFP foci, correlated strongly with the fitness defect conferred by deletion of UBP3 (Fig. 3C). This observation suggests that the proteotoxic stress exacerbated by UBP3 deletion in aneuploid yeast contributes significantly to the fitness impairment brought about by the deletion of this DUB gene, and we postulate that the proteotoxicity conferred by UBP3 deletion on the disomes exceeds a threshold tolerable for normal cell growth and viability.
Figure 3.
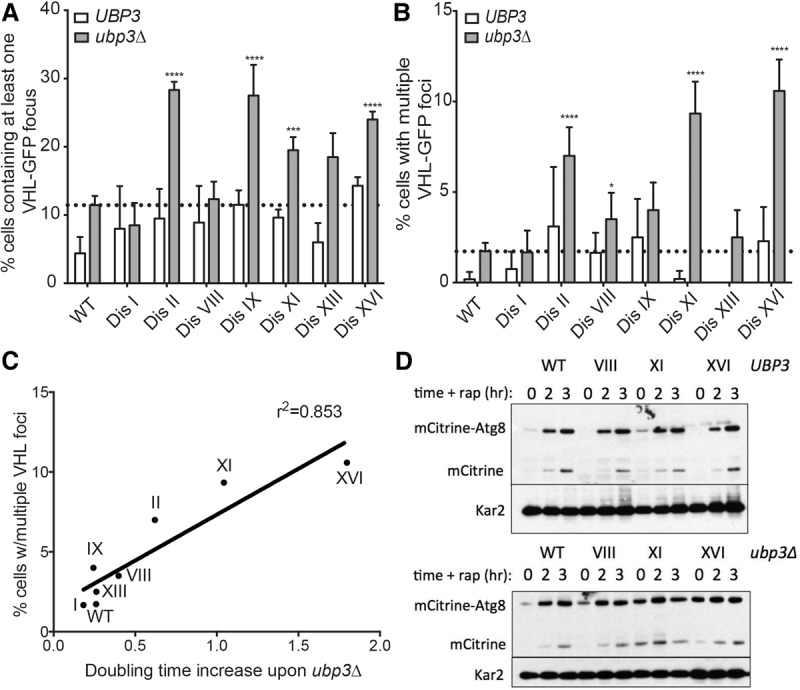
Deletion of UBP3 exacerbates proteotoxicity in disomic yeast strains. (A,B) Euploid (wild type [WT]) and disomic yeast strains wild type or deleted for UBP3 and expressing a VHL-GFP fusion were grown in YEP 2% raffinose and 2% galactose at 25°C, and the percentage of cells with at least one VHL-GFP focus (A) or more than one VHL-GFP focus (B) was determined. n = 3. SEM is shown. n = 200 cells per time point. (*) P < 0.05; (***) P < 0.001; (****) P < 0.0001, Student's t-test. The euploid ubp3Δ focus level is indicated with the dotted line. (C) The correlation between the percentages of disomic ubp3Δ cells harboring more than one VHL-GFP focus and the doubling time increase of each disome upon UBP3 deletion is plotted. (D) Cells were grown to mid-log phase in YPD. Samples were taken before the addition of 0.2 µg/mL rapamycin (t = 0) and after 2 and 3 h of drug treatment. The mobility of mCitrine-Atg8 was determined by Western blot analysis. The slower-migrating form represents the uncleaved mCitrine-Atg8 fusion. The faster-migrating form represents cleaved mCitrine. Kar2 was used as loading control.
In addition to aggregate clearance, Ubp3 has been reported to play roles in multiple types of autophagy (Kraft et al. 2008; Kelly and Bedwell 2015; Müller et al. 2015). Using an mCitrine-Atg8 reporter construct, we assessed the capacity of disomes with and without UBP3 to induce autophagy (Klionsky et al. 2014; Nair et al. 2014). In this assay, cells whose endogenous Atg8 was N-terminally tagged with mCitrine were grown in rich medium, and autophagy was induced by the addition of rapamycin to mimic starvation. Cleavage of mCitrine from the Atg8 protein occurs upon delivery of the fusion to the vacuole, where Atg8 is rapidly degraded, and mCitrine remains relatively stable (Shintani and Klionsky 2004). Therefore, detection of free mCitrine reflects autophagic activity, while the levels of the mCitrine-Atg8 fusion reflect the induction of endogenous ATG8 expression. Importantly, the disomes showed normal induction of autophagy in response to rapamycin addition (Fig. 3D). The wild-type strain lacking UBP3 exhibited mild autophagy defects, as reported previously, but the ubp3Δ disomes remained competent for autophagy (Fig. 3D). In fact, levels of Atg8 were elevated under basal conditions in the ubp3Δ disomes (Fig. 3D, t = 0), indicating that deletion of UBP3 in the disomic strains does not interfere with autophagy but rather induces proteotoxic stress that leads to up-regulation of autophagy. These results support our conclusion that the ubp3Δ disomes are under severe proteotoxic stress and indicate that the autophagy pathway may be increasingly relied on in the presence of significantly compromised proteostasis in yeast. Thus, Ubp3 is a pleiotropic DUB with roles in cellular protection from proteotoxic stress and acts as an aneuploidy-buffering protein whose deletion substantially exacerbates proteotoxicity in aneuploid yeast.
Cells lacking UBP3 show hallmarks of impaired proteasome function
Our results indicate that proteotoxicity is elevated in disomes lacking UBP3. However, the molecular defect underlying this phenotype was unclear. The clearance of misfolded VHL proceeds through both Hsp70 and Hsp90 chaperones followed by degradation by the proteasome (McClellan et al. 2005). Assessment of the in vivo folding of the Hsp90 substrate Src tyrosine kinase can be achieved by inducing expression of either c-src, which does not affect proliferation of yeast cells, or v-src, whose overexpression is lethal in yeast (Xu and Lindquist 1993). In cells with uncompromised Hsp90 activity, expression of v-src is lethal due to proper folding of the toxic protein; when Hsp90 activity is impaired, however, improved growth is seen in v-src-expressing cells upon induction. Disomic yeast are known to have reduced Hsp90 folding capacity (Oromendia et al. 2012); however, UBP3 deletion caused no enhancement of this phenotype (data not shown). These findings indicate that deletion of UBP3 does not impair proliferation of disomic yeast by further hampering Hsp90 activity.
Whether Ubp3 was required for proteasome function had not been explored previously. However, Isasa et al. (2015) showed that cells lacking UBP3 accumulate ubiquitylated proteins. We confirmed that deletion of UBP3 indeed leads to the accumulation of ubiquitylated proteins, in contrast to deletion of other DUBs, exemplified by the proteasome-inhibiting DUB Ubp6 (Fig. 4A; Isasa et al. 2015). To begin to determine whether loss of UBP3 affected proteasome function, we deleted UBP3 in cells carrying a temperature-sensitive allele of the gene encoding a proteasome lid subunit, Rpn6 (Isono et al. 2005). Deletion of UBP3 exhibits a strong synthetic fitness defect when combined with the rpn6-1 allele (Fig. 4B,C). This finding raises the possibility that UBP3 deletion leads to impaired proteasome function.
Figure 4.
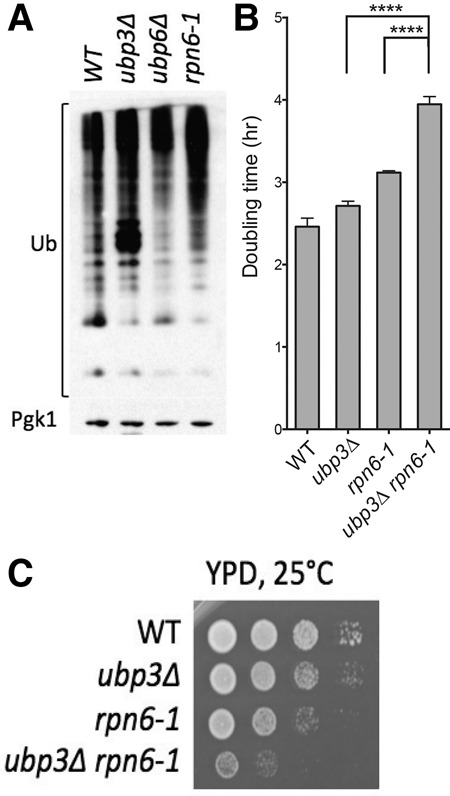
Cells lacking UBP3 accumulate ubiquitylated proteins and exhibit synthetic negative interactions with the hypomorphic proteasome mutant rpn6-1. (A) Cells were grown to mid-log phase in YPD, and total ubiquitin conjugates were detected using anti-ubiquitin antibody in Western blot analysis. Pgk1 was used as a loading control. (B) Doubling times of the indicated strains were determined as described in Figure 1 at 25°C. P < 0.0001, Student's t-test. (C) Cultures of the strains in B were grown overnight in YPD, and 10-fold serial dilutions were plated on YPD plates at 25°C.
Deletion of UBP3 increases the half-lives of proteasome substrates
In order to more directly test the role of Ubp3 in proteasome function, we assessed the half-lives of various ubiquitin–proteasome substrates in the presence and absence of UBP3. For these experiments, genes encoding ubiquitin–proteasome substrates were placed under the control of the galactose-inducible/glucose-repressible GAL1-10 promoter. Transcription was then induced for 45 min, after which time transcription was repressed by glucose addition. At the same time, cycloheximide was added to prevent further translation. Protein levels were then measured by Western blotting to determine the half-lives of various ubiquitin–proteasome substrates.
The first substrate that we examined was URL-Bub2-HA, a fusion between a ubiquitin–arginine–lacZ (URL) degron and the mitotic exit network protein Bub2 (Bachmair et al. 1986; Visintin et al. 2008). Exposure of an arginine at the N terminus of a protein after cleavage of the N-terminal ubiquitin moiety leads to rapid degradation of the protein by the N-end rule pathway (Bachmair et al. 1986). Thus, the URL-Bub2-HA fusion is highly unstable in wild-type cells, with a half-life of ∼1.5 min (Fig. 5A,B). Upon deletion of UBP3, degradation of the fusion protein was substantially delayed, with the half-life of this protein increasing to ∼8.5 min in the ubp3Δ strain (Fig. 5A,B).
Figure 5.
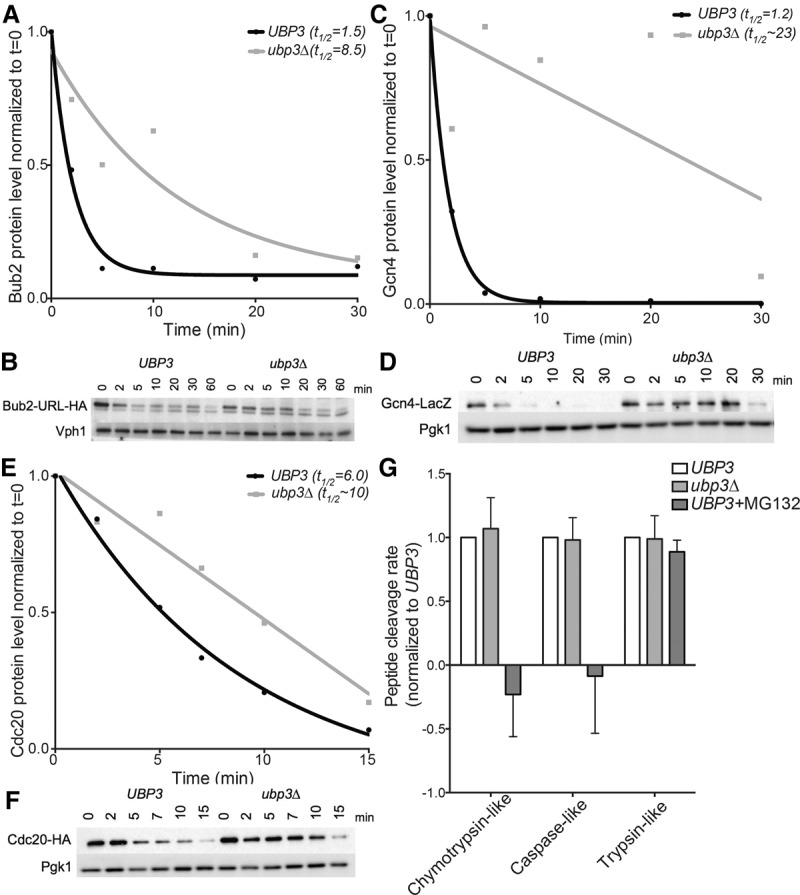
Deletion of UBP3 increases the half-lives of known ubiquitin–proteasome substrates without affecting the in vitro catalytic activity of the proteasome. (A–F) Cells were grown overnight in medium containing raffinose at room temperature, and expression of tagged substrates was induced by the addition of 2% galactose for 45 min. The t = 0 sample was taken before the addition of 2% glucose and 50 µg/mL cycloheximide. Samples were taken at the indicated times after cycloheximide addition, and protein levels were analyzed by Western blot analysis. Quantifications relative to the loading control and normalized to time = 0 are plotted in A, C, and E. (A,B) The half-life of Bub2 in wild-type cells and cells lacking UBP3. Vph1 was used as a loading control. (C,D) The half-life of Gcn4 in wild-type cells and cells lacking UBP3. Pgk1 was used as a loading control. (E,F) The half-life of Cdc20 in wild-type cells and cells lacking UBP3. Pgk1 was used as a loading control. (G) The in vitro proteasome activity of UBP3 and ubp3Δ cell lysates was determined using cleavage of fluorogenic substrates as a readout for protease activity. MG132 was added to wild-type lysates to inhibit the activity of two of the three peptidase activities of the proteasome.
The second ubiquitin–proteasome substrate whose half-life we determined was the transcription factor Gcn4. Gcn4 protein levels are controlled by the SCF ubiquitin ligase (Irniger and Braus 2003). This ubiquitin ligase complex ubiquitylates substrates that have been phosphorylated previously (Willems et al. 2004). Using an inducible Gcn4-lacZ fusion, we observed that deletion of UBP3 significantly delayed the degradation of this construct. Wild-type cells degraded the majority of Gcn4 by 5 min after cycloheximide addition, while ubp3Δ cells required 30 min to reach similar levels (Fig. 5C,D).
Finally, we examined the half-life of Cdc20, whose degradation depends on a ubiquitin ligase known as the anaphase-promoting complex or cyclosome (APC/C) (Prinz et al. 1998; Barford 2010). Degradation of Cdc20-HA was also delayed in cells lacking UBP3, with the estimated half-life of this protein increasing from 6 to ∼10 min in ubp3Δ cells (Fig. 5E,F). We conclude that the deubiquitinase Ubp3 is required for the degradation of multiple different ubiquitin/proteasome substrates in euploid yeast cells.
Although cells lacking UBP3 display delayed degradation of a variety of proteasome substrates, each of which enters the proteasome via distinct ubiquitylation pathways, the fundamental peptidase activities of the catalytic core of the proteasome remain intact. Using fluorogenic substrates, each of which is cleaved by one of the three proteasome-resident peptidase activities, we determined that ubp3Δ cell lysates are competent to cleave these peptides with the same kinetics as wild type (Fig. 5G). This is perhaps unsurprising, as a deubiquitinase is more likely to be involved in the regulation of the proteasome rather than its peptidase activities, but the mechanism by which Ubp3 impinges on the proteasome remains unknown.
Aneuploid cells have impaired proteasome function
Previous work suggested that proteasomal degradation was limiting in disomic yeast strains. Several disomes exhibit sensitivity to the proteasome inhibitor MG132 (Torres et al. 2007), and deletion of the gene encoding the proteasome-inhibiting DUB Ubp6 was shown to improve the proliferation of most disomic strains at high temperatures (Dephoure et al. 2014). Gene expression analysis of disomic strains further supports the conclusion that proteasomal activity is limiting in aneuploid yeast. Under standard growth conditions (batch growth), genes whose expression changes according to the degree of aneuploidy are predominantly involved in the environmental stress response (ESR) or are otherwise known to vary according to growth rate (Sheltzer et al. 2012). However, when the disomes are grown under nutrient limitation in chemostats to equalize proliferation between euploid and aneuploid strains, the ESR gene signature is abolished (Torres et al. 2007). In order to identify transcripts that vary according to the degree of aneuploidy but are independent of culture conditions, we combined gene expression data from aneuploid strains grown in batch culture and under doubling time-equalizing phosphate-limited conditions in chemostats. Genes whose expression increased with an increased degree of aneuploidy were strongly enriched for subunits of the proteasome complex (P < 10−6) and other genes involved in proteolysis (P < 10−3) (Fig. 6A; Supplemental Table 1). Indeed, proteasomal subunit gene expression correlates strongly with degree of aneuploidy, suggesting that proteasome function is limiting in disomic cells (Fig. 6B,C).
Figure 6.
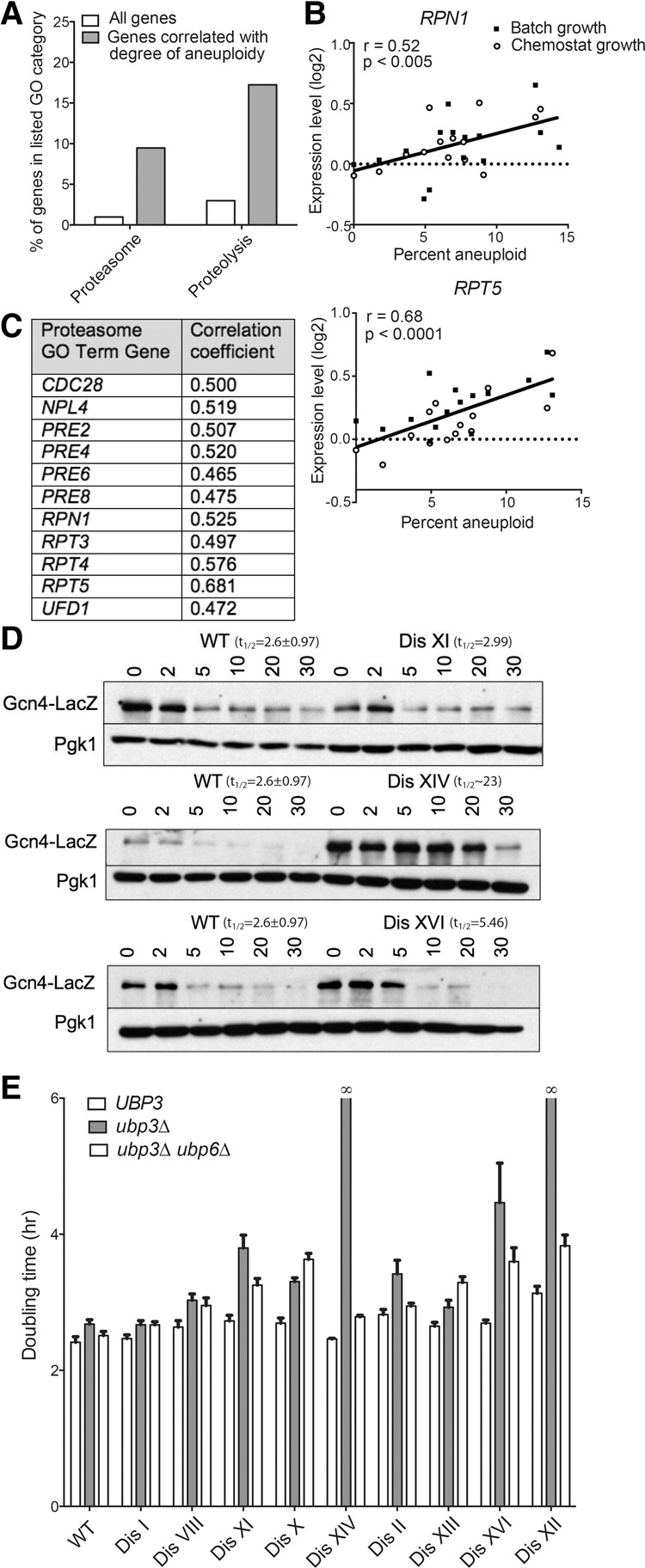
Disomic yeast strains show impaired protein degradation and up-regulate proteasome gene expression. (A) The percentages of the genes whose expression correlates with the degree of aneuploidy (dark gray) and the percentages of all genes across the genome (white) that belong to the gene ontology (GO) term categories “proteasome” and “proteolysis” are plotted. (B) The relative expression levels of proteasome component genes RPT5 and RPN1 in disomic strains grown in either batch culture or chemostats are plotted against the percent aneuploidy of each strain (number of disomic ORFs/total number of yeast ORFs). Data are from Torres et al. (2007). Linear regression values are shown. (C) Genes belonging to the GO term category “proteasome” whose expression is correlated with the degree of aneuploidy are listed along with the correlation coefficient for each gene. (D) The experiment was performed as described for Figure 5, A–F. Western blots show degradation of Gcn4-LacZ in wild type and three disomes. Pgk1 was used as a loading control. (E) Doubling times of the indicated strains were determined as described in Figure 1.
To more directly test whether deletion of UBP3 causes a fitness defect in disomic yeast strains because it enhances an already existing protein degradation defect in aneuploid cells, we determined the half-life of the SCF substrate Gcn4 in two disomes, disomes XIV and XVI, whose proliferation was severely impacted by deleting UBP3, and in one disome, disome XI, whose fitness was moderately affected by UBP3 deletion (Fig. 1C). The half-life of Gcn4-lacZ was greatly increased in cells disomic for either chromosome XIV or XVI while remaining unchanged in disome XI cells (Fig. 6D). These findings suggest that the protein degradation defect in disomes XIV and XVI is so severe as to be apparent even in the presence of a functional UBP3 gene. In contrast, the protein degradation defect of disome XI cells is too subtle to be detected in protein half-life measurements and becomes apparent only in cells lacking UBP3. Consistent with this idea is the observation that the two disomies that are synthetic lethal with deletion of UBP3—XII and XIV—are also synthetic lethal with the rpn6-1 mutation (Torres et al. 2010). Finally, the observation that deletion of UBP6, which causes hyperactivation of the proteasome (Hanna et al. 2006), partially rescues the proliferation defects of disomic strains harboring a UBP3 deletion further supports this hypothesis (Fig. 6E). We conclude that proteasome function is limiting in many disomic yeast strains and propose that a further decrease in proteasome function is the likely basis for much of the fitness defect conferred by deletion of UBP3 on disomic yeast strains.
Depletion of the human homolog of UBP3, USP10, impairs proliferation of aneuploid human cells
Ubp3 is conserved. Its closest homolog in mammals is encoded by the USP10 gene (Soncini et al. 2001). To assess whether the fitness defect conferred by UBP3 deletion on aneuploid yeast is conserved in higher eukaryotes, we treated euploid RPE-1 cells with siRNA to deplete USP10 and induced chromosome missegregation 24 h later by one of two methods. We treated RPE-1 cells with reversine, an inhibitor of the spindle assembly checkpoint (SAC) protein kinase Mps1, or depleted the SAC protein Mad2 via siRNA (Santaguida et al. 2010). These two methods lead to the missegregation of one or a few chromosomes in the vast majority of cells (Santaguida et al. 2010, 2015). Induction of chromosome missegregation in control RNAi-treated cells led to an ∼25% decrease in cell viability (Fig. 7A). Depletion of USP10 led to a significant further decrease in cell proliferation. Fewer than 50% of cells survived the induction of chromosome missegregation in the absence of USP10 (Fig. 7A; Supplemental Fig. S4). Spautin-1, a compound that specifically inhibits USP10 (Liu et al. 2011) caused an effect similar to that of knockdown of the gene (Fig. 7B). Treatment of RPE-1 cells with this drug concurrent with induction of chromosome missegregation by Mad2 depletion led to a significant decrease in cell number compared with cells in which USP10 was not inhibited. This effect was independent of the presence of a functional SAC; when the aneuploid cell population was established by reversine treatment but the drug was removed to reinstate the SAC before spautin-1 treatment, the result was the same (Fig. 7C). Finally, USP10 inhibition also inhibits the proliferation of stable aneuploid cells; mouse embryonic fibroblasts (MEFs) trisomic for chromosome 13 or 16 were more sensitive to spautin-1 treatment than their euploid counterparts (Fig. 7D).
Figure 7.

Depletion and inhibition of USP10 impairs the growth of aneuploid human cells. (A) RPE-1 cells were treated with siRNA targeting USP10 or control siRNA. Twenty-four hours later, cells were treated with either MAD2 siRNA or control siRNA or reversine or solvent. Three days after Mad2 siRNA or reversine treatment, cells were harvested, and the number of cells was determined. n = 3. Mean ± SD shown. P < 0.05, Student's t-test. (B) RPE-1 cells were treated with siRNA targeting Mad2 or control siRNA. Twenty-four hours later, cells were treated with spautin-1 or solvent only. Three days after spautin treatment, cells were harvested, and the number of cells was determined. n = 3. Mean ± SD shown. P < 0.01, Student's t-test. (C) RPE-1 cells were treated with reversine or solvent to induce chromosome missegregation. Twenty-four hours later, the drug was washed out, and cells were treated with spautin-1 or solvent. Three days after spautin treatment, cells were harvested, and the number of cells was determined. n = 3. Mean ± SD shown, P = 0.0015, Student's t-test. (D) MEFs either euploid or trisomic for chromosome 13 or 16 were treated with spautin-1 or solvent only. Three days after treatment, cells were harvested, and the number of cells was determined. n = 3. Mean ± SD shown. P < 0.05, Student's t-test. (E) RPE-1 cells were treated with spautin-1 or solvent. Twenty-four hours later, cells were treated with reversine or solvent to induce chromosome missegregation. Three days after reversine treatment, cells were treated with Bafilomycin A1 (BafA1) or solvent only and harvested 6 h later. LC3-I and LC3-II levels were analyzed by Western blot analysis. GAPDH was used as a loading control. (F) Quantification of LC3-II levels in E were normalized to the untreated control (lane 1). (G) Levels of USP10 gene expression in matched normal and cancer cells were plotted. Data are from the Broad Institute GDAC Firehose. (BRCA) Breast-invasive carcinoma; (COADREAD) colorectal adenocarcinoma; (HNSC) head and neck squamous cell carcinoma; (KIPAN) pan-kidney cohort; (LIHC) liver hepatocellular carcinoma; (LUAD) lung adenocarcinoma; (LUSC) lung squamous cell carcinoma; (PRAD) prostate adenocarcinoma; (STAD) stomach adenocarcinoma. (*) P < 0.0001.
USP10 has been implicated previously in the regulation of proteins involved in autophagy (Liu et al. 2011). Aneuploid cells are under proteotoxic stress and have been found to exhibit defects in autophagy-mediated protein degradation (Ohashi et al. 2015; Santaguida et al. 2015). Specifically, autophagic cargo accumulates within autolysosomes following chromosome missegregation (Santaguida et al. 2015). This autophagic flux defect can be assessed by analyzing the migration of the autophagic vesicle protein LC3. To be incorporated into autophagic vesicles, LC3 is cleaved and lipidated, resulting in the generation of the protein known as LC3-II. Accumulation of LC3-II is thus indicative of the accumulation of autophagosomes (Klionsky et al. 2014). Consistent with previously published results (Santaguida et al. 2015), we found that cells induced to missegregate chromosomes by reversine treatment harbored elevated levels of LC3-II relative to euploid cells (Fig. 7E [lanes 1,2], F). Autophagic flux can be assessed by comparing LC3-II levels between untreated cells and cells treated with the lysosomal inhibitor Bafilomycin A1 (BafA1) 6 h prior to harvesting. In cells with compromised flux, the differential between LC3-II levels treated with and without BafA1 is lower than that of cells with normal flux. Using this metric, we confirmed that autophagic flux is decreased following chromosome missegregation (Fig. 7E,F; Santaguida et al. 2015).
While aneuploidy causes a decrease in autophagic flux, spautin-1 prevents the formation of autophagosomes altogether. Consistent with previously published findings, we did not detect LC3-II in cells treated with the USP10 inhibitor irrespective of whether cells were induced to missegregate chromosomes (Fig. 7E [lanes 3,4], F; Liu et al. 2011). Even inhibition of lysosomal degradation by BafA1 treatment did not lead to the accumulation of LC3-II when USP10 was inhibited (Fig. 7E [lane 8], F). Thus, aneuploid cells in which USP10 is inhibited are unable to generate autophagosomes and are thus blocked at early stages of autophagy. Our results suggest that the inhibition of early stages of autophagosome formation in aneuploid cells, which depend on autophagy for full viability (Tang et al. 2011), contributes to the fitness defect conferred by USP10 depletion or inhibition on aneuploid cells. Consistent with the conclusion that USP10 is important for the survival of aneuploid cells, we found that USP10 expression is elevated in an array of highly aneuploid cancer types, including colorectal adenocarcinoma and lung squamous cell carcinoma among others (Fig. 7G). We speculate that USP10 and other such aneuploidy-tolerating proteins are overexpressed and relied on by a variety of tumor types.
Discussion
In yeast and mammalian cells, increasing chromosome copy number leads to a corresponding elevation in the levels of proteins encoded on the duplicated chromosomes (Williams et al. 2008; Pavelka et al. 2010; Torres et al. 2010; Dephoure et al. 2014). These proteins cause a plethora of cellular phenotypes dependent on both their specific identities and the sum of imbalanced proteins affecting general homeostatic cellular pathways. The deubiquitinase Ubp3 is integral to cellular homeostasis of aneuploid yeast cells, allowing cells to tolerate both karyotype-specific and karyotype-independent effects of aneuploidy. Deletion of UBP3 severely enhanced the secretion defect of disome XVI cells, illustrating that deletion of this DUB gene accentuates chromosome-specific phenotypes of aneuploid cells. General deficiencies associated with aneuploidy are also enhanced by loss of Ubp3 function. Deletion of UBP3 impairs the efficiency of proteasomal degradation and substantially enhances proteotoxicity in aneuploid yeast. Proteotoxicity is not the only general aneuploidy-associated stress enhanced by deletion of UBP3. Whereas deletion of UBP3 in wild-type cells conferred a mild sensitivity to the oxidative agent diamide, all disomes except disome X exhibited significantly increased sensitivity to the drug in the absence of UBP3 (Supplemental Fig. S5). We propose that pleiotropic proteins such as Ubp3 are central to the survival of aneuploid cells. Like aneuploidy, such proteins affect numerous cellular functions, the impairment of which leads to synthetic fitness defects when combined with multiple different aneuploidies.
The deubiquitinase Ubp3 is required for efficient proteasome function in yeast
Our data provide the first evidence for a general role of Ubp3 in ubiquitin-mediated protein degradation. We found that degradation of many different proteins targeted by distinct ubiquitin ligases is stabilized in cells lacking UBP3. While we cannot exclude the possibility that deletion of UBP3 independently affects all ubiquitination systems that we examined (e.g., by modulating the activity of the E1 enzyme Uba1), we favor the idea that UBP3 affects protein degradation at a step following ubiquitylation. Consistent with this hypothesis is the observation that ubiquitylated proteins accumulate in ubp3Δ cells (Fig. 4A; Isasa et al. 2015). How Ubp3 contributes to the degradation of ubiquitylated proteins remains to be determined. UBP3 could deubiquitinate many different substrates. Indeed, previous studies have demonstrated roles for Ubp3 in enhancing or delaying the degradation of particular substrates by direct deubiquitination (Brew and Huffaker 2002; Cohen et al. 2003a; Kraft et al. 2008; Kvint et al. 2008; Chew et al. 2010; Li and Wang 2013). Ubp3 could also affect the proteasome itself. A small fraction of Ubp3 (5%) associates with the proteasome, and a direct interaction between Ubp3 and proteasomal components has been documented both in vitro and in vivo (Mao and Smerdon 2010). It is thus possible that an as yet unidentified substrate of Ubp3 is important for promoting proteasome function and that deletion of the DUB destabilizes this proteasome activator, thereby decreasing overall proteasome function. Indeed, the activity of the Rpn10 subunit of the 19S proteasome regulatory particle has been shown to be modulated by monoubiquitination, controlled by the E3 ligase Rsp5 and its antagonist, the DUB Ubp2 (Isasa et al. 2010). Perhaps Ubp3 plays a similar role for a distinct proteasomal component or proteasomal activator, thereby modulating the activity or stability of an adaptor protein that delivers substrates to the regulatory particle or directly modulating the activity of the regulatory particle itself. Such a mechanism would not cause changes in the in vitro protease activity of the proteasome core particle, providing an explanation for our finding that this enzymatic activity remains unchanged in ubp3Δ cell lysates. It will be important to determine the exact mechanism by which Ubp3 promotes the degradation of proteasome substrates given the central role of this macromolecular machine in myriad cellular processes and disease states (Schmidt and Finley 2014).
It is noteworthy that Ubp3 is not the first DUB implicated in controlling the survival of aneuploid cells. We found previously that the DUB Ubp6 affects the viability of disomic yeast strains, especially at high temperatures (Torres et al. 2010; Dephoure et al. 2014). However, unlike deletion of UBP3, deletion of UBP6 enhanced rather than impaired the viability of disomic yeast strains. This effect was attributed to the role of Ubp6 in antagonizing proteasome function. Ubp3, on the other hand, is required for full proteasome function (this study), providing an explanation for how two members of the same class of enzymes can have opposite effects on the fitness of aneuploid cells. Ultimately, both DUBs impact the UPS, indicating that the proteasome is a highly sensitive molecular machine whose proper regulation is integral to the viability of aneuploid cells.
The Ubp3 homolog USP10 confers aneuploidy tolerance in human cells
The detrimental effects of loss of UBP3 function on aneuploid cells are not confined to budding yeast. We found that aneuploid cells generated by transient inhibition of the SAC are sensitive to loss of function of the UBP3 homolog USP10. Whether loss of USP10 function synergizes with the same aneuploidy-associated stresses in mammalian cells as in yeast cells remains to be determined. To date, USP10 has not been implicated in proteasome function in mammalian cells. However, like UBP3 in yeast, USP10 has been reported to regulate numerous cellular functions, including controlling the induction of autophagy, sensing energy stress via AMPK, and regulating the stability of p53 (Yuan et al. 2010; Liu et al. 2011; Deng et al. 2016). USP10 can thus also be considered a pleiotropic hub protein that, like UBP3, impacts the fitness of aneuploid mammalian cells in multiple ways. One plausible mechanism by which loss of USP10 function negatively affects the fitness of aneuploid mammalian cells is through its role in autophagy. In this study, we demonstrate that inhibition of USP10 impairs the ability of both aneuploid and euploid cells to form autophagosomes. Acutely aneuploid cells are known to experience autophagic flux defects as well as display a lysosomal stress response even under standard growth conditions (Santaguida et al. 2015). Given the increased dependence on autophagy of cells with unbalanced karyotypes (Tang et al. 2011; Stingele et al. 2012; Santaguida et al. 2015), this mechanism likely explains at least part of the growth defect conferred by USP10 depletion on aneuploid cells.
Genome-wide screens in yeast have the potential to identify therapeutic targets in cancer
It is exciting to note that a genome-wide genetic screen in S. cerevisiae was successfully used to identify a key regulator of aneuploid cell homeostasis that is conserved from yeast to human cells. The conservation of the integral role played by such a pleiotropic gene in aneuploid cells suggests that highly pleiotropic hub genes may represent a category of genes whose proper regulation is essential to the ability of cells to tolerate the consequences of chromosome missegregation and the resulting unbalanced karyotypes. This conclusion is intriguing in terms of both illustrating the utility of screens in model organisms to illuminate mammalian cell biology and suggesting a novel group of targets whose manipulation could be exploited therapeutically in aneuploid contexts.
Importantly, aneuploidy is a highly prevalent feature of human cancers; >90% of solid tumors are reported to be aneuploid (Beroukhim et al. 2010). Thus, inhibition of USP10 or other pleiotropic regulatory proteins could be a useful therapeutic strategy to preferentially inhibit the proliferation of aneuploid cancer cells. In this regard, it is exciting to note that USP10 expression levels are elevated across an array of cancer types (Fig. 7G), potentially indicating a reliance of these cells on the activity of this pleiotropic DUB. However, it is important to note that the specific role of USP10 in stabilizing p53 will likely impinge on the utility of targeting USP10; tumors with mutant p53 may be more amenable to USP10 inhibition as a therapeutic approach (Yuan et al. 2010).
Proteotoxic crisis is a key feature of cancer cells, likely due to both an abundance of missense mutations and copy number variation affecting cancer genomes (Deshaies 2014). For this reason, proteasome inhibitors have been used successfully to treat cancers, including multiple myeloma and mantle cell lymphoma, and aneuploid and chromosomally unstable cancer cell lines are preferentially sensitive to drugs interfering with protein folding, autophagy, and HSF1 activation (Tang et al. 2011; Santagata et al. 2013). Finally, inhibitors of the protein quality control AAA ATPase p97 are preferentially cytotoxic to cancer cells (Magnaghi et al. 2013; Chou et al. 2011). Excitingly, the yeast homolog of p97, Cdc48, is an interacting protein of Ubp3 (Ossareh-Nazari et al. 2010a,b), further supporting the idea that the reliance of aneuploid cells on Ubp3 function could be therapeutically relevant. Thus, the identification of additional aneuploidy-tolerating genes will be a crucial approach to illuminate future chemotherapeutic targets.
Materials and methods
Yeast strains and plasmids
Disomes used in this study are derivatives of those published in Torres et al. (2007) or were generated using the same method. De novo gene deletions were generated using published methods (Longtine et al. 1998) in a wild-type W303 yeast strain. Disomes carrying candidate gene deletions were constructed by crosses. Karyotypes of key disomic strains were verified by comparative genomic hybridization as described (Torres et al. 2007) and were analyzed with Java TreeView. All strains are listed in Supplemental Table 2.
Doubling time analysis
Cells were grown overnight at room temperature in yeast extract/peptone medium containing 2% glucose (YPD) and diluted to OD600 = 0.1 the next morning. The growth rate of these cultures at either 25°C or 37°C was measured in triplicate using a BioTek plate reader to take measurements every 15 min for 24 h. Data were analyzed using Gen5 BioTek software. The period of exponential growth was used to calculate doubling time using GraphPad Prism software. Data shown are the average of three to eight biological replicates performed on different days.
Assessment of yeast drug sensitivity and fitness by serial dilution
Overnight cultures in YPD were diluted to OD600 = 1.0, and 1.9 µL of 10-fold serial dilutions was spotted on YPD plates that contained either 50 µg/mL Brefeldin A or 0.75 mM diamide. Drug sensitivity plates were incubated for 2 d at 30°C before imaging.
Analysis of cellular proteotoxicity using the VHL-GFP reporter
The number of VHL-GFP foci was determined as described (Oromendia et al. 2012). Strains harboring the GAL-VHL-GFP fusion were grown at 25°C in YEP 2% raffinose and 2% galactose to mid-log phase. Samples were fixed with 3.7% formaldehyde, and the percentage of cells harboring VHL-GFP foci was determined. At least 300 cells were counted for each strain.
Protein half-life measurements
Cells were grown overnight in medium containing raffinose (selective for the plasmid, if applicable) at room temperature. Expression of tagged substrates was induced by the addition of 2% galactose for 45 min. The t = 0 sample was taken, followed by addition of 2% glucose and 50 µg/mL cycloheximide. Samples were taken at the indicated times after cycloheximide addition and processed as described for Western blot analyses using either 8% or 10% SDS-PAGE.
Protein trafficking assay
Detection of Ccw14p intermediates was performed as described previously (Dodgson et al. 2016). Cell cultures were grown in YPD to mid-exponential phase prior to TCA extraction and Western blot analysis using 10% SDS-PAGE.
Western blot analyses
Yeast cell pellets were suspended in 2 mL of 5% trichloroacetic acid, and, after incubation for 10 min at 4°C, pellets were washed with acetone. Dried pellets were then suspended in 100 µL of breakage buffer (50 mM Tris-HCl at pH 7.5, 1 mM EDTA, 2.75 mM DTT, Roche complete protease inhibitor), glass beads were added, and cells were broken by beating for 5 min on a Biospec minibead beater. SDS sample buffer (3×) was added, and samples were boiled for 5 min and centrifuged briefly. Human cells were lysed in lysis buffer (50 mM Tris- HCl at pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 0.5% NP-40, 10% glycerol, protease inhibitor cocktail [Roche], phosphatase inhibitor cocktail [Roche]) for 30 min on ice. All lysates were loaded on 8%, 10%, or 15% SDS–polyacrylamide gels for electrophoresis and transferred onto nitrocellulose membranes before probing with primary antibody. The primary antibodies used were as follows: mouse anti-GFP (1:1000; Clontech, JL-8), mouse anti-HA (1:1000; Covance, HA.11), rabbit anti-Gcn4 (1:200; Santa Cruz Biotechnology, FL-281), rabbit Ccw14 antiserum (1:1000) (Dodgson et al. 2016), mouse anti-ubiquitin (1:4000; Santa Cruz Biotechnology, P4D1), rabbit anti-LC3 (1:1000; Sigma), rabbit anti-Kar2 antiserum (1:200,000) (Rose et al. 1989), mouse anti-Vph1 (1:4000; Abcam), mouse anti-GAPDH (1:1000; Santa Cruz Biotechnology, G-9), and mouse anti-Pgk1 (1:10,000; Molecular Probes, A-6457). The secondary antibodies used were donkey anti-rabbit conjugated to horseradish peroxidase (HRP) (1:5000; GE Healthcare, NA934) and sheep anti-mouse conjugated to HRP (1:5000; GE Healthcare, NA931). HRP conjugates were detected using Amersham ECL Prime reagent according to the manufacturer's instructions.
Proteasome activity assay
Cells were grown to late exponential phase in YPD before pelleting and snap-freezing. Cell pellets were broken in 200 µL of lysis buffer (50 mM Tris HCl at pH 7.5, 150 mM NaCl, 5 mM MgCl2, 10% glycerol) with zirconia beads on a Biospec minibead beater for 5 min. Lysates were clarified twice by centrifugation at 15,000 rpm for 5 min. Proteasome activity was assayed using the proteasome activity fluorometric assay kit II (UBPBio) according to the manufacturer's instructions. Briefly, 150 µL of warmed substrate (Suc-LLVY-AMC, Z-LLE-AMC, or Boc-LRR-AMC in proteasome assay buffer) was added to the wells of a 96-well plate, and 40 µL of 5 mg/mL cell lysate or lysis buffer was then added. To inhibit proteasome function, 100 µM MG132 was used. Readings were taken every minute for 20 min at 30°C in a BioTek plate reader.
Transcriptional enrichment analysis
Microarray data from aneuploid yeast strains were acquired from Torres et al. (2007). For each strain, genes annotated to disomic chromosomes were excluded from consideration. To determine the degree of aneuploidy in each strain, the number of ORFs present on disomic chromosomes was divided by the total number of ORFs in the yeast genome. A Pearson correlation coefficient (PCC) was calculated between the expression level of each gene and the percent aneuploidy in each strain. A PCC cutoff of r = ±0.4629 (P < 0.01) was applied to identify genes whose expression was correlated or anti-correlated with increasing aneuploidy. Gene ontology terms were identified using GProfiler with a P-value threshold of 10−3 (Reimand et al. 2016).
Mammalian cell culture conditions and drug treatments
Cell lines were cultured in DMEM (Invitrogen) supplemented with 10% FBS, 2 mM L-glutamine, and 100 U/mL penicillin/streptomycin. Cells were grown at 37°C with 5% CO2 in a humidified environment. For experiments involving drug treatments, controls represent cells treated with vehicle alone. Reversine was obtained from Cayman Chemical, and Spautin-1 and BafA1 were purchased from Sigma-Aldrich.
RNAi
Silencer select siRNA oligos to knock down USP10 and MAD2 were purchased from Life Technologies and transfected using RNAiMAX (Invitrogen).
Cell viability counting
To determine the degree of cell proliferation following induction of chromosome missegregation, cells were harvested after 3 d of Mad2 siRNA or reversine treatment to determine cell number using the Cellometer AutoT4 (Nexcelom).
Analysis of USP10 gene expression in large-scale cancer data sets
RNA sequencing data were downloaded from the Broad Institute GDAC Firehose (https://gdac.broadinstitute.org). Normalized read counts from tumor and normal specimens were compared using a Mann-Whitney U-test.
Supplementary Material
Acknowledgments
We thank V. Denic for the mCitrine-Atg8 construct, C. Kaiser for the Ccw14 antiserum, and Frank Solomon, Vlad Denic, and members of the Amon laboratory for suggestions and critical reading of this manuscript. This work was supported by National Institutes of Health CA206157 and GM118066 to A.A. S.E.D. was supported by a Massachusetts Institute of Technology School of Science Fellowship in Cancer Research. S.S. was supported by the American Italian Cancer Foundation (AICF), a Fellowship in Cancer Research from Marie Curie Actions and the Italian Association for Cancer Research (AIRC), and a Koch Institute Quinquennial Cancer Research Fellowship. A.A. is also an investigator of the Howard Hughes Medical Institute and the Glenn Foundation for Biomedical Research.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date ate online at http://www.genesdev.org/cgi/doi/10.1101/gad.287474.116.
References
- Bachmair A, Finley D, Varshavsky A. 1986. In vivo half-life of a protein is a function of its amino-terminal residue. Science 234: 179–186. [DOI] [PubMed] [Google Scholar]
- Baker RT, Tobias JW, Varshavsky A. 1992. Ubiquitin-specific proteases of Saccharomyces cerevisiae. Cloning of UBP2 and UBP3, and functional analysis of the UBP gene family. J Biol Chem 267: 23364–23375. [PubMed] [Google Scholar]
- Barford D. 2010. Structure, function and mechanism of the anaphase promoting complex (APC/C). Quart Rev Biophys 44: 153–190. [DOI] [PubMed] [Google Scholar]
- Baxter BK, Craig EA. 1998. Isolation of UBP3, encoding a de-ubiquitinating enzyme, as a multicopy suppressor of a heat-shock mutant strain of S. cerevisiae. Curr Genet 33: 412–419. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland E, Hult M, Bell SD, Sunnerhagen P, Downs JA. 2007. The Bre5/Ubp3 ubiquitin protease complex from budding yeast contributes to the cellular response to DNA damage. DNA Repair 6: 1471–1484. [DOI] [PubMed] [Google Scholar]
- Bonney ME, Moriya H, Amon A. 2015. Aneuploid proliferation defects in yeast are not driven by copy number changes of a few dosage-sensitive genes. Genes Dev 29: 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew CT, Huffaker TC. 2002. The yeast ubiquitin protease, Ubp3p, promotes protein stability. Genetics 162: 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles JS, Hamilton ML, Petes TD. 2010. Meiotic chromosome segregation in triploid strains of Saccharomyces cerevisiae. Genetics 186: 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Mulla WA, Kucharavy A, Tsai H-J, Rubinstein B, Conkright J, McCroskey S, Bradford WD, Weems L, Haug JS, et al. 2015. Targeting the adaptability of heterogeneous aneuploids. Cell 160: 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew BS, Siew WL, Xiao B, Lehming N. 2010. Transcriptional activation requires protection of the TATA-binding protein Tbp1 by the ubiquitin-specific protease Ubp3. Biochem J 431: 391–399. [DOI] [PubMed] [Google Scholar]
- Chou T-F, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, et al. 2011. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci 108: 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Belgareh N, Haguenauer-Tsapis R, Dargemont C. 2003a. Ubp3 requires a cofactor, Bre5, to specifically de-ubiquitinate the COPII protein, Sec23. Nat Cell Biol 5: 661–667. [DOI] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Dargemont C. 2003b. Deubiquitination, a new player in Golgi to endoplasmic reticulum retrograde transport. J Biol Chem 278: 51989–51992. [DOI] [PubMed] [Google Scholar]
- Deng M, Yang X, Qin B, Liu T, Zhang H, Guo W, Lee SB, Kim JJ, Yuan J, Pei H, et al. 2016. Deubiquitination and activation of AMPK by USP10. Mol Cell 61: 614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Hwang S, O'Sullivan C, Dodgson SE, Gygi SP, Amon A, Torres EM. 2014. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife 3: e03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ. 2014. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol 12: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodgson SE, Kim S, Costanzo M, Baryshnikova A, Morse DL, Kaiser CA, Boone C, Amon A. 2016. Chromosome-specific and global effects of aneuploidy in Saccharomyces cerevisiae. Genetics 202: 1395–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly N, Passerini V, Dürrbaum M, Stingele S, Storchova Z. 2014. HSF1 deficiency and impaired HSP90-dependent protein folding are hallmarks of aneuploid human cells. EMBO J 33: 2374–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairn GD, Curwin AJ, Stefan CJ, McMaster CR. 2007. The oxysterol binding protein Kes1p regulates Golgi apparatus phosphatidylinositol-4-phosphate function. Proc Natl Acad Sci 104: 15352–15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Hathaway NA, Tone Y, Crosas B, Elsasser S, Kirkpatrick DS, Leggett DS, Gygi SP, King RW, Finley D. 2006. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell 127: 99–111. [DOI] [PubMed] [Google Scholar]
- Irniger S, Braus GH. 2003. Controlling transcription by destruction: the regulation of yeast Gcn4p stability. Curr Genet 44: 8–18. [DOI] [PubMed] [Google Scholar]
- Isasa M, Katz EJ, Kim W, Yugo V, González S, Kirkpatrick DS, Thomson TM, Finley D, Gygi SP, Crosas B. 2010. Monoubiquitination of RPN10 regulates substrate recruitment to the proteasome. Mol Cell 38: 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isasa M, Rose CM, Elsasser S, Navarrete-Perea J, Paulo JA, Finley DJ, Gygi SP. 2015. Multiplexed, proteome-wide protein expression profiling: yeast deubiquitylating enzyme knockout strains. J Proteome Res 14: 5306–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isono E, Saito N, Kamata N, Saeki Y, Toh-e A. 2005. Functional analysis of Rpn6p, a lid component of the 26 S proteasome, using temperature-sensitive rpn6 mutants of the yeast Saccharomyces cerevisiae. J Biol Chem 280: 6537–6547. [DOI] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J. 2008. Misfolded proteins partition between two distinct quality control compartments. Nature 454: 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly SP, Bedwell DM. 2015. Both the autophagy and proteasomal pathways facilitate the Ubp3p-dependent depletion of a subset of translation and RNA turnover factors during nitrogen starvation in Saccharomyces cerevisiae. RNA 21: 898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Cuervo AM, Seglen PO. 2014. Methods for monitoring autophagy from yeast to human. Autophagy 3: 181–206. [DOI] [PubMed] [Google Scholar]
- Kraft C, Deplazes A, Sohrmann M, Peter M. 2008. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol 10: 602–610. [DOI] [PubMed] [Google Scholar]
- Kvint K, Uhler JP, Taschner MJ, Sigurdsson S, Erdjument-Bromage H, Tempst P, Svejstrup JQ. 2008. Reversal of RNA polymerase II ubiquitylation by the ubiquitin protease Ubp3. Mol Cell 30: 498–506. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang Y. 2013. Ras protein/cAMP-dependent protein kinase signaling is negatively regulated by a deubiquitinating enzyme, Ubp3, in yeast. J Biol Chem 288: 11358–11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, et al. 2011. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147: 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- Magnaghi P, D'Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, et al. 2013. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9: 548–556. [DOI] [PubMed] [Google Scholar]
- Mao P, Smerdon MJ. 2010. Yeast deubiquitinase Ubp3 interacts with the 26 S proteasome to facilitate Rad4 degradation. J Biol Chem 285: 37542–37550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R, Zielke CL, Ronald Zielke H, Pevsner J. 2003. Global up-regulation of chromosome 21 gene expression in the developing down syndrome brain. Genomics 81: 457–467. [DOI] [PubMed] [Google Scholar]
- McClellan AJ, Scott MD, Frydman J. 2005. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 121: 739–748. [DOI] [PubMed] [Google Scholar]
- Müller M, Kötter P, Behrendt C, Walter E, Scheckhuber CQ, Entian K-D, Reichert AS. 2015. Synthetic quantitative array technology identifies the Ubp3–Bre5 deubiquitinase complex as a negative regulator of mitophagy. Cell Rep 10: 1215–1225. [DOI] [PubMed] [Google Scholar]
- Nagaoka SI, Hassold TJ, Hunt PA. 2012. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet 13: 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair U, Thumm M, Klionsky DJ, Krick R. 2014. GFP-Atg8 protease protection as a tool to monitor autophagosome biogenesis. Autophagy 7: 1546–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nostramo R, Varia SN, Zhang B, Emerson MM, Herman PK. 2016. The catalytic activity of the Ubp3 deubiquitinating protease is required for efficient stress granule assembly in Saccharomyces cerevisiae. Mol Cell Biol 36: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi A, Ohori M, Iwai K, Nakayama Y, Nambu T, Morishita D, Kawamoto T, Miyamoto M, Hirayama T, Okaniwa M, et al. 2015. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat Commun 6: 7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oling D, Eisele F, Kvint K, Nystrom T. 2014. Opposing roles of Ubp3-dependent deubiquitination regulate replicative life span and heat resistance. EMBO J 33: 747–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia AB, Dodgson SE, Amon A. 2012. Aneuploidy causes proteotoxic stress in yeast. Genes Dev 26: 2696–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bonizec MEL, Cohen M, Dokudovskaya S, Delalande FCO, Schaeffer C, Van Dorsselaer A, Dargemont C. 2010a. Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep 11: 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Cohen M, Dargemont C. 2010b. The Rsp5 ubiquitin ligase and the AAA–ATPase Cdc48 control the ubiquitin-mediated degradation of the COPII component Sec23. Exp Cell Res 316: 3351–3357. [DOI] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. 2010. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468: 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz S, Hwang ES, Visintin R, Amon A. 1998. The regulation of Cdc20 proteolysis reveals a role for APC components Cdc23 and Cdc27 during S phase and early mitosis. Curr Biol 8: 750–760. [DOI] [PubMed] [Google Scholar]
- Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J. 2016. g:Profiler—a Web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 44: W83–W89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MD, Misra LM, Vogel JP. 1989. KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 57: 1211–1221. [DOI] [PubMed] [Google Scholar]
- Santagata S, Mendillo ML, Tang YC, Subramanian A, Perley CC, Roche SP, Wong B, Narayan R, Kwon H, Koeva M, et al. 2013. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 341: 1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. 2010. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 190: 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Vasile E, White E, Amon A. 2015. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev 29: 2010–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Finley D. 2014. Regulation of proteasome activity in health and disease. Biochim Biophys Acta 1843: 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. 2011. Aneuploidy drives genomic instability in yeast. Science 333: 1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. 2012. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci 109: 12644–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. 2004. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem 279: 29889–29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncini C, Berdo I, Draetta G. 2001. Ras–GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene 20: 3869–3879. [DOI] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Peplowska K, Cox JUR, Mann M, Storchova Z. 2012. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y-C, Williams BR, Siegel JJ, Amon A. 2011. Identification of aneuploidy-selective antiproliferation compounds. Cell 144: 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924. [DOI] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A. 2010. Identification of aneuploidy-tolerating mutations. Cell 143: 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin C, Tomson BN, Rahal R, Paulson J, Cohen M, Taunton J, Amon A, Visintin R. 2008. APC/C–Cdh1-mediated degradation of the Polo kinase Cdc5 promotes the return of Cdc14 into the nucleolus. Genes Dev 22: 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. 2006. Does aneuploidy cause cancer? Curr Opin Cell Biol 18: 658–667. [DOI] [PubMed] [Google Scholar]
- Willems AR, Schwab M, Tyers M. 2004. A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta 1695: 133–170. [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. 2008. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322: 703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lindquist S. 1993. Heat-shock protein hsp90 governs the activity of pp60v-src kinase. Proc Natl Acad Sci 90: 7074–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. 2010. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 140: 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.