

Abstract
Genome sequence analysis of Middle East respiratory syndrome coronavirus (MERS-CoV) variants from patient specimens has revealed the evolutionary dynamics and mechanisms of pathogenesis of the virus. However, most studies have analyzed the consensus sequences of MERS-CoVs, precluding an investigation of intrapatient heterogeneity. Here, we analyzed non–consensus sequences to characterize intrapatient heterogeneity in cases associated with the 2015 outbreak of MERS in South Korea. Deep-sequencing analysis of MERS-CoV genomes performed on specimens from eight patients revealed significant intrapatient variation; therefore, sequence heterogeneity was further analyzed using targeted deep sequencing. A total of 35 specimens from 24 patients (including a super-spreader) were sequenced to detect and analyze variants displaying intrapatient heterogeneity. Based on the analysis of non–consensus sequences, we demonstrated the intrapatient heterogeneity of MERS-CoVs, with the highest level in the super-spreader specimen. The heterogeneity could be transmitted in a close association with variation in the consensus sequences, suggesting the occurrence of multiple MERS-CoV infections. Analysis of intrapatient heterogeneity revealed a relationship between D510G and I529T mutations in the receptor-binding domain (RBD) of the viral spike glycoprotein. These two mutations have been reported to reduce the affinity of the RBD for human CD26. Notably, although the frequency of both D510G and I529T varied greatly among specimens, the combined frequency of the single mutants was consistently high (87.7% ± 1.9% on average). Concurrently, the frequency of occurrence of the wild type at the two positions was only 6.5% ± 1.7% on average, supporting the hypothesis that selection pressure exerted by the host immune response played a critical role in shaping genetic variants and their interaction in human MERS-CoVs during the outbreak.
Keywords: recurrent upper and lower respiratory tract infections
INTRODUCTION
Middle East respiratory syndrome coronavirus (MERS-CoV) was first isolated from a patient in Saudi Arabia in 2012 and has been shown to cause severe acute respiratory illness, including fever, cough, and shortness of breath (Zaki et al. 2012). As of March 1, 2016, 1638 laboratory-confirmed cases (587 deaths; 36% case fatality rate [CFR]) have been reported to the World Health Organization. A South Korean outbreak of MERS began in May 2015, and its transmission continued until early July, resulting in 186 laboratory-confirmed cases with 38 deaths (20.4% CFR). In contrast to previous studies, which have suggested limited person-to-person transmissibility of MERS-CoV (Breban et al. 2013; Cotten et al. 2013b), many secondary and tertiary cases of transmission occurred during the South Korean outbreak. Importantly, more than half of the tertiary cases were transmitted from one particular super-spreader, called Patient 14 in this study. This unusual transmission pattern raised questions related to transmissibility as well as the potential adaptations of MERS-CoV to the human host. To address these questions, several researchers have investigated MERS-CoV sequences. However, all previous studies on the South Korean outbreak have focused only on the consensus sequences of MERS-CoVs (Wang et al. 2015; Kim et al. 2016a,b; Park et al. 2016; Seong et al. 2016).
The evolutionary dynamics of RNA viruses are complex because of their high mutation rates, rapid replication rates, and large population sizes. Multiple rounds of replication of a given viral genome can generate a cloud of diverse variants or a heterogeneous viral population. Previous reports on other RNA viruses have suggested that quasispecies diversity, rather than the selection of individual variants, correlates with pathogenicity and enables adaptation to new environments (Vignuzzi et al. 2006; Lauring and Andino 2010). The importance of characterizing viral populations as swarms with similar variants has led several groups to use next-generation sequencing (NGS) technologies on clinical samples to explore the complexity of the spectrum of mutant viruses, including HIV-1 and hepatitis B virus (Bushman et al. 2008; Eriksson et al. 2008; Margeridon-Thermet et al. 2009). Although the description of MERS-CoV quasispecies is far from completion, previous studies that recovered whole MERS-CoV genome sequences from dromedary camels revealed the existence of intrahost single-nucleotide variants (Briese et al. 2014; Borucki et al. 2016).
To date, no intrahost MERS-CoV single-nucleotide variants have been identified in humans, except by Cotton et al. (2013a), where data from one specimen showed the presence of non–consensus variants (<5%). Currently, it is hard to discern whether only consensus sequences have been reported or whether human MERS-CoV sequences represent almost clonal virus populations within individual cases. By analyzing non–consensus sequences in 35 specimens from 24 patients infected during the South Korean outbreak (2015), we demonstrated the existence of intrapatient heterogeneity and investigated its functional implications.
RESULTS
Patients and Sample Collection
Among the 24 cases, 14 were patients, four were caregivers, and six were health-care workers (HCWs) (Table 1). Patient 14 was a second-generation case who had been exposed to the index patient. Exposure to Patient 14 led to a second wave of the outbreak, resulting in a total of 81 third-generation infections (Oh et al. 2015). Specimens from 20 of these third-generation cases were analyzed in this study (Supplemental Fig. S1). Patients 162, 164, and 169 were HCWs and fourth-generation case patients who had been exposed to Patient 135.
Table 1.
Characteristics of patients included in this study
| Patient no. | Age/sex | Case type | Disease severity | Underlying disease | Transmission | |
|---|---|---|---|---|---|---|
| Generation | Source patient | |||||
| 14 | 35/M | Patient | Severe | None | 2nd | 1 |
| 48 | 37/M | Caregiver | Moderate | None | 3rd | 14 |
| 50 | 80/F | Patient | Severe | Posterior cerebral artery infarction | 3rd | 14 |
| 61 | 55/M | Caregiver | Severe | None | 3rd | 14 |
| 62 | 30/M | HCW | Mild | None | 3rd | 14 |
| 66 | 42/F | Patient | Moderate | Myelodysplastic syndrome | 3rd | 14 |
| 68 | 54/F | Caregiver | Moderate | None | 3rd | 14 |
| 75 | 62/M | Patient | Moderate | Rectal cancer | 3rd | 14 |
| 77 | 63/M | Patient | Severe | Necrotizing pancreatitis | 3rd | 14 |
| 78 | 41/F | HCW | Moderate | None | 3rd | 14 |
| 80 | 34/M | Patient | Severe | Malignant lymphoma | 3rd | 14 |
| 99 | 47/M | Caregiver | Moderate | None | 3rd | 14 |
| 100 | 32/F | Patient | Moderate | Neuroendocrine tumor | 3rd | 14 |
| 101 | 84/M | Patient | Severe | Renal cell carcinoma | 3rd | 14 |
| 102 | 48/F | Patient | Moderate | None | 3rd | 14 |
| 103 | 65/M | Patient | Moderate | Middle cerebral artery infarction | 3rd | 14 |
| 134 | 67/F | Patient | Mild | None | 3rd | 14 |
| 135 | 32/M | HCW | Severe | None | 3rd | 14 |
| 155 | 42/F | Patient | Mild | Hypertrophic cardiomyopathy | 3rd | 14 |
| 157 | 59/M | Patient | Severe | Lung cancer | 3rd | 14 |
| 162 | 32/M | HCW | Severe | None | 4th | 135 |
| 164 | 35/F | HCW | Moderate | None | 4th | 135 |
| 169 | 33/M | HCW | Moderate | None | 4th | 135 |
| 177 | 49/F | Patient | Severe | Malignant lymphoma | 3rd | 14 |
HCW, health-care worker.
To determine whether viral genomic variability was related to disease severity, patient cases were categorized as mild (symptomatic cases without pneumonia; n = 3), moderate (cases with pneumonia; n = 11), and severe (cases with respiratory failure or death; n = 10) (Table 1). In eight cases, multiple lower respiratory tract specimens were obtained (Supplemental Table S1). Specimens from patients 50, 77, and 135 were obtained before and after respiratory failure.
Characterization of MERS-CoV Genome
Specimens from eight patients who had been identified as MERS-CoV-positive were sequenced. The samples were found positive by both upstream-of-the-envelope gene (upE) and open reading frame 1a (orf1a) real-time polymerase chain reaction (PCR) assays, with cycle threshold (Ct) values averaging 17.3 (15.4–22.6) and 18.3 (15.4–23.0), respectively (Supplemental Table S1). For each patient, a MERS-CoV consensus sequence that indicated the major nucleotide at each genomic position was generated. The genome sequences of the eight isolates differed from each other at only seven positions (Table 2). The nucleotide substitutions were all nonsynonymous and occurred in the orf1ab (n = 3) and S (n = 4) genes (Table 2). The sequences of the eight isolates had high nucleotide identities (ranging from 99.96% to 100%, with 99.98% to 100% sequence identities in ORF 1a and 1b [orf1ab] and 99.85% to 100% identities in the spike glycoprotein gene) with recently published sequences of MERS-CoV isolates from the outbreak in South Korea (Kim et al. 2015; Lu et al. 2015; Seong et al. 2016). The E, M, and N genes were 100% identical to the previously described MERS-CoV isolates from South Korea. The sequence from Patient 14 in this study differed from the sequence from a previous study at two positions (Seong et al. 2016). Because the frequencies of the major nucleotides at these positions were close to 50% in this study, the differences between the consensus sequences may have arisen from small technical variations in measurement.
Table 2.
Summary of genetic variants in the full-genome sequences of Middle East respiratory syndrome coronavirus (MERS-CoVs) isolates from the South Korean outbreak (2015)
| Gene | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| orf1ab | S | orf4a/b | orf5 | ||||||||||||||||||||||||
| Genomic position | 2917 | 5852 | 5917 | 6126 | 6286 | 7933 | 8995 | 10089 | 11257 | 18916 | 19075 | 20411 | 21638 | 21726 | 21864 | 22356 | 22984 | 23041 | 23043 | 23303 | 24383 | 24622 | 25495 | 25903 | 25968 | 26109 | 26979 |
| Protein | nsp3 | nsp | nsp | nsp | nsp | nsp | S | NS4A | NS4B | NS5 | |||||||||||||||||
| 4 | 5 | 6 | 14 | 15 | |||||||||||||||||||||||
| Position at protein | 27 | 1005 | 1027 | 1097 | 1150 | 1699 | 166 | 24 | 107 | 305 | 358 | 280 | 61 | 91 | 137 | 301 | 510 | 529 | 530 | 616 | 976 | 1056 | 1347 | 18 | 39 | 6 | 47 |
| Substitution | N | S | N | N | N | N | N | N | N | S | S | N | S | N | N | N | N | N | N | S | S | N | N | N | S | N | N |
| Nucleotide change (reference/alternative) | A/G | G/A | C/T | G/C | C/T | C/T | G/T | A/G | C/T | G/A | G/A | A/T | C/G | C/T | A/C | C/T | A/G | T/C | G/C | T/C | C/T | A/G | C/T | C/T | A/T | T/C | T/C |
| Amino acid change (reference/alternative) | N/S | S | S/F | A/P | A/V | S/F | R/M | S/G | A/V | L | R | T/S | G | H/Y | S/R | R/C | D/G | I/T | V/L | V | I | Q/R | P/L | P/S | S | M/D | V/A |
| Isolates (GenBank ID) | |||||||||||||||||||||||||||
| ChinaGD01 (KT006149) | C | T | T | A | G | C | T | C | |||||||||||||||||||
| MERS-CoV/KOR/KNIH/002_05_2015 (KT029139) | C | C | T | ||||||||||||||||||||||||
| KOREA/Seoul/163-2-2015 (KT374050) | A | T | T | A | C | T | G | C | |||||||||||||||||||
| KOREA/Seoul/163-1-2015 (KT374051) | A | T | T | A | C | T | G | C | |||||||||||||||||||
| KOREA/Seoul/014-1-2015 (KT374052) | T | T | A | C | C | T | C | ||||||||||||||||||||
| KOREA/Seoul/014-2-2015 (KT374053) | T | T | A | A | C | C | T | C | · | ||||||||||||||||||
| KOREA/Seoul/035-1-2015 (KT374054) | T | G | T | A | C | C | T | C | C | ||||||||||||||||||
| KOREA/Seoul/035-2-2015 (KT374055) | T | G | T | A | C | C | T | C | C | ||||||||||||||||||
| KOREA/Seoul/168-1-2015 (KT374056) | G | T | A | T | G | C | T | T | C | ||||||||||||||||||
| KOREA/Seoul/168-2-2015 (KT374057) | G | T | A | T | G | C | T | T | T | C | |||||||||||||||||
| MERS-CoV/KOR/Seoul/014_2015 (KX034093) | T | A | T | C | C | T | C | ||||||||||||||||||||
| MERS-CoV/KOR/Seoul/050-1-2015 (KX034094) | T | A | G | C | T | C | |||||||||||||||||||||
| MERS-CoV/KOR/Seoul/066-2015 (KX034095) | T | A | G | C | T | C | |||||||||||||||||||||
| MERS-CoV/KOR/Seoul/077-2-2015 (KX034096) | T | T | A | C | C | T | C | ||||||||||||||||||||
| MERS-CoV/KOR/Seoul/080-3-2015 (KX034097) | T | A | T | G | C | T | C | ||||||||||||||||||||
| MERS-CoV/KOR/Seoul/162-1-2015 (KX034098) | T | T | A | C | C | T | C | ||||||||||||||||||||
| MERS-CoV/KOR/Seoul/169-2015 (KX034099) | T | T | T | A | T | C | C | T | C | ||||||||||||||||||
| MERS-CoV/KOR/Seoul/177-3-2015 (KX034100) | T | T | A | C | C | T | C | ||||||||||||||||||||
Consensus sequences of MERS-CoV associated with the South Korean outbreak reported in this study and those from previous studies were compared. KT029139 was reported in Kim et al. 2015 and KT374050—KT374057 were in Kim et al. 2016a. All sequences were aligned with the National Center for Biotechnology Information (NCBI) reference sequence (NC_019843.3). At positions with discordant nucleotides, alternative nucleotides are displayed.
N, nonsynonymous substitution; S, synonymous substitution.
Using 105 previously published genome sequences of MERS-CoVs, including those from recent cases in South Korea, we analyzed the phylogenetic relationships of eight newly sequenced isolates. The new sequences clustered together with the other MERS-CoV isolates from the 2015 South Korean outbreak (Supplemental Fig. S2; Kim et al. 2015; Lu et al. 2015; Seong et al. 2016).
Presence of Intrapatient Heterogeneity in MERS-CoVs
Analyzing the MERS-CoV genome sequences, we noticed that a significant number of nucleotide sites had mixed bases. Because deep sequencing not only generates a consensus sequence but also identifies non–consensus nucleotides at each position, Cotton et al. (2013a) analyzed nucleotide variants present at >1% frequency to determine the variants important for intrahost evolution. In the previous study, the nucleotide variants present at a frequency greater than the sequencing error rate (i.e., 1%) were considered to be true variants, although this estimate may be conservative for most applications. However, errors introduced during reverse transcription and PCR amplification can significantly distort estimates of allele frequencies, especially if a limited amount of RNA is used as an input. Thus, in this study, we evaluated the reproducibility of allele frequencies of non–consensus nucleotides. For this purpose, duplicate libraries generated by independent complementary DNA (cDNA) synthesis using the same RNA samples were sequenced and compared. Allele frequencies of duplicate samples from Patients 80 and 162 were significantly correlated, implying that the majority of mixed bases were a reflection of intrapatient heterogeneity rather than a result of sequencing errors (Fig. 1A,B).
Figure 1.

Reproducibility assessment for the detection of non–consensus variants. Correlation of allele frequencies between sample replicates from (A) Patient 80 and (B) Patient 162. (C) Representative validation by the Sanger sequencing method.
Despite the relatively high reproducibility, we focused on nucleotide variants present at a relatively high frequency (i.e., ≥30%) to minimize false positives among non–consensus variants. Positions where a nucleotide variant was present at a frequency of ≥30% in any of the eight isolates were selected for further investigation using additional samples. Targeted deep sequencing was performed on an additional 27 samples targeting 3003-bp regions that included the variable sites. Eleven libraries from independent cDNA syntheses of five samples, including four libraries in duplicate and one in triplicate, were constructed to evaluate the technical noise in measuring allele frequency. Only variants with frequencies significantly greater than those from technical noise were considered true variants. Technical noise is described in Methods and was based on a 5% significance level after performing Bonferroni adjustment. A total of 16 positions displaying intrapatient heterogeneity were identified through statistical testing. The results from seven specimens were validated using Sanger sequencing. We tested nine sites and confirmed concordant mixed bases at seven positions—namely, positions 6884, 7317, 11257, 21726, 22356, 22984, and 23041 (Fig. 1C), but found discrepancies at positions 7322 and 19075. Although we were not able to validate all variants by Sanger sequencing owing to a limited sample volume, the results from the test set showed a 78% validation rate. In addition, the frequencies of the two variants at positions 22984 and 23041 (i.e., G at position 22984 and T at 23041; A at 22984 and C at 23041) were highly correlated in the specimens (Supplemental Fig. S3), suggesting a tight linkage between these variants instead of random sequencing errors.
Fourteen positions exhibiting mixed bases are presented in Figure 2. These include all seven nucleotide variants identified in the full-genome sequences of the eight MERS-CoV isolates, indicating that intrapatient MERS-CoV heterogeneity was closely associated with isolate-specific variants in the consensus sequences. Taken together, our data demonstrate the intrapatient heterogeneity of MERS-CoVs.
Figure 2.

Summary of variable sites among 35 samples from 24 patients. At each position, a mixed base is displayed and colored if its frequency was >10%. For mixed bases, the font size reflects the frequency of that base. Mod, moderate; Sev, severe.
Interactions Between Genetic Variants
As described above, there was a tight linkage between positions 22984 and 23041—the variant at one position and the wild type at the other position or vice versa (i.e., D510G and I529; D510 and I529T). Based on the genetic linkage, a functional relationship was expected between the variants at these two positions. In fact, these two mutations were located in one of the two major binding patches in the receptor-binding domain (RBD), where escape mutations from neutralizing antibodies were previously reported (Tang et al. 2014).
Based on the tight correlation of base frequencies at positions 22984 and 23041, our data suggested that the double mutant carrying D510G and I529T is rare. Therefore, we selected sequencing reads covering both positions in each specimen to measure the frequencies of the double mutants (i.e., D510G, I529T), the single mutants (i.e., D510G, I529 and D510, I529T), and the wild type (i.e., D510, I529). On average, the frequency of the double mutant was only 1.4% ± 0.3%, whereas the single mutants and the wild type were present at 87.7% ± 1.9% and 6.5% ± 1.7%, respectively (Fig. 3). Recently, both D510G and I529T mutations in RBD were shown to reduce its affinity for human CD26 compared with the wild-type RBD (Kim et al. 2016c). Although the previous study did not test the binding affinity of the double mutant, in our data the two mutations are mutually exclusive, suggesting that the double mutant severely impairs viral fitness. Notably, although the frequency of each single mutant varied greatly among specimens, the combined frequency of both single mutants was consistently high in most samples. At the same time, the frequency of the wild type was no more than 10% in most samples (31 of 35 specimens). Thus, our data suggest that there was strong selection pressure favoring the variants of the spike glycoprotein with reduced affinity for the host receptor.
Figure 3.
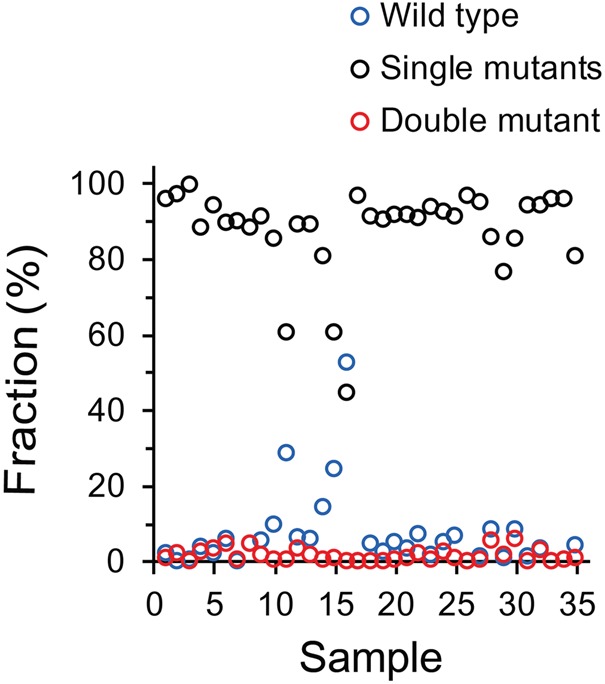
The fraction of mutant and wild-type alleles at positions 510 and 529 of the spike glycoprotein. To measure the frequencies of the mutant and the wild-type alleles, sequencing reads covering positions 510 and 529 were selected. For each specimen, the read counts supporting each type were divided by the total read counts. The frequencies of the double mutants (D510G, I529T), the single mutants (D510G, I529 and D510, I529T), and the wild-type (D510, I529) alleles are plotted for 35 specimens sorted by sample number.
Another notable nonsynonymous substitution was A107V (at position 11257), which occurred in the nonstructural protein 6 (nsp6) coding region within orf1ab. This A107V variation in nsp6 and the I529T substitution in the spike glycoprotein were frequently found in the South Korean isolates and appeared to be genetically linked (Fig. 2; Supplemental Table S2). Nsp6 is a membrane-spanning protein and is an integral component of the viral replication complex involved in double-membrane vesicle formation (Lundin et al. 2014). Although the functional importance of these MERS-CoV variants in viral replication and their interaction with the host immune system remain to be elucidated, our data showed that the selection of these variants may not be independent from each other, suggesting the unit of selection may be a combination of variants rather than individual variants.
DISCUSSION
In this study, we demonstrated the intrapatient heterogeneity of MERS-CoVs, ruling out the possibility of technical noise. Based on the statistical test described above, we first selected 16 non–consensus variant candidates with frequencies significantly higher than those from technical noise. Among those variant candidates, we tested nine candidates by Sanger sequencing and validated seven variants, resulting in a 78% validation rate. After removal of the two nonvalidated candidates, we ultimately listed a total of 14 non–consensus variants, expecting an 89% validation rate. There was a tight correlation between the frequencies of the two variants at positions 22984 and 23041. These results suggest that technical artifacts were highly unlikely to have generated such non–consensus variants. Our analysis was limited to relatively high-frequency variants because the sequencing error rate presents certain limitations for the study of RNA as opposed to DNA viruses. Technical advances such as the use of unique molecular identifiers to remove PCR errors may produce more accurate descriptions of MERS-CoV populations in patients (Kinde et al. 2011). Despite the limitations, we demonstrated the presence of heterogeneous MERS-CoV populations in clinical samples from MERS-CoV-infected patients.
Given the large number of cases that resulted from exposure to Patient 14, it was intriguing that Patient 14 displayed the highest intrapatient heterogeneity. Among the 10 single-nucleotide variants that were observed in more than one patient, six variants were present at frequencies >0% in Patient 14 (Fig. 2). Thus, the majority of intra- and interpatient heterogeneity observed in different patients seemed to originate from the variants present in Patient 14. Our results showed that intrapatient heterogeneity can be transmitted in some cases, indicating that humans can be simultaneously infected with more than one MERS-CoV.
Because most of the mixed bases observed in Patient 14 were not completely fixed in the subsequent generation of cases, the single-nucleotide variants might not have had significantly higher fitness than wild type. For example, the sequence from Patient 14 had a mixed base C/T at position 11257. Among sequences from patients exposed to Patient 14, a subset displayed either C or T alone with little intrapatient heterogeneity, whereas others showed similar intrapatient heterogeneity to Patient 14. In addition, we could not find any significant differences in genetic variants based on disease severity group (Supplemental Tables S3 and S4). Taken together, genetic variant composition in each patient varied, regardless of the transmissibility or disease severity, suggesting that transmission of individual genetic sequences was stochastic rather than selective. Even if the genetic variants had a selective advantage, the advantage of individual genetic sequences might have been weak and/or varied depending on the patient.
Although we could not provide a clear picture of the functional interactions among distinct genetic variants in the population, the high ratio of nonsynonymous to synonymous substitutions offered a clue to their functional impact. The 14 variants consisted of 12 nonsynonymous and two synonymous substitutions. Assuming that the unit of selection might not be an individual variant but the population as a whole, we calculated dN (the number of nonsynonymous changes per nonsynonymous site) and dS (the number of synonymous changes per synonymous site) by the Pamilo–Bianchi–Li method (Li 1993; Pamilo and Bianchi 1993). The dN/dS ratio was 1.03, hinting at the possibility of positive selection. Notably, six of the 12 nonsynonymous substitutions were found in the spike glycoprotein-coding region. As quasispecies theory proposes, the unit of selection might not be an individual virus but the population as a whole. Our data showed that the frequencies of the single mutants (D510G, I529 and D510, I529T) significantly fluctuated among specimens, but the combined frequency of the single mutants was consistently high in most samples, suggesting a combination of variants as the unit of selection.
Recently, in an analysis of 13 MERS-CoV genomes associated with the 2015 outbreak in South Korea, Kim et al. (2016c) reported that 11 of those genomes had an I529T mutation in RBD, and one had a D510G mutation. The study showed that D510G and I529T mutations resulted in reduced affinity of RBD for the human CD26 receptor compared with the wild-type RBD. Based on the spread of a mutant MERS-CoV with reduced affinity for the receptor, the authors suggested that MERS-CoV adaptation during human-to-human spread might be driven by host immunological pressure such as neutralizing antibodies (Kim et al. 2016c). Consistent with the previous analysis, our data showed that the I529T and D510G mutations were observed in the consensus sequences from 29 and 4 of 35 samples, respectively. Furthermore, our analysis at an intrapatient level showed that the frequency of the wild type at both positions was only 6.5% ± 1.7% on average, supporting a strong selection pressure favoring the variants over the wild type.
Thus, we questioned whether the selection pressure was exerted by a host immune response such as neutralizing antibodies, as previously suggested (Kim et al. 2016c). In such cases, a reduction in host immune pressure might increase the frequency of the wild type. Four specimens displaying relatively high frequency (i.e., >10%) of the wild type belonged to Patients 77 and 80. We noticed dramatic changes in routine blood test results such as white blood cell (WBC) counts and C-reactive protein (CRP) levels in these patients. We analyzed 19 serial specimens from eight patients. For this, we used normalized WBC count values expressed as a percentage of the first of a series of samples from each patient. Serial samples from Patients 77 and 80 displayed a dramatic decrease in the normalized WBC count and a simultaneous increase in the frequency of the wild-type allele (Supplemental Fig. S4). Whereas WBC counts significantly decreased in those patients, the CRP level increased during the period of MERS-CoV infection, indicating an impaired immune response. Although these data are limited owing to the small sample size and the lack of direct measurement of host immunological pressure, our results suggest that the selection pressure exerted by the host immune response might favor variants with reduced affinity to the host receptor; therefore, a reduction in this selection pressure resulted in the expansion of viruses with the wild-type allele. The characterization and quantification of neutralizing antibodies in patients over time is required to determine their association with the mutants and to validate the hypothesis. Moreover, more accurate descriptions of MERS-CoV populations within patients will advance our understanding of the complex molecular evolution of MERS-CoVs.
Because the evolution of a virus is a continuous process that takes place during intrapatient infection and interpatient transmission, the evolution of MERS-CoVs should be studied at both intra- and interpatient levels to obtain a better understanding of the principles underlying the complex molecular evolution of MERS-CoVs in natural populations. However, our knowledge of MERS-CoV evolution primarily depends on analysis at the interpatient level, ignoring genetic diversity within individual patients. In this study, we demonstrated intrapatient heterogeneity in human MERS-CoV isolates. Based on the analyses of the genetic diversity of MERS-CoVs at both the intra- and interpatient levels, our results shed light on the evolutionary dynamics of MERS-CoVs associated with the South Korean outbreak.
METHODS
Collection of Clinical Specimens
A total of 35 clinical specimens from 24 patients were determined positive for MERS-CoV and were included in this study. Clinical information and information about possible exposure to other MERS-CoV patients was collected from the electronic medical records and publicly available data from multiple sources including the South Korea Centers for Disease Control and Prevention and the South Korea Ministry of Health and Welfare. These data included age, gender, epidemiologic link, dates of suspected exposure to MERS patients, initial symptoms, date of symptom onset, and clinical courses.
MERS-CoV Real-Time Reverse Transcription PCR Assays
RNA was extracted from clinical specimens using either a QIAamp DSP Viral RNA Mini Kit (Catalog No. 61904, QIAGEN GmbH) or an automated MagNAPure 96 extraction instrument with a total nucleic acid isolation kit (Roche). The extraction was performed according to the manufacturers’ instructions and the extracted RNA was stored at −70°C.
MERS-CoVs were detected in specimens using real-time reverse transcription (rRT)-PCR. Extracted RNAs were screened by targeting the upE region, and positive results were confirmed by a subsequent amplification of orf1a using a PowerChek MERS Real-Time PCR kit (Kogene Biotech, Seoul, South Korea). All rRT-PCR reactions were performed using the 7500 Fast Real-Time PCR System (Applied Biosystems) with a total reaction volume of 20 μL (15 μL of PCR reaction mixture and 5 μL of template RNA). The thermocycling conditions included a reverse transcription reaction for 30 min at 50°C, followed by 10 min at 95°C, and then 40 cycles of 15 sec at 95°C and 60 sec at 60°C. A positive viral template control and a nontemplate control were included in each run. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was amplified simultaneously as an internal control to monitor PCR inhibition. A positive result was identified by a well-defined exponential fluorescence curve that crossed the defined threshold at ≤35 cycles in both the upE and ORF1a assays.
Reverse Transcription and PCR Amplification for Sequencing
Five microliters of viral RNA from each sample was used as a template for cDNA synthesis using a Superscript III First-Strand Synthesis System (Life Technologies). Equal amounts of cDNA product were used to perform PCR with a Herculase II Fusion DNA polymerase (Agilent Technologies). For full-genome sequencing, primers described by Cotten et al. (2013a) were used with minor modifications for efficient amplification. A set of 60 primers was used to generate fifteen 2.5-kb overlapping amplicons (four primers per amplicon) and three additional primers were added for the extreme termini of the genome (Supplemental Table S5). Primers used for the targeted sequencing are listed in Supplemental Table S6. Primers and nucleotides were removed from the final PCR products using a MiniElute PCR purification kit (QIAGEN).
Sequencing
The PCR products from each patient were combined into a pool of approximately equal molarity and subjected to paired-end library construction with a TruSeq Nano DNA Sample prep kit (Illumina). Libraries were sequenced using an Illumina MiSeq sequencer (Illumina), generating 150-bp paired-end reads. On average, 4113× and 9361× raw read depths were achieved for full-genome and targeted deep sequencing, respectively. Sequencing metrics including mean depth of coverage of each sample are summarized in Supplemental Table S7. Bases were called with MiSeq reporter software (ver. 2.4.60). The reads were quality filtered using the command “fastq_quality_filter,” which required a minimum of 90% bases with a quality score of 20 or higher. Paired-end reads were aligned with the National Center for Biotechnology Information (NCBI) MERS-CoV reference sequence (NC_019843.3) using the Burrows–Wheeler Aligner (BWA) (Li and Durbin 2010). SAMtools v0.1.19 (Li et al. 2009), GATK v2.4-7 (McKenna et al. 2010), and Picard v1.93 were used for sorting SAM/BAM files, local realignment, and duplicate markings, respectively. We used the SAMtools “mpileup” command to determine the read depth. The consensus sequence was determined from the most commonly expressed nucleotide at each position.
Phylogenetic Analysis
We downloaded a total of 105 human MERS-CoV genome sequences from the NCBI database (http://www.ncbi.nlm.nih.gov/genomes/VirusVariation/Database/nph-select.cgi?cmd=database&taxid=1335626). Combining the eight MERS-CoV genome sequences from our study with the downloaded sequences, a phylogenetic analysis was performed after aligning the sequences using MUSCLE (http://drive5.com/muscle). A total of 30,130 aligned nucleotides were input into MEGA 7.0 (http://www.megasoftware.net) (Tamura et al. 2013), and a phylogenetic tree was constructed using the neighbor-joining method. The GenBank accession number followed by a brief description of the sequence, including the virus variant name and the country of isolation, is provided for each sequence in Supplemental Figure S2. Evolutionary distances were computed using the Kimura two-parameter model. The scale bar at the bottom of the figure indicates nucleotide substitutions per site.
Determination of Technical Noise and Significance Test for Variants
We used Xijbr to denote the allele frequency for the ith sample, the jth chromosomal position, the base (A, T, G, or C), and the rth replicated experiment. If the total depth was <100× at a given ijr, we treated it as missing, irrespective of the b value. We defined the background noise value (Nijb) as the average of the absolute differences between replicated data for all combinations:
Each variant allele frequency was tested by Z with a mean of
and a variance of
when replicates were available for only n samples.
ADDITIONAL INFORMATION
Data Deposition and Access
The seven full-length and one partial virus genomes were deposited at GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession numbers KX034093–KX034100. Raw sequencing data were deposited in the Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP072910.
Ethics Statement
This study was approved by The Samsung Medical Center (Seoul, South Korea) institutional review board (IRB) (no. 2015-07-005) and all methods were carried out in accordance with the approved guidelines. The IRB approved a waiver to document informed consent because the study planned to (1) use residual viral RNA samples without further collection of clinical samples, (2) analyze only viral RNA without human genome analysis, and (3) anonymize and de-identify the specimen and patient information before the analysis.
Acknowledgments
We thank the technical staff of the Samsung Genome Institute for next-generation sequencing.
Author Contributions
D.P. and H.J.H. managed data processing, designed the experiment, and coordinated the analyses. Y.J.K. designed the experiment. Y.J.K. and H.-J.J. performed the experiments. D.-S.S. performed data processing, analyses, and statistical tests. E.-H.I. analyzed the consensus sequence data. J.-W.K., N.Y.L., E.-S.K., C.I.K., and D.R.C. collected the clinical data and analyzed the data. J.-H.A., S.S.C., and Y.-J.K. advised on analyses and the manuscript. K.R.P., C.-S.K., and W.-Y.P. conceived of the study and participated in its design. D.P., H.J.H., C.-S.K., and W.-Y.P. wrote the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by a research grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI13C2096 to W.-Y.P.).
Competing Interest Statement
The authors have declared no competing interest.
Referees
Mazin Barry
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Borucki MK, Lao V, Hwang M, Gardner S, Adney D, Munster V, Bowen R, Allen JE. 2016. Middle East respiratory syndrome coronavirus intra-host populations are characterized by numerous high frequency variants. PLoS One 11: e0146251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breban R, Riou J, Fontanet A. 2013. Interhuman transmissibility of Middle East respiratory syndrome coronavirus: estimation of pandemic risk. Lancet 382: 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese T, Mishra N, Jain K, Zalmout IS, Jabado OJ, Karesh WB, Daszak P, Mohammed OB, Alagaili AN, Lipkin WI. 2014. Middle East respiratory syndrome coronavirus quasispecies that include homologues of human isolates revealed through whole-genome analysis and virus cultured from dromedary camels in Saudi Arabia. MBio 5: e01146-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman FD, Hoffmann C, Ronen K, Malani N, Minkah N, Rose HM, Tebas P, Wang GP. 2008. Massively parallel pyrosequencing in HIV research. AIDS 22: 1411–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten M, Lam TT, Watson SJ, Palser AL, Petrova V, Grant P, Pybus OG, Rambaut A, Guan Y, Pillay D, et al. 2013a. Full-genome deep sequencing and phylogenetic analysis of novel human betacoronavirus. Emerg Infect Dis 19: 736B–742B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten M, Watson SJ, Kellam P, Al-Rabeeah AA, Makhdoom HQ, Assiri A, Al-Tawfiq JA, Alhakeem RF, Madani H, AlRabiah FA, et al. 2013b. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet 382: 1993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson N, Pachter L, Mitsuya Y, Rhee SY, Wang C, Gharizadeh B, Ronaghi M, Shafer RW, Beerenwinkel N. 2008. Viral population estimation using pyrosequencing. PLoS Comput Biol 4: e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Cho YJ, Kim DW, Yang JS, Kim H, Park S, Han YW, Yun MR, Lee HS, Kim AR, et al. 2015. Complete genome sequence of Middle East respiratory syndrome coronavirus KOR/KNIH/002_05_2015, Isolated in South Korea. Genome Announc 3 10.1128/genomeA.00787-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Kim YJ, Park SH, Yun MR, Yang JS, Kang HJ, Han YW, Lee HS, Man Kim H, Kim H, et al. 2016a. Variations in spike glycoprotein gene of MERS-CoV, South Korea, 2015. Emerg Infect Dis 22: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Kim YJ, Lemey P, Lee I, Park S, Bae JY, Kim D, Kim H, Jang SI, Yang JS, et al. 2016b. The recent ancestry of Middle East respiratory syndrome coronavirus in Korea has been shaped by recombination. Sci Rep 6: 18825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Cheon S, Min CK, Sohn KM, Kang YJ, Cha YJ, Kang JI, Han SK, Ha NY, Kim G, et al. 2016c. Spread of mutant Middle East respiratory syndrome coronavirus with reduced affinity to human CD26 during the South Korean Outbreak. MBio 7: e00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. 2011. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci 108: 9530–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring AS, Andino R. 2010. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 6: e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH. 1993. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol 36: 96–99. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Wang Y, Wang W, Nie K, Zhao Y, Su J, Deng Y, Zhou W, Li Y, Wang H, et al. 2015. Complete genome sequence of Middle East respiratory syndrome coronavirus (MERS-CoV) from the first imported MERS-CoV case in China. Genome Announc 3 10.1128/genomeA.00818-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin A, Dijkman R, Bergstrom T, Kann N, Adamiak B, Hannoun C, Kindler E, Jonsdottir HR, Muth D, Kint J, et al. 2014. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the middle East respiratory syndrome virus. PLoS Pathog 10: e1004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margeridon-Thermet S, Shulman NS, Ahmed A, Shahriar R, Liu T, Wang C, Holmes SP, Babrzadeh F, Gharizadeh B, Hanczaruk B, et al. 2009. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J Infect Dis 199: 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MD, Choe PG, Oh HS, Park WB, Lee SM, Park J, Lee SK, Song JS, Kim NJ. 2015. Middle East respiratory syndrome coronavirus superspreading event involving 81 persons, Korea 2015. J Korean Med Sci 30: 1701–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamilo P, Bianchi NO. 1993. Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol Biol Evol 10: 271–281. [DOI] [PubMed] [Google Scholar]
- Park WB, Kwon NJ, Choe PG, Choi SJ, Oh HS, Lee SM, Chong H, Kim JI, Song KH, Bang JH, et al. 2016. Isolation of Middle East respiratory syndrome coronavirus from a patient of the 2015 Korean Outbreak. J Korean Med Sci 31: 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong MW, Kim SY, Corman VM, Kim TS, Cho SI, Kim MJ, Lee SJ, Lee JS, Seo SH, Ahn JS, et al. 2016. Microevolution of outbreak-associated Middle East respiratory syndrome coronavirus, South Korea, 2015. Emerg Infect Dis 22: 327–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30: 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XC, Agnihothram SS, Jiao Y, Stanhope J, Graham RL, Peterson EC, Avnir Y, Tallarico AS, Sheehan J, Zhu Q, et al. 2014. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc Natl Acad Sci 111: E2018–E2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. 2006. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439: 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu D, Shi W, Lu R, Wang W, Zhao Y, Deng Y, Zhou W, Ren H, Wu J, et al. 2015. Origin and possible genetic recombination of the Middle East respiratory syndrome coronavirus from the first imported case in China: phylogenetics and coalescence analysis. MBio 6: e01280-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367: 1814–1820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.