

Abstract
To comprehensively evaluate a European–American child with severe hypertension, whole-exome sequencing (WES) was performed on the child and parents, which identified causal variation of the proband's early-onset disease. The proband's hypertension was resistant to treatment, requiring a multiple drug regimen including amiloride, spironolactone, and hydrochlorothiazide. We suspected a monogenic form of hypertension because of the persistent hypokalemia with low plasma levels of renin and aldosterone. To address this, we focused on rare functional variants and indels, and performed gene-based tests incorporating linkage scores and allele frequency and filtered on deleterious functional mutations. Drawing upon clinical presentation, 27 genes were selected evidenced to cause monogenic hypertension and matched to the gene-based results. This resulted in the identification of a stop-gain mutation in an epithelial sodium channel (ENaC), SCNN1B, an established Liddle syndrome gene, shared by the child and her father. Interestingly, the father also harbored a missense mutation (p.Trp552Arg) in the α-subunit of the ENaC trimer, SCNN1A, possibly pointing to pseudohypoaldosteronism type I. This case is unique in that we present the early-onset disease and treatment response caused by a canonical stop-gain mutation (p.Arg566*) as well as ENaC digenic hits in the father, emphasizing the utility of WES informing precision medicine.
Keywords: elevated diastolic blood pressure, elevated mean arterial pressure, elevated systolic blood pressure
INTRODUCTION
Primary or essential hypertension (OMIM #145500) is considered a multifactorial disease with a polygenic contribution; however, a small number of those with primary hypertension have been found to have an oligogenic form of hypertension. In diagnosing childhood-onset essential hypertension, secondary etiologies, such as renovascular hypertension, coarctation of the aorta, pheochromocytoma, hyperthyroidism, glomerulopathies, and structural renal conditions with pyelonephritic scarring, are first comprehensively evaluated and ruled out (National High Blood Pressure Education Program 2004; Freundlich and Ludwig 2005; Vehaskari 2009). Once the secondary etiology for hypertension has been ruled out, then genetic basis for hypertension can be evaluated. Monogenic forms of hypertension share a common pathway of excessive reabsorption of sodium and chloride in distal tubules in kidneys leading to volume expansion, hypertension, and low plasma levels of renin activity. A monogenic form of hypertension can sometimes be clinically suspected but difficult to diagnose with ease and often missed as an etiology of hypertension. Several forms of monogenetic, low-renin hypertension have been previously characterized (Toka and Luft 2002; Garovic et al. 2006), such as (a) glucocorticoid remediable aldosteronism (GRA), (b) apparent mineralocorticoid excess (AME), (c) Liddle syndrome (or pseudoaldosteronism) (Vehaskari 2009), and (d) Gordon syndrome, pseudohypoaldosteronism type II (PHAII). Currently, a monogenic form of hypertension is determined by molecular testing in a specialized laboratory and is not readily available commercially for all types.
Whole-exome sequencing (WES) provides a broad assessment to identify pathogenic genetic variants in patients with severe disease, potentially obviating a number of invasive, expensive, and/or time-consuming diagnostic evaluations. The technique of next-generation sequencing with WES has been of pivotal value in molecular diagnosis of a wide spectrum of Mendelian traits, thus improving the therapeutics and counseling of the affected individuals (Hanchard et al. 2013; Gonzaga-Jauregui et al. 2014). WES is also useful in known monogenetic conditions, particularly those with significant locus heterogeneity, to identify lesser-recognized genes or in the detection of a novel gene. In this report, we describe a 5-yr-old European–American child who presented with severe hypertension and was later diagnosed by WES with a stop-gain mutation (p.Arg566*, rs137852704) in SCNN1B known to cause Liddle Syndrome (OMIM #177200) inherited via her father. The utility of WES in the diagnosis of the child and father for this rare mutation with a potential for a tailored therapeutic approach is described.
RESULTS
Clinical Presentation and Family History
Severe hypertension, especially diastolic hypertension, was diagnosed at age 5 yr in a previously healthy Caucasian (non-Hispanic white) girl and followed until 17 yr of age. She presented with a history of persistently elevated blood pressures, easy fatigue, and palpitations. She was on no medications or supplements that could elevate her blood pressure. She was born at 39 wk of gestation with no significant past medical history except for allergic rhinitis. On physical examination, the child had no dysmorphism or obesity but she had a short stature with a height of 117cm at 5 yr of age, which was less than the 5th percentile at diagnosis, and a height of 150 cm at 17 yr of age. Her body weight was 22 kg at diagnosis. She was found to have an average of three measurements of blood pressure in three different clinic visits measuring 154/107 mmHg, which was above the gender, age, and height percentile-specific 99th percentile (119/82 mmHg) per the Fourth Report (National High Blood Pressure Education Program 2004), and the sustained hypertension was confirmed by a 24-h ambulatory blood pressure monitoring with an average of 137/96 mmHg, along with a 24-h systolic blood pressure load of 98% and diastolic blood pressure load of 94%. The systolic nocturnal dip in the blood pressures was 13% and diastolic dip was 20%. She had a soft ejection systolic murmur at the left upper sternal border and the rest of her physical examination was unremarkable.
There was a family history of hypertension (Fig. 1) with both parents diagnosed with hypertension, her mother diagnosed at the age of 35 yr but not taking any antihypertensive medications and father at 19 yr of age, treated with lisinopril. The mother also had a short stature with a height of 150 cm, whereas her father was of normal height at 183 cm. There was no family history of sudden unexplained death but there was a history of stroke in the maternal grandmother.
Figure 1.
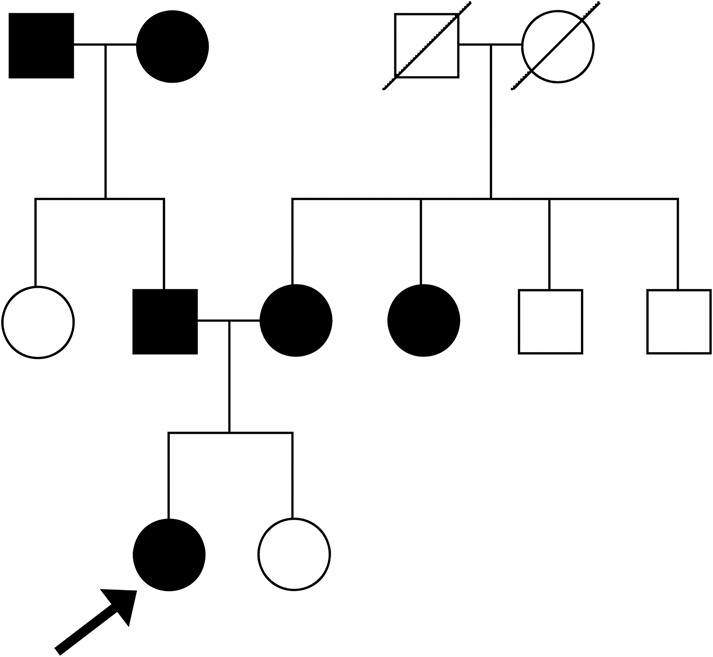
Pedigree of the family with the proband marked by an arrow. The shaded shapes identify individuals with a clinical history of hypertension.
Upon further evaluation at the tertiary hypertension clinic, the child was confirmed to have persistent hypertension at the age of 5 yr per recommendations by the Fourth Working Group (National High Blood Pressure Education Program 2004). She underwent a thorough evaluation for other secondary causes of hypertension per recommendations (National High Blood Pressure Education Program 2004), including a urinary evaluation, blood tests, echocardiogram, sleep study, renal ultrasound, and renal magnetic resonance imaging. She was found to have low levels of serum potassium, serum aldosterone, and plasma renin activity. These measures were serum aldosterone levels of 1.6 ng/dL and plasma renin activity of 0.1 ng/mL/h with a serum aldosterone to renin ratio of 16. She also underwent an echocardiogram and was found to have evidence of end-organ damage with left ventricular hypertrophy and a secundum atrial septal defect. Her lipid profile, glucose, and C-reactive protein were within normal limits. A 24-h measurement of urinary catecholamine metabolites (metanephrine, normetanephrine, homovanillic acid, and vanillylmandelic acid) was normal. Her renal imaging did not reveal any abnormalities. Her electrocardiogram showed normal conduction intervals and the sleep study was normal.
Because of the persistent hypokalemia with low plasma levels of renin and aldosterone, a monogenic form of hypertension was suspected. However, because of family preference, further evaluation by genetic studies was deferred until the age of 17 yr when the family agreed to participate in a study protocol in which the WES was provided without any charge to the family.
Genomic Analyses
The p.Arg566* stop-gain variant (rs137852704, c.1696C>T) shared by the proband and her father was identified in exon 13 of the sodium channel epithelial 1 β-subunit (SCNN1B) gene on Chromosome 16p12.2. A missense mutation (rs5742912, c.1654T>C) was also found in exon 9 of the ENaC α-subunit (SCNN1A) gene and only present in the father (Table 1). pVAAST results for p.Arg566* yielded a P = 9.02 × 10−4 (confidence interval: 5.4 × 10−4–1.3 × 10−3) and computed composite likelihood ratio test p (CLRTp) score of 19.601. Other top CLRTp scoring genes meeting the Bonferroni-corrected threshold included an insertion in retrotransposon-like 1 (RTL1) and coiled-coil domain containing 66 (CCDC66) genes (CLRTp score = 22.285, P = 4.17 × 10−7) and a deletion in the CTAGE family member 5, ER export factor (CTAGE5) gene (CLRTp score = 22.281, P = 4.17 × 10−7).
Table 1.
Variants from whole-exome sequencing by family member
| Gene | Family member affected | Variant (rs#/type/protein change) | Referent/alternate allele (genotype) | HGVS DNA reference | HGVS protein reference | ExAC AF (%) |
|---|---|---|---|---|---|---|
| SCNN1B | Child, father | rs137852704/stop gain/p.Arg566* | C/T (heterozygous) | NM_000336 | NP_000327 | 8.263 × 10−6 |
| SCNN1A | Father | rs5742912/missense/ p.Trp552Arga | A/G (heterozygous) | NM_001159576 | NP_001153048 | 0.018 |
HGVS, Human Genome Variation Society; ExAC, Exome Aggregation Consortium; AF, allele frequency.
aPredicted clinical significance of benign/pathogenenic, SIFT (0), PolyPhen (0.993).
Immediate Treatment Outcomes
At the age of 5 yr she was found to have severe and resistant hypertension requiring multiple drugs to control the blood pressure with a final regimen of amiloride, spironolactone, and hydrochlorothiazide with normalized blood pressures for her height, age, and gender. She was also recommended a heart-healthy diet and lifestyle with a restricted salt intake. Although the Liddle syndrome diagnosis was made later, amiloride was specifically chosen for her at presentation for the potassium-sparing effect, given her low serum potassium and suspicion of monogenic hypertension. Amiloride, a diuretic, is also a drug of choice for Liddle syndrome as it blocks the epithelial sodium channel (ENaC) activity in the collecting tubules. Her initial renal function was normal with a serum creatinine of 0.4 mg/d but her urinalysis showed proteinuria. The proteinuria resolved with antihypertensive therapy, but her low serum potassium levels did not resolve. She was found to have normal serum electrolytes except for low serum potassium of 3.4 mEq/L at initial presentation. The low serum potassium levels persisted despite therapy with a high dose of spironolactone used in the initial therapeutic regimen, with the highest serum potassium recorded over time being 3.5 mEq/L.
The end-organ damage in the form of left ventricular hypertrophy resolved with a low-salt diet and antihypertensive therapy but recurred after a year or so from noncompliance of medications. The sustained hypertension was confirmed by a 24-h ambulatory blood pressure monitoring off medications. The left ventricular hypertrophy resolved within a year of reinstitution of antihypertensive therapy. Her secundum atrial septal defect did not resolve spontaneously with age and was device-closed at the age of 14 yr. Based on the patient's diagnoses, her father was also diagnosed with Liddle syndrome and recommended treatment with potassium-sparing diuretics.
DISCUSSION
Liddle syndrome is an autosomal dominant disease leading to a salt-sensitive systemic hypertension that is usually diagnosed later in life and requires tailored pharmacotherapy to control the blood pressure. Liddle syndrome is inherited in an autosomal dominant fashion with mutations in either the SCNN1B or SCNN1G genes and characterized by increased activity of the amiloride-sensitive ENaC, causing abnormal sodium transport by the renal tubule. ENaC has functional properties of the distal renal sodium channel, with high sodium selectivity, low conductance, and amiloride sensitivity. It is also called pseudoaldosteronism or pseudohyperaldosteronism because of normal or low aldosterone levels and characterized by early and/or severe hypertension and hypokalemia along with low plasma levels of renin and aldosterone. Patients have high blood pressure but may remain completely asymptomatic from Liddle syndrome. However, some children can present with poor growth that can resolve when the hypertension is treated. Older patients often present with symptoms of hypokalemia (i.e., fatigue, palpitations, or muscular weakness leading to dyspnea, constipation, abdominal distention, and exercise intolerance).
We describe a 5-yr-old female proband and her father who harbored a stop-gain mutation in an epithelial sodium channel, SCNN1B, an established Liddle syndrome gene. The case is unique in that it was early-onset disease with severe systemic hypertension with a canonical stop-gain mutation (p.Arg566*). Furthermore, the child had a secundum atrial septal defect and a finding of a congenital heart defect has not been previously reported along with Liddle syndrome. The difficulty in diagnosing monogenic forms of hypertension led to a missed diagnosis, as described in our case whereby the father's diagnosis was made after diagnosing the child. An early diagnosis and therapy of Liddle syndrome in childhood is imperative for the prevention of cardiovascular, renovascular, and neurovascular complications in adult life. The utility of using WES to diagnose a Mendelian disease is also emphasized by this case report.
Several types of mutations have been linked to Liddle syndrome, leading to an increase in ENaC activity. A particular stop codon causing Liddle syndrome (p.Arg566*, R566X, and previously reported as Arg564) was first identified to be in complete linkage in a large pedigree (Shimkets et al. 1994). Molecular characterization of the p.Arg566* protein truncating mutation shows deletion of the last 75 amino acids and removal of the cytoplasmic carboxyl tail. The functional consequences of the mutation studied in Xenopus laevis oocytes reported unaltered biophysical and pharmacological properties of the channel; however, evidence of constitutive channel hyperactivity in the cell membrane was subsequently reported (Schild et al. 1995). Further, in a mouse model, introducing the same mutation by Cre/loxP-mediated recombination, mice with high-salt intake recapitulated the human syndrome of high blood pressure, hypokalemia, cardiac and renal hypertrophy, further confirming causality of the mutation with Liddle syndrome (Pradervand et al. 1999). The p.Arg566* truncating variant has been clinically characterized in a Japanese pedigree (50-yr-old male proband), adult members of a Swedish family, and a Chinese family (Melander et al. 1998; Kyuma et al. 2001; Shi et al. 2010). However, we present an early-onset case in a 5-yr-old girl with a p.Arg566* mutation of the epithelial sodium channel β-subunit. Other mutations reported in Liddle syndrome that delete or alter a conserved proline-rich amino acid sequence, PY motif, have been reported in childhood or adolescent cases and include P616L missense mutation in an 8-yr-old Czech boy (Ciechanowicz et al. 2005) and a 13-yr-old Serbian boy (Bogdanovic et al. 2012) and a Y618H mutation in a 4-yr-old Afro-Haitian girl (Freundlich and Ludwig 2005).
In addition to Liddle syndrome, other disease diagnoses including cystic fibrosis–like disease and pseudohypoaldosteronism have reported associations with ENaC mutations. The ENaC is composed of three subunits, α, β, and γ, with compound heterozygotes in any three of the genes (SCNN1A, SCNN1B, SCNN1G) being associated with a cystic fibrosis–like disease, consisting of lung disease symptoms (Azad et al. 2009; Ramos et al. 2014). Interestingly, the father harboring the p.Arg566* variant in SCNN1B inherited by the girl also harbored a missense variant in SCNN1A but did not carry symptoms or a diagnosis of cystic fibrosis–like disease. Unfortunately, comprehensive clinical history and physical examination were not available for the father to determine his complete phenotype; however, different rare variants within SCNN1A have also been associated with pseudohypoaldosteronism, type I (OMIM: 264350), with the possibility of the compound heterozygotes in different ENaC subunits contributing to his phenotype in a digenic manner (Chang et al. 1996; Bonny et al. 1999; Schaedel et al. 1999).
METHODS
Whole-Exome Sequencing
DNA sequencing of the child proband, mother, and father was performed at Baylor College of Medicine's Human Genome Sequencing Center on Illumina HiSeq after exome capture with NimbleGen's VCRome2.1. Before analysis, data were processed and alleles jointly called using Mercury (Reid et al. 2014). Sequencing yields had an average of 92% of target sites and a depth of coverage of 20× or greater and a mean sequencing depth of 92×. Each single-nucleotide variant (SNV) call was filtered based on the following criteria to produce a high-quality variant list: low SNV posterior probability (<0.95), low variant read count (<3), variant read ratio <0.25 or >0.75, strand bias of >99% variant reads in a single-strand direction, or total coverage <10-fold. All variant calls filtered by these criteria, and reference calls with <10-fold coverage, were set to missing. The variant call filters were the same for indels except that a total coverage <30-fold was used for variant sites. Whole-exome sequencing coverage for individuals of the trio is reported in Table 2. The variant p.Arg566* was confirmed for the child proband by Sanger sequencing in a Clinical Laboratory Improvement Amendment (CLIA)-certified laboratory.
Table 2.
Whole-exome sequencing coverage
| Sample name | Unique aligned (MB) | Total (MB) | Align Read 1 (%) | Align Read 2 (%) | Average coverage | Per 20 coverage bases |
|---|---|---|---|---|---|---|
| Child | 5810 | 6196 | 98 | 98 | 75 | 92 |
| Mother | 9406 | 10,236 | 97 | 96 | 120 | 97 |
| Father | 3762 | 4202 | 94 | 93 | 49 | 83 |
Statistical Analysis
A pedigree was constructed as a PED formatted file to include the sequenced proband, mother, and father for sequenced family members with extended pedigree shown in Figure 1. Based on the affection status throughout the pedigree, a dominant inheritance pattern model was evaluated using the Pedigree Variant Annotation, Analysis, and Search Tool (pVAAST) software (Hu et al. 2014). All functionally annotated SNVs and indels found within the three whole-exome sequenced cases (proband, mother, father) were compared with 1000 Genomes controls available as part of the pVAAST software. The two-point logarithm of odds (LOD) scores as calculated for an entire sequenced gene for the extended pedigree is integrated as a separate log-likelihood ratio along with functional prediction annotation and minor allele frequency comparing cases and controls. The ranked pVAAST results yield a computed composite likelihood ratio test (CLRTp score). Functional SNVs included synonymous and nonsynonymous codon changes, stopcodon loss and gain, indels, and splice-site variants (genome build version hg19). The number of permutations was set to the Bonferroni-corrected threshold for the number of genes tested at P < 2.4 × 10−6.
Based on serum measurements and anthropomorphic measurements collected at clinical presentation, the CLRTp scored results were compared with a candidate gene list reported in OMIM to be associated with Gordon hyperkalemia–hypertension syndrome, AME, hyperaldosteronism familial type I, familial glucocorticoid deficiency, congenital adrenal hyperplasia, and Liddle syndrome. The candidate gene list included PHA2A, PHA2B, WNK4, PHA2C, WNK1, PHA2D, KLHL3, PHA2E, CUL3, HSD11B2, KCNJ5, CYP11B1, CYP11B2 , HBD, HBB, MC2R also known as ACTHR, MRAP, NNT, CYP21A2, CYP11A1, CYP17, ACTHR, CYP21, CYP21B, HLA-B, SCNN1G, and SCNN1B.
ADDITIONAL INFORMATION
Data Deposition and Access
The ClinVar accession number for the SCNN1B variant is SCV000282671 (http://www.ncbi.nlm.nih.gov/clinvar/). Patient consent was given to release the patients’ sequencing data and was submitted by the Baylor Hopkins Center for Mendelian Genomics to the Database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap) under study accession number phs000711.v3.p1.
Ethics Statement
The study was approved by the Institutional Committee for the Protection of Human Subjects at the University of Texas Health Science Center and Children's Memorial Hermann Hospital, Texas Medical Center. All participants and parents gave their informed assent and written consent, respectively, for this study.
Acknowledgments
We thank Dr. Sanjay Shete, Dr. Michelle Barratt, Dr. Dianna Milewicz, Dr. Jon Tyson, Dr. Eric Boerwinkle, and Dr. Jacqueline Hecht for their mentorship of M.G.-M. during her career development award by the National Institutes of Health. We would also like to thank the family who agreed to participate in this study.
Author Contributions
M.G.-M. contributed to patient recruitment and phenotyping; L.M.P. and S.S. contributed to sequence data analysis and interpretation; L.M.P. and M.G.-M. contributed to functional evaluation of variant; D.M. and S.W. contributed to Sanger sequencing; L.M.P. and M.G.-M. contributed to writing of the initial draft of the manuscript. All authors contributed to revising the manuscript and reviewing the final draft.
Funding
The project described was supported by Grant Number K23HL089391 for “Determination of genetics of childhood onset hypertension” (PI Monesha Gupta) from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Competing Interest Statement
The authors have declared no competing interest.
REFERENCES
- Azad AK, Rauh R, Vermeulen F, Jaspers M, Korbmacher J, Boissier B, Bassinet L, Fichou Y, des Georges M, Stanke F, et al. 2009. Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis–like disease. Hum Mutat 30: 1093–1103. [DOI] [PubMed] [Google Scholar]
- Bogdanovic R, Kuburovic V, Stajic N, Mughal SS, Hilger A, Ninic S, Prijic S, Ludwig M. 2012. Liddle syndrome in a Serbian family and literature review of underlying mutations. Eur J Pediatr 171: 471–478. [DOI] [PubMed] [Google Scholar]
- Bonny O, Chraibi A, Loffing J, Jaeger NF, Grunder S, Horisberger JD, Rossier BC. 1999. Functional expression of a pseudohypoaldosteronism type I mutated epithelial Na+ channel lacking the pore-forming region of its α subunit. J Clin Invest 104: 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, et al. 1996. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet 12: 248–253. [DOI] [PubMed] [Google Scholar]
- Ciechanowicz A, Dolezel Z, Placha G, Starha J, Gora J, Gaciong Z, Brodkiewicz A, Adler G. 2005. Liddle syndrome caused by P616R mutation of the epithelial sodium channel β subunit. Pediatr Nephrol 20: 837–838. [DOI] [PubMed] [Google Scholar]
- Freundlich M, Ludwig M. 2005. A novel epithelial sodium channel β-subunit mutation associated with hypertensive Liddle syndrome. Pediatr Nephrol 20: 512–515. [DOI] [PubMed] [Google Scholar]
- Garovic VD, Hilliard AA, Turner ST. 2006. Monogenic forms of low-renin hypertension. Nat Clin Pract Nephrol 2: 624–630. [DOI] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Mir S, Penney S, Jhangiani S, Midgen C, Finegold M, Muzny DM, Wang M, Bacino CA, Gibbs RA, et al. 2014. Whole-exome sequencing reveals GPIHBP1 mutations in infantile colitis with severe hypertriglyceridemia. J Pediatr Gastroenterol Nutr 59: 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchard NA, Murdock DR, Magoulas PL, Bainbridge M, Muzny D, Wu YQ, Wang M, McGuire AL, Lupski JR, Gibbs RA, et al. 2013. Exploring the utility of whole-exome sequencing as a diagnostic tool in a child with atypical episodic muscle weakness. Clin Genet 83: 457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Roach JC, Coon H, Guthery SL, Voelkerding KV, Margraf RL, Durtschi JD, Tavtigian SV, Shankaracharya, Wu W, et al. 2014. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol 32: 663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyuma M, Ura N, Torii T, Takeuchi H, Takizawa H, Kitamura K, Tomita K, Sasaki S, Shimamoto K. 2001. A family with Liddle's syndrome caused by a mutation in the β subunit of the epithelial sodium channel. Clin Exp Hypertens 23: 471–478. [DOI] [PubMed] [Google Scholar]
- Melander O, Orho M, Fagerudd J, Bengtsson K, Groop PH, Mattiasson I, Groop L, Hulthen UL. 1998. Mutations and variants of the epithelial sodium channel gene in Liddle's syndrome and primary hypertension. Hypertension 31: 1118–1124. [DOI] [PubMed] [Google Scholar]
- National High Blood Pressure Education Program. 2004. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics 114: 555–576. [PubMed] [Google Scholar]
- Pradervand S, Wang Q, Burnier M, Beermann F, Horisberger JD, Hummler E, Rossier BC. 1999. A mouse model for Liddle's syndrome. J Am Soc Nephrol 10: 2527–2533. [DOI] [PubMed] [Google Scholar]
- Ramos MD, Trujillano D, Olivar R, Sotillo F, Ossowski S, Manzanares J, Costa J, Gartner S, Oliva C, Quintana E, et al. 2014. Extensive sequence analysis of CFTR, SCNN1A, SCNN1B, SCNN1G and SERPINA1 suggests an oligogenic basis for cystic fibrosis–like phenotypes. Clin Genet 86: 91–95. [DOI] [PubMed] [Google Scholar]
- Reid JG, Carroll A, Veeraraghavan N, Dahdouli M, Sundquist A, English A, Bainbridge M, White S, Salerno W, Buhay C, et al. 2014. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics 15: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaedel C, Marthinsen L, Kristoffersson AC, Kornfalt R, Nilsson KO, Orlenius B, Holmberg L. 1999. Lung symptoms in pseudohypoaldosteronism type 1 are associated with deficiency of the α-subunit of the epithelial sodium channel. J Pediatr 135: 739–745. [DOI] [PubMed] [Google Scholar]
- Schild L, Canessa CM, Shimkets RA, Gautschi I, Lifton RP, Rossier BC. 1995. A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc Natl Acad Sci 92: 5699–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi JY, Chen X, Ren Y, Long Y, Tian HM. 2010. [Liddle's syndrome caused by a novel mutation of the γ-subunit of epithelial sodium channel gene SCNN1G.] (In Chinese.) Zhonghua Yi Xue Yi Chuan Xue Za Zhi 27: 132–135. [DOI] [PubMed] [Google Scholar]
- Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR Jr, Ulick S, Milora RV, Findling JW, et al. 1994. Liddle's syndrome: heritable human hypertension caused by mutations in the β subunit of the epithelial sodium channel. Cell 79: 407–414. [DOI] [PubMed] [Google Scholar]
- Toka HR, Luft FC. 2002. Monogenic forms of human hypertension. Semin Nephrol 22: 81–88. [DOI] [PubMed] [Google Scholar]
- Vehaskari VM. 2009. Heritable forms of hypertension. Pediatr Nephrol 24: 1929–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]