

Abstract
The elongation step in transcription has gained attention for its roles in regulation of eukaryotic gene expression and for its influence on RNA processing. Sophisticated genetic analyses have identified factors and/or conditions that may affect transcription elongation rate or processivity; however, differentiation of direct and indirect effects on transcription is difficult using in vivo strategies. Therefore, effective, reproducible in vitro assays have been developed to test whether a given factor or condition can have a direct effect on the kinetics of transcription elongation. We have adapted a fully reconstituted transcription system for RNA polymerase I (Pol I) for kinetic analysis of transcription elongation rate in vitro. The assay described here has proven to be effective in the characterization of defects or enhancement of wild-type transcription elongation by RNA Pol I. Since transcription elongation by RNA Pol I has only recently gained significant attention, this assay will be a valuable resource for years to come.
Keywords: Transcription, rRNA, Ribosome, Elongation rate, Pause site, RNA polymerase I
1. Introduction
Transcription of ribosomal RNA constitutes more than 60% of total transcription in proliferating eukaryotic cells, and is a target for regulation of ribosome synthesis (1, 2). Due to the intimate link between rRNA synthesis rate and cell proliferation, the molecular mechanisms that regulate polymerase I (Pol I) transcription are directly relevant to our understanding of important human diseases like cancer (3). However, the mechanisms by which rRNA synthesis is controlled have not yet been fully described. Recent studies have identified factors that influence transcription elongation by RNA Pol I to have a role in the regulation of rRNA synthesis rates (4, 5). Thus, a reliable assay for RNA Pol I transcription elongation is essential. We have adapted a previously described multiround transcription elongation system for analysis of transcription elongation rate as well as intrinsic pausing and arrest (5–7).
When developing this system, every effort was made to preserve the context of transcription elongation encountered in vivo. Methods exist for formation of transcription elongation complexes in vitro (e.g., scaffold templates); however, we have chosen to use promoter-dependent transcription initiation and nucleotide limitation to synchronize transcription elongation complexes. As with all experiments in vitro, this assay is subject to the criticism that it does not fully recapitulate in vivo conditions. Nevertheless, we have minimized the potential for artifacts induced by the assay and we have purified all of the factors essential for transcription by RNA Pol I. Furthermore, we have demonstrated that this assay is effective in quantification of both positive and negative effects on transcription elongation by Pol I. Thus, the assay described here is the most robust assay for measurement of direct effects of a factor or condition on the transcription elongation by Pol I to date.
2. Materials
2.1. Cell Growth, Harvest, and Lysis
Luria–Bertani broth (Fisher Scientific; New Jersey) supplemented with 100 μg/ml of ampicillin.
Isopropyl β-D-1-thiogalactopyranoside (IPTG).
YEPD medium: 1% (w/v) Yeast extract (Becton, Dickinson and Company, BD, Maryland), 2% (w/v) peptone, 2% (w/v) dextrose.
Breakage buffer: 500 mM KCl, 50 mM Tris–HCl, pH 7.6, 20% (v/v) glycerol, 5 mM MgCl2, 5 mM imidazole, 1 mM PMSF, 0.1% (v/v) Tween 20.
2.2. Factor Purification
Nickel Sepharose 6 Fast Flow (GE Healthcare Bio-Sciences, GE; Uppsala).
Monoclonal anti-HA agarose conjugate, clone HA-7 (Sigma–Aldrich, Saint Louis).
HA peptide (GenScript, New Jersey).
Nickel wash buffer: 200 mM KCl, 50 mM Tris–HCl, pH 7.6, 5 mM MgCl2, 10 mM imidazole, 0.1% Tween 20, 20% glycerol.
Nickel elution buffer: 200 mM KCl, 50 mM Tris–HCl, pH 7.6, 5 mM MgCl2, 250 mM imidazole, 0.1% Tween 20, 20% glycerol
Columns: HiTrap Heparin HP, HiTrap Q FF, MonoQ 5/50 GL, MonoS 5/50 GL (GE).
Standard gradient buffer: 200 mM (A) or 1 M (B) KCl, 20 mM Tris–HCl, pH 7.8, 20% glycerol, 0.1 mM EDTA (see Note 1).
Upstream activating factor (UAF) extraction buffer: 200 mM Tris acetate, pH 8.0, 400 mM ammonium sulfate, 10% glycerol, 0.1% Tween 20, 20 mM imidazole.
AKTA Purifier, UPC-10 with fraction collector (GE) or suitable similar instrument for liquid chromatography.
2.3. DNA Template Preparation
Plasmid pNOY746: 35S rDNA with promoter, all C residues in nontemplate strand mutated to G between +1 and +56, relative to transcription start site.
Oligonucleotides (Integrated DNA technologies, Iowa): −247 oligo: 5′-gttgtaaaacgacggccagtgc-3′ and +763 oligo: 5′-cttagacat-gcatggcttaatc-3′.
PCR purification kit (Qiagen, Maryland).
2.4. Transcription Assays
2.5× transcription buffer: 50 mM Tris acetate, pH 7.9, 250 mM potassium glutamate, 20 mM magnesium acetate, 5 mM dithiothreitol, RNasin Plus (Promega), 0.5 mg/ml acetylated bovine serum albumin.
Ultrapure NTPs.
α32 P GTP. Radioactive materials must be handled with extreme caution using proper personal protection and disposal.
1 mg/ml heparin, sodium salt.
Phenol, 3 mg/ml (w/v) glycogen, 1 M ammonium acetate in 100% ethanol.
10× TBE buffer: 108 g/l Tris base, 55 g/l boric acid, 7.44 g/l ethylenediaminetetraacetic acid (EDTA).
90% formamide loading dye: 90% deionized formamide, 1× TBE, 0.05% bromophenol blue, 0.05% xylene cyanol.
2.5. Gel Electrophoresis and Quantification
40% (w/v) acrylamide/bis solution (29:1).
Urea.
N, N, N, N′-Tetramethylethylenediamine (TEMED).
10% (w/v) ammonium persulfate.
16.5 × 28-cm vertical gel plates with 0.7-mm spacers and 20-well comb.
Storm phosphorimager with 8 × 10-in. phosphor screens or equivalent imaging instrumentation.
3. Methods
Detailed analysis of transcription elongation in vitro requires highly purified, active protein samples and properly prepared template DNA and reagents. Since this assay uses promoter-dependent transcription initiation and a modified rDNA template, it closely mimics the in vivo state of transcription elongation (as closely as possible for an in vitro assay). As a consequence, all of the transcription initiation factors must be purified as well as the polymerase. Thus, most of the effort in executing this experiment is invested in protein purification. There are other protocols for analysis of short RNA synthesis from scaffold DNA templates using only the polymerase (8), but those methods are not described here.
The transcription elongation assay for RNA Pol I is an adaptation of a previously published assay for multiround transcription. Two previous papers describe the reconstituted transcription assay in detail and can be seen for additional reference (6, 9). To aid reproducibility, the entire procedure is described here. Some steps in the method described here are redundant with those published previously.
Since quantification of transcription elongation requires single-round transcription, the signal-to-noise ratio of these experiments is lower than for more robust multiround assays. Therefore, it is essential that extreme care be devoted to purification of the factors required, since minor contamination can lead to RNA degradation or nonspecific transcription, both of which increase the background in the assay. The following protocol is subdivided by purification of each factor and by the assay description itself.
3.1. Preparation of Cell Extracts
Rrn3p and Tata-binding protein (TBP) are expressed in Escherichia coli (BL21 DE3) from pET plasmid derivatives (His-tagged Rrn3p, pNOY3162 (6), and His-tagged TBP, 6HisT-pET11 (10)).
E. coli cells carrying the appropriate plasmid are grown in Luria–Bertani medium, supplemented with 100 μg/ml ampicillin. Cultures are grown with aeration at 25°C to an A600 = 0.5, and protein expression is induced with 0.5 mM IPTG.
After >12 h of induction, cells are harvested by centrifugation (10,000 × g for 30 min), washed once in breakage buffer, spun down again, and frozen at −80°C.
To purify Pol I, core factor, and UAF, strains expressing 6-his, 3-hemagglutinnin (HA)-tagged proteins are used (strains: NOY760 = tagged A135 to purify Pol I; NOY797 = tagged Rrn7p to purify core factor; and NOY798 = tagged Rrn5p to purify UAF).
Yeast cultures are grown with aeration in YEPD medium at 30°C to an A600 = 0.8.
Cells are harvested by centrifugation (6,000 × g for 20 min), washed with breakage buffer, spun down again, and the pellets are frozen at −80°C.
3.2. Rrn3p Purification
Frozen cell pellets are thawed on ice, suspended in threefold excess (v/w) breakage buffer, and disrupted using a French press (8,000 psi, internal pressure) (see Note 2).
After lysis, the extract is cleared by centrifugation (30,000 × g for 30 min).
The supernatant is mixed with Ni-Sepharose FF that has been prewashed twice with breakage buffer. This mixture is incubated at 4°C with gentle agitation for 2 h. Approximately 1 ml of settled resin is used per 2 g of wet cell mass.
Resin is separated from “flow through” by gentle centrifugation (500 × g for 5 min). Resin is resuspended in equal volume of breakage buffer and poured onto an empty column containing fresh, prewashed Ni-Sepharose FF (10% of volume used in binding) (see Note 3).
Using either gravity flow or pump-assisted flow, resin is washed with five-column volumes of Ni wash buffer.
Protein is eluted with three-column volumes of Ni elution buffer.
Eluted protein is loaded onto a Q-Sepharose column using an AKTA Purifier UPC-10 (or equivalent instrument for liquid chromatography) (see Note 4).
Sample is loaded and flow through is collected in 100% gradient buffer A (200 mM KCl).
After stable baseline is achieved, a 50-ml gradient from 200 mM to 1 M KCl is applied, collecting 1-ml fractions. The flow rate is 0.5 ml/min. Rrn3 elutes at ~400 mM KCl (see Note 5).
Peak fractions are pooled and rebound in batch to Ni-Sepharose FF. Resin volume is reduced to 20% of previous Ni purification.
Ni-binding, washing, and elution are repeated as described above.
Eluted protein is applied to a MonoQ column and eluted as described for the Q-Sepharose column. Pure Rrn3p elutes at ~400 mM KCl.
3.3. Tata-Binding Protein Purification
Cells are thawed and broken and first Ni column is performed as for Rrn3p.
Ni eluate is loaded onto an HiTrap heparin column in gradient buffer A, and flow through is collected.
After achieving a stable baseline, a 180-ml gradient from 0 to 60% gradient B is executed, collecting 1-ml fractions. His-TBP elutes at ~430 mM KCl.
Peak fractions are pooled and dialyzed into 100 mM KCl-modified gradient buffer.
Sample is then loaded onto a MonoS column and after a stable baseline is achieved, an 80-ml gradient from 100 mM to 360 mM KCl is run, collecting 1-ml fractions. TBP elutes at ~200 mM KCl.
3.4. RNA Polymerase I Purification
Cell pellets are thawed on ice as for E. coli cells (see Note 6).
Cells are resuspended in twofold excess (v/w) breakage buffer and disrupted in a French press (25,000 psi internal pressure) (see Note 2).
Extract is cleared by centrifugation (30,000 × g for 30 min).
Supernatant is mixed with prewashed Ni-Sepharose FF (~1 ml settled resin per 2 g of wet cell paste) and incubated at 4°C for 2 h with gentle agitation.
Resin is separated from the flow through by gentle centrifugation (500 × g for 5 min) and then poured into a column containing prewashed Ni resin, ~10% of resin volume used in batch binding.
Using gravity or pump-assisted flow, column is washed with five-column volumes of Ni wash buffer and proteins are eluted with three-column volumes of Ni elution buffer.
Eluted material is loaded onto an HiTrap heparin column in 100% gradient buffer A, and flow through is collected. After reaching a stable baseline, a 50-ml gradient from 200 mM to 1 M KCl is run, collecting 1-ml fractions. Pol I elutes at ~375 mM KCl.
Peak fractions are pooled and diluted with an equal volume of “no-salt” gradient buffer. This sample is then loaded onto a MonoQ column. After baseline is reached in 160 mM buffer A, a 50-ml gradient from 0 to 50% buffer B (500 mM KCl) is run, collecting 1-ml fractions. Pol I elutes at ~330 mM KCl (see Note 7).
3.5. Core Factor Purification
Cell pellets are thawed, broken, and fractionated with Ni-Sepharose and HiTrap heparin as described for Pol I. Core factor elutes from heparin at ~350 mM KCl.
Peak fractions are pooled and mixed with 1 ml of anti-HA affinity resin. The sample is incubated with gentle agitation at 4°C for 2 h (see Note 8).
The mixture is then applied to an empty column and washed with greater than five-column volumes of gradient buffer A.
Before elution, a HiTrap Q column is attached downstream of the column holding the anti-HA resin.
Gradient buffer A plus 1 mg/ml HA peptide is then recirculated over the two columns for greater than 2 h (see Note 9).
The Q column is detached and loaded in the AKTA purifier. A 50-ml gradient from 0 to 60% buffer B is executed, collecting 1-ml fractions. Core factor elutes at ~350 mM KCl.
3.6. UAF Purification
Cells are thawed, broken, and fractionated with Ni-Sepharose as described for Pol I, except that UAF extraction buffer is used to break and wash the beads.
After washing the Ni resin, an HiTrap heparin column (that has been previously washed into the elution buffer) is attached downstream of the nickel column.
Approximately five-column volumes of modified gradient buffer with 450 mM KCl and 250 mM imidazole are passed over the resin to elute UAF from the Ni-Sepharose onto the heparin resin.
The heparin column is then attached to an FPLC and eluted with a 50-min gradient from 450 mM KCl to 1 M KCl (flow rate 0.5 ml/min). UAF elutes at ~650 mM KCl.
The peak fractions are pooled and mixed with sufficient anti-HA immune affinity resin and incubated at 4°C with gentle agitation (see Note 8).
The beads are centrifuged (2 min at 1,000 × g) to pellet the beads and the supernatant is removed and saved. Five resin volumes of 400 mM KCl gradient buffer is added to the beads; they are gently mixed and spun down again at 1,000 × g.
This wash procedure is repeated three more times.
The beads are then resuspended and transferred to a small empty column.
An HiTrap heparin column is attached downstream of the column, and 400 mM gradient buffer + 0.5 mg/ml HA peptide is recirculated over the anti-HA beads and through the heparin column. Eluted UAF remains attached to the heparin, but HA peptide flows through.
After overnight recirculation (at 4°C), the heparin column is detached and mounted onto an FPLC. Fractionation is achieved with a 50-min gradient from 400 mM to 1 M KCl. One-milliliter fractions are collected. Pure UAF elutes at approximately 650 mM KCl.
3.7. DNA Template Preparation
Since only 6 C residues are present in the first 55 nucleotides of the transcribed rRNA, we chose to mutate those Cs to Gs. These mutations conserved the G/C content of the transcript, but permitted transcription initiation and promoter clearance in the absence of CTP. This modified rDNA template is encoded in pNOY746 (7).
Using pNOY746 as a template and −247 and +763 oligonucleotides (sequences provided above), we use standard PCR conditions to amplify the C-less promoter region.
The resulting 1,010 bp PCR product is purified using the Qiagen PCR purification kit (see Note 10).
After elution from the kit, DNA concentration is measured spectrophotometrically at 260-nm wavelength.
3.8. Transcription Elongation Assay
Having purified all of the necessary components, the transcription elongation rate assay is performed at 23°C using all RNase-free reagents.
All nonradioactive reagents are mixed first. These steps can be performed without shielding.
For each condition to be tested, a large master mix is prepared (see Note 11). [For example, if a factor’s effect on transcription elongation rate is to be tested, then two mixes are prepared, one with the appropriate amount of factor, and one with an equal volume of buffer only or heat-inactivated factor.]
Each reaction equals 20 μl; 17 μl of master mix; 1 μl of NTP mix (ATP, GTP, UTP, and α32 P GTP); 1 μl of heparin (1 mg/ml); and 1 μl of CTP.
Recipe per reaction: 1× transcription buffer, 6 ng DNA template, 0.2 μl of active UAF preparation, 50 nM TBP, 0.2 μl of active core factor preparation, 20 nM Pol I/Rrn3 complex (see Note 12), plus any additional factors or variables.
After preparing the master mix, NTP mix is added. The concentration of NTPs is exceptionally important for the elongation rate in vitro (Fig. 1) and can be modified to optimize the reaction for probing faster or slower effects. Since each sample is ultimately 20 μl, we make 20× stocks of everything, and our standard final NTP concentrations are 100 μM ATP, 100 μM UTP, 10 μM GTP, and 10 μCi α32 P GTP. To a 11× master mix, we add 11 μl of our 20× stock (see Note 13).
After 5 min, we add 11 μl of 1 mg/ml heparin (50 μg/ml final concentration). Heparin prevents reinitiation and disrupts initiation complexes that have not cleared the promoter.
After 4 min, we remove a 20-μl sample and add it to a tube containing 60 μl of acid phenol and 50 μl of 3 mg/ml glycogen. This sample is “stopped” and stable for later processing.
After 1 more minute (at t = 0), 10 μl of 20× CTP is added to a final concentration of 100 μM.
As a function of time, 20-μl samples are collected and added directly to tubes containing phenol and glycogen (as in step 8).
Typical time points = 0, 30 s, 60 s, 2 min, 3 min, 4 min, 5 min, 6 min, 8 min, and 10 min. These vary based on your reaction conditions and desired focus of the experiment.
After completion of the time course, each sample is mixed again by pipetting vigorously up and down.
Samples are centrifuged at 17,000 × g for 10 min.
Aqueous phase is removed to a fresh tube containing 330 μl of 1 M ammonium acetate in 95% ethanol (see Note 14).
Samples are incubated on crushed dry ice for 15 min.
Samples are spun at 17,000 × g for 10 min.
Ethanol is carefully and completely removed.
20 μl of formamide loading dye is added to each dried pellet, and the samples are heated to 95°C for 10 min prior to electrophoresis.
Fig. 1.
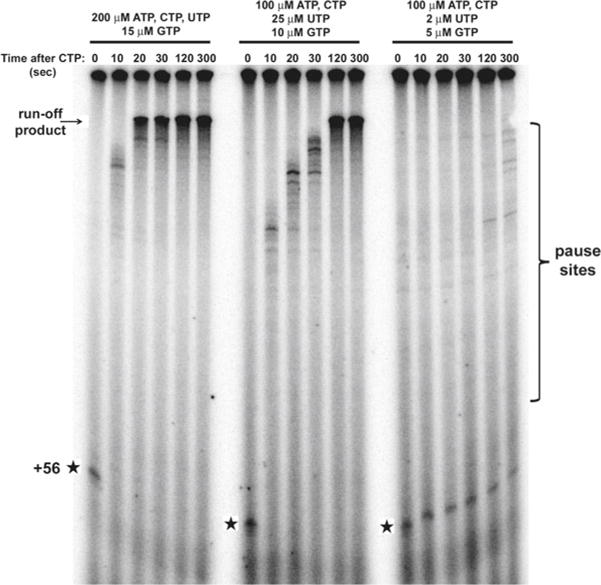
Nucleoside triphosphate concentration dependence of RNA polymerase I transcription elongation rate in vitro. Elongation assays were performed as described herein with three different NTP concentrations (indicated on top of gel). Samples were collected at identical time points under all three conditions, purified, and run on an 8% denaturing polyacrylamide gel. The gel was dried and analyzed by phosphorimaging. The +56 CTP arrest (stars), intrinsic pause sites, and runoff product positions are indicated.
3.9. Electrophoresis and Quantification
The gel contains 1× TBE, 8% polyacrylamide (29:1, acrylamide to bis), and 7 M urea. 50 ml of this solution is dissolved with stirring at room temperature. The urea requires >15 min to dissolve.
The spacers and plates are assembled and clipped with medium binder clips in place and a small amount of Vaseline is used at each of the two bottom corners of the spacers.
When mix is dissolved, 250 μl of 10% APS and 50 μl of TEMED are added and stirred for ~1 min.
This mix is then poured into the plates avoiding the introduction of any bubbles.
Once the mix is poured into the plates and bubbles are removed (typically by “vigorously” tapping the glass), the comb is inserted, being careful to avoid bubbles in the wells.
In ~30 min, the gel is sufficiently solidified to be wrapped on top (over the comb) with cellophane.
Gel is kept at room temperature overnight.
The next day, the gel is pre-run for 45 min at 700 V in 1× TBE.
During the pre-run, RNA samples are incubated at 95°C for 10 min.
10 μl of each RNA sample is loaded (directly from the heat block) into the gel (see Note 15).
Once loaded, the gel is run at 700 V until the dark blue band (bromophenol blue) reaches the bottom of the gel (see Note 16).
The gel is removed and dried onto a double layer of Whatman 3-mm paper under a sheet of cellophane at 80°C under vacuum for 1 h.
The dried gel is exposed ~16 h to a phosphorimager cassette (depending on the age of the 32P and the strength of the transcription).
After generation of the image with a phosphorimager, the amount of runoff product is quantified with the imager’s packaged software (see Note 17).
If runoff product accumulation is plotted as a function of time, a sigmoidal plot is typically generated.
There are several defensible methods for calculating elongation rates. One easy and reproducible method is to divide the length (in nucleotides) of the transcript (downstream of the +55 pause) by the time (in seconds) at which the sigmoidal curve crosses 100% of runoff accumulation. This yields an elongation rate in nucleotides/second.
In addition to quantification of the net elongation rate in your sample, visual analysis of your gel yields qualitative detection of intrinsic pause/arrest sites (Fig. 1).
Acknowledgments
The author wishes to thank Professor Masayasu Nomura for his pioneering development of the reconstituted transcription assay for RNA Pol I and for guidance in the adaptation of the system to its current form. This work is supported by the National Institutes of Health grant #GM84946.
Footnotes
We use the same base buffer for all gradient elutions. In some cases, as noted in the method, we use a different starting and finishing salt concentration. This can be achieved by making fresh buffers with different KCl concentrations or by using the gradient mixer in the FPLC to adjust the starting and final concentrations of KCl. Either method works. We also make a stock of gradient buffer with no salt that is used to dilute salty fractions for additional fractionation.
After one pass through the press, extracts tend to be viscous. A second pass reduces viscosity making subsequent steps more efficient. Also note that PMSF is added to a final concentration of 1 mM just before cell disruption.
We use the XK16 empty column (GE) since it can accommodate variable bed heights and has proper fittings for use with the AKTA Purifier FPLC. Any manufacturer of empty columns can be used.
HiTrap columns are sold in 1- or 5-ml sizes. The binding capacity of these resins is high, so the 1 ml variety is usually sufficient, but if a very large preparation is performed, one can use the 5-ml cartridges.
Concentrations for elution are estimates from the resulting chromatograph. Slight variations in pH or buffer preparation can affect retention time of a given protein on the column; thus, elution should be confirmed by Western Blot using commercially available antibodies.
Larger mass of cells is required for factors purified from yeast than from E. coli. Typical preparative mass of cells used for Pol I is ~100 g of wet cell mass versus ~20 g for proteins produced in E. coli.
Pol I is relatively pure after MonoQ; however, WT Pol I also binds to MonoS and elutes with a standard gradient. However, purification through MonoQ is standard and sufficient for enzymology.
The anti-HA resin has a very high binding capacity, but for retention on small disposable columns for recirculation, usually >500 μl of settled resin is used.
Recirculation of HA peptide for elution onto downstream columns is generally performed at 4°C using a peristaltic pump overnight for highest yield.
Qiagen has begun including pH indicators in their DNA purification kits. Kits without this indicator are available and must be used for efficient transcription in vitro.
Each reaction is arbitrarily 20 μl; thus, for ten time points, we typically make a 11× (220 μl) mix.
Molar quantities are provided for all proteins, but core factor and UAF. Since these complexes are purified in very low quantities, accurate quantification of the samples is not feasible. Thus, the activity of each preparation is determined empirically by titration into transcription assays. The volumes described are representative for fractions purified as described herein. Pol I and a slight molar excess of Rrn3p are incubated together at room temperature for 4 h to preform the initiation-competent complex. Without preincubation, transcription initiation efficiency is reduced.
After addition of the NTPs, everything is timed. It is useful to write out the exact time at which every supplement or sample is taken and then keep a timer running in plain sight throughout the experiment.
For quantification, it is important that the volume of aqueous phase removed be identical between samples. Thus, it is useful to set the pipette to a low volume (e.g., 60 μl) to keep error low.
Urea gels accumulate debris in the wells rapidly which affects the RNA migration. Use a fine needle and 1× TBE to “spray” each well and remove debris prior to loading.
Radioactive GTP has migrated into the bottom of buffer tank, so great care should be taken in removing and disposing of this buffer after running the gel.
We have used GE or Bio-Rad imagers as well as ImageQuant or Quantity One software. There is very little difference in data quality; it is just a matter of becoming familiar with one program/instrument or the other.
References
- 1.Nomura M, Nogi Y, Oakes M. Transcription of rDNA in the Yeast. In: Olson MOJ, editor. Saccharomyces cerevisiae. The Nucleolus, Kluwer Academic/Plenum Publishers; London: 2004. pp. 128–153. [Google Scholar]
- 2.Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 3.Drygin D, Rice WG, Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu Rev Pharmacol. 2010;50:131–156. doi: 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- 4.Stefanovsky V, et al. Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol Cell. 2006;21:629–639. doi: 10.1016/j.molcel.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Smith ADT, Renfrow MB, Schneider DA. The RNA polymerase-associated factor 1 complex (Paf1C) directly increases the elongation rate of RNA polymerase I and is required for efficient regulation of rRNA synthesis. J Biol Chem. 2010;285:14152–14159. doi: 10.1074/jbc.M110.115220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keener J, Josaitis CA, Dodd JA, Nomura M. Reconstitution of yeast RNA polymerase I transcription in vitro from purified components. TATA-binding protein is not required for basal transcription. J Biol Chem. 1998;273:33795–33802. doi: 10.1074/jbc.273.50.33795. [DOI] [PubMed] [Google Scholar]
- 7.Schneider DA, et al. Transcription elongation by RNA polymerase I is linked to efficient rRNA processing and ribosome assembly. Mol Cell. 2007;26:217–229. doi: 10.1016/j.molcel.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuhn CD, et al. Functional architecture of RNA polymerase I. Cell. 2007;131:1260–1272. doi: 10.1016/j.cell.2007.10.051. [DOI] [PubMed] [Google Scholar]
- 9.Tongaonkar P, Dodd JA, Nomura M. Purification and assay of upstream activation factor, core factor, Rrn3p, and yeast RNA polymerase I. Methods Enzymol. 2003;370:109–120. doi: 10.1016/S0076-6879(03)70010-X. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann A, Roeder RG. Purification of his-tagged proteins in non-denaturing conditions suggests a convenient method for protein interaction studies. Nucleic Acids Res. 1991;19:6337–6338. doi: 10.1093/nar/19.22.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]