

Abstract
Deletion of proprotein convertase Mbtps1 in bone osteocytes leads to a significant postnatal increase in skeletal muscle size and contractile function, while causing only a 25% increase in stiffness in long bones. Concerns about leakiness in skeletal muscle were discounted since Cre recombinase expression does not account for our findings, and, Mbtps1 protein and mRNA is not deleted. Interestingly, the response of normal skeletal muscle to exercise and the regenerative response of skeletal muscle to the deletion of Mbtps1 in bone share some key regulatory features including a preference for slow twitch muscle fibers. In addition, transcriptional regulators PPAR, PGC-1α, LXR, and repressors DEC1 and DEC2 all occupy central positions within these two pathways. We hypothesize that the age-dependent muscle phenotype in Dmp1-Cre Mbtps1 cKO mice is due to bone→muscle crosstalk. Many of the myogenic genes altered in this larger and functionally improved muscle are regulated by circadian core transcriptional repressors DEC1 and DEC2, and furthermore, display a temporal coordination with Dec1 and Dec2 expression consistent with a regulatory co-dependency. These considerations lead us to propose that Dmp1-Cre Mbtps1 cKO osteocytes activate myogenesis by increased release of an activator of muscle PPAR-gamma, for example, PGE2 or sphingosine-1-P, or, by diminished release of an inhibitor of LXR, for example, long-chain polyunsaturated fatty acids. We hope that further investigation of these interacting pathways in the Dmp1-Cre Mbtps1 cKO model will lead to clinically translatable findings applicable to age-related sarcopenia and other muscle wasting syndromes.
Introduction
We recently demonstrated dramatic new musculoskeletal functions for ubiquitously expressed proprotein convertase MBTPS1 (SKI-1, S1p, Pcsk8) which cover the vertebrate lifespan. In the embryo, MBTPS1 is required for normal segmentation of somites during spinal development and Mbtps1 deficient embryos exhibit fused vertebrae and shortened tails as well as hindlimb paralysis.1 Also, deletion of Mbtps1 in bone osteocytes leads to a delayed muscle phenotype which is expressed only in mature (or older, JP Gorski, unpublished result) 10- to 12-month-old mice but not at 3 months of age2 (Figure 1). In contrast to aged littermate controls, the muscles of Dmp1-Cre Mbtps1 cKO mice, which we have termed ‘sumo' mice, are larger in size, display a 30% increase in contractile performance relative to control littermates even when normalized for size, and exhibit morphological characteristics of actively regenerating muscle.2 Although absolute contractile force for control muscles was lower than wild-type C57Bl6 background strain mice, we believe that the reason may be due to hyperglycemia, which was recently detected in the floxed strain (see below). Thus our findings indicate that deletion of Mbtps1 in bone causes a relative increase of 30% in contractile force. The muscle phenotype was localized to slow twitch (soleus, SOL) muscles expressing type I myosin heavy chain but not to fast twitch (EDL) muscles. As Mbtps1 is deleted in bone osteocytes and not in skeletal muscle tissues,2 we have proposed that bone→muscle crosstalk signaling is responsible for the latter age-related muscle phenotype. In an effort to identify the bone-derived crosstalk factor and to understand more fully the nature of the induced metabolic changes, we analyzed the transcriptome of soleus muscles from ‘sumo' mice and littermate controls (GSE69985, NCBI).
Figure 1.

Deletion of Mbtps1 in bone (red symbols) increases muscle mass after skeletal maturity (arrow, 15–20 weeks.). Left panel, two representative litters are depicted. Right panel, two male littermates at 10 months of age. Fat content (% body wt.) is not substantially different (not shown).
Gene profiling revealed increased expression in pathways mediating EGFR1 signaling, circadian exercise, striated muscle contraction, glycolysis, fatty acid oxidation and adipogenesis (Table 1). Briefly, actinin-α3 (ACTN3) was elevated in cKO SOL; ACTN3 is a biomarker for increased muscle performance in elite athletes.3 Other striated muscle contraction genes increased included Mg29, vimentin, desmin and slow twitch muscle restricted troponin. Embryonic and perinatal myosin heavy chains (MYH3 and MYH8), which have been used as biomarkers to identify muscle regeneration,4,5,6 were also increased. MYF6, which is associated with satellite cell activation during muscle regeneration,7 and PAX7, MYOD1 and MYOG, which are required for postnatal growth and regeneration of adult skeletal muscle,8,9,10 were also elevated in Dmp1-Cre Mbtps1 cKO SOL. Metabolically, enhanced expression of triglyceride lipase in Mbtps1 cKO SOL should facilitate release and oxidation of fatty acids. Interestingly, expression of basic helix-loop-helix domain containing transcriptional regulators DEC1 (BHLHE40, STRA-13 and BHLHB3) and DEC2 (BHLHE41, SHARP1 and BHLHB3) was decreased significantly in Mbtps1 cKO SOL compared with controls at the time the mice were killed.2
Table 1. Gene profiling reveals signaling pathways activated in Dmp1 cre Mbtps1 cKO soleus muscle.
| EGFR1 |
| Striated muscle contraction |
| Membrane trafficking |
| Fatty acid oxidation |
| Glycolysis |
| Circadian exercise |
| Adipogenesis |
Abbreviation: EGFR, epidermal growth factor receptor.
DEC1 and DEC2 are core circadian clock genes which like PER and CRY are positively regulated by BMAL and CLOCK;11,12 in return, expression of DEC1 and DEC2 are subject to negative feedback by transcription factor SREBP-1.12,13 DEC1 and DEC2 expression in muscle displays a rhythmic 24–28 h cycle14 which because of negative feedback regulation would be predicted to be offset by ∼12–24 h compared with that for BMAL and CLOCK. BMAL expression is required for satellite cell activation in muscle regeneration.15 DEC1/DEC2, in contrast, were shown previously to repress myogenesis in vitro and in vivo by blocking transcription of MYOD1, as well by altering expression of a number of contractile and mitochondrial proteins by skeletal muscle.16,17,18 By analogy with the age-related onset of the Dmp1-Cre Mbtps1 cKO phenotype, DEC2 null mice show no deficit with respect to repair of embryonic muscle, however, as predicted for a repressor of myogenesis, regeneration after injury in post-natal DEC2(−/−) mice is increased.18 As SOL in Dmp1-Cre Mbtps1 cKO mice display characteristics of regenerating muscle, we hypothesized that a cycling circadian expression pattern for myogenic repressors DEC1/DEC2 could stimulate or reduce expression of pro- or anti-myogenic genes, respectively, during their ‘down cycle'. Since Lecomte and colleagues have already identified (+) and (−) transcriptional muscle-specific targets of DEC1/DEC2,16 we asked whether expression of these target genes is temporally correlated with that of transcriptional regulators DEC1/DEC2 in Dmp1-Cre Mbtps1 cKO muscle? Importantly, the majority of muscle-specific target genes (79 out of 110) displayed the expected responses16 as when DEC1/DEC2 are downregulated.2 As expression of both DEC1 and DEC2 is low in Dmp1-Cre Mbtps1 cKO SOL (array data deposited at NCBI, #GSE69985), this is the expected outcome. Although our transcriptional data represents a single time point, and not a systematic timed study of circadian regulation, we still believe the high degree of correlation between predicted and observed target gene expression observed supports a role for DEC1/DEC2 in the regeneration and growth of Dmp1-Cre Mbtps1 cKO SOL.2 The purpose of this article is to justify a hypothetical mechanistic bone→muscle crosstalk signaling pathway that explains the ‘sumo mouse' muscle phenotype while incorporating what is known about the control of myogenesis following exercise.
Physical exercise, muscle mass gain and the effect with age
Exercise is currently the only proven method to both inhibit age-related muscle loss and to at least partially restore muscle mass lost via inactivity and age-related sarcopenia.19,20 In terms of exercise, the two main types of physical exercise affect skeletal muscles differently, for example, endurance and resistance (strength) training. Resistance training works both fast twitch and slow twitch muscles while endurance training primarily works the slow twitch fibers and changes these fibers in terms of increased efficiency and resistance to fatigue.20,21 Exercise-induced changes in muscles include an increase mitochondrial number and size and in their metabolic capacity to store glycogen and fat, as well as to catabolically use fat and glycogen stores as an energy source. A key similarity between the effect of endurance training and the effect of conditional deletion of Mbtps1 in bone is that they both impact predominantly slow twitch muscle fibers. For example, Dmp-1-Cre Mbtps1 cKO SOL muscles (slow twitch), but not EDL muscles, gain mass, alter their metabolic profile, and contract more forcefully. Within soleus muscles, only type I (slow twitch) myosin heavy chain expressing muscle fibers contain centralized nuclei, a morphological characteristic of regeneration. However, not all features are shared with entrained muscle since SOL and EDL from Dmp1-Cre Mbtps1 cKO do not display increased resistance to fatigue nor cKO EDL exhibit immunochemical evidence of a fast twitch to slow twitch transition.2,22
Because historically there is a significant positive correlation of muscle strength and cross-sectional area (CSA), we recently have measured myofiber CSA for our ‘sumo' mice (Table 2). The data show that average myofiber CSA for the larger SOL muscles in DMP1-Cre Mbtps1 cKO mice are not statistically different from control littermates. Actual values, however, are very similar to that for published values for murine SOL muscles (Table 2). However, when myofibers within SOL with central nuclei (∼10–15% of total), which we have suggested is a marker for muscle regeneration in our model, were measured it is apparent that the average CSA was significantly smaller than that for total cells (Table 2). We believe that this finding is consistent with findings of a large number of publications23,24,25 and supports ongoing regeneration within Dmp1-Cre Mbtps1 cKO muscle. Specifically, during regeneration of muscle following an injury stimulus, the average CSA of myofibers is initially lower. However, as time elapses during the repair phase, the average CSA increases to reach that of the muscle prior to injury. We believe the rationale is that new myogenic progenitors, stimulated by the injury stimulus, are formed and proceed to ‘differentiate through fusion with each other or to damaged fibers to reconstitute fiber integrity and function'.26
Table 2. Comparison of cross-sectional area of myofibers from Dmp1-Cre Mbtps1 cKO and control SOL muscles.
| Control (all myofibers) | Dmp1-Cre Mbtps1 cKO (all myofibers) | Dmp1-Cre Mbtps1 cKO (myofibers only with centralized nuclei) |
|---|---|---|
| 1,744±377 μm2 STD | 1,557±357 μm2 STD P=0.274 (two tailed t-test) | 1,162±192 μm2 STD P=0.006 (two tailed t-test) |
Abbreviations: SOL, soleus; STD, standard deviation.
Although age was once presumed to cause dramatic reductions in optimal muscle performance after endurance training, further research has shown that the rate of decline may be much slower than once believed.27 Recently, Karsenty and colleagues28 showed that osteocalcin signaling in myofibers is necessary for maximal adaption to exercise. They also showed that exogenous osteocalcin can dramatically restore the exercise capacity of old mice to that of 3 months old mice.28 Since no significant difference exists between the osteocalcin content of Dmp1-Cre Mbtps1 cKO mice and controls at 12 months of age (23.4±7.1 vs 30.8±11.6 ng ml−1, respectively; (Gorski, JP, unpublished result) and osteocalcin did not stimulate muscle growth and regeneration,28 we do not believe osteocalcin has a prominent role in the Mbtps1 cKO muscle phenotype (Figure 1).
Exercise and the signaling pathway regulating slow twitch muscle regeneration
Much work has gone into identifying the signaling pathway that regulates the response of skeletal muscle to endurance training as outlined above. While the individual genes required for exercise-induced increases in myogenesis and in oxidative fuel usage, as well as fiber type transition are generally known, the physiological signals or triggers remain uncertain. However, several key genes mediate changes in myogenesis, in oxidative fuel usage and in fiber type transition: PGC-1α, PPAR-β/δ, MGF (a splice variant of IGF-1), and Rev-erb-α (Figure 2).
Figure 2.

Hypothetical pathway regulating the response of skeletal slow twitch muscle to endurance training: myogenesis, fuel usage, and myosin fiber type change.
PGC-1α (PPARγ co-activator 1α) is a transcriptional co-activator that interacts with and regulates the activities of cAMP response element binding protein (CREB). PGC-1α is directly regulated by myogenic regulatory factors (MEF2c and MyoD1).29 It provides a direct link between external physiological agents and is a master regulator of mitochondrial biogenesis and muscle fiber type determination.30 One such physiological agent, hypoxia, is a consequence of physical exercise and leads to increased HIF-1α and DEC1 expression31 (Figure 2). DEC1 represses transcription by directly competing with MyoD1 for the same PGC-1α promoter-binding site.32 Importantly, DEC1 is reciprocally regulated by SREBP1 which requires proteolytic activation by MBTPS1.16 SREBP-1 is a required transcription factor for most enzymes and receptors mediating lipolysis and lipogenesis and mitochondrial biogenesis, as well as myogenesis.33 The light/dark entrained mammalian circadian clock is known to control glucose and lipid metabolism and mitochondrial oxidative metabolism. Although widely appreciated, the fact that PGC-1α, SREBP1, DEC1 and MyoD1 are all cyclically expressed in skeletal muscle would seem to provide an explanation.34
Peroxisome proliferator activated nuclear receptors (PPARs) function as transcription factors and are so named because they are activated by ligands that induce proliferation of peroxisomes, organelles that mediate oxidation of fatty acids. PPARs can be activated both by dietary and endogenous lipid compounds. Once activated, PPARs can redirect a cell's metabolism. In skeletal muscle, activated PPAR-β/δ induces a switch to form increased numbers of type I muscle fibers.35 Targeted expression of PPAR-β/δ in skeletal muscle produces a mouse capable of running twice the distance of a wild-type littermate as well conferring resistance to obesity.35 Lipid ligands capable of activating PPARs include long-chain fatty acids, eicosanoids, and prostaglandins, although no consensus agreement exists as to the identity of the primary physiological activator.36,37
MGF (mechano growth factor) is a splice variant of IGF-1.38 IGF-1 and MGF are upregulated in exercised39 and damaged skeletal muscle where MGF has been shown to stimulate proliferation and inhibit differentiation.40 In general, animal studies have confirmed cell culture studies and confirmed that MGF is a local tissue repair factor which responds to mechanical demands.38 In a broader sense, different splice variants of IGF-1 display distinct functions within the myogenic pathway wherein isoform IGF-1Eb stimulates satellite cell activation, while isoform IGF-1Ea promotes myoblast proliferation and differentiation into myotubes.41
In summary, the above rationale of exercise effects on skeletal muscle suggests several conclusions. The response of skeletal muscle to exercise and its regenerative response to deletion of Mbtps1 in bone shares several key regulatory components. Transcriptional repressors DEC1 and DEC2, PPAR-delta, and PGC-1a are all prominently expressed after exercise and in Dmp1-Cre Mbtps1 cKO muscle. While the myogenic actions of PPAR-delta and PGC-1a are well documented, the robust regulatory actions of DEC1 and DEC2 on the proliferation of myogenic satellite cells and the expression of myokines and mitochondrial genes is under appreciated. Implicitly our analysis indicates that bone→muscle crosstalk can modify the metabolism of muscle independent of physical exercise. Although the actual bone-derived crosstalk factor is still under investigation, we have learned much about the regenerative response of skeletal muscle to exercise and to deletion of Mbtps1 in bone as detailed in the next section. As a result, we wish to emphasize the role of DEC1 and DEC2 in bone muscle crosstalk and exercise-induced muscle regeneration.
Circadian core genes DEC1/DEC2 regulate myogenesis (and adipogenesis)
Transcription factor SREBP-1, also termed ADD-1 or adipocyte determination and differentiation-dependent factor 1, is a major positive determinant of lipogenesis in mammals and avian species.42 SREBP-1 is known to directly regulate the expression of over 200 genes involved in de novo cholesterol and fatty synthesis as well as their transport and cellular uptake33 (Figure 3). In view of their common stem cell, the myocyte and adipocyte lineages represent alternative differentiation pathways. Importantly, the work of Lecomte,16 indicates that while SREBP-1 actively promotes adipocyte differentiation, overexpression of SREBP1 in muscle in vivo causes muscle atrophy16 while in vitro, SREBP-1 inhibits myoblast to myotube differentiation. Specifically, overexpression of activated SREBP1 isoforms 1a and 1c in muscle cells altered the expression of over 1300 genes in differentiated human myotubes.17 Genes experiencing substantial change encoded early myogenic transcription factors MYOD1, MYOG, and MEF2C as well as many muscle contractile apparatus proteins (myosin heavy and light chains, troponins and titin; Figure 3). Interestingly, most genes were downregulated in the presence of SREBP-1a or SREBP-1c, however, transcriptional repressors Dec1 and Dec2 were induced. Subsequent work demonstrated that Dec1 and Dec2 are direct transcriptional targets of SREBP-1a and SREBP-1c and that DEC1 and DEC2 then together mediate the repression of hundreds of muscle genes (Figure 3).16,17 Taken together, this work illustrates an under-appreciated function of transcription factor SREBP-1 (and its target surrogate repressors DEC1 and DEC2), for example, to regulate the differentiation of mesenchymal stem cells to fat and muscle. When present, activated SREBBP-1 stimulates the formation of adipocytes and blocks the differentiation into myocytes.43 When SREBP-1 is absent, the default fate of mesenchymal stem cells is to differentiate into myocytes. As illustrated in the next sections, we discuss how systemic hormones, environmental factors, circadian cycling, and post-translation processing reactions can determine the nuclear content of activated SREBP-1 (and its surrogates DEC1 and DEC2).
Figure 3.

Summary of proposed relationships involving SREBP1 and MBTPS1 and DEC1/DEC2 and their regulation of myogenesis in skeletal muscle.
DEC1/DEC2 are subject to regulation at the transcriptional level by hormones (insulin and retinoic acid), environmental factors (hypoxia, oxysterols and hyperglycemia), and indirectly via SREBP1
These regulatory pathways are summarized in Figure 4 and illustrate that expression of Dec1 and Dec2 is intimately linked with the metabolic well-being of the host, its access to nutritional sources of fats and glucose, circadian cycling, and its exposure to specific environmental factors. Among these direct regulators, hormones insulin and retinoic acid are prominent. Specifically, Yamada K et al.44 showed that insulin induces Dec1 via a phosphoinositide-3-kinase pathway. Rexinoid bexarotene induces transcription of Dec2, which then can repress cell proliferation by blocking expression of cyclin D145 (Figures 3 and 4). Interestingly, retinoic acid also indirectly regulates Dec1 and Dec2 through SREBP1. Yoshikawa T et al.46 demonstrated that the liver X receptor–retinoid X receptor is an activator of the SREBP1c promoter and that ligands 22-hydroxycholesterol and 9-cis-retinoic acid are able to induce both SREBP-1c mRNA and protein production in cultured cells. As shown in Figure 4, SREBP1 and DEC1/DEC2 reciprocally regulate each other at the transcriptional level.16,47
Figure 4.
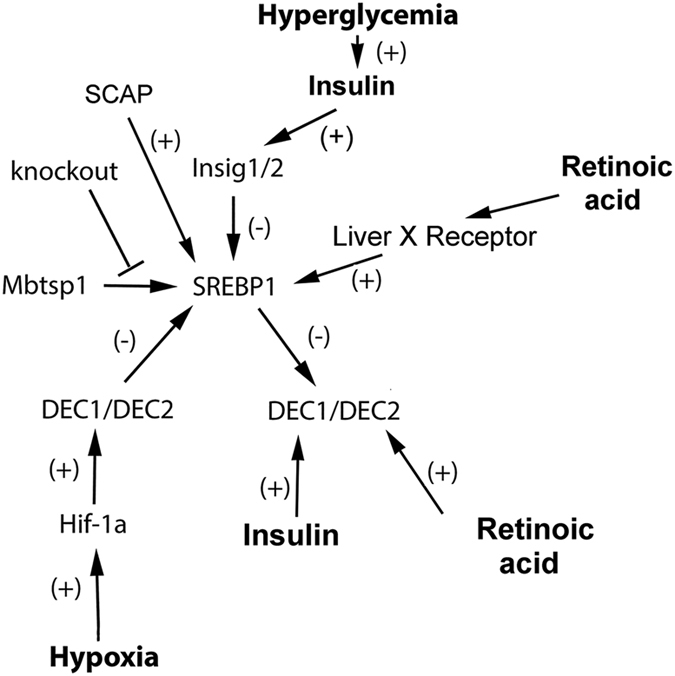
Dec1 and Dec2 are subject to transcriptional regulation by environmental agents: systemic hormones, environmental conditions, and SREBP1.
As well as hormones, expression of Dec 1 and Dec2 is also controlled by environmental factors (Figure 4). Hypoxia stimulates a general response in all cells that ultimately produces transcription factor HIF-1α which then initiates the hypoxic response. One of the important targets of HIF-1α is the transcriptional repressor pair DEC1 and DEC2. HIF-1α activates transcription of both Dec1 and Dec2 by binding to hypoxia response elements in the transcriptional regulatory regions of both of these genes.31 Teleologically, HIF-1α induction of Dec1 and Dec2 (which would also inhibit cyclin D1 expression) can be viewed as beneficial in terms of conserving resources since a low oxygen environment would not be conducive to cell proliferation. Environmentally, hyperglycemia represents another indirect regulator of Dec1 and Dec2 expression. Specifically, hyperglycemia was recently shown to promote N-glycosylation of SCAP, a SREBP-1 transport protein. Under these conditions, the N-glycosylated SCAP protein displays a reduced association with its regulatory protein INSIG-1 which facilitates SREBP1 transport to the Golgi and its subsequent activation/release from the Golgi membrane and nuclear import of the transcriptionally active domain.48 Finally, oxysterols can also activate SREBP1c transcription by binding to the liver X receptor49 whereas long chain polyunsaturated fatty acids suppress SREBP1c transcription by inhibiting liver X receptor binding to LXR response elements.50 An increase in activated nuclear SREBP1 would be predicted to lead to an increase in Dec1 and Dec2 transcription (Figures 3 and 4).
DEC1 and DEC2 are subject to regulation at the protein level by sumoylation, ubiquitination and phosphorylation
DEC1 is subject to sumoylation at lysine residues 159 and 279.51 Interestingly, sumoylation had several consequences evident in cultured cells. First, sumoylation of DEC1 stabilized it's turnover since the alternate post-translational reaction of ubiquitination was inhibited.52 Second, sumoylation facilitated its repression of circadian core clock genes Clock and Bmal by recruitment of histone deacetylase1. DEC2 is similarly sumoylated on lysine residues 240 and 25553 which enhances recruitment of co-repressor G9a (a histone H3 lysine 9 dimethylase). Since mutation of these two SUMO acceptor sites and its degradation by SUMO protease SENP1 both limit the ability of DEC2 to block myogenesis,18 we assume that sumoylated DEC2 (and DEC1) represent the active penultimate functional forms of these myogenic repressors.
Interestingly, SREBP1a and SREBP1c, but not SREBP2, are hyperphosphorylated within mitotic cells.54 Hyperphosphorylated SREBP1 was stabilized in G2/M phase and this leads to an increase in its transcriptional activity. However, in general, phosphorylation of SREBP1 in its ‘phosphodegron' (a portion of a protein that is important in regulating its degradation) facilitates its degradation by the ubiquitin-proteasome system where Ser-434 may function as a molecular switch.55
DEC1 and DEC2 are subject to regulation by circadian core proteins
Circadian rhythms are outputs of an internal biological clock that in isolation from environmental cues maintains a highly reproducible cycle with a period of about 24 h. The period and phase of the rhythm are adjusted in nature to those of the earth's rotation by input pathways like the daily light cycle or ingestion of food. The mammalian circadian clock is a circadian transcriptional-translational oscillator driven by rhythmic activation and repression of the CLK/BMAL transcription factor (Figure 2).56 The CLK/BMAL transcription factor binds to an E-box to activate transcription of both core circadian genes as well as many genes that are outputs of the circadian clock. Alternatively, other core circadian elements include period (Per) and cryptochromes (Cry) mRNAs which encode repressor proteins that accumulate with a phase delay relative to their mRNAs because PER protein is phosphorylated by kinases, particularly casein kinases I and II, and then degraded. However, PER and CRY become stabilized when they form a complex that moves into the nucleus, where they inactivate CLK/BMAL, thereby turning off their own transcription which, for each, is activated by CLK/BMAL. This negative feedback loop produces circadian rhythms in CLK/BMAL-dependent gene expression that produce the numerous circadian outputs in muscle and other tissues.56
Regulation of transcription factor SREBP1 activation by cholesterol57 is a well known environmental input to the circadian clock transcriptional circuit. Cholesterol regulates its own synthesis via a homeostatic negative feedback pathway involving SREBP1. Two Golgi-resident proteases are involved in this process, MBTPS1 and Site-2 protease, that sequentially cleave and finally release activated SREBP1 from the Golgi membrane. Once cleaved, the helix-loop-helix motif-containing SREBP1 fragment is transported to the nucleus where it activates numerous genes controlling the synthesis of cholesterol. As the level of cholesterol rises, it eventually downregulates the cleavage of SREBP1 by inducing an INSIG/SCAP interaction, and cholesterol homeostasis is maintained.57
The cholesterol homeostatic mechanism interacts with the circadian clock in several ways. First, SREBP1c activates expression of Dec1,16 which represses circadian gene expression by binding to the same E box site that recruits the CLK/BMAL circadian activator protein.11 As noted below, DEC1 also represses the Notch pathway, thereby providing a link between metabolism, the clock and muscle stem cell activation (Figure 3). Hence, SREBP1 induces a repressor that down regulates transcription of genes controlled by the CLK/BMAL activator. In turn, DEC1 levels oscillate in a circadian manner11 because they are induced by CLK/BMAL,12 and DEC1 represses SREBP1 transcription.47,58 In addition, the circadian nuclear receptor REV-ERBα, that is activated by CLK/BMAL and represses BMAL, also regulates the production of activated SREBP1 because it produces rhythmic transcription of INSIG, a factor that inhibits transport of SREBP1 to the Golgi59 (Figure 2).
Metabolic input to the circadian clock also comes from several metabolic nuclear hormone receptors. The retinoic acid receptors RAR and RXR interact with CLK and its paralog MOP4 in response to binding of retinoic acid to inhibit activation of genes by CLK and MOP4. The Retinoic X Receptor (RXR) and Liver X Receptor (LXR)-RXR heterodimer both bind DEC1 and DEC2, which thereby inhibit the genes that they activate, including sterol-activated genes.60
An additional level of metabolic control interfaces with these transcriptional circuits, e.g., control by the insulin receptor and its related downstream kinase signaling pathways.61 Dec1 mRNA is induced by insulin receptor signaling via a PI3 kinase pathway, and a high carbohydrate diet induces high levels of Dec1.44 The AMPK kinase interacts with the insulin-signaling pathway to stimulate catabolism. One of the AMPK regulatory subunits (PRKBAB2) shows a circadian oscillation, suggesting that AMPK kinase activity also oscillates in a circadian manner. Catalytically, AMPK phosphorylates the CLK/BMAL repressor CRY, resulting in CRY's degradation;62 however, AMPK also activates PGC-1a, providing another regulatory interaction with the circadian transcriptional network.
Rationalization of the role of DEC1 and DEC2 in the ‘Muscle Phenotype' of Dmp1-Cre Mbtps1 cKO mice
Having reviewed the regulatory pathways above which control expression and protein processing of DEC1, DEC2, and SREBP1, we now conclude with a rationalization of how a SREBP1 dependent myogenic pathway in skeletal muscle could be activated transcriptionally by osteocytes. Furthermore, assuming bone→muscle signaling operates systemically, our rationalization will also address how skeletal muscle could respond selectively to a SREBP1 activator while fat tissue does not. At the outset, we also need to provide an explanation for existing data which show the opposite outcome (inhibition of myogenesis) when Dec1 or Dec2 are over-expressed in muscle.
We envision that overexpression of Srebp1 in muscle by an adenoviral vector, as was done by Lecomte et al.16 should result in a continuous high level of expression avoiding on and off cycles, as the adenoviral promoter is not regulated by feedback from SREBP1 or DEC1 or DEC2. As a result, when SREBP1 is over-expressed, we also expect that it will induce persistent expression of Dec1 or Dec2 at a high level. Under these conditions, assuming commensurate efficient translation, high levels of DEC1 or DEC2 protein are expected to effectively repress their target genes which include many myogenic growth factors (MyoD1, MyoG and Mef2c), satellite cell markers (Pax7, Myhc3 and Myhc6), and muscle performance genes.16 We propose that this is the reason that overexpression of SREBP1 in muscle via adenoviral vector avoids feedback inhibition and leads to complete blockage of myogenesis. On the other hand, Srebp1, Dec1 and Dec2 are normally expressed on a repeating 24 h on and off circadian cycle.60 In view of the fact that DEC1 and DEC2 are repressors of Srebp1 and that SREBP1 is required for transcription of Dec1, we envision a reciprocal feedback loop wherein, in the absence of adenoviral mediated overexpression of Srebp1, the relative cyclic expression pattern for Srebp1 should be offset relative to that of its Dec1/Dec2 repressors by approximately 12 h.16 Because of the complex activation pathway needed to transport SREBP1 protein to the cis-medial Golgi, we presume that Dmp1-Cre Mbtps1 cKO mice must necessarily express an activator of the SCAP transport system, for example, hyperglycemia, oxysterol, and/or insulin. Interestingly, recent unpublished data show that Dmp1-Cre Mbtps1 cKO male mice are both hyperinsulinemic, as well as hyperleptinemic (Gorski, JP, unpublished data; Figure 5 and Table 3). Although explanations for how remain speculative, concurrent hyperinsulemia and hyperleptinemia conditions would provide a means to both activate Srebp1 expression systemically while limiting its actions to regeneration of skeletal muscle rather than growth and differentiation of adipocytes and fat. We present our rationale below.
Figure 5.

Elevated serum insulin levels appear to be permissive for weight (lean muscle mass) gain in 12-month-old Dmp1-Cre Mbtps1 cKO mice.
Table 3. Dmp-1-Cre Mbtps1 cKO mice have twofold higher serum leptin concentration than littermate controls.
| Dmp1-cre Mbtps1 cKO | Control littermates | |
|---|---|---|
| Leptin | 7176±2446, pg ml−1 | 3575±1725, pg ml−1 |
| P=0.018 |
As insulin is an inducer of Srebp1 transcription,63,64,65 we believe this provides a ready explanation for how SREBP1 expression could be activated in hyperinsulemic Dmp1-Cre Mbtps1 cKO mice. The latter group showed that insulin action required the presence of SRE (sterol response elements) within the Srebp1 promoter, while Cagen et al.65 showed that full insulin action required the combinatorial participation of Sp-1, SREBP1, LXR, and NF-Y cis-acting elements. Our preliminary evidence suggests that hyperinsulinemia is permissive for increased skeletal muscle growth. Specifically, serum insulin concentrations above the normal range (∼0.4 ng ml−1) are associated with larger body weight (muscle mass) in Dmp1-Cre Mbtps1 cKO mice at 10–12 months of age although body weight seems to plateau at ∼50 g (Figure 5). Since full activation of the SREBP1 promoter requires participation by LXR, we speculate that bone osteocytes secrete a lipid-like activator of LXR.46,66 Osteocytes are known to produce oxysterols which could bind to and activate the LXR receptor. Alternatively, as noted above, PPAR-γ receptor regulates several aspects of muscle regeneration following exercise. Among these ligands sphingosine-1-P and PGE2 are known to stimulate myogenesis by blocking expression of myogenic repressor myostatin and to induce FOXO1 which then transcriptionally activates fiber type transition and mitochondrial biogenesis (Figure 2 and Table 1). Importantly, sphingosine-1-P and PGE2 are produced by osteocytes and the amount released can be increased dramatically in response to oscillatory fluid flow (cell stress).67,68 In this way, we propose that Dmp1-Cre Mbtps1 cKO osteocytes are stimulated to release elevated amounts of sphingosine-1-P or PGE2, and after making its way to the circulation, this bone-derived signaling agent would then activate PPAR-γ receptors expressed on skeletal muscle cells. As noted above, activated PPAR-γ receptors are believed to stimulate myogenesis after exercise according to well established pathways.
Because we have proposed that activation of SREBP1 is required for the muscle phenotype, we have also questioned why SREBP1, also known as ADD-1 (adipocyte development and differentiation factor one) and a master gene of lipid metabolism, does not also stimulate fat production in Dmp1-Cre Mbtps1 cKO mice? We hypothesize that an observed 2-fold elevated content of serum leptin (Table 3) blocks adipogenesis. Interestingly, when both 10- to 12-month-old controls and knockout mouse data were plotted against body weight, we observed a strong linear relationship (r2=0.89) between leptin concentration in serum and total body weight (Figure 6). Noticeably, Dmp1-Cre Mbtps1 cKO values generally segregated separately from controls, although both seem to lie on the same 95% regression line. Leptin, generally thought to be produced primarily by adipocytes, regulates satiety while also controlling energy expenditure. However, we presume that the most likely source for leptin in Dmp1-Cre Mbtps1 cKO mice is skeletal muscle69,70,71 since piximus analyses for body fat and direct dissection could not account for the weight gain observed2 (Figure 1) and since a linear relationship exists between body mass (e.g., skeletal muscle) and serum leptin (Figure 6). Plasma leptin suppresses AMPK activity in the hypothalamus and restricts food intake.72 Body weight is determined by the balance between food intake and energy expenditure. It is noteworthy that 10- to 12-month-old male Dmp1-Cre Mbtps1 cKO mice consumed the same amount of food as littermate controls over a two day period after acclimatization (Table 4) despite the former group weighing an average 3.4 gm more.2 We interpret this finding as indicating that Dmp1-Cre Mbtps1 cKO mice may use energy more efficiently than controls. Consistent with this finding, whole-genome array analyses of SOL from Dmp1-Cre Mbtps1 cKO mice have already documented a metabolic shift towards more oxidative metabolic pathways of fat and glucose use (array data deposited at NCBI under accession number GSE69985; Gorski et al.2).
Figure 6.

Serum leptin is strongly correlated with total body weight for adult male Mbtps1 cKO and control mice. Red line roughly separates control data points from those for Mbtps1 cKO values. Black line represents 95% confidence regression line. r2 value was determined by linear regression analysis using SigmaStat program.
Table 4. Comparison of food intake and blood glucose values for adult knockout and control littermates.
| Food consumption per 2 days | Blood glucose | |
|---|---|---|
| DMP1 cre SKI-1 cKO (males) n=8 | 9.58 g±1.28 STD | 239.0±51.5 STD |
| Control littermates (males) n=5 | 9.88 g±1.45 STD | 236.2±52.6 STD |
| Statistics | P=0.697 | P=0.911 (two-tailed t-test) |
Abbreviation: STD, standard deviation.
Hamrick et al.73 showed that aged mice (24 months), but not 12-month-old mice, responded to injection with recombinant leptin by increasing the size and mass of muscles (including EDL), and induction in muscle of several miRNAs known to be associated with regeneration and repair. While it is tempting to presume that increased muscle in Dmp1-Cre Mbtps1 cKO mice is solely the result of paracrine signaling by leptin (Figure 6), our phenotype is not superimposable since it is evident at 12 months as well as 26 months of age (unpublished result, JP Gorski) and is restricted to SOL (not EDL) muscles.2 Thus, while the elevated leptin content of Dmp-Cre Mbtps1 cKO mice likely contributes positively to the observed muscle phenotype, it does not appear to be the sole determinant.
Summary points
Conditional deletion of proprotein convertase Mbtps1 in bone osteocytes leads to a dramatic change in skeletal muscle size and contractile function while causing only limited change in the structure and function of long bones. However, we believe that a significant 25% increase in stiffness2 could be a direct response of knockout long bones to the increases in body weight and muscle mass.
The muscle phenotype is not evident in 3 month old mice, but is detectable at >10 months of age.
Soleus muscle fibers exhibited significantly more centralized nuclei which were restricted exclusively to type I myosin heavy chain (slow twitch) expressing cells. When combined with increased expression of early myogenic growth factors (MyoD1, MyoG, Mef2c), of embryonic and prenatal myosin heavy chains, of enhanced performance genes (Actinin-3), and of satellite cell biomarkers (Pax7), we conclude that Dmp1-Cre Mbtps1 cKO muscle exhibits morphological and transcriptional characteristics of regenerating muscle.
Concerns about leakiness of the Dmp1-Cre in skeletal muscle were discounted since Cre recombinase expression does not account for our findings, and, Mbtps1 protein and mRNA are not deleted. Based on our findings, the most likely explanation for the muscle phenotype is age-dependent bone→muscle crosstalk, for example, a bone derived factor that causes skeletal muscle myogenesis.
Many of the myogenic genes altered in muscle are regulated by circadian core transcriptional repressors DEC1 and DEC2, and, furthermore, display a temporal coordination with Dec1 and Dec2 expression which implies a regulatory co-dependency. Importantly, DEC1 and DEC2 are themselves regulated at the transcriptional level by reciprocal feedback loops with SREBP1.
In order to rationalize a mechanism responsible for the muscle phenotype, we considered both the pattern of gene expression, its shared features with that for entrained muscle, and the identity of known osteocyte-derived mediators of myogenesis. However, in contrast to entrained muscle, SOL and EDL from Dmp1-Cre Mbtps1 cKO do not display increased resistance to fatigue nor cKO EDL exhibit immunochemical evidence of a fast twitch to slow twitch transition.2 These considerations lead us to propose that Dmp1-Cre Mbtps1 cKO osteocytes activate myogenesis by release an activator of muscle PPAR-γ, for example, PGE2 or sphingosine-1-P. PPAR-γ plays a similar prominent role in mediating muscle regeneration after exercise. Alternatively, deletion of Mbtps1 in osteocytes may lead to reduced release of an inhibitor of myogenesis.
In addition, we believe activation of Srebp1, Dec1, and Dec2 expression in skeletal muscle is also required to obtain the muscle phenotype. At present, we do not have clear explanations for the elevated serum levels of insulin and leptin in Dmp1-Cre Mbtps1 cKO mice but we envision that these systemic hormones are critical to establishment and maintenance of the muscle phenotype. Specifically, elevated insulin levels in Mbtps1 cKO mice are proposed to induce Srebp1 expression in skeletal muscle thus leading indirectly to activation over 100 myogenic genes. We propose that elevated levels of leptin serve to block adipogenesis which would normally be a consequence of increased systemic Srebp1 expression and to contribute positively to the myogenic and regenerative muscle phenotype in Dmp1-Cre Mbtps1 cKO mice.
Taken together, we believe this rationale provides a reasonable explanation for the age-dependent Dmp1-Cre Mbtps1 cKO muscle phenotype. However, achieving muscle regeneration and growth with age is a complex process involving signaling pathways controlling satellite cell differentiation, energy utilization, the balance of fat and muscle, and circadian cycling. We hope that further investigation of these interacting pathways identified in the Dmp1-Cre Mbtps1 cKO mouse will lead to clinically translatable findings applicable to age-related sarcopenia and other muscle wasting syndromes.
Acknowledgments
This work was supported by grants to JPG from the Scoliosis Research Society and the University of Missouri Research Board.
Footnotes
The authors declare no conflict of interest.
References
- Achilleos A, Huffman NT, Marcinkiewicyz E, Seidah NG, Chen Q, Dallas SL et al. MBTPS1/SKI-1/S1P proprotein convertase is required for ECM signaling and axial elongation during somitogenesis and vertebral developmentdagger. Hum Mol Genet 2015; 24: 2884–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JP, Huffman NT, Vallejo J, Brotto L, Chittur SV, Breggia A et al. Deletion of Mbtps1 (Pcsk8, S1p, Ski-1) gene in osteocytes stimulates soleus muscle regeneration and increased size and contractile force with age. J Biol Chem 2016; 291: 4308–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Garton F, North K. alpha-actinin-3 and performance. Med Sport Sci 2009; 54: 88–101. [DOI] [PubMed] [Google Scholar]
- Weiss A, Schiaffino S, Leinwand LA. Comparative sequence analysis of the complete human sarcomeric myosin heavy chain family: implications for functional diversity. J Mol Biol 1999; 290: 61–75. [DOI] [PubMed] [Google Scholar]
- Mathes AL, Lafyatis R. Role for Toll-like receptor 3 in muscle regeneration after cardiotoxin injury. Muscle Nerve 2011; 43: 733–740. [DOI] [PubMed] [Google Scholar]
- Sartore S, Gorza L, Schiaffino S. Fetal myosin heavy chains in regenerating muscle. Nature 1982; 298: 294–296. [DOI] [PubMed] [Google Scholar]
- Goetsch SC, Hawke TJ, Gallardo TD, Richardson JA, Garry DJ. Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol Genomics 2003; 14: 261–271. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Le Grand F, McKinnell I, Kuang S. The molecular regulation of muscle stem cell function. Cold Spring Harb Symp Quant Biol 2008; 73: 323–331. [DOI] [PubMed] [Google Scholar]
- Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell 2000; 102: 777–786. [DOI] [PubMed] [Google Scholar]
- Olguin HC, Yang Z, Tapscott SJ, Olwin BB. Reciprocal inhibition between Pax7 and muscle regulatory factors modulates myogenic cell fate determination. J Cell Biol 2007; 177: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 2002; 419: 841–844. [DOI] [PubMed] [Google Scholar]
- Hamaguchi H, Fujimoto K, Kawamoto T, Noshiro M, Maemura K, Takeda N et al. Expression of the gene for Dec2, a basic helix-loop-helix transcription factor, is regulated by a molecular clock system. Biochem J 2004; 382: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto T, Noshiro M, Sato F, Maemura K, Takeda N, Nagai R. A novel autofeedback loop of Dec1 transcription involved in circadian rhythm regulation. Biochem Biophys Res Commun 2004; 313: 117–124. [DOI] [PubMed] [Google Scholar]
- Wu T, Ni Y, Zhuge F, Fu Z. Resetting process of peripheral circadian gene expression after the combined reversal of feeding schedule and light/dark cycle via a 24-h light period transition in rats. Physiol Res 2010; 59: 581–590. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Yin H, Nam D, Li Y, Ma K. Brain and muscle Arnt-like 1 promotes skeletal muscle regeneration through satellite cell expansion. Exp Cell Res 2015; 331: 200–210. [DOI] [PubMed] [Google Scholar]
- Lecomte V, Meugnier E, Euthine V, Durand C, Freyssenet D, Nemoz G et al. A new role for sterol regulatory element binding protein 1 transcription factors in the regulation of muscle mass and muscle cell differentiation. Mol Cell Biol 2010; 30: 1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rome S, Lecomte V, Meugnier E, Rieusset J, Debard C, Euthine V et al. Microarray analyses of SREBP-1a and SREBP-1c target genes identify new regulatory pathways in muscle. Physiol Genomics 2008; 34: 327–337. [DOI] [PubMed] [Google Scholar]
- Acharjee S, Chung TK, Gopinadhan S, Shankar SR, Wang Y, Li L et al. Sharp-1 regulates TGF-beta signaling and skeletal muscle regeneration. J Cell Sci 2014; 127: 599–608. [DOI] [PubMed] [Google Scholar]
- Roth SM, Martel GF, Ivey FM, Lemmer JT, Metter EJ, Hurley BF et al. High-volume, heavy-resistance strength training and muscle damage in young and older women. J Appl Physiol 2000; 88: 1112–1118. [DOI] [PubMed] [Google Scholar]
- Shefer G, Rauner G, Yablonka-Reuveni Z, Benayahu D. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS ONE 2010; 5: e13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LV. Effects of age and training on skeletal muscle physiology and performance. Phys Ther 1994; 74: 71–81. [DOI] [PubMed] [Google Scholar]
- Coffey VG, Hawley JA. The molecular bases of training adaptation. Sports Med 2007; 37: 737–763. [DOI] [PubMed] [Google Scholar]
- Neves Jde C, Rizzato VR, Fappi A, Garcia MM, Chadi G, van de Vlekkert D et al. Neuraminidase-1 mediates skeletal muscle regeneration. Biochim Biophys Acta 2015; 1852: 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church JE, Gehrig SM, Chee A, Naim T, Trieu J, McConell GK et al. Early functional muscle regeneration after myotoxic injury in mice is unaffected by nNOS absence. Am J Physiol Regul Integr Comp Physiol 2011; 301: R1358–R1366. [DOI] [PubMed] [Google Scholar]
- Itai Y, Kariya Y, Hoshino Y. Morphological changes in rat hindlimb muscle fibres during recovery from disuse atrophy. Acta Physiol Scand 2004; 181: 217–224. [DOI] [PubMed] [Google Scholar]
- Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. Satellite cells and skeletal muscle regeneration. Compr Physiol 2015; 5: 1027–1059. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Seals DR. Endurance exercise performance in Masters athletes: age-associated changes and underlying physiological mechanisms. J Physiol 2008; 586: 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mera P, Laue K, Ferron M, Confavreux C, Wei J, Galan-Diez ML et al. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab 2016; 23: 1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha ) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA 2003; 100: 1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem 2002; 277: 1645–1648. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Kawamoto T, Tanimoto K, Nishiyama M, Honda H, Kato Y. Identification of functional hypoxia response elements in the promoter region of the DEC1 and DEC2 genes. J Biol Chem 2002; 277: 47014–47021. [DOI] [PubMed] [Google Scholar]
- Hsiao SP, Huang KM, Chang HY, Chen SL. P/CAF rescues the Bhlhe40-mediated repression of MyoD transactivation. Biochem J 2009; 422: 343–352. [DOI] [PubMed] [Google Scholar]
- Shimano H. Sterol regulatory element-binding protein family as global regulators of lipid synthetic genes in energy metabolism. Vitam Horm 2002; 65: 167–194. [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Schroder E, Edelmann SE, Hughes ME, Kornacker K, Balke CW et al. Age-associated disruption of molecular clock expression in skeletal muscle of the spontaneously hypertensive rat. PLoS ONE 2011; 6: e27168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol 2004; 2: e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista FA, Trivella DB, Bernardes A, Gratieri J, Oliveira PS, Figueira AC et al. Structural insights into human peroxisome proliferator activated receptor delta (PPAR-delta) selective ligand binding. PLoS ONE 2012; 7: e33643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahremany S, Livne A, Gruzman A, Senderowitz H, Sasson S. Activation of PPARdelta: from computer modelling to biological effects. Br J Pharmacol 2015; 172: 754–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel W, Raimann A, Halbauer D, Scharmer D, Sagmeister S, Wessner B et al. Insulin-like growth factor I (IGF-1) Ec/Mechano Growth factor--a splice variant of IGF-1 within the growth plate. PLoS ONE 2013; 8: e76133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Alnaqeeb M, Simpson H, Goldspink G. Cloning and characterization of an IGF-1 isoform expressed in skeletal muscle subjected to stretch. J Muscle Res Cell Motil 1996; 17: 487–495. [DOI] [PubMed] [Google Scholar]
- McKoy G, Ashley W, Mander J, Yang SY, Williams N, Russell B et al. Expression of insulin growth factor-1 splice variants and structural genes in rabbit skeletal muscle induced by stretch and stimulation. J Physiol 1999; 516: (Pt 2): 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheny RW Jr., Nindl BC, Adamo ML. Minireview: Mechano-growth factor: a putative product of IGF-I gene expression involved in tissue repair and regeneration. Endocrinology 2010; 151: 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondret F, Ferre P, Dugail I. ADD-1/SREBP-1 is a major determinant of tissue differential lipogenic capacity in mammalian and avian species. J Lipid Res 2001; 42: 106–113. [PubMed] [Google Scholar]
- Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 1996; 10: 1096–1107. [DOI] [PubMed] [Google Scholar]
- Yamada K, Kawata H, Shou Z, Mizutani T, Noguchi T, Miyamoto K. Insulin induces the expression of the SHARP-2/Stra13/DEC1 gene via a phosphoinositide 3-kinase pathway. J Biol Chem 2003; 278: 30719–30724. [DOI] [PubMed] [Google Scholar]
- Li Y, Shen Q, Kim HT, Bissonnette RP, Lamph WW, Yan B et al. The rexinoid bexarotene represses cyclin D1 transcription by inducing the DEC2 transcriptional repressor. Breast Cancer Res Treat 2011; 128: 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa T, Shimano H, Amemiya-Kudo M, Yahagi N, Hasty AH, Matsuzaka T et al. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol 2001; 21: 2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SM, Cho HJ, Cho H, Kim KH, Kim JB, Park H. Stra13/DEC1 and DEC2 inhibit sterol regulatory element binding protein-1c in a hypoxia-inducible factor-dependent mechanism. Nucleic Acids Res 2008; 36: 6372–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015; 28: 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 2000; 14: 2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa T, Shimano H, Yahagi N, Ide T, Amemiya-Kudo M, Matsuzaka T et al. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J Biol Chem 2002; 277: 1705–1711. [DOI] [PubMed] [Google Scholar]
- Hong Y, Xing X, Li S, Bi H, Yang C, Zhao F et al. SUMOylation of DEC1 protein regulates its transcriptional activity and enhances its stability. PLoS ONE 2011; 6: e23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova AV, Ivanov SV, Danilkovitch-Miagkova A, Lerman MI. Regulation of STRA13 by the von Hippel-Lindau tumor suppressor protein, hypoxia, and the UBC9/ubiquitin proteasome degradation pathway. J Biol Chem 2001; 276: 15306–15315. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shankar SR, Kher D, Ling BM, Taneja R. Sumoylation of the basic helix-loop-helix transcription factor sharp-1 regulates recruitment of the histone methyltransferase G9a and function in myogenesis. J Biol Chem 2013; 288: 17654–17662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoechea-Alonso MT, Punga T, Ericsson J. Hyperphosphorylation regulates the activity of SREBP1 during mitosis. Proc Natl Acad Sci USA 2005; 102: 11681–11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoechea-Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem 2009; 284: 5885–5895. [DOI] [PubMed] [Google Scholar]
- Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol 2014; 24: 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res 2009; 50(Suppl): S15–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Cui A, Xue Y, Cui Y, Dong X, Gao Y et al. Hepatic differentiated embryo-chondrocyte-expressed gene 1 (Dec1) inhibits sterol regulatory element-binding protein-1c (Srebp-1c) expression and alleviates fatty liver phenotype. J Biol Chem 2014; 289: 23332–23342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Martelot G, Claudel T, Gatfield D, Schaad O, Kornmann B, Lo Sasso G et al. REV-ERBalpha participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol 2009; 7: e1000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Noshiro M, Choi M, Morita K, Kawamoto T, Fujimoto K et al. The basic helix-loop-helix proteins differentiated embryo chondrocyte (DEC) 1 and DEC2 function as corepressors of retinoid X receptors. Mol Pharmacol 2009; 76: 1360–1369. [DOI] [PubMed] [Google Scholar]
- Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 2015; 25: 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009; 326: 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonsong T, Norton L, Chokkalingam K, Jewell K, Macdonald I, Bennett A et al. Effect of exercise and insulin on SREBP-1c expression in human skeletal muscle: potential roles for the ERK1/2 and Akt signalling pathways. Biochem Soc Trans 2007; 35: 1310–1311. [DOI] [PubMed] [Google Scholar]
- Dif N, Euthine V, Gonnet E, Laville M, Vidal H, Lefai E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J 2006; 400: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagen LM, Deng X, Wilcox HG, Park EA, Raghow R, Elam MB. Insulin activates the rat sterol-regulatory-element-binding protein 1c (SREBP-1c) promoter through the combinatorial actions of SREBP, LXR, Sp-1 and NF-Y cis-acting elements. Biochem J 2005; 385: 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleyer A, Scholtysek C, Bottesch E, Hillienhof U, Beyer C, Distler JH et al. Liver X receptors orchestrate osteoblast/osteoclast crosstalk and counteract pathologic bone loss. J Bone Miner Res 2012; 27: 2442–2451. [DOI] [PubMed] [Google Scholar]
- Zhang JN, Zhao Y, Liu C, Han ES, Yu X, Lidington D et al. The role of the sphingosine-1-phosphate signaling pathway in osteocyte mechanotransduction. Bone 2015; 79: 71–78. [DOI] [PubMed] [Google Scholar]
- Kamel MA, Picconi JL, Lara-Castillo N, Johnson ML. Activation of beta-catenin signaling in MLO-Y4 osteocytic cells versus 2T3 osteoblastic cells by fluid flow shear stress and PGE2: Implications for the study of mechanosensation in bone. Bone 2010; 47: 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappas M, Yee K, Permezel M, Rice GE. Release and regulation of leptin, resistin and adiponectin from human placenta, fetal membranes, and maternal adipose tissue and skeletal muscle from normal and gestational diabetes mellitus-complicated pregnancies. J Endocrinol 2005; 186: 457–465. [DOI] [PubMed] [Google Scholar]
- Solberg R, Aas V, Thoresen GH, Kase ET, Drevon CA, Rustan AC et al. Leptin expression in human primary skeletal muscle cells is reduced during differentiation. J Cell Biochem 2005; 96: 89–96. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Dukes A, Arounleut P, Davis C, Periyasamy-Thandavan S, Mork S et al. The adipokine leptin mediates muscle- and liver-derived IGF-1 in aged mice. Exp Gerontol 2015; 70: 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995; 269: 543–546. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Herberg S, Arounleut P, He HZ, Shiver A, Qi RQ et al. The adipokine leptin increases skeletal muscle mass and significantly alters skeletal muscle miRNA expression profile in aged mice. Biochem Biophys Res Commun 2010; 400: 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]