

Abstract
Mounting evidence has suggested that G protein-coupled receptors can be stabilized in multiple conformations in response to distinct ligands, which exert discrete functions through selective activation of various downstream signaling events. In accordance with this concept, we report biased signaling of one C6-heterocyclic substituted naltrexamine derivative, namely 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(4′-pyridylcarboxamido)morphinan (NAP) at the mu opioid receptor (MOR). NAP acted as a low efficacy MOR partial agonist in the G protein-mediated [35S]GTPγS binding assay, whereas it did not significantly induce calcium flux or β-arrestin2 recruitment. In contrast, it potently blocked MOR full agonist-induced β-arrestin2 recruitment and translocation. Additionally, NAP dose-dependently antagonized MOR full agonist-induced intracellular calcium flux and β-arrestin2 recruitment. Further results in an isolated organ bath preparation confirmed that NAP reversed the morphine-induced reduction in colon motility. Ligand docking and dynamics simulation studies of NAP at the MOR provided more supporting evidence for biased signaling of NAP at an atomic level. Due to the fact that NAP is MOR selective and preferentially distributed peripherally upon systemic administration while β-arrestin2 is reportedly required for impairment of intestinal motility by morphine, biased antagonism of β-arrestin2 recruitment by NAP further supports its utility as a treatment for opioid-induced constipation.
Keywords: mu opioid receptor, functional selectivity, biased signaling, β-arrestin2, NAP, nalbuphine
Introduction
G protein-coupled receptors (GPCRs) are transmembrane receptors and account for approximately 4% of the human genome.1 As such, nearly 30% of FDA-approved drugs target GPCRs.2 Historically, GPCRs were assumed to exist in equilibrium between active and inactive states and activation of GPCRs would equally affect all downstream signaling pathways.3, 4 However, accumulating evidence has suggested that a single GPCR can assume multiple conformations that are differentially stabilized by distinct ligands, which exert discrete functions through selective activation of various downstream effectors, a phenomenon termed “functional selectivity”, “biased agonism”, “ligand directed trafficking”, “stimulus trafficking”, or “pluridimensional efficacy”.5–8 In this regard, a biased ligand may behave as an agonist in one signaling pathway while acting as an antagonist in another; or the rank order of potency or efficacy of two distinct ligands may be reversed in one pathway compared to another.
The mu opioid receptor (MOR) belongs to the Class A GPCR superfamily and the majority of opioids exert their analgesic activity primarily via activating the MOR.9 MOR activation has also been associated with the adverse effects of opioids, such as dependence, and gastrointestinal and respiratory dysfunction.10 Upon activation, MOR predominately couples to Gαi/o, two subtypes of the Gα subunit, which together with Gβ/γ orchestrate downstream signaling cascades including those contributing to antinociception. During this process, β-arrestins, including β-arrestin1 and β-arrestin2, may bind to the phosphorylated MOR (which occurs rapidly following agonist exposure) and diminish G protein-mediated signaling. In particular, studies have indicated that MOR internalization and desensitization may primarily be mediated by β-arrestin2. For example, in β-arrestin2 knockout or knockdown mice a significantly enhanced and prolonged analgesic effect of morphine was observed. Moreover, morphine also produced less tolerance, dependence, constipation, and respiratory suppressive side effects in the absence of β-arrestin2.9 It is therefore conceivable that a biased MOR agonist that selectively activates G protein over β-arrestin2 holds greater therapeutic potential compared to an unbiased one. In fact, a G protein-biased MOR ligand TRV130 ([(3-methoxythiophen-2-yl)methyl](2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro[4.5]decan-9-yl]ethyl)amine, Figure 1) has recently been reported to show potent analgesic effects with reduced gastrointestinal and respiratory dysfunction compared to morphine.11 Based on this premise, it follows that biased ligands that are selective agonists at one pathway over another by virtue of differential efficacy could also act as biased competitive antagonists for the non-preferred pathway,, assuming an orthosteric mode of receptor binding. In general for MOR ligands, those with low efficacy for G protein activation also have low efficacy for β-arrestin2 recruitment, and in fact partial agonists such as buprenorphine do not significantly recruit β-arrestin2 in cell models.12
Figure 1.
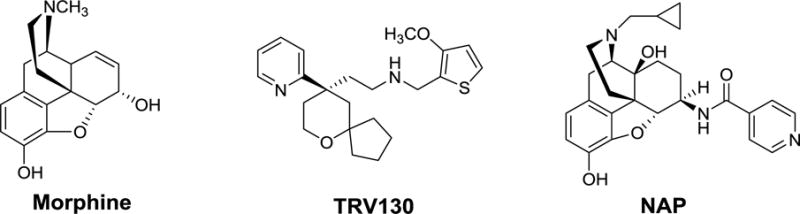
Chemical structures of the MOR unbiased ligand morphine, G protein biased ligand TRV130, and our lead compound NAP.
Our pursuit of highly selective and potent non-peptide MOR ligands (preferential G protein-coupling antagonists) have yielded several promising lead compounds, including 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(4′-pyridylcarboxamido)morphinan (NAP), which acted as a peripherally selective MOR modulator with potential to treat opioid-induced constipation (Figure 1).13–16 Briefly, NAP behaved as a low efficacy MOR partial agonist in the [35S]GTPγS binding assay whereas it also competitively inhibited MOR full agonist [D-Ala2-MePhe4-Gly(ol)5]enkephalin (DAMGO)-stimulated [35S]GTPγS binding.13, 15 Interestingly, NAP produced no apparent analgesic effects at doses up to 100 mg/kg, while it could antagonize the antinociceptive effect of morphine in the warm water tail-immersion assay with moderate potency.13 Furthermore, being a P-glycoprotein substrate, NAP has limited access to the central nervous system and dose-dependently restored morphine-impaired intestinal motility without precipitating significant withdrawal symptoms (jumps, wet-dog shakes, or locomotion).14, 16 While originally NAP was defined as a potent antagonist on the MOR due to its lack of efficacy from our in vivo studies, its partial agonism on the MOR from the in vitro experiment results really intrigued us. Inspired and encouraged by these results and the aforementioned “proof-of-concept” in development of the G protein-biased MOR agonist TRV130, and based on the concept of biased antagonism, we hypothesized that NAP would act as a neutral antagonist for β-arrestin2 recruitment and low efficacy partial agonist for G-protein activation. We further propose that this pharmacodynamics profile would result in net antagonism of opioid agonist-impaired gut motility. Therefore, we investigated herein the ability of NAP to activate or antagonize both G protein activation and β-arrestin2 recruitment to the MOR. Chimeric Gαqi5-mediated Ca2+ mobilization assays were first employed to study G protein-mediated signaling and β-arrestin2 recruitment and translocation assays were performed to examine potential β-arrestin2 interactions upon ligand binding to the MOR. The low efficacy opioid receptor partial agonist nalbuphine16 was tested along with NAP for comparison in these two studies. We also examined isolated intestinal tissue preparations to determine the influence of NAP on the inhibitory effect of morphine on colonic motility. In addition, molecular docking studies based on a recently available crystal structure of the MOR in a membrane system were pursued to investigate possible mechanisms of the observed modulation at an atomic level.
Results and Discussion
Intracellular Ca2+ concentration undergoes a rapid and transient increase upon Gq activation.18 Cytosolic Ca2+ measurement has thus become a routine assay to assess GPCR function. While the MOR is mainly Gi/o mediated, it would be interesting to further profile NAP in this G protein-mediated signaling cascade by comparing with some other low efficacy agonist of the MOR. Because the GTPγS assay directly measures G-protein activation, the initial step in MOR signal transduction, we decided to use a more downstream measure of G-protein-mediated effector activity in order to allow amplification of the signal and potentially reveal a higher level of partial agonism. Moreover, because we intended to compare these results to a CHO cell-based assay for β-arrestin recruitment (presented below), we therefore performed cell-based Ca2+ flux assays for NAP in hMOR-CHO cells transfected with chimeric Gαqi5. Intriguingly, results showed that whereas DAMGO dose-dependently increased the intracellular Ca2+ level with an EC50 of 7.5 ± 1.6 nM,19 no apparent agonism was observed for NAP or nalbuphine, another partial agonist of the MOR, in the Ca2+ mobilization assay (up to the highest concentration (100 μM) tested for NAP, the average of observed efficacy was 5±3% of the maximal effect relative to DAMGO, a full agonist as our reference compound, Figure 2A). On the contrary, both drugs inhibited Ca2+ flux induced by DAMGO (Figure 2B), similar to the MOR antagonist naltrexone (NTX). The order of Ca2+ flux inhibition potency is: NTX > NAP > nalbuphine. Several factors might contribute to the lack of agonism of NAP and nalbuphine in the Ca2+ flux assay. First, these two compounds may act as neutral MOR antagonists in this signaling pathway, although our previous studies found that NAP acted as low efficacy partial agonist in GTPγS binding assays. Second, introduction of chimeric or promiscuous G proteins may cause such a phenomenon. A similar scenario has been reported whereby ipsapirone, a highly potent and efficacious 5-HT1A receptor agonist in cAMP inhibition assays, failed to show any agonist effect in the Ca2+ mobilization assay.20, 21 Third, Ca2+ flux of slow binding agonists18 may not be detectable within our initial experimental time course (i.e. 2 min). However, no apparent agonist activity was observed for either NAP or nalbuphine with extended detection time (from initially 2 min to 10 min, see details in the Supporting Information). Apparently, as a low efficacy MOR ligand, activation of Gi/o by NAP seemed insufficiently efficacious to induce significant downstream Ca2+ flux compared to DAMGO, a full agonist of MOR, even when facilitated by a chimeric G-protein. In addition, the characteristic inhibitory modulation by NAP of the MOR from our in vivo studies (gastrointestinal transit and/or warm water tail immersion) matched well with the findings from the Ca2+ flux assays.
Figure 2.
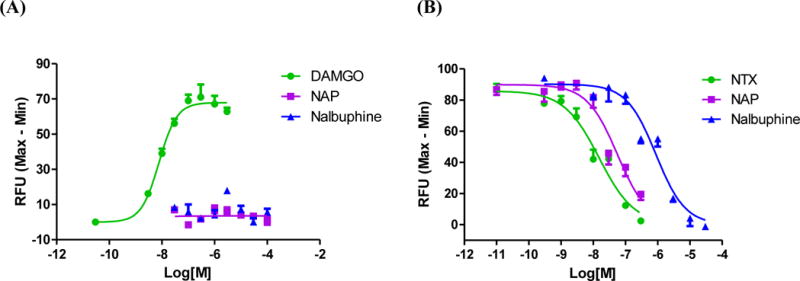
Ca2+ flux assays in Gαqi5 transfected hMOR-CHO cells. The results shown are representative of at least three independent experiments. (A) The MOR full agonist DAMGO dose-dependently increased intracellular Ca2+ level, whereas no apparent agonism was observed for NAP and nalbuphine. (B) NAP and nalbuphine significantly antagonized DAMGO-induced intracellular Ca2+ increase. The IC50 ± S.E.M. values of NTX, NAP and nalbuphine are: 15.5 ± 0.1 nM, 52.1 ± 9.4 nM, and 887.6 ± 59.7 nM, respectively.
To further elucidate the pharmacological profile of NAP for modulation of different downstream signaling/regulatory pathways, it was then subjected to β-arrestin2 recruitment assessment using the PathHunter™ assay in CHO-K1 cells (DiscoveRx). The MOR full agonist DAMGO produced dose-dependent β-arrestin2 recruitment with an EC50 (95% CI) of 114.4 (30.9–423.3) nM, which is consistent with the reported data by Burfold and colleagues.22 In comparison, activation of MOR by NAP or nalbuphine did not recruit β-arrestin2 (up to the highest concentration (10 μM) tested for NAP, the average of observed effect was 2±2% of the maximal effect relative to DAMGO, Figure 3A). In contrast, both ligands blocked DAMGO-induced β-arrestin2 recruitment with IC50 values in the low nanomolar level (Figure 3B), which is about an order of magnitude more potent than those for inhibiting DAMGO-induced Ca2+ flux. This discrepancy may be because Ca2+ flux is further downstream than β-arrestin2 recruitment following receptor activation, or it could be due to differing sensitivities of the two assays, which is reflected in the differential potencies for DAMGO (7.5 nM for Ca2+ flux vs. 114.4 nM for β-arrestin2 recruitment).
Figure 3.
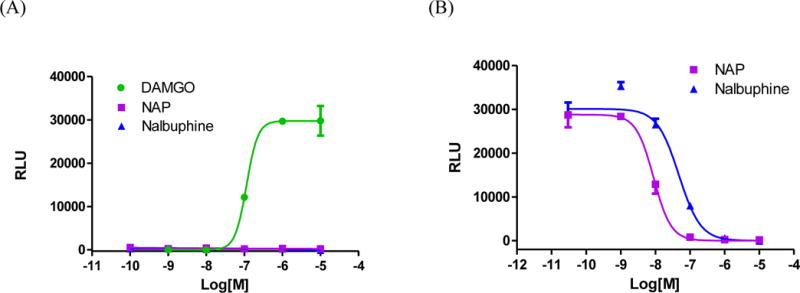
β-Arrestin2 recruitment assay using PathHunter eXpress OPRM1 CHO-K1 βarrestin2 assay kit. (A) The MOR full agonist DAMGO dose-dependently increased βarrestin2 association, whereas NAP and nalbuphine displayed no apparent agonism. (B) NAP and nalbuphine significantly antagonized DAMGO-induced βarrestin2 recruitment in a dose-dependent fashion. The IC50 (95% CI) values of NAP and nalbuphine are: 8.8 (4.0–19.3) nM and 47.2 (14.4–155.0) nM, respectively.
The MOR-β-arrestin2 recruitment assay platform uses an enzyme fragment complementation approach with modification of the MOR C-terminus, adjacent to a region containing phosphorylation sites that are known to be crucial for recruitment of β-arrestin2 to the MOR. Therefore, we also employed an additional approach to visualize interactions between activated receptor and β-arrestin2: ligand-induced translocation of green fluorescence protein (GFP)-tagged β-arrestin2 to the MOR was assessed using MOR-βarr2eGFP-U2OS (MBU). DAMGO-induced β-arrestin2 translocation has been previously reported.23 The results from NAP confocal microscopy were in good agreement with the PathHunter™ β-arrestin2 recruitment assay (Figure 4). NAP did not induce visible β-arrestin2 recruitment in MBU cells. Moreover, pretreatment of NAP significantly blocked DAMGO-induced β-arrestin2 translocation (Figure 4). Compared to the previous results with [35S]GTPγS at the MOR, the β-arrestin2 results further suggest that NAP may be a biased antagonist at the MOR against β-arrestin2 recruitment.
Figure 4.
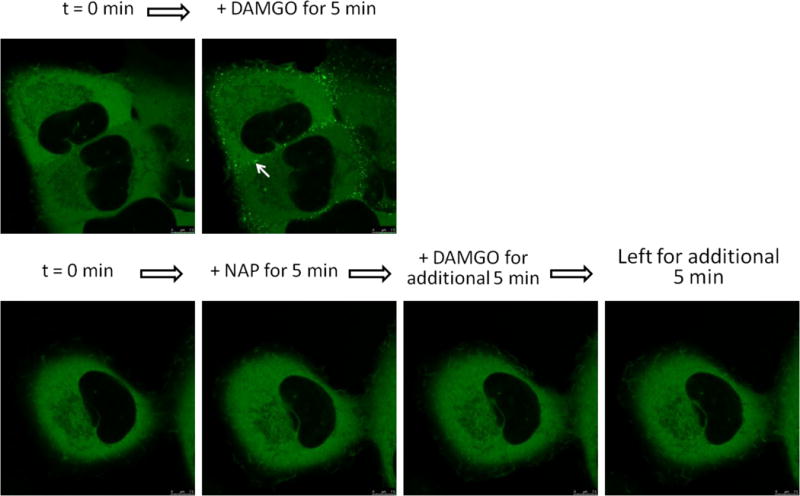
β-Arrestin2 translocation assay of NAP in opioid receptor-expressing βarr2eGFP-U2OS cells. β-arrestin2 translocation to cell membrane (bright green punctae as indicated by white arrows) evoked by ligand-induced receptor activation in live cells were imaged by confocal microscopy. The MOR full agonist DAMGO was tested concurrently as a positive control. MBU cells were treated with DAMGO, NAP, or NAP plus DAMGO (both at 10 μM) as indicated. NAP did not induce βarrestin2 recruitment. Moreover, pre-treatment with NAP abolished DAMGO-induced βarrestin2 recruitment.
Opioid induced constipation (OIC) is a complex phenomenon that is still not fully understood. It is generally accepted that OIC is promoted primarily by the activation of the MOR. In the gastrointestinal tract, activation of the MOR on enteric neurons within the myenteric plexus causes relaxation of the propulsive longitudinal muscles and increased contractions of the non-propulsive circular muscle.24 Both of these responses can be readily observed in isolated tissue preparations in an organ bath. Previous studies have shown that NAP is able to reverse the reduced intestinal motility caused by morphine in mice.16 To gain further insight into its pharmacology we examined the effects of NAP on morphine-induced circular muscle contractions of isolated distal and proximal colon from mice (colon was chosen because tolerance to morphine does not develop in the colon24). First of all, no significant tissue contraction was observed even at very high concentrations of NAP, in contrast to its low to moderate agonism at all three opioid receptors in the [35S]GTPγS assay. This finding is in agreement with its insignificant effect on calcium flux and β-arrestin2 recruitment. In comparison, morphine induced contractions in both distal and proximal colon, with a higher potency in the proximal colon. This regional difference has also been observed for endomorphin 1 and 2.25 Secondly, within the distal colon a significant inhibition by NAP of the stimulatory action of morphine was observed at 100 nM. This inhibition is demonstrated by the rightward shift in the morphine concentration-effect curve and the decrease in pD2 value from 6.1 ± 0.01 to 5.3 ± 0.1 (Figure 5A). In contrast, within the proximal colon NAP shows significant antagonism of the effects of morphine at 10 nM as seen in the change of pD2 from 6.7 ± 0.01 to 5.9 ± 0.1 after NAP exposure (Figure 5B). Further increases in NAP concentration within the proximal colon did not lead to any further significant changes in the concentration-effect curve (pD2 = 5.4 ± 0.2). These results suggest difference in the sensitivity of the proximal colon to NAP antagonism against morphine compared with the distal colon. Several factors could contribute to the observed region specificity of ligand effects. First, different opioid receptor expression levels have been identified in proximal and distal colon in other rodents.26 Second, difference in the expression of MOR splice variants might also contribute to these results, as recent studies have identified at least 31 splice variants of the OPRM-1 gene which encodes for the MOR.27
Figure 5.
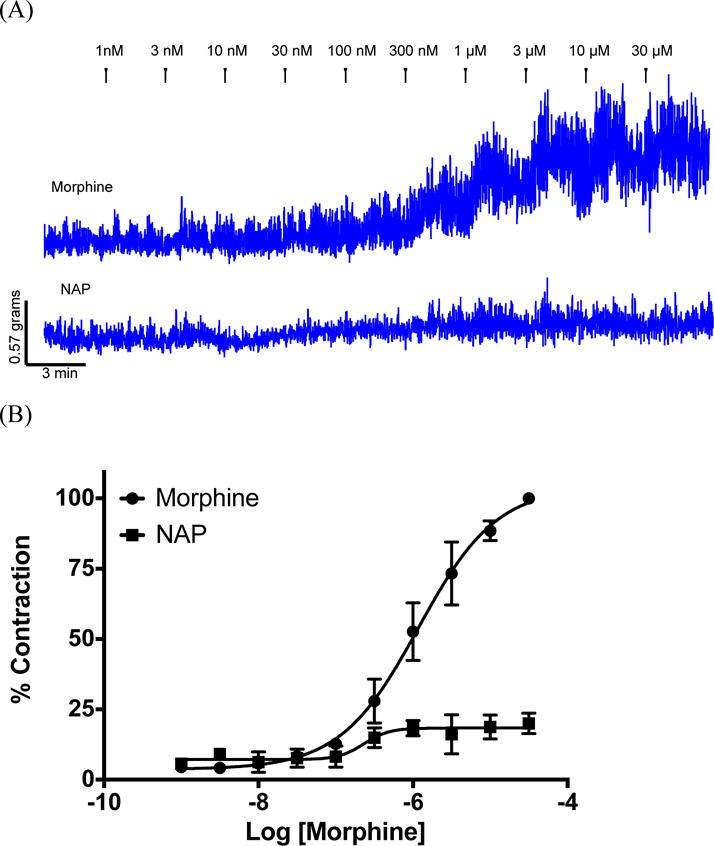
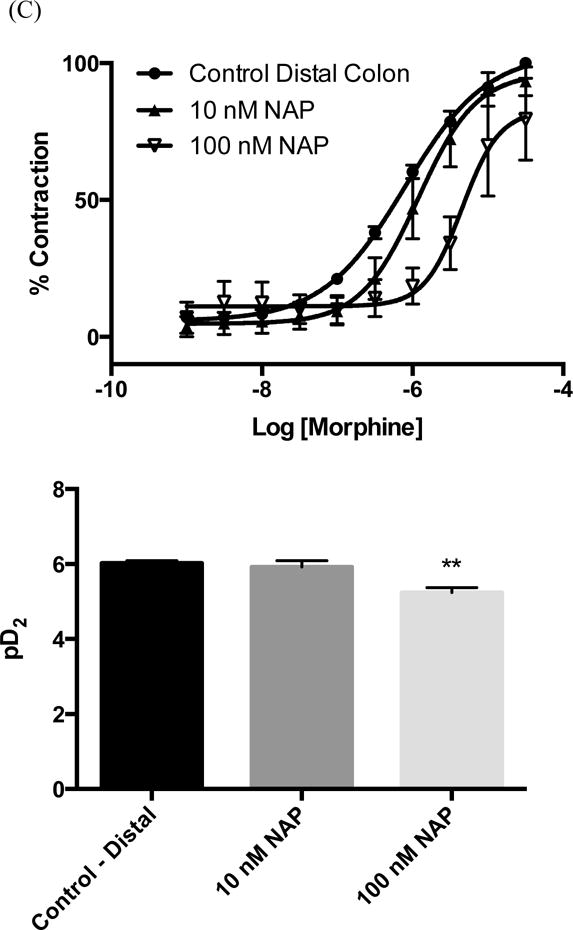
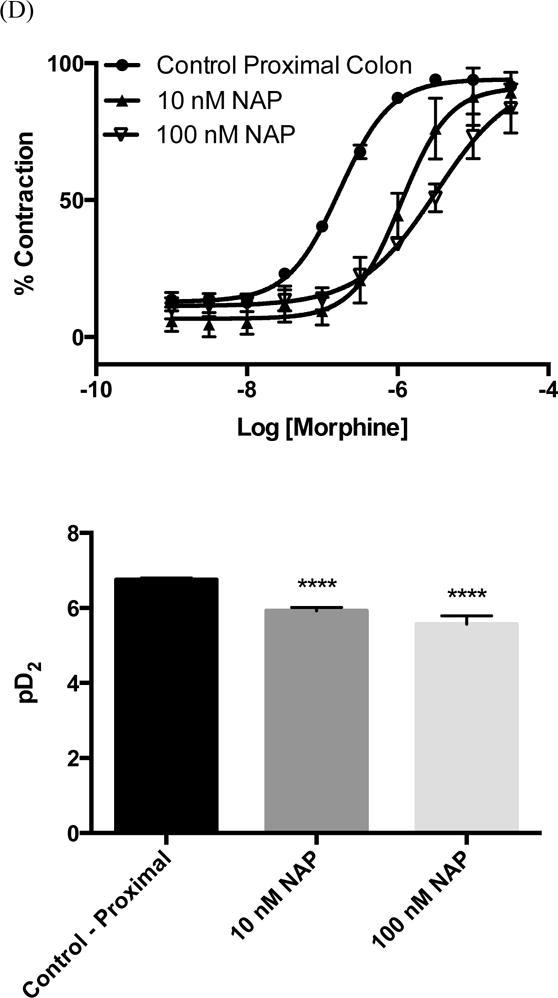
(A) Representative traces from mouse distal colon of circular muscle contractions to increasing concentrations of morphine (top) or NAP (bottom). Isometric tension recordings for morphine and NAP were obtained from the same animal. After an initial concentration-effect curve with morphine the tissue was washed extensively until basal levels returned (>2.5h) and then a concentration-effect experiment with NAP was conducted. Data for NAP were represented as a % of the maximum response obtained with morphine. These traces were from the distal colon; however the proximal colon gave similar results. (B). Quantification of raw traces was done by integrating the total responses between concentrations using the built-in software. Morphine in this assay behaved as a high potency high efficacy agonist pD2 = 5.9 ± 0.1. NAP did display low efficacy partial agonism in this assay (pD2 = 6.6 ± 0.2) however the statistical difference using a t-test between the response at 1nM and 30 μm was marginal (P = 0.02). Data were expressed as mean response ± S.E.M from at least three different animals. NAP Inhibitory effect on morphine-induced tissue contractions were shown in the distal (C) and proximal (D) colon tissue contraction.
Recent structural biology efforts have provided clues to the mechanisms at an atomic level by which biased ligands may modulate the target receptor. Particularly, conformational changes in transmembrane (TM) helix 7, helix 8, and extracellular loop 2 may play some role in β-arrestin-medicated signaling while conformational changes in TM3, 5 and 6, in addition to intracellular loop 2 may be related to G-protein mediated signaling.28 From our collected studies, NAP seems to modulate the MOR in a biased fashion by activating G protein coupling without significant recruitment of β-arrestin2 or induction of calcium flux, at least when measured downstream of Gαqi chimeric G protein activation. To seek possible mechanisms of such biased modulation of the MOR at an atomic level, docking studies of NAP using the recently available crystal structure of MOR29 were conducted. In this crystal structure the MOR was covalently bound to an antagonist β-FNA and, therefore, this structure represents a snapshot of stabilized receptor conformation. In consideration of NAP as a low-efficacy partial agonist at the MOR, this conformation should serve as a reasonable starting point for our docking and simulation studies. The conformation of the co-crystallized antagonist (β-FNA) in the receptor was used as a reference for the docking because NAP shared the same morphinan skeleton of β-FNA. After energy minimization, molecular dynamics simulation was then applied in a membrane system. The results indicated that the epoxymorphinan skeleton of NAP resided in an identical binding pocket to that of β-FNA, while their side chains might have insignificantly different orientations in the binding site (Figure 6). In particular, the side chains of NAP interacted with the originally identified unique “address” domain of the MOR to engender selectivity.13 This domain is mainly composed of the non-conserved amino acid residues Trp318 and Lys303 along with a number of conserved residues. This observation is supported by our site-directed mutagenesis studies.30 Further analysis of this binding domain indicated aromatic stacking interaction of the ligand side chains with Trp318; hydrogen bonding interaction of the ligands with Lys303, and Lys233/Glu229, and hydrophobic interaction of the ligands with Val300. Notably, the interaction with these residues typically does not induce activation of the receptor based on our current knowledge of opioid receptor activation mechanisms.31 More importantly, none of these residues are located on ECL2 and only one is on TM7. On the other hand, most of them are located on TM3, 5, and 6. Such observations seem to be in compliance with the biased modulation properties of NAP on the MOR.
Figure 6.
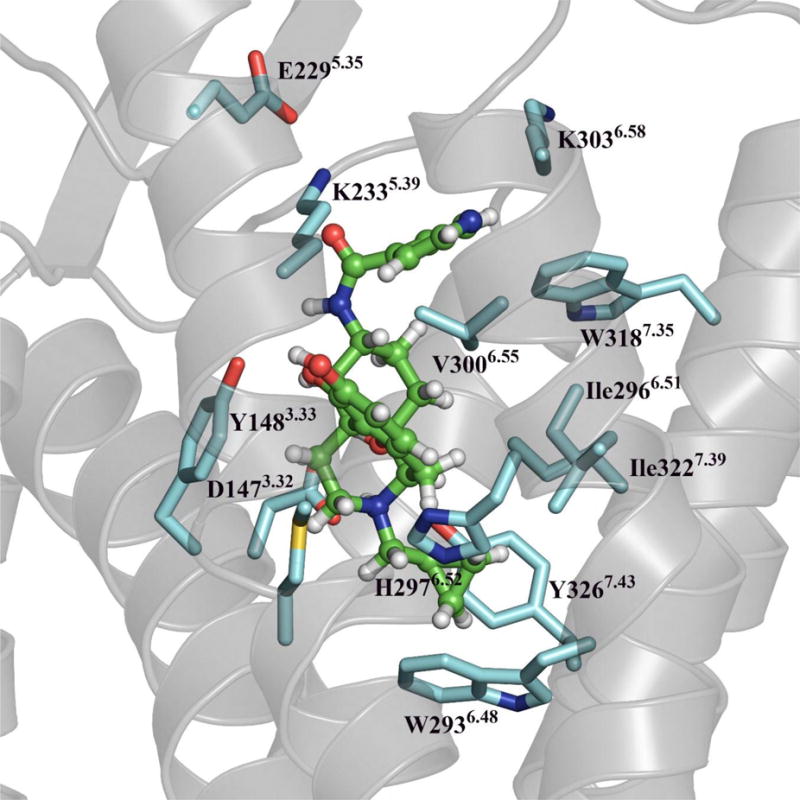
NAP docking pose analysis in the MOR crystal structure after molecular dynamics simulation in a membrane system.
In conclusion, further molecular pharmacological characterization of NAP in the G protein-mediated signaling cascade ([35S]GTPγS binding and Ca2+ flux) and β-arrestin2 recruitment revealed that NAP seemed to carry certain degree of “functional selectivity” between G-protein activation (represented by its partial agonism on [35S]GTPγS binding) and β-arrestin2 recruitment while its insignificant calcium flux efficacy seemed to be intriguing. NAP also exerted high potency in antagonizing morphine-induced circular muscle contraction in distal and proximal colon. Ligand docking and molecular dynamics simulation studies indicated that NAP mostly interacts with residues from TM 3, 5, and 6, where conformational changes are mainly linked to the G protein-mediated signaling pathway. Thus, the observation that NAP failed to recruit β-arrestin2 in addition to its significant inhibition of morphine-reduced colon motility makes NAP a potentially safe option to treat opioid induced constipation as an opioid with mixed partial agonist and antagonist activity, with a bias towards antagonism of the β-arrestin2 pathway. The finding that nalbuphine exhibits similar bias suggests that other low efficacy MOR partial agonists could also be effective against opioid induced constipation, but NAP has the advantages of both peripheral restriction and MOR selectivity.
METHODS
Chemistry
NAP was synthesized as described in reference 12.
Pharmacology
Ca2+ Flux Assay
hMOR-CHO cells were maintained as described previously.18 Four hours after Gαqi5 transfection, cells were plated at 30,000 cells per well into a clear bottom black walled 96-well plate (Greiner Bio-one) and incubated for 24 h. The growth media was then decanted and the wells were washed with 100 μL of 50:1 HBSS:HEPES assay buffer. Cells were then incubated with either 80 μL (agonism study) or 55 μL (antagonism study) of Fluo4 loading buffer (40 μL 2 μM Fluo4-AM (Invitrogen), 84 μL 2.5 mM probenacid, in 8 mL or 5.5 mL assay buffer) for 30 min. For antagonism studies, 25 μL of varying concentrations of test compounds were added in triplicate and the plate was incubated for an additional 15 min. Plates were then read on a FlexStation3 microplate reader (Molecular Devices) at 494/516 ex/em for a total of 120 s. After 15 s of reading, 20 μL of varying concentrations of test compounds in triplicate (agonism study) or 500 nM of DAMGO (NIDA, antagonism study) in assay buffer, or assay buffer alone (control), were added. Changes in Ca2+ flux were monitored and peak height values were recorded. The obtained values were then subjected to nonlinear regression analysis to determine EC50 or IC50 values using Prism 3.0 (GraphPad Software, San Diego, CA).
β-Arrestin2 Recruitment Assay
Experiment performed as described in the manufacturer’s protocol. Briefly, 100 μL of cells were plated into a 96-well tissue culture plate and then incubated for 24 h at 37 °C, 5% CO2. For agonist studies, 10 μLof test compounds were added in duplicate and incubated for 90 min at 37 °C. For antagonism studies, 5 μL of test compounds were first added in duplicate and incubated for 30 min at 37 °C. Then 5 μL of DAMGO at 1 μM (~22 fold EC80 of DAMGO in the agonism study) were added and the cells were incubated for 90 min at 37 °C. After the 90-min incubation time, 55 μL of detection reagent was added to each well and incubated for 60 min at ambient temperature (25 °C). The plate was then read on a FlexStation3 microplate reader (Molecular Devices) for 200 ms with luminescence mode. The obtained values were then subjected to nonlinear regression analysis to determine EC50 or IC50 values of test compounds using Prism 3.0 (GraphPad Software, San Diego, CA).
βArrestin2 Confocal Microscopy
As described in reference 22. Briefly, cells were plated on collagen-coated glass confocal dishes (MatTek, Ashland, MA). Prior to imaging, cells were starved for 60 min in serum-free MEM without phenol red (Life Technologies, Grand Island, NY). Compound was then added at the indicated dose and live cell images were obtained by confocal microscopy (Leica SP5) at the specified time points.
Mouse Organ Bath assays
Morphine (morphine sulfate pentahydrate salt) was procured from the National Institute of Drug Abuse (NIDA), Bethesda, MD and made into a 10 μM stock solution by dissolving in distilled water which was further diluted with distilled water to make the targeted concentrations. NAP was synthesized according to previous reports as the HCl salt and dissolved in distilled water to a stock concentration of 10 μM, which was further diluted with distilled water to make the targeted concentrations. Male Swiss Webster mice (Harlan Laboratories, Indianapolis, IN) weighing 25–30 g were housed 5 to a cage in animal care quarters and maintained at 22 ± 2 °C on a 12 h light-dark cycle. Food and water were available ad libitum. The mice were brought to a test room (22 ± 2 °C, 12 h light-dark cycle), marked for identification and allowed 18 h to recover from transport and handling. Protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Commonwealth University Medical Center and comply with the recommendations of the IASP (International Association for the Study of Pain).
Preparation of Colon Circular Muscle for Isometric Tension Recordings
Mice were first euthanized by cervical dislocation. The colon was then dissected, flushed of its contents, and trimmed of mesentery. Segments of the distal colon (approximately 1 cm from anus) and proximal colon (approximately 1.5 cm from distal portion) were removed and placed in a dissecting dish containing Krebs solution (118 mM NaCl, 4.6 mM KCl, 1.3 mM NaH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, 11 mM glucose, and 2.5 mM CaCl2) bubbled with 95% O2 and 5% CO2. The tissues, 0.5 cm in length, were suspended vertically along the axis of the circular muscle with a metal triangle tied to a hook under 1 g of passive tension in 15 mL siliconized organ baths. The tissues were allowed to equilibrate for 90 min prior to drug exposure, with the Krebs solution changed every 10 min for the first 40 min.
After the tissue was equilibrated an initial dose response curve to morphine was conducted to act as the control. Any tissues that did not respond to morphine were discarded and not used. For runs in which the antagonist was used, following the initial dose response curve to morphine the tissues was washed repeatedly every 10 min for 90–120 min until contractile responses returned to basal levels. Following this NAP was added and allowed to incubate for 15 min after which a second dose response curve to morphine was run. Isometric contractions were recorded by a force transducer (GR-FT03; Radnoti, Monrovia, CA) connected to a personal computer using Acknowledge 382 software (BIO-PAC Systems, Inc., Santa Barbara, CA).
Data Analysis
Contractile responses after repeated administration of morphine were analyzed by taking the integrated response between doses. Data are presented as the mean ± S.E.M for no less than three independent runs. Values of P ≤ 0.05 when compared to controls were considered significant. Effective concentration of agonists to produce 50%-maximal response, reported as negative log (EC50) or pD2, was calculated by nonlinear regression, and data were analyzed by appropriate statistical tools using GraphPad Prism software (Graph-Pad Software Inc.).
Molecular modeling
The molecular structure of the ligand NAP was sketched in SYBYL-X 2.0, and energy minimization of the structure was performed after assigning Gasteiger–Hückel charges (10,000 iterations) with the Tripos force field (TFF). The X-ray crystal structure for MOR (4DKL) was retrieved from the PDB Data Bank. SYBYL-X 2.0 was also used to prepare the obtained protein coordinates for ligand docking by extracting the crystallized ligand and the fusion protein at intracellular loop 3. However, crystallographic waters were preserved. This was followed by addition of hydrogen atoms and subsequent energy minimization of only the added hydrogen atoms.
GOLD5.296, a genetic algorithm-based automated docking program was employed to dock the ligand onto these “cleaned” receptor structures. The binding site was defined to include all atoms within 10 Å of the γ-carbon atom of Asp3.32 for the mu opioid receptor crystal structure, along with a hydrogen bond constraint between the basic nitrogen atom and the carboxylate group oxygen atoms of Asp3.32. The best CHEM-PLP-scored solutions were chosen for molecular dynamics (MD) studies. Gaps in the protein sequence including those due to incorporation of the fusion proteins in the crystal structure (Leu259-Arg273) in the MOR and gaps due to missing electron density in the crystal structure of the receptors were modeled and refined by MODELLER 9v10. Density functional theory (DFT) calculations at the 6-31G* level were employed to calculate partial atomic charges of the NAP atoms using NWChem 6.0. Force field parameters and topology files for NAP were generated utilizing SwissParam. The atomic charges obtained from NWChem were added to the ligand topology file. The topology and parameter files were further edited, accordingly.
Coordinates for the spatial arrangement of the receptors within the lipid bilayer were retrieved from the Orientations of Proteins in Membranes (OPM) database. OPM estimates arrangement of transmembrane protein inside the lipid bilayer by minimizing the transfer energy of the protein from water to the lipid membrane.
System Preparation for MD Simulation
VMD 1.9.1 was used to prepare the system for MD simulations. Coordinate (pdb) and connectivity files (psf) were generated for receptor-ligand complex using the psfgen module. The VMD membrane module was employed to create a lipid bilayer of POPC (1-Palmitoyl-2-oleoylphosphatidylcholine). This was followed by addition of 30 Å of water layers to both sides of membrane at the vertical axis, using Solvate plugin. All the waters and POPC molecules at a distance of 0.65 Å or less from the receptor-ligand complex were then deleted followed by deletion of waters within the POPC membrane. The water system was then ionized to 0.15 M of NaCl by the Autoionize plugin.
Melting Lipids
All molecular modeling simulations were performed using NAMD 2.8. MD simulations were carried out in four stages. In the first stage, equilibration of the fluid like lipid bi-layer was performed via minimization (1000 iterations) followed by NPT equilibration (pressure equilibration) of the lipid tails for a period of 0.5 ns. Simulations were carried out using the CHARMM force field with CHARMM22 parameters for protein, CHARMM27 parameters for lipids and CMAP corrections for proline, glycine and alanine dipeptides with a time-step of 2 femtoseconds (fs). Periodic boundary conditions were employed, and Particle Mesh Ewald (PME) summation was used to calculate long-range electrostatic interactions. Non-bonded interactions were calculated with a smooth cutoff between 10 to 12 Å with a frequency of 1 fs. Constant pressure and temperature at 310 K was maintained via Langevin dynamics.
Equilibration with Constrained Receptor-Protein Complex
In the second stage, an NPT equilibration of the system was run for a period of 1 ns with harmonic constraints placed on protein, NAP and crystallographic water atoms (5 kcal/(mol-Å)) while keeping all the parameters same as earlier.
Equilibration with Unconstrained Receptor-Protein Complex
The harmonic restraint was released in stage 3 and the entire system was equilibrated using the NVT canonical ensemble for a further 1 ns.
Production Run
The final production run was conducted using an NVT ensemble where the whole system was equilibrated for 15 ns.
Energy Analysis
Energy landscape analysis was performed using the NAMD Energy 1.4 plug-in; non bonded interaction analyses were performed at various distances with a dielectric constant of 6.5. All the atoms, including protein and water molecules within a certain cutoff distance from the ligand, were included in the energy analyses. The binding modes with highest non-bonded interactions were selected for further analysis.
Supplementary Material
Acknowledgments
The authors thank NIH/NIDA for financial support. The authors also thank Dr. Lawrence Barak at Duke University for the generous gift of opioid receptor-expressing βarr2eGFP-U2OS cells. The authors would also like to acknowledge DiscoveRx for the gift of the PathHunter eXpress OPRM1 CHO-K1 βarrestin2 assay kit.
Funding Sources
This work was partially supported by NIH/NIDA DA024022 (Y.Z.).
Footnotes
SUPPORTING INFORMATION: Results for NAP Long term Ca2+ flux assay in Gαqi5 transfected hMOR-CHO cells. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contribution
Y.Z. conceived and oversaw the project. Y.Z., H.I.A, and E.J.B. designed the experiments. Y.Y. conducted Ca2+ flux assay, and β-arrestin2 recruitment assay. S.A.Z. finished the modeling studies. Organ bath work was finished by D.A.W. under supervision of H.I.A and W.L.D. β-Arrestin2 confocal microscopy was carried out by J.M.S. and A.B. under supervision of J.M.S. Y.Z., D.E.S., and J.M.S. analyzed the data and discussed the results. Y.Z., J.M.S., and D.E.S. drafted and revised the manuscript.
References
- 1.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 2.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 3.Leff P. The two-state model of receptor activation. Trends Pharmacol Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- 4.Flordellis CS. The plasticity of the 7TMR signaling machinery and the search for pharmacological selectivity. Curr Pharm Des. 2012;18:145–160. doi: 10.2174/138161212799040556. [DOI] [PubMed] [Google Scholar]
- 5.Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- 6.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 8.Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 9.Raehal KM, Bohn LM. beta-arrestins: regulatory role and therapeutic potential in opioid and cannabinoid receptor-mediated analgesia. Handb Exp Pharmacol. 2014;219:427–443. doi: 10.1007/978-3-642-41199-1_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNicol E, Horowicz-Mehler N, Fisk RA, Bennett K, Gialeli-Goudas M, Chew PW, Lau J, Carr D. Management of opioid side effects in cancer-related and chronic noncancer pain: a systematic review. J Pain. 2003;4:231–256. doi: 10.1016/s1526-5900(03)00556-x. [DOI] [PubMed] [Google Scholar]
- 11.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 12.McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, Henderson G, Kelly E. μ-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol. 2010;78:756–66. doi: 10.1124/mol.110.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y. Design, synthesis, and biological evaluation of 6alpha- and 6beta-N-heterocyclic substituted naltrexamine derivatives as mu opioid receptor selective antagonists. J Med Chem. 2009;52:1416–1427. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitra P, Venitz J, Yuan Y, Zhang Y, Gerk PM. Preclinical disposition (in vitro) of novel mu-opioid receptor selective antagonists. Drug Metab Dispos. 2011;39:1589–1596. doi: 10.1124/dmd.111.038588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan Y, Li G, He H, Stevens DL, Kozak P, Scoggins KL, Mitra P, Gerk PM, Selley DE, Dewey WL, Zhang Y. Characterization of 6alpha- and 6beta-N-heterocyclic substituted naltrexamine derivatives as novel leads to development of mu opioid receptor selective antagonists. ACS Chem Neurosci. 2011;2:346–351. doi: 10.1021/cn2000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan Y, Stevens DL, Braithwaite A, Scoggins KL, Bilsky EJ, Akbarali HI, Dewey WL, Zhang Y. 6beta-N-heterocyclic substituted naltrexamine derivative NAP as a potential lead to develop peripheral mu opioid receptor selective antagonists. Bioorg Med Chem Lett. 2012;22:4731–4734. doi: 10.1016/j.bmcl.2012.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sell SL, McMahon LR, France CP. Relative efficacy of buprenorphine, nalbuphine and morphine in opioid-treated rhesus monkeys discriminating naltrexone. J Pharmacol Exp Ther. 2003;306:1167–1173. doi: 10.1124/jpet.103.051425. [DOI] [PubMed] [Google Scholar]
- 18.Zhang R, Xie X. Tools for GPCR drug discovery. Acta Pharmacol Sin. 2012;33:372–384. doi: 10.1038/aps.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan Y, Zaidi SA, Stevens DL, Scoggins KL, Mosier PD, Kellogg GE, Dewey WL, Selley DE, Zhang Y. Design, Syntheses, and Pharmacological Characterization of 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3-carboxamido)morphinan Analogues as Opioid Receptor Ligands. Bioorg Med Chem. 2015;23:1701–15. doi: 10.1016/j.bmc.2015.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kostenis E, Waelbroeck M, Milligan G. Techniques: promiscuous Galpha proteins in basic research and drug discovery. Trends Pharmacol Sci. 2005;26:595–602. doi: 10.1016/j.tips.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Kowal D, Zhang J, Nawoschik S, Ochalski R, Vlattas A, Shan Q, Schechter L, Dunlop J. The C-terminus of Gi family G-proteins as a determinant of 5-HT(1A) receptor coupling. Biochem Biophys Res Commun. 2002;294:655–659. doi: 10.1016/S0006-291X(02)00535-1. [DOI] [PubMed] [Google Scholar]
- 22.Burford NT, Wehrman T, Bassoni D, O’Connell J, Banks M, Zhang L, Alt A. Identification of selective agonists and positive allosteric modulators for micro- and delta-opioid receptors from a single high-throughput screen. J Biomol Screen. 2014;19:1255–1265. doi: 10.1177/1087057114542975. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Braithwaite A, Yuan Y, Streicher JM, Bilsky EJ. Behavioral and cellular pharmacology characterization of 17-cyclopropylmethyl-3,14beta-dihydroxy-4,5alpha-epoxy-6alpha-(isoquinoline-3′-ca rboxamido)morphinan (NAQ) as a mu opioid receptor selective ligand. Eur J Pharmacol. 2014;736:124–130. doi: 10.1016/j.ejphar.2014.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang M, Maguma HT, Smith TH, Ross GR, Dewey WL, Akbarali HI. The role of beta-arrestin2 in the mechanism of morphine tolerance in the mouse and guinea pig gastrointestinal tract. J Pharmacol Exp Ther. 2012;340:567–576. doi: 10.1124/jpet.111.186320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu Y, Cui Y, Wang X, Lai LH, Wang CL, Fan YZ, Liu J, Wang R. In vitro characterization of the effects of endomorphin 1 and 2, endogenous ligands for mu-opioid receptors, on mouse colonic motility. Biochem Pharmacol. 2007;73:1384–1393. doi: 10.1016/j.bcp.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 26.Fickel J, Bagnol D, Watson SJ, Akil H. Opioid receptor expression in the rat gastrointestinal tract: a quantitative study with comparison to the brain. Brain Res Mol Brain Res. 1997;46:1–8. doi: 10.1016/s0169-328x(96)00266-5. [DOI] [PubMed] [Google Scholar]
- 27.Pan YX. Diversity and complexity of the mu opioid receptor gene: alternative pre-mRNA splicing and promoters. DNA Cell Biol. 2005;24:736–750. doi: 10.1089/dna.2005.24.736. [DOI] [PubMed] [Google Scholar]
- 28.Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaidi SA, Arnatt CK, He H, Selley DE, Mosier PD, Kellogg GE, Zhang Y. Binding mode characterization of 6alpha- and 6beta-N-heterocyclic substituted naltrexamine derivatives via docking in opioid receptor crystal structures and site-directed mutagenesis studies: application of the ‘message-address’ concept in development of mu opioid receptor selective antagonists. Bioorg Med Chem. 2013;21:6405–6413. doi: 10.1016/j.bmc.2013.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tehan BG, Bortolato A, Blaney FE, Weir MP, Mason JS. Unifying family A GPCR theories of activation. Pharmacol Ther. 2014;143:51–60. doi: 10.1016/j.pharmthera.2014.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.