

Abstract
The creation of a humanized chimeric mouse nervous system permits the study of human neural development and disease pathogenesis using human cells in vivo. Humanized glial chimeric mice with the brain and spinal cord being colonized by human glial cells have been successfully generated. However, generation of humanized chimeric mouse brains repopulated by human neurons to possess a high degree of chimerism have not been well studied. Here we created humanized neuronal chimeric mouse brains by neonatally engrafting the distinct and highly neurogenic human induced pluripotent stem cell (hiPSC)–derived rosette-type primitive neural progenitors. These neural progenitors predominantly differentiate to neurons, which disperse widely throughout the mouse brain with infiltration of the cerebral cortex and hippocampus at 6 and 13 months after transplantation. Building upon the hiPSC technology, we propose that this potentially unique humanized neuronal chimeric mouse model will provide profound opportunities to define the structure, function, and plasticity of neural networks containing human neurons derived from a broad variety of neurological disorders.
A unique humanized neuronal chimeric mouse model was generated by engrafting highly neurogenic human induced pluripotent stem cell-derived progenitor cells in neonatal mice.
Introduction
The advent of human induced pluripotent stem cell (hiPSC) technology has led to the generation of patient- and disease-specific hiPSCs, which presents an unprecedented opportunity for studying the pathogenesis of neurodevelopmental and neurodegenerative diseases with unlimited human neural cells (1, 2). With iPSC technology, previous studies have established in vitro human cellular models to reveal mechanisms of a variety of neurological disorders. While basic aspects of the disease phenotypes such as differentiation of neural progenitor cells (NPCs), functional properties of the NPC-derived neurons and glia, and the neuron-glia interactions can be examined using the hiPSC-based in vitro model, the consequences of these events towards the formation or disruption of neural circuits in the developing or aging central nervous system (CNS) can only be studied in vivo. Therefore, generation of an experimental model that permits the analysis of neural cells derived from patients with neurological disorders in an unperturbed nervous system would provide important insights into disease pathology, progression, and mechanism.
Recent studies have tested using glial progenitor cells that are derived from human fetal brain tissue or human pluripotent stem cells (hPSCs) to generate a chimeric mouse brain and its utility in disease modeling (3–7). Neonatally engrafted glial progenitor cells have been shown to efficiently integrate and widely disperse in the normal adult mouse brains, generating chimeric mouse brains. These chimeric mouse brains possess a high degree of chimerism and are colonized by human glial progenitor cells and their derived astroglia; these mice are thereby called humanized glial chimeric mice (6, 7). However, the generation of humanized chimeric mouse brains that are largely repopulated by human neurons is far less studied. Previous transplantation studies mainly engrafted fetal human brain cells, purified human CNS stem cells, or hPSC-derived NPCs that were expanded with fibroblast growth factor 2 (FGF-2) and epidermal growth factor (EGF) (8–14). These human neural cells were able to integrate, migrate, and differentiate to neurons after transplantation, but the degree of human neuronal brain chimerism generated from these transplanted human neural cells is incomparable to the human glial brain chimerism from the transplanted human glial progenitor cells. In this study, we explore the generation of a humanized chimeric mouse brain by using hiPSC-derived rosette-type primitive NPCs (pNPCs). The pNPCs are highly neurogenic and responsive to instructive neural patterning cues in vitro (15, 16). Using immunodeficient mice as host, we demonstrate that a simple procedure using neonatal bilateral ventricle delivery results in widespread distribution of the pNPC-derived human neural cells, predominantly neurons, with infiltration of the cerebral cortex and hippocampus at 6 and 13 months after transplantation. We propose that this approach can be used to further model and study the disease mechanisms for a broad variety of neurological disorders, by using disease-specific hiPSC-derived human neurons in vivo.
Results
Neonatally engrafted hiPSC-derived pNPCs progressively expand and migrate in murine forebrain.
To explore whether hiPSC-derived pNPCs (hiPSC-pNPCs) that were neonatally grafted into the mouse brain could expand, migrate, and largely repopulate the mouse brain in adulthood, we first derived pNPCs from 2 hiPSC lines generated from healthy individuals in the absence of morphogens, as described in previous studies from us and others (15–20). The differentiation procedure is shown in Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/jci.insight.88632DS1. As shown in Figure 1A, the pNPCs were derived from hiPSCs in high purity, as indicated by nearly all the cells expressing the NPC markers nestin (97.3% ± 2.1%) and Pax6 (96.8% ± 2.7%, n = 4 from each hiPSC line). Then, we transplanted the hiPSC-pNPCs into the lateral ventricles of postnatal day 0 (P0) rag1–/– immunodeficient mouse brains (2 sites). The hiPSC-pNPC–engrafted mice were assessed by histology at different ages up to 13 months, and the transplanted human cells were tracked by human nuclear antigen (hN) staining.
Figure 1. Transplantation of hiPSC-pNPCs into the mouse brain.

(A) A schematic diagram showing the generation of humanized neuronal chimeric mouse brains. From left to right: neural rosettes derived from hiPSCs, nestin- and Pax6-expressing rosette-type hiPSC-pNPCs, stereotaxic injection of hiPSC-pNPCs into the neonatal mouse brain, and representative dot map showing the distribution of human nuclear antigen–positive (hN+) cells (in red) in 2 sagittal sections from 6-month- and 13-month-old mice. Notably, a small portion of the engrafted human cells is seen in the cerebellum. (B–E) Enlarged images from a sagittal section from a 13-month-old mouse showing the wide distribution of hN+ human cells in the hippocampus (HIP), cerebral cortex (CTX), striatum (STR), and the area near the injection site. Scale bars: 500 μm (B) and 100 μm (C–E). hiPSCs, human induced pluripotent stem cells; hiPSC-pNPCs, hiPSC-derived primitive neural progenitor cells.
Similar to the previous studies on transplantation of human brain tissue–derived glial progenitor cells (6, 7), we found that the hiPSC-pNPCs significantly expanded and migrated widely in the mouse brain. By 6 months of age, hiPSC-pNPCs had infiltrated into both gray and white matter, and largely repopulated the mouse forebrain. The human cells were also seen in the cerebellum (Figure 1A). As shown in Figure 1, B–D, by 13 months of age, the transplanted hiPSC-NPCs were distributed in a relatively uniform manner throughout both white and gray matter, including the hippocampus, cerebral cortex, and striatum. We also noticed a dense hN-positive engraftment close to the lateral ventricle where the cells were initially transplanted (Figure 1E). Moreover, we examined proliferation of the human donor cells by double staining of hN and Ki67. As shown in Supplemental Figure 2, the percentage of Ki67+ proliferating human cells decreased over time from 3 weeks to 6 months and 13 months. Because hiPSC-derived neural cells are reflective of very early human development (<6 weeks) (21, 22), similar to the neonatally transplanted glial progenitor cells derived from fetal brain tissue (late second-trimester subventricular zone) (7), these cells are also expected to divide for over 9 months in the mouse host. We also propose that the large number of human donor cells in the mouse brain in part result from the sustained proliferation of the human progenitor cells (6, 7). Previous studies reported that transplanted hPSC-derived NPCs could be tumorigenic, particularly when contaminated with residual pluripotent stem cells (23, 24). By inhibiting SMAD signaling (15, 25), the hiPSCs were efficiently differentiated into pNPCs in high purity (Figure 1A). In our pNPC cultures, we did not find any cells that expressed OCT4, which is a marker for pluripotent stem cells and is also expressed in tumor-initiating cells (26). Moreover, a previous study demonstrated that although pNPCs were highly proliferative and had clonogenic capacity, they were not tumorigenic in immunodeficient mice (15). Consistently, we did not observe any tumor formation of the transplanted cells at all time points examined.
Engrafted hiPSC-pNPCs predominantly differentiate to excitatory neurons in the gray matter.
To analyze the fate of hiPSC-pNPCs dispersed in the mouse brain, we first examined the neuronal fate of these cells. At 3 weeks after transplantation, the engrafted cells clustered in the ventricles and were restricted to the injection sites (Figure 2A). We found that the vast majority of hN+ cells (>95%) were positive for the neuronal marker βIII-tubulin and doublecortin (Dcx), a marker for migrating neuroblasts, indicating the predominant differentiation of transplanted hiPSC-pNPCs to neurons (Figures 2A and 3A). At this point, none of the cells around the injection site expressed the mature-neuron marker, NeuN (Figures 2A and 3B). At 6 and 13 months after transplantation, the engrafted cells were found far beyond the injection sites, demonstrating migration and integration of the transplanted cells into both the white and gray matter. The engrafted hiPSC-pNPCs differentiated to neurons in the gray matter but not in the white matter, as no Dcx- or NeuN-positive human cells were seen in the corpus callosum. We next analyzed the percentage of Dcx+/hN+ cells among the hN+ cells in the striatum. At 6 months, the percentage of Dcx+ cells significantly decreased, and by 13 months, few Dcx+ cells were seen in the striatum (Figures 2A and 3A). As shown in Figures 2B and 3B, the percentage of NeuN+ neurons among total hN+ cells increased from 3 weeks (<0.1%) to 6 months (73.5% ± 7.3%, n = 6) and 13 months (68.1% ± 7.0%, n = 6). Few cells were positive for GABA in the striatum (Figures 2B and 3C; 3.3% ± 1.5%, n = 6), suggesting that the hiPSC-pNPCs rarely gave rise to inhibitory neurons. In order to further demonstrate that these human neurons are functionally active in the mouse brain, we first double stained for human-specific MAP2 (hMAP2), which selectively labels dendrites of donor-derived human neurons, and postsynaptic density protein 95 (PSD-95), which labels all the postsynaptic compartments from host mouse neurons and donor-derived human neurons. As shown in Figure 2C, the PSD-95+ puncta were found to distribute along the hMAP2+ dendrites, demonstrating that the human neurons formed synapses in the mouse brain. Furthermore, we examined the expression of c-Fos, an activity-dependent immediate early gene that is expressed in neurons following depolarization and often used as a marker for mapping neuronal activity (27). As shown in Figure 2C, a large portion of the hN+ neurons at 13 months were positive for c-Fos, suggesting that they received synaptic neurotransmission and were active in the mouse brain. Moreover, by performing electron microscopic analysis, we also observed synaptic terminals formed between human neurons labeled by diaminobenzidine (DAB) staining against human-specific neural cell adhesion molecule (hN-CAM) and mouse neurons that were not labeled by the DAB staining (Supplemental Figure 3). Taken together, all these results demonstrate that the hiPSC-pNPCs predominately differentiated to excitatory neurons, which matured and integrated into the mouse brain.
Figure 2. Predominant neuronal differentiation of neonatally engrafted hiPSC-pNPCs in the mouse brain.

(A and B) Representative images of βIII-tubulin– (βIIIT-), Dcx-, and NeuN-expressing neurons in the human nuclear antigen–positive (hN+) cells near the injection site 3 weeks after transplantation, and in the striatum (STR) at 6 and 13 months after transplantation. At 13 months, there are few hN+ cells that express GABA. Arrowheads in B indicate hN+/GABA+ neurons. The area in the white box is shown enlarged in the bottom-left corner. Scale bars: 50 μm (original) and 25 μm (enlarged). (C) The left panels show representative images of human-specific MAP2 (hMAP2)–expressing dendrites showing localization of foci of the excitatory synapse–specific PSD-95 protein in the STR. Scale bars: 50, 20, and 10 μm from top to bottom images, respectively. The right panels show representative images of c-Fos–expressing active neurons in the hN+ cells in the STR. Scale bar: 50 μm. (D) Representative images of Tbr1-expressing excitatory neurons in the hN+ cells in the superficial and deep layers of the cerebral cortex (CTX), and in the CA3 and dentate gyrus (DG) regions in the hippocampus at 13 months. The human neurons in the DG region are surrounded by synapsin-1 (Syn) staining. The area in the white box is enlarged in the 2 images below. Arrows indicate hN+/Tbr1– cells, and arrowheads indicate hN+/Tbr1+ cells. Scale bars: 50 μm (both original and enlarged images). Dcx, doublecortin; PSD-95, postsynaptic density-95.
Figure 3. Quantification of the neurons differentiated from the engrafted hiPSC-pNPCs.
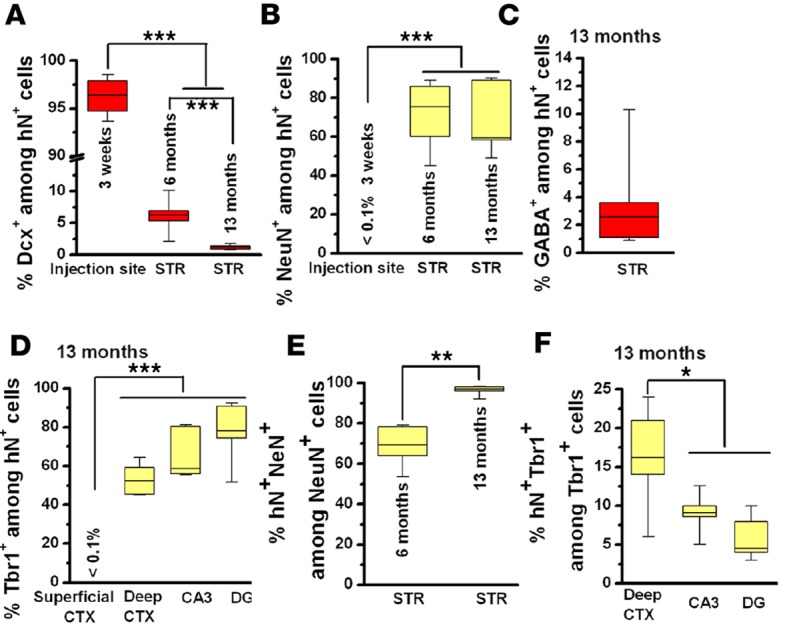
(A–D) The percentage of Dcx-, NeuN-, GABA-, and Tbr1-expressing cells in the hN+ cells in different brain regions from 3 weeks to 13 months (n = 6 for each time point). All quantitative data are analyses of pooled data collected from transplantation of hiPSC1- and hiPSC2-pNPCs. (E and F) The percentage of donor-derived hN+/NeuN+ and hN+/Tbr1+ cells in the total resident NeuN+ and Tbr1+ neurons, respectively, in different brain regions at 6 and 13 months (n = 6 for each time point). Data are presented as box-and-whisker plots. The central horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate the range of the data points. Student’s t tests and 1-way ANOVA were performed to assess the significance. *P < 0.05, **P < 0.01, and ***P < 0.001. Dcx, doublecortin; hiPSC-pNPCs, human induced pluripotent stem cell–derived primitive neural progenitor cells.
At 13 months, hN+ cells were seen in the cerebral cortex and the hippocampus, but in the superficial layers of the cerebral cortex, none of the hN+ cells were positive for the neuron marker NeuN. We further double stained for hN and T-brain 1 (Tbr1), a T-box transcription factor that labels excitatory neurons in the adult cerebral cortex and hippocampus (28, 29). As expected, we did not observe any hN+ cells that were positive for Tbr1 in the superficial layers of the cerebral cortex, whereas about 50% of the hN+ cells were positive for Tbr1 in the deep layers of the cerebral cortex (Figures 2D and 3D). In the hippocampus, an even higher percentage of hN+/Tbr1+ cells (around 70%) was seen among the hN+ cells (Figures 2D and 3D). We also did not observe any hN+/GABA+ neurons in the cerebral cortex and the hippocampus. Many of the hN+ neurons in the hippocampus were surrounded by synapsin-1 staining (Figure 2D), suggesting that the human neurons formed synapses with other neurons. In addition, we analyzed the proportions of donor-derived neurons among the resident neurons. In the striatum, the percentage of hN+/NeuN+ neurons among total NeuN+ neurons significantly increased from 6 months to 13 months (Figure 3E), reaching a high degree of chimerism at 13 months in the striatum. However, the degree of chimerism is much lower in the deep cortex and the hippocampus, as indicated by the percentage of hN+/Tbr1+ neurons among total Tbr1+ neurons at 13 months (Figure 3F).
Engrafted hiPSC-pNPCs differentiate to astroglial and oligodendroglial cells.
We next examined the glial fate of the engrafted hiPSC-pNPCs. At 3 weeks, few of the hN+ cells differentiated to astroglia, as identified by glial fibrillary acidic protein (GFAP) staining (Figure 4, A and C, 2.5% ± 2.3% of the total hN+ cells, n = 6). At 6 months after transplantation, the engrafted cells differentiated into astroglia in both the white and gray matter (Figure 4C). We analyzed the percentage of GFAP+ cells among the hN+ cells in the corpus callosum and the striatum. As shown in Figure 4, A and C, there were many more hN+ cells in the corpus callosum that were positive for GFAP (63.1% ± 5.8%, n = 6) than in the striatum (11.1% ± 2.3%, n = 6). By 13 months, the transplanted cells were widely dispersed in the cerebral cortex. Around 30% of the total hN+ cells expressed GFAP in the corpus callosum, whereas far fewer hN+ cells expressed GFAP in the gray matter, including the cerebral cortex (11.2% ± 4.5%, n = 6) and striatum (9.5% ± 3.6%, n = 6). At 13 months of age, the astroglia differentiated from hiPSC-pNPCs particularly in the corpus callosum and the cerebral cortex, appeared to be large in size, compared with the host murine astrocytes, and extended long and tortuous processes (Figure 4B), which is consistent with previous studies on transplanting hPSC-derived immature astroglia or human tissue–derived glial progenitor cells (6, 17, 30). However, unlike transplantation of hPSC-derived neural progenitors into the spinal cords of immunodeficient mice (31), the human astroglia did not significantly replace the endogenous astrocytes in the mouse brain (Figure 4, B and C).
Figure 4. Astroglial differentiation of neonatally engrafted hiPSC-pNPCs.

(A) Representative images of glial fibrillary acidic protein–expressing (GFAP-expressing) astroglia in the human nuclear antigen–positive (hN+) cells near the injection site at 3 weeks after transplantation, and in the corpus callosum (CC), striatum (STR), and cerebral cortex (CTX) at 6 and 13 months after transplantation. Arrowheads indicate the hN+/GFAP+ human astroglia, and arrows indicate the hN–/GFAP+ endogenous host astroglia. Scale bars: 50 μm. The dotted lines highlight the CC region. (B) Representative image showing colocalization of hN and GFAP, and the long and tortuous processes of human astroglia in the mouse brain. (C) The percentage of GFAP-expressing cells in the hN+ cells at different brain regions from 3 weeks to 13 months (n = 6 for each time point). All quantitative data are analyses of pooled data collected from transplantation of hiPSC1- and hiPSC2-pNPCs. Data are presented as box-and-whisker plots. The central horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate the range of the data points. Student’s t tests and 1-way ANOVA were performed to assess the significance. **P < 0.01 and ***P < 0.001. hiPSC-pNPCs, human induced pluripotent stem cell–derived primitive neural progenitor cells.
The engrafted hiPSC-pNPCs also generated oligodendroglial cells in the mouse brain. As shown in Figure 5A, at 3 weeks, very few cells were positive for NG2, a marker for glial progenitor cells that are committed to oligodendroglia progenitor cells (OPCs) in the developing and adult CNS (32). At 6 months, the percentage of NG2+/hN+ cells in the corpus callosum significantly increased (Figure 6A; 26.4% ± 2.4%, n = 6) and was much higher than that in the striatum (Figure 6A; 1.8% ± 0.5%, n = 6). By 13 months, the percentage of NG2+/hN+ cells in the corpus callosum increased to around 60%, whereas the percentage of NG2+/hN+ cells in the striatum remained low (Figure 6A; 3.6% ± 0.6%, n = 6). We also observed that, in the superficial layers of the cerebral cortex, the majority of the hN+ cells (about 80%) expressed NG2. To further examine the population of oligodendroglia lineage cells at 13 months, we stained for the pan-oligodendroglial marker OLIG2 (Figures 5B and 6B). We found that in the corpus callosum, striatum, and superficial layers of the cerebral cortex, the percentage of OLIG2+/hN+ cells was similar to that of NG2+/hN+ cells, suggesting that the oligodendroglial lineage cells differentiated from the engrafted hiPSC-pNPCs mainly stayed at the progenitor stage. We also examined the expression of myelin basic protein (MBP), a marker for mature oligodendrocytes and myelin. We found that some of the hN+ cells in the corpus callosum (4.3% ± 2.0%, n = 6) and striatum (3.5% ± 0.9%, n = 6), and few (0.9% ± 0.6%, n = 6) in the superficial layers of the cerebral cortex were positive for MBP (Figures 5B and 6C).
Figure 5. Oligodendroglial differentiation of neonatally engrafted hiPSC-pNPCs.

(A) Representative images of NG2-expressing cells in the human nuclear antigen–positive (hN+) cells near the injection site at 3 weeks after transplantation, and in the corpus callosum (CC), striatum (STR), and superficial cerebral cortex (CTX) at 6 and 13 months after transplantation. Arrowheads indicate the hN+/NG2+ human cells. Scale bars: 50 μm. The dotted lines highlight the CC region. (B) Representative images of OLIG2- and MBP-expressing oligodendroglial cells in the hN+ cells in the CC, STR, and superficial CTX at 13 months after transplantation. Arrowheads indicate the hN+/OLIG2+ human oligodendroglia or hN+/MBP+ mature oligodendrocytes. The arrow indicates the hN–/MBP+ endogenous host mature oligodendrocytes. Areas outlined in white boxes are enlarged. Scale bars: 50 μm (original) and 25 μm (enlarged). hiPSC-pNPCs, human induced pluripotent stem cell–derived primitive neural progenitor cells.
Figure 6. Quantification of the oligodendroglial cells differentiated from the engrafted hiPSC-pNPCs.

(A–C) The percentage of NG2-, OLIG2-, and MBP-expressing cells in the hN+ cells at different brain regions from 3 weeks to 13 months (n = 6 for each time point). All quantitative data are analyses of pooled data collected from transplantation of hiPSC1- and hiPSC2-pNPCs. Data are presented as box-and-whisker plots. The central horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate the range of the data points. Student’s t tests and 1-way ANOVA were performed to assess the significance. *P < 0.05 and ***P < 0.001. hiPSC-pNPCs, human induced pluripotent stem cell–derived primitive neural progenitor cells.
Discussion
In this study, by engrafting neonatal mice with hiPSC-pNPCs, we demonstrate the generation of humanized chimeric mouse brains that are largely repopulated by human neurons. The transplanted hiPSC-pNPCs are highly neurogenic and give rise to a large proportion of neurons, unlike engrafted glial progenitor cells that exclusively give rise to glial cells within the recipient mice. The engrafted hiPSC-pNPCs also appeared to respond to region-specific signals, differentiating to glial cells in the white matter. In the gray matter including the cerebral cortex, striatum, and hippocampus, the vast majority of the neurons derived from hiPSC-pNPCs are excitatory neurons, with few GABA+ inhibitory neurons being identified. Interestingly, we also find that the human cells widely disperse in the hippocampus, with a majority of them differentiating into Tbr1+ excitatory neurons. The human neurons are able to mature and integrate into host neural circuits, as indicated by expression of synaptic proteins and the neuronal activity marker c-Fos.
Previous studies have documented the transplantation of human NPCs into rodent brains in different contexts. However, the generation of humanized chimeric mice with a high degree of human neuronal brain chimerism has not been well studied. We propose that 2 critical improvements in this study allow the generation of such humanized neuronal chimeric mouse brains. First, we transplanted NPCs at a distinct primitive stage, named pNPCs (15, 16), into the brains of immunodeficient mice. Previous transplantation studies mainly engrafted fetal human brain cells, purified human CNS stem cells (8–12), or hPSC-derived NPCs that were expanded with growth factors, FGF-2 and EGF (13, 14). Compared with pNPCs, these CNS stem cells or NPCs usually do not respond to developmental patterning cues, and after expansion in the presence of FGF-2 and EGF, these cells progress in a few passages toward a definitive stage with increased gliogenic bias and concomitant loss of neurogenic potential (15, 16). Indeed, a recent study (31) shows that hPSC-derived NPCs that are patterned with spinal cord identity and further expanded in vitro nearly exclusively give rise to astrocytes after being engrafted into the spinal cord, generating humanized astroglial chimeric mouse spinal cord. In our study, as shown in Figures 2 and 3, the engrafted pNPCs highly efficiently generate βIII-tubulin+ and DCX+ immature neurons at 3 weeks after transplantation and generate NeuN+ and c-Fos+ functional mature neurons at 13 months after transplantation. Moreover, using immunodeficient mice as recipient also significantly promoted the survival of xenografted human neural cells (6, 7, 31). Previous transplantation studies using nonimmunocompromised rodents were not able to achieve a high degree of brain chimerism, even with injections of immunosuppressant (11, 14, 33, 34). Thus, in this study, transplantation of pNPCs with great neurogenic potential into the immunodeficient mouse brain facilitates the generation of the humanized neuronal chimeric mouse brains. Second, we transplanted pNPCs into the mouse brain at the earliest postnatal age, P0, as there is an age-related decline of the neurogenic niche in the brain (35, 36). A previous study demonstrated that only a small portion (1%–3%) of human neural stem cells transplanted into the adult rodent brain expressed the neuronal marker MAP-2 and exhibited neuron-like morphology (37). The neurogenic niche in the neonatal mouse brain may promote neurogenesis from the transplanted pNPCs, further facilitating the generation of humanized neuronal chimeric mouse brains. Moreover, the migration capacity of transplanted human cells appears to decrease dramatically along with age of the recipient animals. Human neural cells transplanted into the adult rodent brain were not able to widely disperse in the brain (38–40), unlike the humanized glial chimeric mouse brains generated by engrafting the cells within a day of birth (6, 7). Previous studies have also explored transplantation into the fetal brain, but likely owing to the use of human fetal brain tissue–derived stem cells, generation of humanized neuronal chimeric brains was not achieved (10, 11). In the future, it would be interesting to test whether transplantation of pNPCs into the fetal brain where neurogenesis is actively ongoing would result in chimeric brains with an even higher degree of human neuron chimerism than neonatal transplantation of pNPCs does. In this study, the transplanted pNPCs nearly exclusively differentiated into excitatory neurons, and we propose that this might be because of the lack of intrinsic patterning cues that direct inhibitory neuron production in the neonatal brain. Transplantation of pNPCs into the fetal brain may lead to the generation of different types of human neurons in the mouse brain, particularly inhibitory neurons, as a result of responding to the intrinsic cues that promote inhibitory neuron development in the embryonic brain (41).
Another great advance of this study is the utility of hiPSCs. hiPSCs derived from individuals with neurological disorders have proven to be valid human cellular models that could enable detailed functional studies of the pathogenesis of neurodevelopmental and neurodegenerative diseases. However, our brain is a complex network in which information is continuously processed and transported between spatially distributed but functionally linked regions. Obviously, the current hiPSC model of disease-in-a-dish is far too primitive to model complex neural network formation of the human brain. Although the engrafted pNPCs give rise to different types of neural cells in the mouse brain, the majority of the pNPCs generate neurons, particularly in the gray matter. Thus, because of the extensive neuronal chimerism and circuit integration of the human neurons, these hiPSC-based neuronal chimeric mice will provide a unique model to investigate how diseased human neurons participate in the formation or disruption of neural circuits in the developing or aging CNS in vivo. Recent studies have developed cell type–specific differentiation protocols to derive different types of glial cells including astroglia and oligodendroglia, from hPSCs (18, 30, 42). In the future, complemented by human glial chimeric mice generated with the subtype-specific glial cells, we may be able to dissect the contribution of different cell types to the observed disease phenotypes.
Furthermore, intellectual performance is an accumulated outcome of functional brain networks. Remarkably, recent studies show that humanized glial chimeric mice can manifest significant enhanced long-term potentiation and learning, demonstrating the possibility of using the humanized chimeric mouse model to study the contribution of human neural cells to the cognitive performance (3, 6). Therefore, in combination with iPSC technology, future studies are warranted to examine the functionality of the normal and diseased human neurons in the in vivo system, and its connectivity with cognitive behavioral performance.
Methods
Culture of hiPSCs.
The 2 hiPSC lines, hiPSC1 and hiPSC2, were generated from healthy individuals’ fibroblasts as described in our previous study (17). The hiPSCs were cultured on plates coated with hESC-qualified Matrigel (Corning) with mTeSR1 media (STEMCELL Technologies), and passaged approximately once per week using dispase (1 mg/ml; STEMCELL Technologies) (17, 20).
Differentiation of hiPSCs to pNPCs.
Small molecules were used to induce differentiation of hiPSCs to pNPCs, as reported in previous studies (15, 25). As shown in Supplemental Figure 1, to induce neural differentiation, embryoid bodies (EBs) were treated with SB431542 (5 μM; Stemgent) or noggin (50 ng/ml; Peprotech), inhibitors of the SMAD signaling pathway, in neural induction medium containing DMEM/F12 (HyClone) and 1× N2 (Thermo Fisher Scientific) for 1 week. EBs were then grown on plates coated with growth factor–reduced Matrigel (BD Biosciences) in medium consisting of DMEM/F12, 1× N2, and laminin (1 μg/ml; Sigma-Aldrich). NPCs in the form of rosettes developed for another 7 days (week 2). Next, rosettes were manually isolated from surrounding cells and expanded for 7 days (week 3) in suspension in NPC medium, composed of a 1:1 mixture of Neurobasal (Thermo Fisher Scientific) and DMEM/F12 and supplemented with 1× N2, 1× B27-RA (Thermo Fisher Scientific), 20 ng/ml FGF2 (Peprotech), human leukemia inhibitory factor (hLIF, 10 ng/ml, Millipore), CHIR99021 (3 μM, Stemgent), SB431542 (2 μM), and ROCK inhibitor Y-27632 (10 μM, Tocris). CHIR99021 is a glycogen synthase kinase 3 (GSK3) inhibitor. Synergistic inhibition of GSK3 promotes capture/maintenance of pNPCs (15, 43). Then, the pNPCs were replated for 1 week (week 4), and the expanded neural rosettes were dissociated into single cells using TrypLE Express (Thermo Fisher Scientific) for transplantation.
Cell transplantation.
hiPSC-pNPCs were suspended as single cells at a final concentration of 100,000 cells per μl in PBS. The pNPCs were then injected into the brains of P0 rag1–/– immunodeficient mice (B6.129S7-Rag1tm1Mom/J on a C57BL/6 background, Jackson Laboratories). The mouse pups were anesthetized by placing them on ice for 5 minutes. Once cryoanesthetized, the pups were placed on a digital stereotaxic device (David KOPF Instruments), equipped with a neonatal mouse adaptor (Stoeling). The pups were then injected with 1.5 μl of cells into both lateral ventricles. The precise transplantation sites were 1 mm from the midline between the Bregman and Lambda and 1.2 mm deep. Hamilton syringes and needles were used to deliver cells by inserting directly through the skull into the target sites (17). The pups were weaned at 3 weeks, and were kept up to 13 months before they were tested for the engraftment of human cells.
Immunostaining.
Cells fixed with 4% paraformaldehyde and brain sections (18 μm thick) from 3-week-, 6-month-, and 13-month-old mice were processed for immunofluorescence staining (19). The information for primary antibodies and dilutions are listed in Supplemental Table 1. Secondary antibodies (all from Invitrogen) were all used at 1:1,000. Slides or coverslips were mounted with the anti-fade Fluoromount-G medium containing 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Southern Biotechnology). Diaminobenzidine (DAB) immunostaining was performed with antibody against hN-CAM as described in our previous study (18).
Transplant mapping and cell counts.
Montages for whole-section mapping of donor cells were generated on a Nikon A1 or a Zeiss laser-scanning microscope. Quantification of donor cell phenotype in mice was based on counts of different brain regions in at least 4 consecutive sections of each mouse. The number of positive cells from each section was counted after a Z projection. The cells were counted with ImageJ software (NIH). Six mice were used for each time point.
Electron microscopy.
The selected vibratome sections that contained DAB-labeled cells were postfixed and processed for electron microscopy (EM) as described in our previous studies (20, 44). EM images were captured using a high-resolution charge-coupled device (CCD) camera (FEI).
Data analysis.
Values are represented as the mean ± SEM. All assessments were analyzed using 1-tailed Student’s t test when only 2 groups were compared or 1-way ANOVA when 3 or more groups were compared. A P value less than 0.05 was considered significant.
Study approval.
All experiments conducted on hiPSCs were approved by the Stem Cell Research Oversight Committee at the University of Nebraska Medical Center. Animal experiments were performed following protocols approved by the University of Nebraska Medical Center.
Author contributions
PJ conceived and supervised the study. CC and PJ carried out experiments and interpreted data. WK provided critical input to the research design. PJ wrote the paper with input from all coauthors.
Supplementary Material
Acknowledgments
We thank Howard Gendelman from the University of Nebraska Medical Center for helpful comments. We also thank Ranjie Xu for technical assistance and Andrew Brawner for the help with editing the manuscript. This work was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the NIH under grant number P30GM110768, and UNMC lab startup fund (both to PJ).
Footnotes
Conflict of interest: The authors declare that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(19):e88632. doi:10.1172/jci.insight.88632.
Contributor Information
Chen Chen, Email: chenchen.ucdavis@gmail.com.
Woo-Yang Kim, Email: wooyang.kim@unmc.edu.
Peng Jiang, Email: peng.jiang@unmc.edu.
References
- 1.Marchetto MC, Brennand KJ, Boyer LF, Gage FH. Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum Mol Genet. 2011;20(R2):R109–R115. doi: 10.1093/hmg/ddr336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17(3):170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- 3.Goldman SA, Nedergaard M, Windrem MS. Modeling cognition and disease using human glial chimeric mice. Glia. 2015;63(8):1483–1493. doi: 10.1002/glia.22862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondo Y, et al. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. J Clin Invest. 2014;124(12):5323–5336. doi: 10.1172/JCI76629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benraiss A, et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat Commun. 2016;7:11758. doi: 10.1038/ncomms11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han X, et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell. 2013;12(3):342–353. doi: 10.1016/j.stem.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Windrem MS, et al. A competitive advantage by neonatally engrafted human glial progenitors yields mice whose brains are chimeric for human glia. J Neurosci. 2014;34(48):16153–16161. doi: 10.1523/JNEUROSCI.1510-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamaki S, et al. Engraftment of sorted/expanded human central nervous system stem cells from fetal brain. J Neurosci Res. 2002;69(6):976–986. doi: 10.1002/jnr.10412. [DOI] [PubMed] [Google Scholar]
- 9.Uchida N, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97(26):14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ourednik V, et al. Segregation of human neural stem cells in the developing primate forebrain. Science. 2001;293(5536):1820–1824. doi: 10.1126/science.1060580. [DOI] [PubMed] [Google Scholar]
- 11.Brüstle O, et al. Chimeric brains generated by intraventricular transplantation of fetal human brain cells into embryonic rats. Nat Biotechnol. 1998;16(11):1040–1044. doi: 10.1038/3481. [DOI] [PubMed] [Google Scholar]
- 12.Zhou FW, et al. Functional integration of human neural precursor cells in mouse cortex. PLoS One. 2015;10(3):e0120281. doi: 10.1371/journal.pone.0120281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reubinoff BE, et al. Neural progenitors from human embryonic stem cells. Nat Biotechnol. 2001;19(12):1134–1140. doi: 10.1038/nbt1201-1134. [DOI] [PubMed] [Google Scholar]
- 14.Zhang SC, Wernig M, Duncan ID, Brüstle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19(12):1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- 15.Li W, et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci U S A. 2011;108(20):8299–8304. doi: 10.1073/pnas.1014041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22(2):152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen C, et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat Commun. 2014;5:4430. doi: 10.1038/ncomms5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang P, et al. hESC-derived Olig2+ progenitors generate a subtype of astroglia with protective effects against ischaemic brain injury. Nat Commun. 2013;4:2196. doi: 10.1038/ncomms3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Jiang P, Deng W. OLIG gene targeting in human pluripotent stem cells for motor neuron and oligodendrocyte differentiation. Nat Protoc. 2011;6(5):640–655. doi: 10.1038/nprot.2011.310. [DOI] [PubMed] [Google Scholar]
- 20.Jiang P, Chen C, Liu XB, Pleasure DE, Liu Y, Deng W. Human iPSC-derived immature astroglia promote oligodendrogenesis by increasing TIMP-1 secretion. Cell Rep. 2016;15(6):1303–1315. doi: 10.1016/j.celrep.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson M, et al. Defining the nature of human pluripotent stem cell progeny. Cell Res. 2012;22(1):178–193. doi: 10.1038/cr.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paşca AM, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12(7):671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy NS, Cleren C, Singh SK, Yang L, Beal MF, Goldman SA. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat Med. 2006;12(11):1259–1268. doi: 10.1038/nm1495. [DOI] [PubMed] [Google Scholar]
- 24.Chung S, et al. Genetic selection of sox1GFP-expressing neural precursors removes residual tumorigenic pluripotent stem cells and attenuates tumor formation after transplantation. J Neurochem. 2006;97(5):1467–1480. doi: 10.1111/j.1471-4159.2006.03841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan GJ, Chang ZY, Schöler HR, Pei D. Stem cell pluripotency and transcription factor Oct4. Cell Res. 2002;12(5-6):321–329. doi: 10.1038/sj.cr.7290134. [DOI] [PubMed] [Google Scholar]
- 27.Loebrich S, Nedivi E. The function of activity-regulated genes in the nervous system. Physiol Rev. 2009;89(4):1079–1103. doi: 10.1152/physrev.00013.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodge RD, Hevner RF. Expression and actions of transcription factors in adult hippocampal neurogenesis. Dev Neurobiol. 2011;71(8):680–689. doi: 10.1002/dneu.20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang GS, et al. Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron. 2004;42(1):113–128. doi: 10.1016/S0896-6273(04)00139-4. [DOI] [PubMed] [Google Scholar]
- 30.Krencik R, Weick JP, Liu Y, Zhang ZJ, Zhang SC. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat Biotechnol. 2011;29(6):528–534. doi: 10.1038/nbt.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, et al. Human-derived neural progenitors functionally replace astrocytes in adult mice. J Clin Invest. 2015;125(3):1033–1042. doi: 10.1172/JCI69097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68(4):668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flax JD, et al. Engraftable human neural stem cells respond to developmental cues, replace neurons, and express foreign genes. Nat Biotechnol. 1998;16(11):1033–1039. doi: 10.1038/3473. [DOI] [PubMed] [Google Scholar]
- 34.Keyoung HM, et al. High-yield selection and extraction of two promoter-defined phenotypes of neural stem cells from the fetal human brain. Nat Biotechnol. 2001;19(9):843–850. doi: 10.1038/nbt0901-843. [DOI] [PubMed] [Google Scholar]
- 35.Katsimpardi L, et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344(6184):630–634. doi: 10.1126/science.1251141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villeda SA, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477(7362):90–94. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundberg C, Martínez-Serrano A, Cattaneo E, McKay RD, Björklund A. Survival, integration, and differentiation of neural stem cell lines after transplantation to the adult rat striatum. Exp Neurol. 1997;145(2 Pt 1):342–360. doi: 10.1006/exnr.1997.6503. [DOI] [PubMed] [Google Scholar]
- 38.Fricker RA, Carpenter MK, Winkler C, Greco C, Gates MA, Björklund A. Site-specific migration and neuronal differentiation of human neural progenitor cells after transplantation in the adult rat brain. J Neurosci. 1999;19(14):5990–6005. doi: 10.1523/JNEUROSCI.19-14-05990.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olstorn H, Moe MC, Røste GK, Bueters T, Langmoen IA. Transplantation of stem cells from the adult human brain to the adult rat brain. Neurosurgery. 2007;60(6):1089–98.:;discussion 1098. doi: 10.1227/01.NEU.0000255461.91892.0D. [DOI] [PubMed] [Google Scholar]
- 40.Englund U, Björklund A, Wictorin K. Migration patterns and phenotypic differentiation of long-term expanded human neural progenitor cells after transplantation into the adult rat brain. Brain Res Dev Brain Res. 2002;134(1-2):123–141. doi: 10.1016/S0165-3806(01)00330-3. [DOI] [PubMed] [Google Scholar]
- 41.Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5(10):793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- 42.Douvaras P, Fossati V. Generation and isolation of oligodendrocyte progenitor cells from human pluripotent stem cells. Nat Protoc. 2015;10(8):1143–1154. doi: 10.1038/nprot.2015.075. [DOI] [PubMed] [Google Scholar]
- 43.Kim WY, et al. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009;12(11):1390–1397. doi: 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang P, et al. Generation and characterization of spiking and nonspiking oligodendroglial progenitor cells from embryonic stem cells. Stem Cells. 2013;31(12):2620–2631. doi: 10.1002/stem.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.