

Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease and one of the leading inherited causes of infant mortality. SMA results from insufficient levels of the survival motor neuron (SMN) protein, and studies in animal models of the disease have shown that increasing SMN protein levels ameliorates the disease phenotype. Our group previously identified and optimized a new series of small molecules, with good potency and toxicity profiles and reasonable pharmacokinetics, that were able to increase SMN protein levels in SMA patient–derived cells. We show here that ML372, a representative of this series, almost doubles the half-life of residual SMN protein expressed from the SMN2 locus by blocking its ubiquitination and subsequent degradation by the proteasome. ML372 increased SMN protein levels in muscle, spinal cord, and brain tissue of SMA mice. Importantly, ML372 treatment improved the righting reflex and extended survival of a severe mouse model of SMA. These results demonstrate that slowing SMN degradation by selectively inhibiting its ubiquitination can improve the motor phenotype and lifespan of SMA model mice.
Inhibiting the ubiquitination and degradation of survival motor neuron protein improves motor function and extends lifespan in a severe mouse model of spinal muscular atrophy.
Introduction
Spinal muscular atrophy (SMA), an inherited autosomal neurodegenerative disease, is the most prevalent genetic disorder causing infant mortality. SMA afflicts approximately 1 in 11,000 infants, and about 1 in 40 of the Eurasian population are carriers for the disease (1). In severe cases, death ensues within the first 2 years of life. There are no approved treatments for SMA other than supportive care (2–4). SMA results from insufficient levels of the survival motor neuron (SMN) protein (5), which is produced by expression of the SMN1 and SMN2 genes. A single nucleotide difference between SMN1 and SMN2 in exon 7 causes this exon to be skipped. While SMN1 primarily produces full-length SMN protein, the predominant protein product of SMN2 is SMNΔ7, which is unstable (6, 7). SMA is the consequence of loss of the SMN1 gene, with the number of copies of SMN2 acting as a disease modifier (6, 8–10). Current investigational approaches to enhance SMN protein levels include increasing SMN2 exon 7 inclusion, increasing overall SMN2 transcript expression, and stabilizing the residual SMN protein in SMA patients (11, 12). While the mechanisms that regulate SMN2 splicing have been well documented, less is known about the factors that govern SMN protein stability.
Therapeutically, increasing levels of SMN should prevent motor neuron dysfunction in SMA. This treatment concept has been validated in mouse models of SMA by several approaches including targeted transgenic expression of SMN1 in motor neurons, gene replacement with adeno-associated viruses, oligonucleotides that promote inclusion of exon 7, and histone deacetylase (HDAC) inhibitors that increase SMN expression (13–15). Additionally, SMA severity in human patients and SMA mouse models correlates with the number of SMN2 copies (16, 17). Currently, there are about 18 therapeutic programs for SMA in various stages of preclinical and clinical development (18). These programs face challenges including efficiency of delivery, toxicity, and variable efficacy. Together, these challenges highlight the need for the development of additional therapeutic targets for SMA.
In a cell-based phenotypic screen for enhancers of SMN protein production, our group discovered a series of arylpiperidines from a qHTS screen of the 210,386-compound NIH Molecular Libraries Small Molecule Repository (MLSMR). After a systematic hit-to-lead medicinal chemistry effort on this series, a pool of lead compounds with good potency, physical properties, tolerance, and CNS penetration able to increase levels of SMN protein were identified (19). In this paper, we present a detailed characterization of one of the lead compounds, ML372, including the mechanism by which it elevates levels of SMN protein and its in vivo efficacy in a severe model of SMA, where it increases motor function and improves survival.
Results
ML372 is brain penetrant and has a reasonable exposure and half-life in vivo.
We previously discovered a novel aryl-thiazol-piperidine series as modulators of SMN protein through a campaign of quantitative high-throughput screening (qHTS) of a 210,386-compound MLSMR diversity set using cell-based SMN2-luciferase reporter assay (19). Through an extensive structure-activity relationship (SAR) investigation, more than 160 analogs of this series were synthesized and tested (schematically summarized in Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/jci.insight.88427DS1). Several potential backup series, such as the thiadiazole or the pyrimidine core, were identified as having potencies in the same range. The details of the SAR can be found in our previous publication about this series (19). ML372 was chosen for advanced studies because of its potency, efficacy, permeability, metabolic stability, and pharmacokinetic properties (Table 1). ML372 has a reasonable half-life in brain (t1/2 = 2.6 hours) as well as in plasma (t1/2 = 2.2 hours) after a single intraperitoneal administration in male FVB mice of a dose of 50 mg/kg. Despite its high plasma protein binding (94.9%), ML372 concentration in brain reaches 5.07 μmol/kg (Cmax) in 30 minutes.
Table 1. Pharmacokinetic properties of ML372 following oral gavage administration to mice.

ML372 increases SMN steady-state protein levels without affecting mRNA expression.
Full-length SMN protein levels can be enhanced by increasing expression from the SMN2 locus, augmenting exon 7 inclusion in SMN2 mRNA, or affecting posttranslational modifications that result in SMN protein stability. To assess the mechanism by which ML372 modulates SMN, we treated SMA patient–derived fibroblast 3813 cells with varying concentrations of the compound for 48 hours and measured SMN2 mRNA expression, changes in the splicing ratio of SMN2 transcript, and SMN protein levels. ML372 did not modify the overall expression of the SMN2 gene (Figure 1A) or the splicing ratio of the SMN2 gene transcript (Figure 1B). On the other hand, similar to what was previously described in the initial studies, ML372 significantly increased SMN protein levels in SMA patient fibroblast cells. ML372 treatment resulted in an average SMN protein level increase of 1.85 ± 0.2-fold at a dose of 300 nM (Figure 1C). In order to investigate the duration of the ML372 effect, a series of compound washout experiments was performed and cells collected at the indicated times. SMN protein levels were enhanced up to 48 hours after the compound was removed (Figure 1D).
Figure 1. ML372 enhances SMN protein level and function.

Spinal muscular atrophy patient fibroblast 3813 cells were treated with vehicle or ML372 at indicated concentrations for 48 hours. (A) SMN2 transcript expression was measured by qPCR. (B) The relative ratio of full-length SMN to the Δ7 mutant (FL/Δ7) was used to determine the exon 7 inclusion in SMN2 mRNA. (C) Western blot was used to determined SMN protein levels (left panel). Densitometry analysis is shown as the mean ± SEM (n = 3, **P < 0.01, ***P < 0.001) (right panel). (D) A series of wash-out experiments were performed and SMN protein level was determined by Western blot. Densitometry analysis is shown as the mean ± SEM (n = 3, *P < 0.05, **P < 0.01) (right panel). All P values determined by unpaired 2-tailed Student’s t test.
ML372 increases SMN protein stability.
As ML372 increases SMN protein levels without altering mRNA levels or splicing of the SMN2 transcript, we further evaluated the effect of ML372 on SMN protein half-life. Cells were treated with ML372 or vehicle and the rates of SMN protein turnover were measured by pulse-chase analyses. We found that treatment with ML372 decreased the rate of turnover of the newly synthesized SMN protein and prolonged the protein half-life from 3.9 hours to 18.4 hours in cells treated with the compound (Figure 2A). ML372 also increased the half-life of the SMNΔ7 protein, the product of the major SMN2 splice variant in SMA patients (Figure 2A). A key mechanism by which SMN protein is degraded is through the ubiquitin proteasome system (9). We first sought to determine if ML372 inhibits proteasome activity. SMA patient–derived fibroblast 3813 and HEK-293T cells were incubated with vehicle or different concentrations of ML372 (0.1–3 μM), and chymotrypsin-like activity of the proteasome was assessed using a fluorogenic cell-based chymotrypsin-like activity assay. Our results demonstrate that ML372 has no effect on the 20S catalytic activity of the proteasome (Supplemental Figure 2).
Figure 2. SMN ubiquitination is modulated by ML372.

(A) Pulse-chase analysis of myc-SMN (left panel) and myc-SMNΔ7 (right panel) transiently expressed in HEK-293T cells in the presence and absence of 0.3 μM ML372. (B) Recombinant SMN was incubated with the ubiquitin-activating enzyme (E1), the ubiquitin-conjugating enzyme (UBCH5b), and Mib1 with or without ML372 at the indicated concentrations. Densitometry analysis is shown as the mean ± SEM (n = 3, ***P < 0.001) (bottom panel). (C) HEK-293T cells were transiently transfected with 1 μg of HA-ubiquitin. Cells were treated with various concentrations of ML372 for 48 hours and ubiquitinated SMN was immunoprecipitated. Densitometry analysis is shown as the mean ± SEM (n = 3, ***P < 0.001) (bottom panel). (D) HEK-293T cells were transiently transfected with 1 μg of myc-Mib1. Cells were treated with various concentrations of ML372 for 48 hours, and SMN was immunoprecipitated. Western blot was used to determine SMN-associated Mib1. Densitometry analysis is shown as the mean ± SEM (n = 3, *P < 0.05) (right panel). P values were determined by 1-way ANOVA followed by Dunnett’s multiple comparisons test.
ML372 inhibits SMN protein ubiquitination.
We previously reported that the E3 ubiquitin-protein ligase mind bomb-1 (Mib1) ubiquitinates SMN, targeting it for degradation (20). In addition Usp9x (also known as fat facets, FAM), a deubiquitinating enzyme (DUB), stabilizes SMN by reducing its ubiquitination (21). Usp9x/FAM also interacts with Mib1 (22), suggesting that they act together to regulate SMN protein levels. Given that SMN ubiquitination precedes its degradation, we next assessed the effect of ML372 on SMN ubiquitination. Thus, we reconstituted in vitro the ubiquitin-E1-E2-E3 complex using ubiquitin-like modifier activating enzyme 1 (UBA1, E1) and the ubiquitin-conjugating enzymes H5b (UBCH5b, E2) and Mib1 (E3) to evaluate its capacity to ubiquitinate SMN in the presence and absence of ML372. ML372 exhibits dose-dependent inhibition of SMN ubiquitination at 0.3–3 μM (Figure 2B). We confirmed that other candidate drugs for SMA therapeutics such as the PTC, Inc. read-through compound PTC124 and the NSAID indoprofen do not inhibit ubiquitination of SMN by Mib1 (Supplemental Figure 3). Indeed, ML372 presents the first evidence of a small molecule increasing SMN protein level by blocking its ubiquitination.
To validate the mechanism of action of ML372 in cultured cells, HEK-293T cells were transfected with HA-tagged ubiquitin and endogenous SMN was immunoprecipitated following compound treatment. Our results indicate that ML372 attenuated ubiquitination of endogenous SMN (Figure 2C). To confirm that the effect of ML372 on SMN ubiquitination was due to Mib1 activity, we cotransfected HEK-293T cells with HA-tagged ubiquitin and myc-tagged Mib1. Overexpression of Mib1 resulted in increased SMN ubiquitination, and consistent with our in vitro data, 300 nM ML372 blocked Mib1-dependent SMN ubiquitination (Supplemental Figure 4). ML372 could potentially alter SMN ubiquitination by either hindering activation of ubiquitin and thioester bond formation with the conjugating enzyme, or preventing Mib1-mediated ligation of ubiquitin to SMN. To determine which one of the 3 enzymes in the ubiquitination cascade exhibits affinity for ML372, we measured binding capacity using a modified surface plasmon resonance imaging (SPRi) technique calibrated for small molecules (23). We found that ML372 only displays binding to the E3 ligase Mib1 (Supplemental Table 2). Additionally, using an in vitro ubiquitin-activation assay, we confirmed that ML372 did not inhibit ubiquitin activation by E1 or transfer to an E2 through a thioester bond intermediate (Supplemental Figure 5). We then assessed if ML372 prevents association of Mib1 with SMN in cultured cells. HEK-293T cells were transfected with myc-tagged Mib1 and coimmunoprecipitation with endogenous SMN was measured in the absence and presence of the compound. ML372 attenuated the interaction of Mib1 and SMN (Figure 2D). Together, these data strongly suggest that ML372 blocks SMN ubiquitination by blocking its interaction with Mib1.
ML372 augments SMN levels in SMNΔ7 SMA mice. To evaluate the effect of ML372 in vivo, we used SMNΔ7 mice (Smn–/–, SMN2+/+, SMNΔ7+/+). Although ML372 is orally bioavailable, to accurately dose the newborn pups, we decided to use i.p. injection as the preferable route of administration. To ensure the continuous maintenance of the compound in the CNS well above its EC50, we initiated our in vivo studies using an i.p. dose of ML372 of 50 mg/kg twice per day with an interval of 8 hours. No adverse events were observed in WT mice administered ML372 throughout the study period. With positive tolerability and reasonable pharmacokinetic data of ML372, we decided to test the efficacy of the compound in SMNΔ7 mice. Neonatal SMNΔ7 mice were treated for 5 days, starting from postnatal day 5 (PND5) (PND1 = birth). On PND9, the mice were sacrificed and tissues were harvested for further analysis. Similar to what was observed in WT mice, ML372 did not induce any toxicity in the SMNΔ7 mice. The levels of SMN protein in brain, spinal cord, and muscle tissues from treated animals were compared with tissues from vehicle-treated animals. ML372 increased levels of SMN in SMA brain tissues approximately 2-fold by Western blot analysis compared with vehicle control samples (Figure 3A). Importantly, similar to our pharmacokinetic study using WT mice, we detected a significant amount of ML372 in brain tissue by liquid chromatography/mass spectrometry (LC/MS) and a consequent increase in the level of SMN protein (Supplemental Table 1). Western blot analysis of spinal cord and muscle tissues from ML372-treated animals also indicate an average 2-fold increase in SMN protein levels (Figure 3, B and C).
Figure 3. ML372 increases SMN protein levels in SMNΔ7 spinal muscular atrophy (SMA) mice.
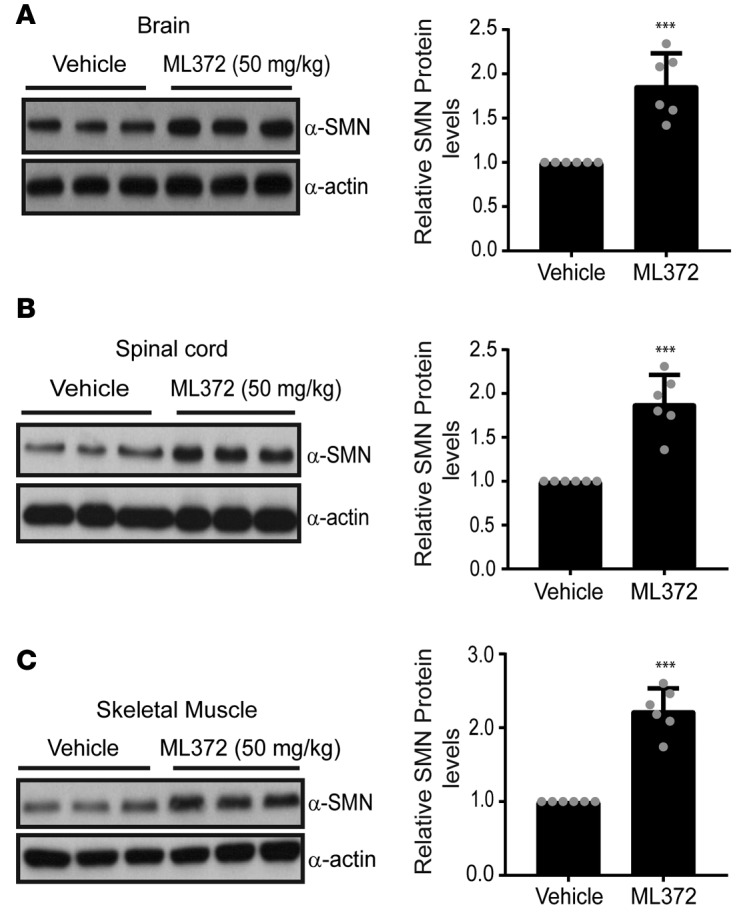
Protein lysates of (A) brain, (B) spinal cord, and (C) muscle tissues (50–100 μg) from vehicle- or ML372-treated SMNΔ7 SMA mice were resolved by 10% SDS-PAGE. Densitometry analysis is shown as the mean ± SEM (n = 6, ***P < 0.001). P values were determined by unpaired 2-tailed Student’s t test.
ML372 improves motor unit architecture.
SMA is characterized by deterioration of anterior horn motor neurons and atrophy of the muscle cells that they innervate. To assess the ability of ML372 to improve the structural arrangement of the motor unit, we performed histological examination of muscle (tibialis anterior [TA]) and spinal cord (lumbar segments) in unaffected, vehicle-, and ML372-treated SMA mice sacrificed at PND13 (n = 5, each cohort). Figure 4A shows images of H&E-stained TA muscle. ML372 significantly increased the mean myofiber diameter from 11.1 μm in SMA mice to 16.8 μm in ML372-treated mice (Figure 4B). Similarly, myofiber number was also increased (1,690 to 2,411 myofibers per section) with ML372 treatment (Figure 4C). Loss of motor neurons is very rapid in the early postnatal stages of this SMA mouse model, with limited motor neuron loss after PND7 (24, 25). Nissl staining of lumbar spinal cord segments (Figure 4D) showed that ML372 increased motor neuron size (Figure 4E, mean somatic diameter of 37.2 μm compared with 32.9 μm in vehicle-treated animals), with no change in the number of motor neurons (Figure 4F). These data are consistent with previous studies demonstrating that SMN-dependent interventions started after disease manifestations may improve the motor unit pathology but not stop motor unit loss (13, 26–28).
Figure 4. ML372 increases myofiber size and number and augments ventral horn neuron size.

SMNΔ7 spinal muscular atrophy (SMA) mice were treated with vehicle or ML372 from postnatal day 5 (PND5) to PND9. Black, unaffected; Red, SMNΔ7 SMA mice treated with vehicle; Blue, SMNΔ7 SMA mice treated with ML372. (A) H&E staining of tibialis anterior (TA) muscles from unaffected, SMA, and drug-treated SMA mice. Scale bars: 100 μm. (B) Mean myofiber diameter was compared in SMA and ML372-treated SMA mice. The results are indicated as the mean ± SEM (n = 20, ****P < 0.0001). (C) Myofiber numbers were counted in the 3 groups. The analysis is shown as the mean ± SEM (n = 20, *P < 0.05). (D) Nissl-stained cross-sections of spinal cord showing ventral horn neurons. Scale bars: 100 μm (upper and lower panels magnification ×10 and ×20, respectively). (E) The size of motor neurons was analyzed in the 3 groups and the mean ventral horn neuron size was compared in SMA mice treated with vehicle or ML372. The data are shown as the mean ± SEM (n = 20, *P < 0.05). (F) The number of motor neurons was determined in the 3 groups, and the analysis is shown as the mean ± SEM (n = 20). P values were determined by unpaired 2-tailed Student’s t test.
ML372 rescues motor function and extends lifespan of SMNΔ7 SMA mice. Affected SMNΔ7 mice live for an average of 14 days and develop clear SMA disease phenotypes by PND5, which include underweight and delayed righting reflex (29, 30). We performed a blinded study in litters that were randomized into vehicle- and compound-treated groups. In weight-matched litters (2.99 ± 0.24 g versus 2.94 ± 0.26 g), the ML372-treated cohort displayed substantially greater weight gain compared with vehicle-treated controls (maximal weight of 5.63 g versus 3.87 g) (Figure 5A). Weight gain in compound-treated littermates has previously been shown to correlate with the potential efficacy of a compound under therapeutic consideration for SMA (11). Moreover, the onset of weight loss was delayed in ML372-treated animals. To evaluate motor function, we assessed the righting reflex (i.e., the average time for mice to right themselves once placed on their backs). The righting times of ML372-treated animals significantly improved starting at PDN10 with an average righting time of 8.4 ± 1.8 seconds compared with 14.8 ± 1.9 seconds in the vehicle-treated cohort. By PND14, many ML372-treated animals were able to right in as little as 4.6 seconds, while all vehicle-treated pups took more than 14 seconds (Figure 5B). ML372 also markedly improved survival (18 ± 0.8 days, n = 12) compared with vehicle-treated controls (14 ± 0.4 days, n = 11). (Figure 5C). Together, these results demonstrate that ML372 significantly improves the motor function and survival rate of SMNΔ7 mice.
Figure 5. ML372 rescues motor function and improves life span.
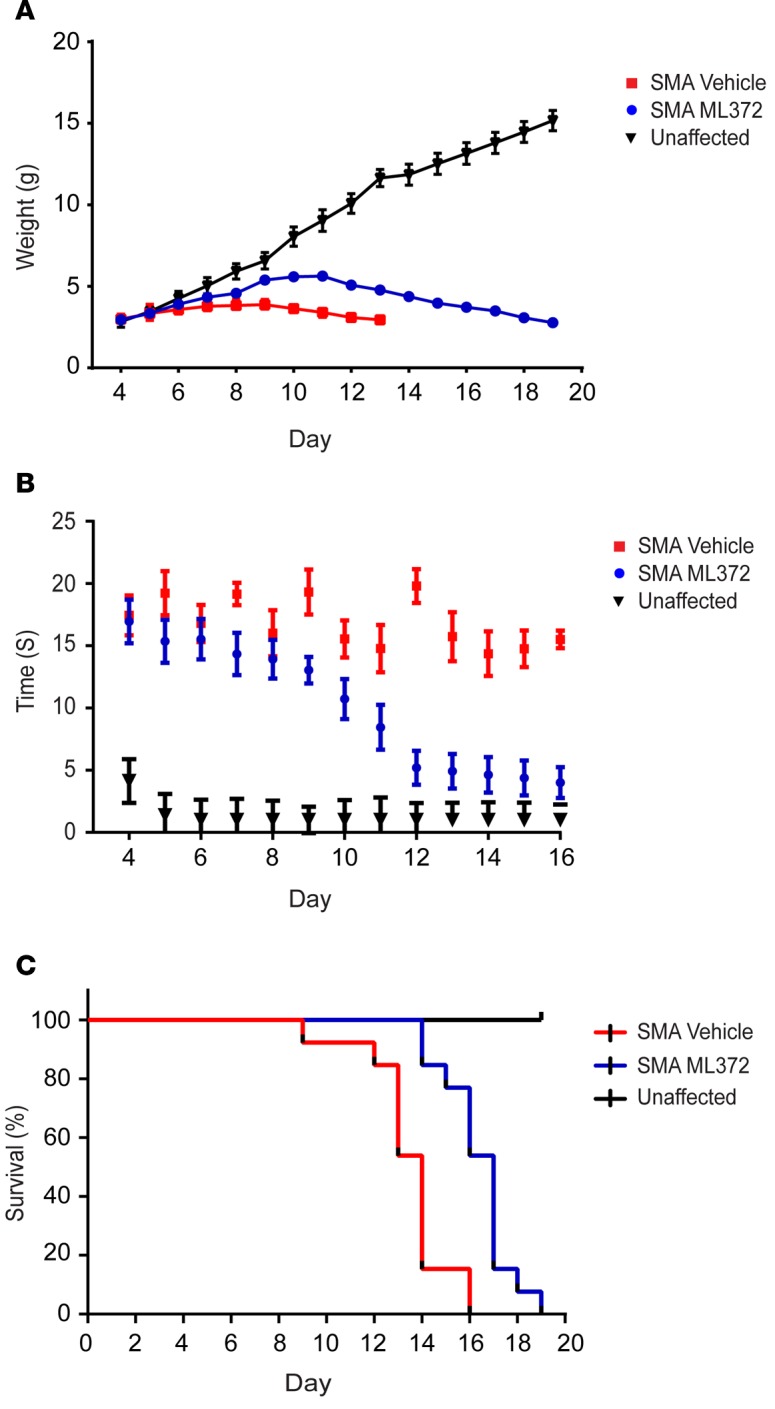
Blinded study was performed in litters that were randomized into vehicle- and compound-treated groups. Red, SMNΔ7 spinal muscular atrophy (SMA) mice treated with vehicle; Blue, SMNΔ7 SMA mice treated with ML372. (A) SMNΔ7 SMA mice were weighed to assess changes in body weight. (B) Motor function was determined by measuring the time to right in unaffected (Black), vehicle-, or ML372-treated SMNΔ7 SMA mice. (C) Kaplan-Meier survival analysis of SMNΔ7 SMA mice treated with vehicle or ML372; P < 0.003, log-rank test.
Discussion
Therapeutic benefit of selectively targeting increasing SMN protein stability.
The fundamental problem in SMA patients is insufficient SMN protein expression. Therefore, therapeutic solutions that increase the amount of SMN protein remain at the forefront of SMA research. Many of the strategies attempt to increase SMN synthesis by increasing mRNA production from the SMN2 gene. Our group has approached this problem from the opposite end of the spectrum — targeting protein degradation. In proof of principle experiments, we previously demonstrated that inhibiting proteasome-mediated degradation improved SMN protein levels in SMA patient–derived cells and model mice (9, 13, 31); however, the ubiquitous requirement of the proteasome in maintaining cellular homeostasis precludes further clinical consideration of proteasome inhibitors. Nevertheless, specific targeting of the SMN degradation pathway remains a viable option for treating SMA.
Protein degradation is a multistep process that requires labeling of the proteins for degradation followed by the selective breakdown of labeled proteins. Proteins are typically marked for degradation by the addition of ubiquitin moieties to sites within the protein by E3 ligases. Unlike the ubiquitous proteasome, the E3 ligase–protein interaction is more specific and can be targeted with fewer off-target effects. This type of approach had been limited by 2 factors — the identity of the E3 ligase that targets SMN for degradation and inhibitors for that ligase. We recently showed that Mib1 is an E3 ligase that selectively targets SMN for degradation. Now in the present study, we show that a first-in-class Mib1 inhibitor, ML372, inhibits SMN ubiquitination, increases SMN stability, and abrogates the disease phenotype in a mouse model of SMA.
ML372 represents a unique therapeutic approach.
The objective of all therapies is to restore SMN protein expression in cells where it is required. SMN expression can be restored through various mechanisms. Compounds targeting increased expression of SMN protein from the SMN2 gene with HDAC inhibitors, antisense oligonucleotides (ASOs), quinazoline, and tetracycline derivatives have all been employed in animal models of the disease (13, 27, 32–41). In the present study, ML372 induced an ~2-fold increase in SMN protein levels in all tissues analyzed and extended survival by 28%. These results are comparable with results with the HDAC inhibitor trichostatin A (TSA), which increases SMN levels through induction of the SMN2 gene (13, 35). Similarly, small molecules and ASOs that modify SMN2 splicing cause a similar 2-fold increase in SMN protein levels (42, 43). Therefore, ML372 seems to be as effective as these other treatments at restoring SMN protein levels.
The goal of any SMA therapeutic is to extend survival and improve motor function. We show that ML372 markedly increased muscle and motor neuron pathology. The modest 28% increase in survival is less than optimal, especially when compared with reports that SMN2 splicing modifiers provide a more substantial survival benefit. Because ML372 and splicing modifiers have similar effects on SMN protein levels, this difference in survival benefit can most likely be attributed to the timing of treatment. When splicing modifiers are given early in the disease progression (PND1) the survival benefit is significantly greater than when splicing-correcting ASOs are given at PND5 or PND7 (42). Similarly, we observed improved survival when TSA was administered at PND2 compared with PND5 (27). In this study we delivered ML372 at PND5 — when the mice have already developed symptoms — to more faithfully replicate the typical clinical presentation.
ML372 may synergize with other approaches.
It would be ideal if a single compound or treatment could be identified that would effectively reverse the course of SMA, and most studies (like this one) are focused on a monotherapeutic approach to the disease. HDAC inhibitors, ASOs, quinazoline, and tetracycline derivatives all target the same process — to increase expression of SMN from the SMN2 gene. The approach that we have employed targets an entirely different biological process. We cannot rule out the possibility that the unique mechanism of action employed by ML372 will prove to be superior to the other approaches. Given that the increase in SMN protein levels in ML372-treated animals is on par with the other approaches, this may not be likely. However, because ML372 targets the degradation pathway instead of the synthetic pathway, it is likely that ML372 will have additive, or even synergistic, effects when combined with splicing modifiers or HDAC inhibitors. Indeed, we have provided initial evidence that simultaneously increasing SMN expression and inhibiting its degradation has synergistic benefit in SMA model mice (35). Therefore, ML372 has the potential to enhance the therapeutic efficacy of most all other options currently under consideration for treating SMA.
Targeting the ubiquitin/proteasome system with E3 ligase inhibitors in other diseases.
Developing specific inhibitors of the ubiquitin proteasome system is difficult because there are no clear ubiquitination consensus domains or motifs. The majority of E3 ligases derive specificity from a complex of protein scaffolds, suggesting that selective inhibitors would have to target protein-protein interactions rather than an enzyme active site. However, small-molecule libraries are not usually replete with compounds that lend themselves to blocking these types of protein-protein interactions. With recent advances in our understanding of the E3-protein interface, nutlin was identified as a potent and selective small-molecule inhibitor of the p53-Mdm2 interaction (44, 45). Nutlin stabilizes p53 by inhibiting its MDM2-dependent ubiquitination and is currently under clinical investigation. More recently a small molecule was identified that blocks the interaction between E3 ligase component SKP2 and the cell cycle regulator p27KIP1. The lead compound was shown to selectively block ubiquitination of SCFSKP2 target proteins, but not other SCF E3 ligase targets (46, 47). Our current study provides new evidence and advances the idea that selective regulation of protein turnover can be a powerful therapeutic tool for treating human disorders.
In conclusion, we have identified a new chemical scaffold, represented by ML372, as potentially novel modulators of SMN protein stability. The lead compound ML372 possesses good potency, ADME (absorption, distribution, metabolism, and excretion) physical properties, tolerance, and CNS penetration. ML372 has demonstrated its ability to elevate SMN protein levels both in vitro and in vivo. Furthermore, ML372 is shown to be able to increase the body weight, rescue motor function, and prolong lifespan in a severe model of SMA, validating Mib1 as a drug target for SMA and making this series a promising chemotype for the development of potential therapeutic agents for the treatment SMA.
Methods
General chemical methods.
All air- or moisture-sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Anhydrous solvents such as dichloromethane (DCM), N,N-dimethylformamide (DMF), acetonitrile, methanol, tetrahydrofuran (THF), and triethylamine (TEA) were obtained from Sigma-Aldrich. Preparative purification was performed on a Waters semipreparative HPLC. The column used was a Phenomenex Luna C18 (5 micron, 30 × 75 mm) at a flow rate of 45.0 ml/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10% to 50% acetonitrile over 8 minutes was used during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies). Method 1: A 7-minute gradient of 4% to 100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8-minute run time at a flow rate of 1.0 ml/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50°C. Method 2: A 3-minute gradient of 4% to 100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5-minute run time at a flow rate of 1.0 ml/min. A Phenomenex Gemini Phenyl column (3 micron, 3 × 100 mm) was used at a temperature of 50°C. Purity determination was performed using an Agilent Diode Array Detector in both Method 1 and Method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical shifts are reported in ppm with undeuterated solvent (DMSO at 2.49 ppm) as the reference for DMSO-d6 solutions. All analogs for assays have purity greater than 95% based on both analytical methods. High-resolution MS was recorded on an Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
Pharmacokinetics.
Pharmacokinetics of ML372 was determined in male FVB mice after single i.p. administration of 50 mg/kg. Mice (n = 33, body weight 22–30 g from Charles River Labs) were housed at the centralized animal facilities in the NIH with a 12-hour light/dark cycle. The animals had free access to water and food. All experimental procedures were approved by the Animal Care and Use Committee (ACUC) of the NIH Division of Veterinary Resources (DVR). The formulation used in this study was 30% 2-hydoxypropyl-β-cyclodextrin (HP-β-CD) in water, and the dosing solution was freshly prepared on the day of dosing. All animals were dosed i.p. with an injection volume of 10 ml/kg. The blood samples (~200 μl) were collected in K2EDTA tubes prior to the dosing (t = 0) and at 0.083, 0.25, 0.5, 1, 2, 4, 8, 12, 16, and 24 hours after drug administration. Plasma samples were harvested after centrifugation at 5,000 g for 10 minutes. Brain samples were collected from each mouse after blood collection. All plasma and brain samples were stored at –80°C until the analysis. The drug concentrations in plasma and brain homogenate were determined with a qualified LC/MS/MS method.
Pharmacokinetics of ML372 in plasma and brain were calculated using the noncompartmental approach (Model 200) of the pharmacokinetic software Phoenix WinNonlin, version 6 (Certara). The area under the plasma concentration-time curve (AUC) was calculated using the linear trapezoidal method. The slope of the apparent terminal phase was estimated by log linear regression using at least 3 data points and the terminal rate constant (λ) was derived from the slope. AUC0–¥ was estimated as the sum of the AUC0–t (where t is the time of the last measurable concentration) and Ct/λ. The apparent terminal half-life (t½) was calculated as 0.693/λ.
Cell culture and compound treatment.
HEK-293T cells and SMA fibroblast 3813 cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS and penicillin/streptomycin/glutamine (Invitrogen) and maintained at 37°C in a humidified 5% CO2 atmosphere. For compound treatment, cells seeded into 6-well plates were treated with different concentrations of ML372 at different times, as indicated.
RNA extraction and quantification.
RNA extraction and real-time quantitative PCR (qPCR) were performed as previously described (48). RNA was extracted from subconfluent cell cultures using TRIzol reagent (Invitrogen). Total RNA (1 μg) was reverse transcribed to cDNA using the Taqman RT reagent kit (Applied Biosystems). qPCR was performed using an ABI Stepone Plus detection system. The reaction was carried out in triplicate samples containing TaqMan universal PCR MasterMix (Applied Biosystems), target primers (300 nM), fluorescent probe (200 nM), and 1 μl of transcribed cDNA (6-fold diluted). Taqman primers 5′ end labeled with 6-carboxyfluorescein (FAM) for SMN and PGK1 (housekeeping gene) were purchased from Applied Biosystems. PCR product amplification was continuously monitored using the Sequence Detection System software version 1.7 (Applied Biosystems). Triplicate cycle threshold (Ct) values were averaged and normalized to an average PGK1 Ct value to calculate the ΔCt. The Δ(ΔCt) was determined by subtracting the control ΔCt value from the experimental ΔCt value. Fold changes were expressed as 2–Δ(ΔCt).
Transfection, Western blotting, immunoprecipitation, and pull-down assays.
Cells were plated into a 6-well culture plate and transiently transfected with 1 μg of plasmid DNA per well using Lipofectamine 3000 reagent (Roche Applied Science) according to the manufacturer’s protocol. Cells were harvested 48 hours after transfection.
Transfected cells were lysed in RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, 0.1% SDS, pH 8.0) and for immunoprecipitation experiments, cell lysates were clarified by centrifugation and precleared with pan-mouse immunoglobulin G (IgG) Dynabeads (Invitrogen) for 1 hour at 4°C. The lysates were then incubated with 1 μg of antibody for 2 hours to overnight and then incubated with pan-mouse IgG Dynabeads for 1 hour on a rotator at 4°C. The beads were washed with NP-40 buffer 2 to 3 times and boiled in SDS gel-loading buffer. Eluted proteins were run in SDS-PAGE for Western blotting. Western blot analysis was carried out essentially as previously described (48). Briefly, samples were resolved in 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore). After blocking with 5% milk in 1% Tween 20/PBS, membranes were incubated with an anti-HA antibody (Sigma-Aldrich, clone HA-7). As secondary antibodies, we used anti-rabbit antibody conjugated with horseradish peroxidase diluted 1:4,000 (Bio-Rad). Bands were visualized using a chemiluminescence detection system (Syagene). Densitometric analysis of the protein bands was carried out using ImageJ software (NIH). For pull-down assays, 1 μg recombinant glutathione S-transferase–tagged Mib1 (GST-Mib1) (Abnova) immobilized on Glutathione Sepharose beads (GE Healthcare) was incubated with recombinant SMN purchased from Enzo Life Sciences at 4°C on a rotator overnight in the presence or absence of ML372. Recombinant GST was used as a control. Beads were spun down, washed 4 times with NP-40 buffer, and boiled in SDS gel-loading buffer for Western blot analysis.
Ubiquitination assay.
In vitro ubiquitination assays were performed as previously described (9). Briefly, recombinant SMN protein was incubated in a reaction mixture containing 10× ubiquitination buffer, 2 mM DTT, 10× ATP regeneration solution, an E1 ubiquitin-activating enzyme, the ubiquitin-conjugating enzyme UBCH5B, and recombinant Mib1 for 1 hour at 37°C. All reagents except recombinant Mib1 were procured from Enzo Life Sciences. Recombinant Mib1 was purchased from (OriGene Technologies). SMN ubiquitination reactions were analyzed by Western blotting, using antibody against polyubiquitin (FK1; Enzo Life Sciences).
Proteasome activity assay.
To determine if ML372 impairs proteasome catalytic activity, proteasome activity assays were performed as previously described (31). Briefly, 1.25 μg of purified h20S proteasome was incubated with DMSO, ML372 (0.3 μM), or epoxomicin (10 μM, BioMol) at 37°C for 30 minutes. Fluorogenic substrate was then added to samples to measure chymotrypsin-like activity (Suc-Leu-Leu-Val-Tyr-AMC, G1100, Ubiquitin-Proteasome Biotechnologies). 20S reaction buffer (20 mM Tris-Cl, 1 mM EDTA, 1 mM NaN3, 1 mM DTT, pH 7.5) was added to a final volume of 100 μl and samples were transferred to a black 96-well plate. All experiments were conducted in triplicate. The plate was then read at 355ex/460em at 37°C with gain set at 783 (based on pilot data) with a FLUOROstar Omega (BMG Labtech. Data were collected immediately after substrate addition and continued for 30 minutes. All data were automatically corrected based on blank wells that contained only the fluorescent substrate and 20S reaction buffer.
Pulse-chase protein labeling.
Pulse-chase analysis was performed as previously described (9). Briefly, HEK-293T cells transiently expressing myc-tagged full-length SMN or SMNΔ7 and in the presence or absence of 0.3 μM ML372 were incubated in cysteine-methionine–free medium (Sigma-Aldrich) for 2 hours; this was followed by incubation in cysteine-methionine–free medium containing 100 μCi of [35S]-labeled cysteine-methionine (GE Healthcare) for 1 hour. After labeling, the cells were washed once with culture medium containing a 10-fold excess of unlabeled methionine and cysteine (5 mM each) and then incubated further in the same medium. The cells were collected at the indicated time points and processed for immunoprecipitation with an anti-SMN antibody (BD Transduction Laboratories, clone 8). Immunoprecipitations were carried out for 6 hours at 4°C with antibodies bound to protein G-Sepharose (Sigma-Aldrich). After 3 washes with lysis buffer (100 mM NaCl, 10 mM Tris-HCl, pH 7.4, 2.5 mM MgCl2, 0.1% NP-40, and protease [Roche]) and phosphatase inhibitor cocktails [Sigma-Aldrich]), bound proteins were eluted by boiling in SDS-PAGE sample buffer. Immunocomplexes were resolved by 10% SDS-PAGE. The gels were dried and exposed to a phosphorimager screen, and the signal was visualized with a Storm phosphorimager system (Molecular Dynamics).
SMA mice drug treatment and survival studies.
The original breeding pairs for the SMA mice (Smn+/–SMN2+/+SMNΔ7+/+) on the FVB background were purchased from Jackson Laboratories. The breeding colony was maintained by interbreeding Smn+/–SMN2+/+SMNΔ7+/+ mice, and offspring were genotyped using PCR assays on tail DNA. The primers used for genotyping were 5′-CAAACACCTGGTATGGTCAGTC-3′ and 5′-GCACCACTGCACAACAGCCTG-3′ for the SMN2 transgene, 5′-GCCTGCGATGTCGGTTTCTGTGAGG-3′ and 5′-CCAGCGCGGATCGGTCAGACG-3′ for the mouse knockout allele, and 5′-GTGTCTGGGCTGTAGGCATTGC-3′ and 5′-GGCTGTGCCTTTTGGCTTATCTG-3′ for the Smn gene (13).
ML372 was dissolved in 30% w/v HP-β-CD/water to a concentration of 3 mg/ml and pups at PND5 were administered i.p., twice daily at 50 mg/kg using a 33-guage needle. Control animals received equal volumes of vehicle.
For biochemical studies, the mice were anesthetized with isoflurane and sacrificed by cervical dislocation as described (13, 27, 35). Brains, spinal cords, and distal hind-limb skeletal muscle were dissected and flash-frozen in liquid nitrogen. The tissues were stored at –80°C.
For 1-time measures, pups were weighed and tested for their ability to right themselves (16). Righting time was defined as the average of 2 trials of the time required for a pup to turn over after being placed on its back (maximum 30 seconds). Mice that lost 30% of their body weight and were unable to right themselves were euthanized.
For survival studies, litters were maintained and kept with the dam until the clinical endpoint.
Compound level analysis in SMA brain tissues.
Compound level analysis in SMA brain tissues was performed at ChemoGenics under a fee-for-contract service. SMA brain tissue samples were mixed with PBS (pH 7.2) (1:1 ratio) and the samples were homogenized using a pestle-type homogenizer. The homogenates were treated with 3 volumes of acetonitrile to precipitate the proteins, and the supernatants were analyzed by LC/MS/MS. A working dilution of test agent in acetonitrile at 50 times the final concentration was prepared and serially diluted samples were prepared. These samples were diluted 50-fold into plasma or blank tissue extract and processed as above. The signal was optimized for each compound by electrospray ionization (ESI) positive or negative ionization mode. An MS2 SIM scan was used to optimize the precursor ion and a product ion analysis was used to identify the best fragment for analysis and to optimize the collision energy. Samples were analyzed by LC/MS/MS using an ABI3000 mass spectrometer coupled with a Shimadzu HPLC and a Sil-HTc chilled autosampler, all controlled by Analyst software (ABI), after separation in a C18 reverse-phase HPLC column (Agilent, Waters, or equivalent). Mobile phase was 10 mM ammonium acetate as mobile phase A and acetonitrile as mobile phase B.
Pathological analysis.
Mice were transcardially perfused with 4% paraformaldehyde. Lumbar spinal cords and distal hind limbs were dissected and postfixed overnight. Hind limb tissues were decalcified, embedded in paraffin, and cross-sectioned at the midpoint of the muscle. All analyses were done blinded. Sections (10 μm) were mounted on slides and stained with H&E. Digital images were captured using a Zeiss Axiovert 100M microscope and analyzed with NIH ImageJ software for total TA cross-sectional area (original magnification, ×20), total TA myofiber number (original magnification, ×20), and myofiber diameter (original magnification, ×20). Myofiber diameter was determined by measuring the largest diameter of at least 300 neighboring myofibers per animal. Paraffin-embedded lumbar spinal cord was serially sectioned in 10-μm steps, mounted on slides, and stained with Nissl stain. Images of 15 contiguous sections, 150 μm apart (original magnification, ×20) were analyzed with NIH ImageJ software. The diameter and number of all neurons greater than 25 μm in the region below a line drawn horizontally at the level of the spinal canal were determined.
Statistics.
All statistical analyses were done using GraphPad Prism v6 (GraphPad Software, Inc.). Data were analyzed using a 2-way ANOVA. A P value less than 0.05 was considered statistically significant.
Study approval.
Studies were approved by the NINDS Animal Care Committee and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. FVB mice were purchased from Jackson Laboratories.
Author contributions
MBA, BGB, KHF, NS, and JJM designed and performed experiments, analyzed the data, and contributed to the writing of the manuscript. JJC and EJA analyzed the data and provided reagents/materials. KEM, JX, JN, ST, WJ, and CA performed experiments and analyzed data.
Supplementary Material
Acknowledgments
This work was supported by grants from an Emerging Investigator award from Fight SMA and the Gwendolyn Strong Foundation (to BGB). KHF is supported by intramural research funds from NINDS (1ZIANS003038-09).
Footnotes
Conflict of interest: The authors declare that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(19):e88427. doi:10.1172/jci.insight.88427.
Contributor Information
Jingbo Xiao, Email: Jingbo.Xiao@fda.hhs.gov.
Jonathan Nofziger, Email: jhnofziger@gmail.com.
Noel Southall, Email: noel.southall@nih.gov.
Wei Zheng, Email: wei.zheng@nih.gov.
Marc Ferrer, Email: marc.ferrer@nih.gov.
Jonathan J. Cherry, Email: jcherry@ranarx.com.
Amy Wang, Email: AMY.WANG@NIH.GOV.
Xin Xu, Email: xin.xu3@nih.gov.
References
- 1.Sugarman EA, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20(1):27–32. doi: 10.1038/ejhg.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Awano T, Kim JK, Monani UR. Spinal muscular atrophy: journeying from bench to bedside. Neurotherapeutics. 2014;11(4):786–795. doi: 10.1007/s13311-014-0293-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D’Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis. 2011;6:71. doi: 10.1186/1750-1172-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51(2):157–167. doi: 10.1002/mus.24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 6.Monani UR, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 7.Lorson CL, et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19(1):63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 8.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burnett BG, Muñoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of SMN protein stability. Mol Cell Biol. 2009;29(5):1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carré A, Empey C. Review of spinal muscular atrophy (SMA) for prenatal and pediatric genetic counselors. J Genet Couns. 2016;25(1):32–43. doi: 10.1007/s10897-015-9859-z. [DOI] [PubMed] [Google Scholar]
- 11.Seo J, Howell MD, Singh NN, Singh RN. Spinal muscular atrophy: an update on therapeutic progress. Biochim Biophys Acta. 2013;1832(12):2180–2190. doi: 10.1016/j.bbadis.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burnett BG, Crawford TO, Sumner CJ. Emerging treatment options for spinal muscular atrophy. Curr Treat Options Neurol. 2009;11(2):90–101. doi: 10.1007/s11940-009-0012-x. [DOI] [PubMed] [Google Scholar]
- 13.Avila AM, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117(3):659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passini MA, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3(72):72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iascone DM, Henderson CE, Lee JC. Spinal muscular atrophy: from tissue specificity to therapeutic strategies. F1000Prime Rep. 2015;7:04. doi: 10.12703/P7-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le TT, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14(6):845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 17.Ling KK, Gibbs RM, Feng Z, Ko CP. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2012;21(1):185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drug Discovery. Cure SMA. http://www.curesma.org/research/our-strategy/drug-discovery/ Accessed October 21, 2016.
- 19.Xiao J, et al. Discovery, synthesis, and biological evaluation of novel SMN protein modulators. J Med Chem. 2011;54(18):6215–6233. doi: 10.1021/jm200497t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon DY, et al. The E3 ubiquitin ligase mind bomb 1 ubiquitinates and promotes the degradation of survival of motor neuron protein. Mol Biol Cell. 2013;24(12):1863–1871. doi: 10.1091/mbc.E13-01-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han KJ, et al. Ubiquitin-specific protease 9x deubiquitinates and stabilizes the spinal muscular atrophy protein-survival motor neuron. J Biol Chem. 2012;287(52):43741–43752. doi: 10.1074/jbc.M112.372318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choe EA, et al. Neuronal morphogenesis is regulated by the interplay between cyclin-dependent kinase 5 and the ubiquitin ligase mind bomb 1. J Neurosci. 2007;27(35):9503–9512. doi: 10.1523/JNEUROSCI.1408-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung SO, Ro HS, Kho BH, Shin YB, Kim MG, Chung BH. Surface plasmon resonance imaging-based protein arrays for high-throughput screening of protein-protein interaction inhibitors. Proteomics. 2005;5(17):4427–4431. doi: 10.1002/pmic.200500001. [DOI] [PubMed] [Google Scholar]
- 24.d’Errico P, et al. Selective vulnerability of spinal and cortical motor neuron subpopulations in delta7 SMA mice. PLoS One. 2013;8(12):e82654. doi: 10.1371/journal.pone.0082654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong L, et al. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29(3):842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lutz CM, et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest. 2011;121(8):3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narver HL, et al. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol. 2008;64(4):465–470. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robbins KL, Glascock JJ, Osman EY, Miller MR, Lorson CL. Defining the therapeutic window in a severe animal model of spinal muscular atrophy. Hum Mol Genet. 2014;23(17):4559–4568. doi: 10.1093/hmg/ddu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gogliotti RG, Quinlan KA, Barlow CB, Heier CR, Heckman CJ, Didonato CJ. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci. 2012;32(11):3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foust KD, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28(3):271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Foran E, et al. CNS uptake of bortezomib is enhanced by P-glycoprotein inhibition: implications for spinal muscular atrophy. Neurobiol Dis. 2016;88:118–124. doi: 10.1016/j.nbd.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riessland M, et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19(8):1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 33.Osman EY, et al. Morpholino antisense oligonucleotides targeting intronic repressor Element1 improve phenotype in SMA mouse models. Hum Mol Genet. 2014;23(18):4832–4845. doi: 10.1093/hmg/ddu198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hastings ML, et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci Transl Med. 2009;1(5):5ra12. doi: 10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwon DY, Motley WW, Fischbeck KH, Burnett BG. Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum Mol Genet. 2011;20(18):3667–3677. doi: 10.1093/hmg/ddr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farooq F, et al. Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Hum Mol Genet. 2013;22(17):3415–3424. doi: 10.1093/hmg/ddt191. [DOI] [PubMed] [Google Scholar]
- 37.Dominguez E, et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2011;20(4):681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 38.Naryshkin NA, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345(6197):688–693. doi: 10.1126/science.1250127. [DOI] [PubMed] [Google Scholar]
- 39.Palacino J, et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol. 2015;11(7):511–517. doi: 10.1038/nchembio.1837. [DOI] [PubMed] [Google Scholar]
- 40.Valori CF, et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;2(35):35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- 41.Passini MA, et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010;120(4):1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hua Y, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478(7367):123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Narayanan U, Ospina JK, Frey MR, Hebert MD, Matera AG. SMN, the spinal muscular atrophy protein, forms a pre-import snRNP complex with snurportin1 and importin beta. Hum Mol Genet. 2002;11(15):1785–1795. doi: 10.1093/hmg/11.15.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel S, Player MR. Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin Investig Drugs. 2008;17(12):1865–1882. doi: 10.1517/13543780802493366. [DOI] [PubMed] [Google Scholar]
- 45.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol. 2012;19(12):1515–1524. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan CH, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154(3):556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abera MB, Kazanietz MG. Protein kinase Cα mediates erlotinib resistance in lung cancer cells. Mol Pharmacol. 2015;87(5):832–841. doi: 10.1124/mol.115.097725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.