

Abstract
Parkinson disease (PD) is the second most common neurodegenerative disorder after Alzheimer disease, whereas Gaucher disease (GD) is the most frequent lysosomal storage disorder caused by homozygous mutations in the glucocerebrosidase (GBA1) gene. Increased risk of developing PD has been observed in both GD patients and carriers. It has been estimated that GBA1 mutations confer a 20‐ to 30‐fold increased risk for the development of PD, and that at least 7–10% of PD patients have a GBA1 mutation. To date, mutations in the GBA1 gene constitute numerically the most important risk factor for PD. The type of PD associated with GBA1 mutations (PD‐GBA1) is almost identical to idiopathic PD, except for a slightly younger age of onset and a tendency to more cognitive impairment. Importantly, the pathology of PD‐GBA1 is identical to idiopathic PD, with nigral dopamine cell loss, Lewy bodies, and neurites containing alpha‐synuclein. The mechanism by which GBA1 mutations increase the risk for PD is still unknown. However, given that clinical manifestation and pathological findings in PD‐GBA1 patients are almost identical to those in idiopathic PD individuals, it is likely that, as in idiopathic PD, alpha‐synuclein accumulation, mitochondrial dysfunction, autophagic impairment, oxidative and endoplasmic reticulum stress may contribute to the development and progression of PD‐GBA1. Here, we review the GBA1 gene, its role in GD, and its link with PD.
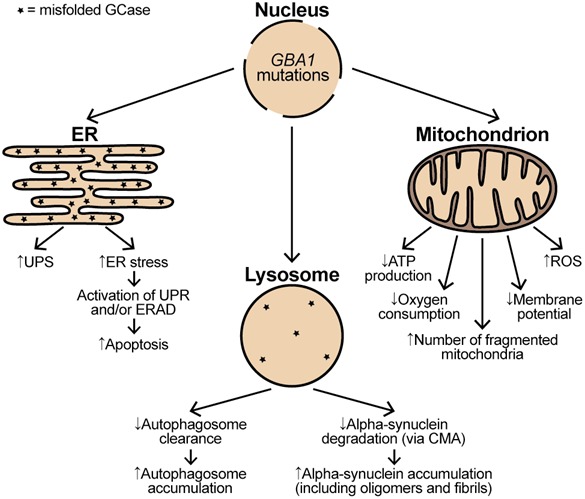
The impact of glucocerebrosidase 1 (GBA1) mutations on functioning of endoplasmic reticulum (ER), lysosomes, and mitochondria. GBA1 mutations resulting in production of misfolded glucocerebrosidase (GCase) significantly affect the ER functioning. Misfolded GCase trapped in the ER leads to both an increase in the ubiquitin–proteasome system (UPS) and the ER stress. The presence of ER stress triggers the unfolded protein response (UPR) and/or endoplasmic reticulum‐associated degradation (ERAD). The prolonged activation of UPR and ERAD subsequently leads to increased apoptosis. The presence of misfolded GCase in the lysosomes together with a reduction in wild‐type GCase levels lead to a retardation of alpha‐synuclein degradation via chaperone‐mediated autophagy (CMA), which subsequently results in alpha‐synuclein accumulation and aggregation. Impaired lysosomal functioning also causes a decrease in the clearance of autophagosomes, and so their accumulation. GBA1 mutations perturb normal mitochondria functioning by increasing generation of free radical species (ROS) and decreasing adenosine triphosphate (ATP) production, oxygen consumption, and membrane potential. GBA1 mutations also lead to accumulation of dysfunctional and fragmented mitochondria.
This article is part of a special issue on Parkinson disease .
Keywords: alpha‐synuclein, Gaucher disease, glucocerebrosidase 1 (GBA1), lysosome, mitochondria, Parkinson disease
Abbreviations used
- Alu
Arthrobacter luteus
- ATG
methionine
- BiP
binding immunoglobulin protein
- CBE
condruitol β‐epoxide
- CHOP
C/EBP homologous protein
- CLEAR
coordinated lysosomal expression and regulation
- CMA
chaperone‐mediated autophagy
- CNS
central nervous system
- DJ‐1
Parkinson protein 7
- ER
endoplasmic reticulum
- ERAD
ER‐associated degradation
- FDA
Food and Drug Administration
- GD
Gaucher disease
- GBA1
glucocerebrosidase
- GCase
glucocerebrosidase enzyme
- LIMP2
lysosomal membrane protein 2
- LRRK2
leucine‐rich repeat kinase 2
- MPR
mannose‐6‐phosphate receptor
- mRNA
messenger RNA
- PARK2
parkin RBR E3 ubiquitin protein ligase
- PCR
polymerase chain reaction
- PC12
pheochromocytoma
- PD
Parkinson disease
- PD‐GBA1
Parkinson‐GBA1
- pH
power of hydrogen
- PINK1
PTEN‐induced putative kinase 1
- PI4K
phosphatidylinositol 4‐kinase
- PTEN
phosphatase and tensin homolog
- SNCA
alpha‐synuclein
- TFEB
transcription factor EB
- UPR
unfolded protein response
Glucocerebrosidase 1 gene
The human glucocerebrosidase 1 (GBA1) gene maps to the 1q21 region and consists of 11 exons within a 7.6 kb sequence (Horowitz et al. 1989; Cormand et al. 1997). Approximately, 16 kb downstream of GBA1 lies an almost identical sequence consisting of 11 exons within a 5.7 kb sequence (Horowitz et al. 1989; Sorge et al. 1990). Despite large deletions of Arthrobacter luteus sequences flanked by direct repeats in the introns, this pseudogene shares 96% coding sequence similarity with the functional gene (Horowitz et al. 1989). As some mutations found in the functional gene can naturally occur within the pseudogene, the presence of the highly homologous pseudogene in close proximity to the functional gene complicates molecular identification of known and novel GBA1 mutations. Therefore, a method allowing the GBA1 gene to be distinguished from the pseudogene is crucial for reliable molecular diagnosis. One such method utilizes a long‐template polymerase chain reaction (PCR) where genomic fragments of differing lengths from both the gene and the pseudogene are amplified simultaneously, before being purified and subsequently used for mutation identification (Tayebi et al. 1996).
Northern blot analysis revealed the existence of at least two GBA1 mRNAs (2.2 and 2.6 kb in length) that arise as a result of alternate polyadenylation sites (Horowitz et al. 1989).The GBA1 mRNA has two in‐frame methionine start codons located in exons 1 and 2, and both methionines are translated to produce functional protein in vitro (Sorge et al. 1985, 1987a). The protein using the start codon in exon 1 contains a 39‐amino acid signal peptide, while the protein arising from the start codon in exon 2 contains only a 19‐amino acid signal peptide. Both are processed to a 496‐amino acid long mature protein after their signal peptide sequences are removed, which is a common occurrence for secreted proteins like glucocerebrosidase (Sorge et al. 1987b).
GBA1 encodes glucocerebrosidase (GCase), a lysosomal enzyme that catalyses the hydrolysis of glycolipid glucocerebroside to ceramide and glucose (Beutler 1992). Glucocerebrosidase is ubiquitously expressed in all types of tissues (The Human Protein Atlas). As with other lysosomal proteins, GCase is synthetized in the rough endoplasmic reticulum (ER). However, in contrast to the majority of other lysosomal proteins, transport of GCase from the ER to lysosomes is not mediated by mannose‐6‐phosphate receptors, but via lysosomal membrane protein 2 (LIMP2). GCase binds to a coiled‐coil domain in the lumenal region of LIMP2 at neutral pH of the ER, and both proteins persist together through the Golgi apparatus and endosomes into the lysosome, where acidic pH facilitates their dissociation (Reczek et al. 2007). Sequential traversing of GCase and the LIMP2 complex is dependent on two phosphatidylinositol 4‐kinases (PI4Ks), with the catalytic activity of the PI4K type IIIβ (PI4KIIIβ) kinase required for exit of the complex from the Golgi apparatus, and the PI4K type IIα (PI4KIIα) kinase needed for correct sorting of the complex from endosomes to lysosomes (Jovic et al. 2012).
The crystal structure of GCase, first obtained in 2003, revealed that GCase has three non‐continuous domains. Domain I consists of a three‐stranded anti‐parallel β‐sheet. It contains two disulphide bridges, which may be involved in GCase folding. Domain II consists of two β‐sheets that form an immunoglobulin‐like domain. Finally, domain III consists of an eight‐stranded β/α triose phosphate isomerase (TIM) barrel and contains the catalytic site of GCase (Dvir et al. 2003).
The human GCase is glycosylated at four out of five available asparagine residues (N19, N59, N146, and N270, but not N462), and glycosylation is required for its catalytic function. It has been shown that mutations of asparagine residues N59, N146, and N270 do not affect GCase catalytic function and GCase interaction with active site‐directed inhibitors and activators. However, mutations of asparagine residue N19 result in production of catalytically inactive enzyme (Berg‐Fussman et al. 1993).
Gaucher disease
Gaucher disease (GD) is named after Dr Philippe Gaucher who, in 1882, first described a young woman with an enlarged spleen containing unusual looking cells (Gaucher 1882). GD is the most common autosomal recessive lysosomal storage disorder. Its most characteristic hallmark is the presence of glucocerebroside‐laden macrophages in the liver, spleen, and bone marrow, known as Gaucher cells. The most common clinical features of GD include hepatosplenomegaly, thrombocytopenia, anemia, bone involvement with osteopenia, osteoporosis, and bone pain because of bone infarcts or pathological fractures (Beutler and Grabowski 2001). GD has been traditionally divided into three distinct clinical forms based on age of onset and involvement of the central nervous system (CNS) (Cox and Schofield 1997). Type 1 GD (OMIM, #230800) is by far the most common form and is classified as non‐neuronopathic since it does not affect the CNS. The disease course of type 1 GD is very heterogeneous with some patients developing first symptoms in early childhood and others not manifesting any symptoms until well into adulthood. Type 2 and type 3 GD are classified as neuronopathic forms. Type 2 GD (OMIM, #230900) is the acute neuronopathic form with onset in infancy. It is characterized by rapid progression of neurological symptoms leading to death within 2 years of age. Infants are apparently normal for the first few months of life, after which they display hepatosplenomegaly, developmental regression, growth arrest, and rapid neurological decline (Stone et al. 2000). Type 3 GD (OMIM, #231000) is a chronic neuronopathic form with onset in early childhood. Unlike type 2, it is characterized by slow progression of neurological symptoms with parallel manifestation of all clinical symptoms diagnosed in type 1 GD leading to death in early adulthood (Beutler and Grabowski 2001). The prevalence of GD in the general population is about 1/40 000–1/50 000 live births, while the incidence of GD among Jews of Ashkenazi origin is up to 1/450 live births with a carrier frequency of about 6% (Grabowski 2008; National Organization of Rare Disorders 2013; Bronstein et al. 2014).
GD is caused by mutations in the GBA1 gene that lead to deficiency of the lysosomal enzyme glucocerebrosidase (GCase). Nearly, 300 pathogenic changes in GBA1, including point mutations, splice‐site mutations, deletions, insertions, and recombinant alleles containing genomic sequences of both the gene and pseudogene, have been identified (Hruska et al. 2008). These alterations lead to production of misfolded mutant enzymes with significantly reduced activity. GCase activity in GD patients is typically only 10–20% of that in normal individuals, while GCase activity in carriers is about 50%. It is thought that, not only GCase deficiency, but also ER stress triggered by the presence of misfolded GCase contribute to the pathogenesis of GD. The two most common GBA1 mutations identified in GD patients are N370S and L444P (Sidransky and Lopez 2012). Interestingly, the type of mutation is broadly predictive of GD form, as patients homozygous or compound heterozygous for the N370S mutation exclusively develop type 1 GD, patients homozygous for the L444P mutation are most likely to develop type 3 GD, while patients identified with a complex allele and a heterozygous L444P mutation are most likely to develop type 2 GD (Grabowski 2008; Sidransky 2012). The severity of the GD phenotype in relation to the observed mutation might be explained by the effect, which the particular mutation has on the GCase structure. Namely, the N370S mutation, which is situated on the longest α‐helix of GCase at the interface of domain II and III, does not directly affect the catalytic activity of GCase since it located too far from the catalytic site of domain III. The L444P mutation, which is situated at the hydrophobic core of domain II, causes conformational changes to the core and to domain II, which might result in the generation of unstable GCase (Dvir et al. 2003).
Currently, two types of treatment are available for patients with GD: enzyme replacement therapy (using imiglucerase, velaglucerase alfa, or taliglucerase alfa) and substrate reduction therapy (using misglustat) (Bennett and Mohan 2013). Although these treatments greatly improve the peripheral (non‐neuronopathic) features of the disease, they are ineffective for treatment of neuronopathic symptoms of type 2 and type 3 GD as they do not cross the blood–brain barrier (Bennett and Mohan 2013). Emerging treatments in the form of small molecular chaperones designed to cross the blood–brain barrier, which are able to bind misfolded mutant GCase in the endoplasmic reticulum, help its correct folding and subsequently assist its transport to the lysosomes, give great promise for treatment of the neuronopathic features of GD. The ability of small molecular chaperones to bind to misfolded GCase (and subsequently to induce proper folding of mutant GCase that in turn would increase functional GCase levels in the lysosomes) is particularly important, as it has been shown that majority of GBA1 mutations lead to the production of misfolded GCase (Sawkar et al. 2002; Bernier et al. 2004; Suzuki et al. 2009; Patnaik et al. 2012). Several such small molecular chaperones that lead to an increase in GCase activity have been investigated in both cell and animal models (Sawkar et al. 2002; Bernier et al. 2004; Steet et al. 2006; Maegawa et al. 2009; Bendikov‐Bar et al. 2011; Patnaik et al. 2012; Bendikov‐Bar et al. 2013; Luan et al. 2013). GCase inhibitors (e.g. N‐(n‐nonyl)deoxynojirimycin, and isofagomine) were the first chaperones shown to increase GCase activity. Both N‐(n‐nonyl)deoxynojirimycin and isofagomine increased GCase activity by increasing GCase trafficking to the lysosomes through specific, but reversible binding of GCase in the ER. Although they led to an increase in GCase activity in fibroblasts derived from Gaucher patients, it has been demonstrated that their clinical application is compromised because of difficulties in balancing chaperone activity with the direct inhibition of GCase activity (Sawkar et al. 2002; Bernier et al. 2004; Steet et al. 2005). Subsequently, a class of pyrazolopyrimidines, the first non‐inhibitory small molecular chaperones, was discovered. The pyrazolopyrimidines act as GCase activators that lead to both an increase in GCase translocation to the lysosomes and in GCase activity in wild‐type and mutant (N370S/N370S and L444P/L444P) human fibroblasts (Patnaik et al. 2012). Another promising small molecular chaperone, ambroxol hydrochloride (from now on commonly referred to as ambroxol), was identified after screening the library of Food and Drug Administration‐approved drugs with a thermal denaturation assay using wild‐type GCase (Maegawa et al. 2009). Ambroxol has long been used to treat airway mucus hypersecretion and hyaline membrane disease in newborns, which demonstrates its non‐toxicity to humans. Ambroxol acts as a GCase mixed‐type inhibitor with its inhibitory property toward GCase being highest at the neutral pH of the endoplasmic reticulum and void at the acidic pH of the lysosomes where functional GCase is required (Maegawa et al. 2009). Several studies have demonstrated that ambroxol treatment of different lines of cultured human GD fibroblasts results in a significant increase in GCase activity (Maegawa et al. 2009; Bendikov‐Bar et al. 2011, 2013; Luan et al. 2013; McNeill et al. 2014). However, whether an increase in GCase activity is because of ambroxol's chaperone activity alone remains to be clarified, as it has been shown in a fibroblast model that ambroxol increased GCase activity by activating the coordinated lysosomal expression and regulation network (coordinated lysosomal expression and regulation) via the action of transcription factor EB, which in turn led to an increase in lysosomal biogenesis. An enhancement of lysosomal mass would most likely lead to an increase in GCase activity, not necessarily via the elevated chaperone activity of ambroxol (McNeill et al. 2014). Currently available data from ambroxol‐treated wild‐type and transgenic mice carrying human GBA1 mutations has convincingly shown increase in GCase activity in the peripheral organs, but variable change in the brain. Hence, more studies are required to determine the effect of ambroxol on GCase activity in the CNS (Luan et al. 2013; Sanders et al. 2013).
The link between GBA1 and Parkinson disease
The link between GD and Parkinson disease (PD) was initially regarded as incidental, thus the first publications describing Gaucher patients with Parkinsonian features originated from individual clinics (McKeran et al. 1985; Turpin et al. 1988; Tayebi et al. 2001). The increasing number of reports suggesting the association between mutations in the GBA1 gene and PD led to more comprehensive studies focusing on several Gaucher patients with PD (Bembi et al. 2003; Tayebi et al. 2003; Varkonyi et al. 2003). Moreover, an increased proportion of PD cases in GD carriers compared to the general population further indicated the association of the GBA1 gene with PD (Goker‐Alpan et al. 2004; Halperin et al. 2006). This finding led researchers to investigate whether the frequency of GBA1 mutations is increased in PD. The first such study identified GBA1 mutations in 12 of 57 (21%) PD postmortem brains (Lwin et al. 2004). This study not only further supported the link between GBA1 and PD, but also indicated that both heterozygous and homozygous mutations in GBA1 might be associated with PD. Subsequently, multiple PD patients were investigated to establish the prevalence of GBA1 mutations, which highlighted the importance of routine GBA1 mutation screening when diagnosing PD individuals. The approach taken by different research groups varied, as some screened only for the most common GBA1 mutations, while others sequenced all exons of the gene (Tables 1, 2, 3). Overall, data acquired from multiple studies showed that the frequency of heterozygous GBA1 mutations varied between 2.3–9.8% in the European population of non‐Ashkenazi Jewish origin, 16.9–31.3% in the European population of Ashkenazi Jewish origin, 1.8–8.7% in the Asian population, and 2.9–8.0% in the combined North–South American population (Tables 1, 2, 3). The most common mutations identified in the European population of non‐Ashkenazi Jewish origin were L444P and N370S, and both mutations were found with similar frequency (Table 1). Interestingly, in the European population of Ashkenazi Jewish origin, the N370S mutation was the most predominant, while in the Asian and North–South American populations, the L444P mutation was the most prevalent (Tables 1, 2, 3). Moreover, most studies conducted on European PD individuals of Ashkenazi Jewish origin did not detect any L444P mutations, while the majority of studies carried out on PD patients from the Asian population did not identify any N370S mutations (Tables 1 and 2). Nevertheless, given the observed discrepancies in incidence of GBA1 mutations within the same population, and the limited number of cases and controls available for individual studies, a much bigger study was required to provide a conclusive answer about the frequency of GBA1 mutations among people of different origin. A multicenter meta‐analysis collected a total of 5691 PD patients and 4898 controls (Sidransky et al. 2009). They analyzed 780 PD individuals and 387 controls of Ashkenazi Jewish origin for both the N370S and L444P mutations, and found that 15% of patients and 3% of controls had one of the two mutations. They also screened 4911 PD individuals and 4511 controls of non‐Ashkenazi Jewish origin and found that 3% of patients and less than 1% of controls had at least one of the two mutations. Finally, they sequenced the entire GBA1 gene in 1883 PD individuals of non‐Ashkenazi Jewish origin and found that 7% of patients carried GBA1 mutations. To summarize, mutations in the GBA1 gene constitute numerically the most important predisposing risk factor for developing PD. Both homozygous and heterozygous GBA1 confer a 20‐ to 30‐fold increased risk for the development of PD, and it is estimated that approximately 5–10% of PD patients have a GBA1 mutation (Sidransky et al. 2009; Bultron et al. 2010; McNeill et al. 2012a,b). However, not all GBA1 mutant carriers will develop PD, and it is currently estimated that 30% will develop the disease by age 80 years (Lesage and Brice 2009; Lesage et al. 2011).
Table 1.
Frequency and type of GBA1 mutations found in the European population
| Number | % with mutation | Method | Alterations found | Most common mutation(s) | Origin | Reference | ||
|---|---|---|---|---|---|---|---|---|
| PD cases | Control cases | PD cases | Control cases | |||||
| 2350 | 1111 | 4.5 | 0.63 | Screening of GBA1 exon 9 and 10 | N370S, L444P, D443N, and IVS10+1G>T | – | Italian | Asselta et al. 2014 |
| 259 | – | 3.5 | – | Sequencing of GBA1 exons | L444P, N370S, N462K, R463C, and R257Q |
N370S (33.33%) L444P (33.33%) |
British | Winder‐Rhodes et al. 2013 |
| 360 | 348 | 5.8 | 1.4 | Sequencing of GBA1 exon 8 to 11 | N370S, D409H, H255Q, L444P, A456P, R463C, and RecNciI | N370S (33.33%) | Serbian | Kumar et al. 2013 |
| 311 | 474 | 2.3 | 1.7 | Genotyping for L444P and N370S | L444P and N370S |
N370S (57.14%) L444P (42.86%) |
Norwegian | Toft et al. 2006 |
| 225 | 186 | 9.8 | 0.5 | Sequencing of GBA1 exons | M123T, L144V, G202R, I260T, T369M, N370S, W393R, D409H, L444P, RecNciI, and S488T |
L444P (27.27%) N370S (22.72%) |
Spanish | Setó‐Salvia et al. 2012 |
| 330 | 240 | 2.7 | 0.4 | Genotyping for L444P and N370S | L444P and N370S | L444P (66.67%) | Russian | Emelyanov et al. 2012 |
| 1391 | 391 | 6.7 | 1.0 | Sequencing of GBA1 exons | Lots of different mutations | N370S (47.40%) | French | Lesage et al. 2011 |
| 205 | 206 | 10.24 | 3.39 | Genotyping for N370S, D409H, L444P, H255Q, R120W, Y108C, IVS6‐2A>G, and IVS10‐1G>A | N370S, L444P, D409H; H255Q, D409H, Y108C, IVS10‐1G>A |
N370S (28.57%) L444P (28.57%) |
Greek | Moraitou et al. 2011 |
| 230 | 430 | 6.1 | 0.7 | Sequencing of GBA1 exons | N370S, N396T, D409H, and L444P |
N370S (33.33%) N396T (33.33%) |
Portuguese | Bras et al. 2009 |
| 172 | 132 | 3.4 | 0.3 | Sequencing of GBA1 exons | L445P, D409H, E326K, H255Q, R329H, L268L, S271G, T428K, and V460L |
H255Q (36.36%) L444P (18.18%) |
Greek | Kalinderi et al. 2009 |
| 790 | 257 | 4.18 | 1.17 | Sequencing of GBA1 exons | L444P, D443N, R463C, RecNciI, RecA456P, N370S, D409H, D380A, c.1263‐1317del55, R257Q, G193E, R131C, K7E, and V458L |
L444P (33.33%) N370S (24.24%) |
British | Neumann et al. 2009 |
| 420 | 4138 | 17.9 | 4.2 | Genotyping for N370S, R496H, 84GG, IVS2+1, V394L, D409H, L444P, and RecTL | N370S, R496H, 84GG, IVS2+1, V394L, L444P, and RecTL | N370S (61.33%) | Ashkenazi Jewish (Israeli) | Gan‐Or et al. 2008 |
| 395 | 483 | 2.8 | 0.2 | Genotyping for L444P and N370S | L444P and N370S | L444P (72.73%) | Italian | De Marco et al. 2008 |
| 178 | 85 | 16.9 | 7.1 | Sequencing of GBA1 exons | N370S, R496H, E326K, T369M, P175P, and 84insGG | N370S (78.33%) | Ashkenazi Jewish (American) | Clark et al. 2007 |
| 99 | 1543 | 31.3 | 6.2 | Genotyping for N370S, L444P, 84GG, IVS2+1G>A, V394L, and R496H | N370S and 84GG | N370S (83.87%) | Ashkenazi Jewish (Israeli) | Aharon‐Peretz et al. 2004 |
Table 2.
Frequency and type of GBA1 mutations found in the Asian population
| Number | % with mutation | Method | Alterations found | Most common mutation(s) | Origin | Reference | ||
|---|---|---|---|---|---|---|---|---|
| PD cases | Control cases | PD cases | Control cases | |||||
| 184 | 130 | 8.7 | 5.4 | Sequencing of GBA1 exons |
c.334_338delCAGAA L264I, L314V, R163Q, F213I, E326K, S364S, F347L, V375L, L444P, RecNciI, and Q497R |
L444P (31.25%) | Chinese | Yu et al. 2015 |
| 480 | 395 | 5 | 0.5 | Sequencing of GBA1 exons | L444P, N386K, P428S, IVS2þ1G>A, IVS9þ3G>C, IVS10‐9_10GT>AG, and c.1309delG | L444P (58%) | Thai | Pulkes et al. 2014 |
| 195 | 443 | 3.08 | 0.0 | Genotyping for L444P, N370S, and R120W mutations | L444P | L444P (100%) | Chinese | Zhang et al. 2012 |
| 277 | 291 | 3.2 | 0.0 | Sequencing of GBA1 exons | N188S, P201H, R257Q, S271G, and L444P |
R257Q (33.33%) L444P (22.22%) |
Korean | Choi et al. 2012 |
| 208 | 298 | 3.4 | 0.3 | Genotyping for L444P, N370S, and R120W mutations | L444P | L444P (100%) | Chinese | Wang et al. 2012 |
| 967 | 780 | 3.72 | 0.26 | Sequencing of GBA1 exons (30 cases) Genotyping for L444P, D409H, R120W, L174P, and Q497R mutations (all individuals) | L444P, RecNciI, and D409H | L444P (75%) | Chinese | Huang et al. 2011 |
| 328 | 300 | 1.8 | 0.7 | Genotyping for N370S mutation | N370S | N370S (100%) | Chinese | Hu et al. 2010 |
| 616 | 411 | 3.2 | 0.2 | Genotyping for L444P mutation | L444P | L444P (100%) | Chinese | Mao et al. 2010 |
| 402 | 412 | 2.74 | 0.0 | Genotyping for L444P, N370S, F213I, and R353W mutations | L444P | L444P (100%) | Chinese | Sun et al. 2010a,b |
| 331 | 347 | 8 | 0.0 | Genotyping for L444P and N370S mutations | L444P | L444P (100%) | Chinese | Tan et al. 2007 |
| 518 | 339 | 3.1 | 1.2 | Genotyping for L444P, RecNciI, and R120W mutations | L444P, RecNciI and R120W | L444P (81.25%) | Taiwanese | Wu et al. 2007 |
| 92 | 92 | 4.1 | 1.1 | Sequencing of GBA1 exons | L444P, D409H, L174P, and Q497R | Each mutation (25%) | Taiwanese | Ziegler et al. 2007 |
Table 3.
Frequency and type of glucocerebrosidase (GBA1) mutations found in the combined North–South American population
| Number | % with mutation | Method | Alterations found | Most common mutation(s) | Origin | Reference | ||
|---|---|---|---|---|---|---|---|---|
| PD cases | Control cases | PD cases | Control cases | |||||
| 128 | 252 | 5.47 | 0.0 | Genotyping for L444P and N370S mutations | L444P | L444P (100%) | Mexican | González‐Del Rincón Mde et al. 2013 |
| 65 | 267 | 3.08 | 0.0 | Genotyping for N370S, L444P and G377S | L444P | L444P (100%) | Brazilian | Spitz et al. 2008 |
| 721 | 554 | 2.9 | 0.4 | Genotyping for L444P and N370S mutations | L444P and N370S |
N370S (52.38%) L444P (47.62%) |
American | Mata et al. 2008 |
| 100 | 94 | 8 | 2.1 | Sequencing of GBA1 exons | N370S, T369M, D409H, and RecNciI |
L444P (25%) T369M (25%) |
American | Clark et al. 2007 |
| 88 | 102 | 5.68 | 0.98 |
Genotyping for N370S, L444P, IVS2≦1 K198T, R329C, 84insGG, and RecNciI |
L444P, N370S, and RecNciI | RecNciI (60%) | Canadian | Sato et al. 2005 |
GBA1‐associated PD (PD‐GBA1) – clinical and biochemical presentation
Individual PD patients with GBA1 mutations cannot be distinguished at the clinical level from idiopathic PD patients without GBA1 mutations. Parkinson‐GBA1 (PD‐GBA1) patients exhibit the classic triad of bradykinesia, rigidity, and tremor, with asymmetric onset (Goker‐Alpan et al. 2008). However, age of onset tends to be slightly younger, an incidence of neuropsychiatric features (such as depression, anxiety, hallucination, and sleep disturbance) is higher, and there is a greater risk for earlier and more prevalent cognitive impairment in PD‐GBA1 patients (Tan et al. 2007; Neumann et al. 2009; Sidransky et al. 2009; Brockmann et al. 2011; McNeill et al. 2012b; Winder‐Rhodes et al. 2013). The pattern of cognitive dysfunction in GBA1‐positive carriers is subtly different, present in those even without PD at the time of investigation (Zokaei et al. 2014). Nigrostriatal imaging with fluorodopa positron emission tomography or single photon emission tomography with dopamine sensitive ligands in PD‐GBA1 demonstrate an asymmetric pattern of abnormality indistinguishable from idiopathic PD (Goker‐Alpan et al. 2012; McNeill et al. 2013b). This contrasts with the imaging of parkin (parkin RBR E3 ubiquitin protein ligase, PARK2) or phosphatase and tensin homolog‐induced putative kinase 1 (PINK1) mutant PD, where abnormalities are usually symmetrical. Patients with GBA1 mutations also exhibit retinal thinning as determined by optical coherence tomography compared to matched controls and similar to that seen in PD patients (McNeill et al. 2013a).
Current evidence suggests that GBA1 mutant homozygote and heterozygote carriers without clinical evidence of PD, exhibit the prodromal features of the disease. Olfactory function and cognitive assessment were significantly reduced, and motor testing abnormal in GBA1‐positive cases without features of PD compared to controls (McNeill et al. 2012b). A 2 year follow‐up showed significant deterioration in scores for depression, rapid eye movement sleep behavior disorder, cognition, olfaction, and motor scores (Beavan et al. 2015). The GBA1 cohort exhibits a relatively rapid evolution of non‐motor and motor features.
The response to dopaminergic therapy in PD‐GBA1 appears to be the same as that seen in idiopathic PD (Ziegler et al. 2007)., including the development of motor complications. In one center, retrospective genetic analysis identified GBA1 mutations in 17% of those who had undergone deep‐brain stimulation, and in whom clinical effect was as good as those without mutations (Angeli et al. 2013).
Importantly, the pathology of PD‐GBA1 is identical to that of idiopathic PD with nigral dopamine loss and Lewy bodies and neurites containing alpha‐synuclein (Westbroek et al. 2011; Swan and Saunders‐Pullman 2013).
Pathogenesis of PD‐GBA1
The mechanism by which GBA1 mutations increase the risk of PD is still unknown. However, given that clinical manifestation and pathological findings are almost identical in PD‐GBA1 and idiopathic PD patients, it is thought that, as in idiopathic PD, alpha‐synuclein accumulation, mitochondrial impairment, autophagic dysfunction, inflammation, and oxidative and endoplasmic reticulum stress may play an important role in both the development and progression of PD‐GBA1 (Schapira and Tolosa 2010).
In contrast to autosomal recessive forms of PD (caused by homozygous mutations in PARK2, PINK1, or DJ‐1) and autosomal dominant forms of PD (caused by heterozygous mutations in alpha‐synuclein (SNCA) or leucine‐rich repeat kinase 2), PD‐GBA1 supports both autosomal recessive and autosomal dominant modes of inheritance (Sidransky 2012).
Autosomal recessive inheritance is generally associated with loss‐of‐function of mutated proteins, and several characteristics of GBA1 suggest loss‐of‐function of mutated GCase.
A subset of PD‐GBA1 patients is identified with null GBA1 alleles, such as 84GG and IVS2+1G>A, which do not encode GCase (Aharon‐Peretz et al. 2004; Gan‐Or et al. 2008; Clark et al. 2009).
Inhibition of GBA1 with condruitol β‐epoxide (CBE) in cell and animal models leads to alpha‐synuclein accumulation (Manning‐Boğ et al. 2009; Cleeter et al. 2013).
Homozygous knock‐out of the Gba1 gene in mice leads to mitochondrial dysfunction, and lipid and alpha‐synuclein accumulation (Enquist et al. 2007; Osellame et al. 2013).
Autosomal dominant inheritance is often associated with gain‐of‐function of mutated proteins, and several features of GBA1 suggest gain‐of‐function of mutated GCase.
Most PD‐GBA1 patients are identified with heterozygous GBA1 mutations.
The missense mutations resulting in production of misfolded GCase account for the majority of GBA1 mutations.
Misfolded GCase interacts directly with alpha‐synuclein, which subsequently leads to increased alpha‐synuclein accumulation (Sidransky and Lopez 2012).
Even though there is evidence supporting loss‐ and gain‐of‐function of GCase, both hypotheses have limitations, as presence of null mutations in some PD‐GBA1 patients contradicts the gain‐of‐function theory, while development of PD‐GBA1 in individuals carrying heterozygous GBA1 mutations contradicts the loss‐of‐function theory. Furthermore, misfolded GCase and loss‐of‐function may result in a secondary gain‐of‐function‐type effect through ER trapping and ER‐associated degradation process (ERAD). Even more importantly, neither the loss‐ nor gain‐of‐function hypotheses explain why the majority of individuals with GBA1 mutations do not develop PD.
GCase and alpha‐synuclein
PD‐GBA1 belongs to a group of diseases collectively known as synucleinopathies, which are characterized by the presence of Lewy bodies and neurites containing SNCA. The importance of SNCA in the pathology of PD‐GBA1 prompted a question about the GCase and SNCA relationship. To date, three main hypotheses linking GCase with alpha‐synuclein have been suggested.
The first proposes that a gain‐of‐function by the misfolded GCase results in its direct interaction with alpha‐synuclein, which then leads to increased SNCA accumulation and aggregation (Sidransky and Lopez 2012). This would require that GCase is able to interact directly with SNCA, and that GCase is present in abnormal protein aggregates containing SNCA, such as Lewy bodies. Indeed, direct interaction between GCase and the C‐terminus of SNCA was shown to occur in lysosomes (Yap et al. 2011). The presence of GCase in the brain tissues samples from PD‐GBA1 patients was detected in 32–90% of Lewy bodies and neurites, showing that mutant GCase and SNCA co‐localize in vivo (Goker‐Alpan et al. 2010). One piece of evidence supporting a gain‐of‐function by the misfolded GCase was provided by a cell model study, where over‐expression of mutant GCase (containing N370S, L444P, D409H, D409V, E235A, or E340A) in neural MES23.6 and pheochromocytoma (PC12) cells led to a 21 to 148% increase in SNCA levels (Cullen et al. 2011). The relationship between misfolded GCase and SNCA was further strengthened by animal model studies. Namely, a progressive increase in SNCA levels was shown in the hippocampus of a homozygous D409V Gba1 mouse model, and a significant increase in SNCA levels was detected in the forebrain and cerebellum of hypomorphic prosaposin mice carrying the homozygous V394L Gba1 mutation (Cullen et al. 2011; Sardi et al. 2013). Also, a progressive increase in SNCA levels, from low levels to substantial SNCA aggregates, was observed in the cortex of hypomorphic prosaposin mice carrying the homozygous D409H Gba1 mutation (Xu et al. 2014).
The second hypothesis proposes that a loss‐of‐function of GCase (GCase deficiency because of, for example, degradation of the misfolded enzyme) leads to accumulation of a substrate (glucocerebroside) that in turn perturbs lipid homeostasis and subsequently affects alpha‐synuclein trafficking, processing, and clearance. This eventually promotes alpha‐synuclein aggregation and facilitates alpha‐synuclein oligomer formation (Mazzulli et al. 2011; Westbroek et al. 2011; Sidransky and Lopez 2012). Further evidence supporting this hypothesis and its consequences on SNCA accumulation was provided from cell and animal model studies examining the effect of GBA1 inhibition by CBE (Manning‐Boğ et al. 2009; Cleeter et al. 2013). Namely, treatment of differentiated SH‐SY5Y cells with CBE resulted in increased levels of SNCA (Manning‐Boğ et al. 2009; Cleeter et al. 2013). Single injection of CBE led to about 20% increase in SNCA levels in the ventral mesencephalon of normal mice. Also, enhanced SNCA immunoreactivity was observed within the cell bodies of the substantia nigra pars compacta and within the cytoplasm and cell nuclei of A9 neurons of CBE‐treated mice (Manning‐Boğ et al. 2009). Finally, SNCA accumulation and SNCA oligomer formation was shown in a mouse model carrying homozygous knock‐out of Gba1, i.e. in mice modeling loss‐of‐function of GCase (Osellame et al. 2013).
The third hypothesis proposes the existence of a bidirectional feedback loop in which GCase deficiency facilitates formation of alpha‐synuclein oligomers, with the subsequent increase in alpha‐synuclein oligomers leading to further decrease in normal GCase activity, which in turn promotes formation of additional alpha‐synuclein oligomers (Mazzulli et al. 2011). It has also been demonstrated in cell models that increased alpha‐synuclein causes a decrease in GCase activity and protein levels, as over‐expression of exogenous SNCA in SH‐SY5Y cell lines resulted in about 44–70% decrease in GCase activity, and about 33–87% decrease in GCase protein levels (Gegg et al. 2012).
Finally, although a majority of the studies conducted to date provide a link between GCase and SNCA via loss‐, gain‐of function, or bidirectional feedback loop, some studies failed to support the relationship between SNCA and mutant GCase. The first such study showed that GBA1 inhibition by CBE in both differentiated SH‐SY5Y cells and rat cortical neuronal cultures did not significantly increase SNCA accumulation (Dermentzaki et al. 2013). This contradicts the results obtained by others, which showed an increase in SNCA levels in CBE‐treated differentiated SH‐SY5Y cells (Manning‐Boğ et al. 2009; Cleeter et al. 2013). This difference may simply have been due to the shorter exposure in the Dermentzaki study. The second such study demonstrated that GBA1 inhibition by CBE in PC12 cell line did not alter SNCA levels, and that over‐expression of human wild‐type GBA1 did not lead to an increase in SNCA levels in neural MES23.5 cell line. The same study, however, showed that over‐expression of wild‐type GBA1 in HEK293‐SNCA [A53T] and PC12 cell lines resulted in decrease of SNCA levels, and that over‐expression of different mutant GCase in MES23.6 and PC12 cells led to a significant increase in SNCA levels (Cullen et al. 2011). The cell model specificity might explain the observed discrepancy in the effect of over‐expression of wild‐type GBA1 on SNCA levels.
Altogether, although convincing evidence supporting the relationship between SNCA and both gain‐ and loss‐of‐function of mutant GCase exists, neither of them explains why only a proportion of individuals with GBA1 mutations develop PD. One plausible explanation might be that in order to develop PD‐GBA1, in addition to the GBA1 mutation, there must be additional genetic alternations. Another credible explanation might be that mutated GCase on its own is not sufficient to induce alpha‐synuclein pathology: perhaps only when other changes occur (e.g. a perturbation of a component of the lysosomal degradation pathway resulting in defective SNCA clearance) can PD‐GBA1 develop.
GCase and mitochondria
Mitochondria not only play a central role in energy production by oxidative phosphorylation but are also involved in many other cellular processes, such as synthesis of steroids and regulation of calcium homeostasis, membrane potential, apoptosis, and stress response. Taking into account the plethora of mitochondrial functions, perhaps not surprisingly, mitochondrial impairment plays an important role in the pathogenesis of PD (Schapira et al. 1989, 1990). In neurons, mechanistically, accumulation of dysfunctional mitochondria results in generation of reactive oxygen species and free radicals leading eventually to neuronal death. Mutations in PARK2, PINK1, and Parkinson protein 7 (PARK7 or DJ‐1) genes, which affect mitochondrial morphology and function, have been identified as causing familial PD (Schapira 2008). However, the role of mitochondrial dysfunction in the pathogenesis of PD‐GBA1 remains elusive and, to date, only three studies have investigated the impact of GBA1 mutations on mitochondrial function. The first study showed that inhibition of GBA1 by CBE in SHSY‐5Y cells led to a decrease in adenosine diphosphate phosphorylation, decline in mitochondrial membrane potential, and increase in free radical generation, so demonstrating that GCase loss‐of‐function causes oxidative stress and mitochondrial dysfunction (Cleeter et al. 2013). The second study showed that loss‐of‐function of GCase in a mouse model carrying homozygous knock‐out of GBA1 resulted in accumulation of dysfunctional and fragmented mitochondria, and reduction in respiratory chain complex activities, membrane potential, and oxygen consumption (Osellame et al. 2013). The third study showed that gain‐of‐function of GCase in hypomorphic prosaposin mice carrying homozygous D409H or V394L Gba1 mutation led to significant reduction in oxygen consumption and mitochondrial adenosine triphosphate (ATP) production. The same findings were also observed in wild‐type cerebral cortical neural cells treated with CBE (Xu et al. 2014). Altogether, data from cell and animal model studies demonstrated that both loss‐ and gain‐of‐function GBA1 mutations cause mitochondrial impairment, and so it would be worth investigating whether mitochondrial dysfunction is also present in PD‐GBA1 brains.
GCase and autophagy
Autophagy is a lysosomal pathway that is involved in degradation of damaged organelles, such as mitochondria and endoplasmic reticulum, and clearance of long‐lived misfolded or aggregated proteins, such as alpha‐synuclein. Three distinct types of autophagy have been identified: macroautophagy, microautophagy, and chaperone‐mediated autophagy (CMA). Increasing evidence indicates the existence of a strong link between impairment of autophagy and PD. Dysfunction of autophagy has been shown both in brain tissue samples from idiopathic PD patients and toxic mouse models of PD (Chu et al. 2009; Alvarez‐Erviti et al. 2010; Dehay et al. 2010; Vila et al. 2011). Although involvement of autophagic impairment in the development of GD and especially PD is not yet fully understood, emerging data clearly suggest its importance.
Lysosomal dysfunction resulting in progressive accu‐mulation of glucocerebroside plays a central role in GD pathogenesis. Accumulation of sphingolipids, to which glucocerebroside belongs, has been shown to alter autophagy by both inducing cell death and reducing autophagosome clearance, and so promoting their accumulation (Tamboli et al. 2011). Indeed, induction of autophagy has been demonstrated in GD human fibroblasts that showed accumulation of glucocerebroside, while increased number of autophagosomes has been shown in hypomorphic prosaposin mice carrying the homozygous V394L Gba1 mutation that showed accumulation of glucocerebroside (Sun et al. 2010a,b; Vaccaro et al. 2010). However, it seems unlikely that the autophagic dysfunction in PD‐GBA1 is because of accumulation of glucocerebroside (and glucosphingosine, a deacetylated glucocerebroside), since substrate accumulation is believed to require both GBA1 alleles to carry a genetic alteration, and this is not the case in the majority of PD‐GBA1 individuals. Analysis of putamen and cerebellum samples from PD‐GBA1 patients supports that notion, as no accumulation of glucocerebroside and glucosphingosine was observed in these brain regions (Gegg et al. 2015). In contrast, significantly increased glucosphingosine levels (and significantly reduced GCase activity) were detected in the substantia nigra and hippocampus of idiopathic PD patients during sixth and seventh decade of life, respectively (Rocha et al. 2015). However, no changes in glucosphingosine levels were observed in the putamen and cerebellum of idiopathic PD patients, although a significant reduction in GCase activity was observed in these regions (Gegg et al. 2015; Rocha et al. 2015). The observed discrepancy in glucosphingosine accumulation among different brain regions should prompt further analysis in both of PD‐GBA1 and idiopathic PD patients to establish whether the relationship between reduction of GCase activity and accumulation of glucosphingosine exists, and, if indeed there is such a link whether autophagic dysfunction should be at least partially attributed to GCase substrate accumulation.
The relationship between SNCA and both gain‐ and loss‐of‐function of mutant GCase provides a plausible explanation linking autophagic impairment with PD‐GBA1. It has been shown that SNCA is preferentially degraded by CMA, and that impairment of CMA leads to accumulation and aggregation of SNCA in SH‐SY5Y cell lines (Alvarez‐Erviti et al. 2010). Conversely, it has been demonstrated that expression of mutant SNCA leads to CMA impairment in proliferating PC12 and SH‐SY5Y cells, while over‐expression of wild‐type SNCA results in CMA dysfunction in differentiated SH‐SY5Y cells (Xilouri et al. 2009). The latter observation is especially interesting, as misfolded GCase has been shown to facilitate SNCA accumulation and aggregation (Mazzulli et al. 2011; Westbroek et al. 2011; Sidransky and Lopez 2012), and so it can be speculated that SNCA increase because of GCase dysfunction might lead to CMA impairment. Thus, finding a therapeutic agent that is able to increase levels of functional, properly folded GCase would enhance SNCA degradation by CMA, and consequently lead to reduction in SNCA levels.
GCase and endoplasmic reticulum stress
Secretory and membrane‐associated proteins are synthetized in the endoplasmic reticulum and newly synthetized proteins are folded in the ER with the help of ER chaperones. Proteins that fail to fold correctly are recognized by the ER quality control system and retained for refolding. Misfolded proteins that fail to refold by ER chaperones are disposed of by ERAD via the ubiquitin–proteasome system. When the number of misfolded proteins exceeds the capacity of the ERAD system, misfolded proteins accumulate in the ER causing stress. In response to such stress, the unfolded protein response (UPR) is activated to help restore normal cell function. If this fails, and ER stress continues, malfunctioning cells are eliminated by apoptosis (Yoshida 2007). Emerging evidence shows the involvement of ER stress in the pathogenesis of PD (Imai et al. 2001; Ryu et al. 2002). The direct link between ER stress and PD comes from studies of parkin (PARK2), which in mutated form causes early onset of PD. Parkin, an E3 ubiquitin ligase is a member of the UPR, where it is responsible for ubiquitination of misfolded proteins for degradation. Clearance of misfolded proteins is impaired when parkin is mutated, which eventually results in ER stress (Imai et al. 2000, 2001). The link between misfolded GCase and ER stress is very plausible, as prolonged accumulation of misfolded GCase in the ER would eventually cause ER stress and UPR activation. Misfolded GCases, including N370S and L444P mutants, have been shown to be retained in the ER and undergo ERAD in human GD fibroblasts (Ron and Horowitz 2005; Bendikov‐Bar et al. 2011). Moreover, it has been reported that GD severity shows correlation with levels of misfolded GCase retention in the ER and so, speculatively, with ER stress (Ron and Horowitz 2005). Biochemical analysis of PD‐GBA1 putamen samples for the presence of UPR markers showed a 63% increase in C/EBP homologous protein levels and a 26% increase in binding immunoglobulin protein. The observed UPR/ERAD could be caused by misfolded GCase, but other causes such as oxidative stress, mitochondrial dysfunction, proteolysis, or altered calcium homeostasis cannot be excluded (Gegg et al. 2012). The direct interaction of misfolded GCase with parkin, and its subsequent ERAD degradation, has been observed in human COS7 and HEK293 cells, providing evidence for the existence of a direct link between PD‐GBA1 and ERAD. Misfolded GCase‐parkin interaction can block parkin interaction with other protein that are degraded via parkin‐mediated ERAD, which would result in further accumulation of proteins in the ER and a further increase in ER stress that would consequently result in neuronal death, and so PD development (Ron et al. 2010). In contrast to human studies, analysis of UPR markers in the neuronal cultures and tissues obtained from transgenic GD mouse models and CBE‐treated normal mice showed no alternations in C/EBP homologous protein, binding immunoglobulin protein, and X‐box binding protein 1 (XBP1) (Farfel‐Becker et al. 2009). Further studies are required to establish whether ER stress really contributes to the pathogenesis of PD‐GBA1.
PD‐GBA1 treatment
There is no specific treatment available for PD‐GBA1 patients to modify the course of the disease. Since nigral dopamine loss observed in individuals with PD‐GBA1 is identical to that observed in individuals with idiopathic PD, PD‐GBA1 patients are normally given dopaminergic therapy to alleviate the dopamine deficit in the striatum. Therefore, the development of new therapies that can slow down disease progression is urgently required. The increasing evidence linking GCase with alpha‐synuclein and mitochondrial pathways has relevance to a potential novel therapeutic strategy for both PD‐GBA1 and idiopathic PD patients. Moreover, even patients without GBA1 mutations show significant reduction in GCase activity in the substantia nigra (Gegg et al. 2012). This highlights the importance of GCase function, not only in PD‐GBA1 but also in idiopathic PD and suggests that GBA1 treatments may potentially prove to be effective with many, if not all, PD patients (Schapira and Gegg 2013). The use of small molecular chaperones such as N‐(n‐nonyl)deoxynojirimycin, isofagomine, pyrazolopyrimidines, or ambroxol (i.e. the same chaperones that are currently being tested for their application in treating GD) may be of interest as a novel therapy for PD, as a means to decrease alpha‐synuclein levels and improve mitochondrial function (Sawkar et al. 2002; Steet et al. 2006; Maegawa et al. 2009; Bendikov‐Bar et al. 2011, 2013; Patnaik et al. 2012; Luan et al. 2013; Sanders et al. 2013; McNeill et al. 2014). Interestingly, the benefits of increasing wild‐type GCase stability, trafficking, and activity in idiopathic PD have been demonstrated through the study of mice over‐expressing human alpha‐synuclein under the murine Thy‐1 promoter (Thy1‐aSyn). Administration of a molecular chaperone AT2101 (afegostat‐tartrate, isofagomine) led to GCase increase in the brain and resulted in improvement of both motor function and neuropathological manifestations (such as elimination of microglial inflammatory response and reduction in alpha‐synuclein immunoreactivity in the substania nigra) in Thy1‐aSyn mice (Richter et al. 2014). These results show that an increase in GCase activity achieved by administration of molecular chaperones might significantly improve clinical and biochemical manifestations of synucleinopathies, even those without GBA1 mutations, and so further development of small molecular chaperones gives a great promise for finding a successful treatment not only for PD‐GBA1, but also for idiopathic PD and other synucleinopathies.
Conclusions
After Alzheimer, Parkinson disease is the second most common neurodegenerative disorder. The most common risk factor for PD involves mutations in the GBA1 gene, which occurs in 5–10% of PD patients (Sidransky et al. 2009; Bultron et al. 2010; McNeill et al. 2012a). This compares with leucine‐rich repeat kinase 2 mutations that are estimated to cause 0.5% of sporadic PD. Further, even in patients without GBA1 mutations, a decrease in GCase activity is still found in all studied cases, suggesting that this protein plays a much more important role in PD than previously appreciated. As such, further studies of GBA1, in particular of treatments that increase levels of GCase, are likely to be one of the most promising future avenues for tackling PD.
Acknowledgments and conflict of interest disclosure
This work was supported by the Medical Research Council (MRC) grant (MR/M006646/1), Javon Trust grant, Parkinson's UK (PUK) grant (G1403) and National Institute of Healthcare Research (NIHR) grant (RCF30AS2012, RCF73TS20145989 and RCF103/AS/2014). AHVS is a National Institute of Healthcare Research (NIHR) Senior Investigator. The authors declare that there are no conflicts of interest.
This article is part of a special issue on Parkinson disease .
References
- Aharon‐Peretz J., Rosenbaum H. and Gershoni‐Baruch R. (2004) Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med. 351, 1972–1977. [DOI] [PubMed] [Google Scholar]
- Alvarez‐Erviti L., Rodriguez‐Oroz M. C., Cooper J. M., Caballero C., Ferrer I., Obeso J. A. and Schapira A. H. (2010) Chaperone‐mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 67, 1464–1472. [DOI] [PubMed] [Google Scholar]
- Angeli A., Mencacci N. E., Duran R. et al (2013) Genotype and phenotype in Parkinson's disease: lessons in heterogeneity from deep brain stimulation. Mov. Disord. 28, 1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselta R., Rimoldi V., Siri C., Cilia R., Guella I., Tesei S., Soldà G., Pezzoli G., Duga S. and Goldwurm S. (2014) Glucocerebrosidase mutations in primary parkinsonism. Parkinsonism Relat. Disord. 20, 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavan M., McNeill A., Proukakis C., Hughes D. A., Mehta A. and Schapira A. H. (2015) Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation‐positive cohort. JAMA Neurol. 72, 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bembi B., Zambito Marsala S., Sidransky E., Ciana G., Carrozzi M., Zorzon M., Martini C., Gioulis M., Pittis M. G. and Capus L. (2003) Gaucher's disease with Parkinson's disease: clinical and pathological aspects. Neurology 61, 99–101. [DOI] [PubMed] [Google Scholar]
- Bendikov‐Bar I., Ron I., Filocamo M. and Horowitz M. (2011) Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 46, 4–10. [DOI] [PubMed] [Google Scholar]
- Bendikov‐Bar I., Maor G., Filocamo M. and Horowitz M. (2013) Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 50, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett L. L. and Mohan D. (2013) Gaucher disease and its treatment options. Ann. Pharmacother. 47, 1182–1193. [DOI] [PubMed] [Google Scholar]
- Berg‐Fussman A., Grace M. E., Ioannou Y. and Grabowski G. A. (1993) Human acid beta‐glucosidase. N‐glycosylation site occupancy and the effect of glycosylation on enzymatic activity. J. Biol. Chem. 268, 14861–14866. [PubMed] [Google Scholar]
- Bernier V., Lagacé M., Bichet D. G. and Bouvier M. (2004) Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol. Metab. 15, 222–228. [DOI] [PubMed] [Google Scholar]
- Beutler E. (1992) Gaucher disease: new molecular approaches to diagnosis and treatment. Science 256, 794–799. [DOI] [PubMed] [Google Scholar]
- Beutler E. and Grabowski G. A. (2001) Glucosylceramide lipidosis–Gaucher disease, in The metabolic and molecular bases of inherited diseases, 8th edn (Scriver C. R., Beaudet A. L., Sly W. S. and Valle D., eds), pp. 3635–3668. McGraw‐Hill, New York. [Google Scholar]
- Bras J., Paisan‐Ruiz C., Guerreiro R., Ribeiro M. H., Morgadinho A., Januario C., Sidransky E., Oliveira C. and Singleton A. (2009) Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Portugal. Neurobiol. Aging 30, 1515–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann K., Srulijes K., Hauser A. K., Schulte C., Csoti I., Gasser T. and Berg D. (2011) GBA‐associated PD presents with nonmotor characteristics. Neurology 77, 276–280. [DOI] [PubMed] [Google Scholar]
- Bronstein S., Karpati M. and Peleg L. (2014) An update of Gaucher mutations distribution in the Ashkenazi Jewish population: prevalence and country of origin of the mutation R496H. Isr. Med. Assoc. J. 16, 683–685. [PubMed] [Google Scholar]
- Bultron G., Kacena K., Pearson D., Boxer M., Yang R., Sathe S., Pastores G. and Mistry P. K. (2010) The risk of Parkinson's disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 33, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. M., Kim W. C., Lyoo C. H. et al (2012) Association of mutations in the glucocerebrosidase gene with Parkinson disease in a Korean population. Neurosci. Lett. 514, 12–15. [DOI] [PubMed] [Google Scholar]
- Chu Y., Dodiya H. A., Ebischer P. O., lanow C. W. and Kordower J. H. (2009) Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha‐synuclein inclusions. Neurobiol. Dis. 35, 385–398. [DOI] [PubMed] [Google Scholar]
- Clark L. N., Ross B. M., Wang Y., Mejia‐Santana H., Harris J., Louis E. D., Cote L. J., Andrews H., Fahn S., Waters C., Ford B., Frucht S., Ottman R. and Marder K. (2007) Mutations in the glucocerebrosidase gene are associated with early‐onset Parkinson disease. Neurol. 69, 1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark L. N., Kartsaklis L. A., Wolf Gilbert R. et al (2009) Association of glucocerebrosidase mutations with dementia with Lewy bodies. Arch. Neurol. 66, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleeter M. W., Chau K. Y., Gluck C., Mehta A., Hughes D. A., Duchen M., Wood N. W., Hardy J., Mark Cooper J. and Schapira A. H. (2013) Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 62, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormand B., Montfort M., Chabas A., Vilageliu L. and Grinberg D. (1997) Genetic fine localization of the beta‐glucocerebrosidase (GBA) and prosaposin (PSAP) genes: implications for Gaucher disease. Hum. Genet. 100, 75–79. [DOI] [PubMed] [Google Scholar]
- Cox T. M. and Schofield J. P. (1997) Gaucher’ s disease: clinical features and natural history. Baillieres Clin. Haematol. 10, 657–689. [DOI] [PubMed] [Google Scholar]
- Cullen V., Sardi S. P., Ng J. et al (2011) Acid β‐glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α‐synuclein processing. Ann. Neurol. 69, 940–953. [DOI] [PubMed] [Google Scholar]
- De Marco E. V., Annesi G., Tarantino P. et al (2008) Glucocerebrosidase gene mutations are associated with Parkinson's disease in southern Italy. Mov. Disord. 23, 460–463. [DOI] [PubMed] [Google Scholar]
- Dehay B., Bové J., Rodríguez‐Muela N., Perier C., Recasens A., Boya P. and Vila M. (2010) Pathogenic lysosomal depletion in Parkinson ‘ s disease. J. Neurosci. 30, 12535–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermentzaki G., Dimitriou E., Xilouri M., Michelakakis H. and Stefanis L. (2013) Loss of β‐glucocerebrosidase activity does not affect alpha‐synuclein levels or lysosomal function in neuronal cells. PLoS ONE 8, e60674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir H., Harel M., McCarthy A. A., Toker L., Silman I., Futerman A. H. and Sussman J. L. (2003) X‐ray structure of human acid‐beta‐glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 4, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emelyanov A., Boukina T., Yakimovskii A., Usenko T., Drosdova A., Zakharchuk A., Andoskin P., Dubina M., Schwarzman A. and Pchelina S. (2012) Glucocerebrosidase gene mutations are associated with Parkinson's disease in Russia. Mov. Disord. 27, 158–159. [DOI] [PubMed] [Google Scholar]
- Enquist I. B., Lo Bianco C., Ooka A., Nilsson E., Månsson J. E., Ehinger M., Richter J., Brady R. O., Kirik D. and Karlsson S. (2007) Murine models of acute neuronopathic Gaucher disease. Proc. Natl Acad. Sci. USA 104, 17483–17488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfel‐Becker T., Vitner E., Dekel H., Leshem N., Enquist I. B., Karlsson S. and Futerman A. H. (2009) No evidence for activation of the unfolded protein response in neuronopathic models of Gaucher disease. Hum. Mol. Genet. 18, 1482–1488. [DOI] [PubMed] [Google Scholar]
- Gan‐Or Z., Giladi N., Rozovski U., Shifrin C., Rosner S., Gurevich T., Bar‐Shira A. and Orr‐Urtreger A. (2008) Genotype‐phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70, 2277–2283. [DOI] [PubMed] [Google Scholar]
- Gaucher P. C. E. (1882) De l’ epithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leucemie. Academic thesis Paris, France. [Google Scholar]
- Gegg M. E., Burke D., Heales S. J., Cooper J. M., Hardy J., Wood N. W. and Schapira A. H. (2012) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 72, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg M. E., Sweet L., Wang B. H., Shihabuddin L. S., Sardi S. P. and Schapira A. H. V. (2015) No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 30, 1085–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker‐Alpan O., Schiffmann R., Lamarca M. E., Nussbaum R. L., Mcinerney‐Leo A. and Sidransky E. (2004) Parkinsonism among Gaucher disease carriers. J. Med. Genet. 41, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker‐Alpan O., Lopez G., Vithayathil J., Davis J., Hallett M. and Sidransky E. (2008) The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch. Neurol. 65, 1353–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker‐Alpan O., Stubblefield B. K., Giasson B. I. and Sidransky E. (2010) Glucocerebrosidase is present in α‐synuclein inclusions in Lewy body disorders. Acta Neuropathol. 120, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker‐Alpan O., Masdeu J. C., Kohn P. D. et al (2012) The neurobiology of glucocerebrosidase‐associated parkinsonism: a positron emission tomography study of dopamine synthesis and regional cerebral blood flow. Brain 135, 2440–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Del Rincón Mde L., Monroy Jaramillo N., Suárez Martínez A. I., Yescas Gómez P., Boll Woehrlen M. C., López López M. and Alonso Vilatela M. E. (2013) The L444P GBA mutation is associated with early‐onset Parkinson's disease in Mexican Mestizos. Clin. Genet. 84, 386–387. [DOI] [PubMed] [Google Scholar]
- Grabowski G. A. (2008) Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 372, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Halperin A., Elstein D. and Zimran A. (2006) Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol. Dis. 36, 426–428. [DOI] [PubMed] [Google Scholar]
- Horowitz M., Wilder S., Horowitz Z., Reiner O., Gelbart T. and Beutler E. (1989) The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics 4, 87–96. [DOI] [PubMed] [Google Scholar]
- Hruska K. S., LaMarca M. E., Scott C. R. and Sidransky E. (2008) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 29, 567–583. [DOI] [PubMed] [Google Scholar]
- Hu F. Y., Xi J., Guo J., Yu L. H., Liu L., He X. H., Liu Z. L., Zou X. Y. and Xu Y. M. (2010) Association of the glucocerebrosidase N370S allele with Parkinson's disease in two separate Chinese Han populations of mainland China. Eur. J. Neurol. 17, 1476–1478. [DOI] [PubMed] [Google Scholar]
- Huang C. L., Wu‐Chou Y. H., Lai S. C., Chang H. C., Yeh T. H., Weng Y. H., Chen R. S., Huang Y. Z. and Lu C. S. (2011) Contribution of glucocerebrosidase mutation in a large cohort of sporadic Parkinson's disease in Taiwan. Eur. J. Neurol. 18, 1227–1232. [DOI] [PubMed] [Google Scholar]
- Imai Y., Soda M. and Takahashi R. (2000) Parkin suppresses unfolded protein stress‐induced cell death through its E3 ubiquitin‐protein ligase activity. J. Biol. Chem. 205, 35661–35664. [DOI] [PubMed] [Google Scholar]
- Imai Y., Soda M., Inoue H., Hattori N., Mizuno Y. and Takahashi R. (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105, 891–902. [DOI] [PubMed] [Google Scholar]
- Jovic M., Kean M. J., Szentpetery Z., Polevoy G., Gingras A. C., Brill J. A. and Balla T. (2012) Two phosphatidylinositol 4‐kinases control lysosomal delivery of the Gaucher disease enzyme, beta‐glucocerebrosidase. Mol. Biol. Cell 23, 1533–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinderi K., Bostantjopoulou S., Paisan‐Ruiz C., Katsarou Z., Hardy J. and Fidani L. (2009) Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Greece. Neurosci. Lett. 452, 87–89. [DOI] [PubMed] [Google Scholar]
- Kumar K. R., Ramirez A., Göbel A. et al (2013) Glucocerebrosidase mutations in a Serbian Parkinson's disease population. Eur. J. Neurol. 20, 402–405. [DOI] [PubMed] [Google Scholar]
- Lesage S. and Brice A. (2009) Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18, R48–R59. [DOI] [PubMed] [Google Scholar]
- Lesage S., Anheim M., Condroyer C. et al ; French Parkinson's Disease Genetics Study Group (2011) Large‐scale screening of the Gaucher's disease‐related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum. Mol. Genet. 20, 202–210. [DOI] [PubMed] [Google Scholar]
- Luan Z., Li L., Higaki K., Nanba E., Suzuki Y. and Ohno K. (2013) The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev. 35, 317–322. [DOI] [PubMed] [Google Scholar]
- Lwin A., Orvisky E., Goker‐Alpan O., Lamarca M. E. and Sidransky E. (2004) Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 81, 70–73. [DOI] [PubMed] [Google Scholar]
- Maegawa G. H., Tropak M. B., Buttner J. D. et al (2009) Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 284, 23502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning‐Boğ A. B., Schüle B. and Langston J. W. (2009) Alpha‐synuclein‐glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 30, 1127–1132. [DOI] [PubMed] [Google Scholar]
- Mao X. Y., Burgunder J. M., Zhang Z. J., An X. K., Zhang J. H., Yang Y., Li T., Wang Y. C., Chang X. L. and Peng R. (2010) Association between GBA L444P mutation and sporadic Parkinson's disease from Mainland China. Neurosci. Lett. 469, 256–259. [DOI] [PubMed] [Google Scholar]
- Mata I. F., Samii A., Schneer S. H. et al (2008) Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch. Neurol. 65, 379–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli J. R., Xu Y. H., Sun Y., Knight A. L., McLean P. J., Caldwell G. A., Sidransky E., Grabowski G. A. and Krainc D. (2011) Gaucher disease glucocerebrosidase and α‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeran R. O., Bradbury P., Taylor D. and Stern G. (1985) Neurological involvement of type 1 (adult) Gaucher's disease. J. Neurol. Neurosurg. Psychiatry 48, 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A., Duran R., Hughes D. A., Mehta A. and Schapira A. H. (2012a) A clinical and family history study of Parkinson's disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 83, 853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A., Duran R., Proukakis C., Bras J., Hughes D., Mehta A., Hardy J., Wood N. W. and Schapira A. H. (2012b) Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov. Disord. 27, 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A., Roberti G., Lascaratos G., Hughes D., Mehta A., Garway‐Heath D. F. and Schapira A. H. (2013a) Retinal thinning in Gaucher disease patients and carriers: results of a pilot study. Mol. Genet. Metab. 109, 221–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A., Wu R. M., Tzen K. Y. et al (2013b) Dopaminergic neuronal imaging in genetic Parkinson's disease: insights into pathogenesis. PLoS ONE 8, e69190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A., Magalhaes J., Shen C. et al (2014) Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation‐linked Parkinson disease cells. Brain 137, 1481–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraitou M., Hadjigeorgiou G., Monopolis I. et al (2011) β‐glucocerebrosidase gene mutations in two cohorts of Greek patients with sporadic Parkinson's disease. Mol. Genet. Metab. 104, 149–152. [DOI] [PubMed] [Google Scholar]
- National Organization of Rare Disorders (2013) Gaucher disease. http://www.rarediseases.org/rare-disease-information/rarediseases/byID/12/viewFullReport. [accessed on July 25, 2013].
- Neumann J., Bras J., Deas E. et al (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 132, 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osellame L. D., Rahim A. A., Hargreaves I. P., Gegg M. E., Richard‐Londt A., Brandner S., Waddington S. N., Schapira A. H. and Duchen M. R. (2013) Mitochondria and quality control defects in a mouse model of Gaucher disease — links to Parkinson's disease. Cell Metab. 17, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik S., Zheng W., Choi J. H. et al (2012) Discovery, structure‐activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J. Med. Chem. 55, 5734–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkes T., Choubtum L., Chitphuk S., Thakkinstian A., Pongpakdee S., Kulkantrakorn K., Hanchaiphiboolkul S., Tiamkao S. and Boonkongchuen P. (2014) Glucocerebrosidase mutations in Thai patients with Parkinson's disease. Parkinsonism Relat. Disord. 20, 986–991. [DOI] [PubMed] [Google Scholar]
- Reczek D., Schwake M., Schroder J., Hughes H., Blanz J., Jin X., Brondyk W., Van Patten S., Edmunds T. and Saftig P. (2007) LIMP‐2 is a receptor for lysosomal mannose‐6‐phosphate‐independent targeting of beta‐glucocerebrosidase. Cell 131, 770–783. [DOI] [PubMed] [Google Scholar]
- Richter F., Fleming S. M., Watson M. et al (2014) A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 11, 840–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha E. M., Smith G. A., Park E., Cao H., Brown E., Hallett P. and Isacson O. (2015) Progressive decline of glucocerebrosidase in aging and Parkinson's disease. Ann. Clin. Transl. Neurol. 2, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron I. and Horowitz M. (2005) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 14, 2387–2398. [DOI] [PubMed] [Google Scholar]
- Ron I., Rapaport D. and Horowitz M. (2010) Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum. Mol. Genet. 19, 3771–3781. [DOI] [PubMed] [Google Scholar]
- Ryu E. J., Harding H. P., Angelastro J. M., Vitolo O. V., Ron D. and Greene L. A. (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J. Neurosci. 22, 10690–10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders A., Hemmelgarn H., Melrose H. L., Hein L., Fuller M. and Clarke L. A. (2013) Transgenic mice expressing human glucocerebrosidase variants: utility for the study of Gaucher disease. Blood Cells Mol. Dis. 51, 109–115. [DOI] [PubMed] [Google Scholar]
- Sardi S. P., Clarke J., Viel C. et al (2013) Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other gaucher‐related synucleinopathies. Proc. Natl Acad. Sci. USA 110, 3537–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato C., Morgan A., Lang A. E., Salehi‐Rad S., Kawarai T., Meng Y., Ray P. N., Farrer L. A., St George‐Hyslop P. and Rogaeva E. (2005) Analysis of the glucocerebrosidase gene in Parkinson's disease. Mov. Disord. 20, 367–370. [DOI] [PubMed] [Google Scholar]
- Sawkar A. R., Cheng W. C., Beutler E., Wong C. H., Balch W. E. and Kelly J. W. (2002) Chemical chaperones increase the cellular activity of N370S beta‐glucosidase: a therapeutic strategy for Gaucher disease. Proc. Natl Acad. Sci. USA 99, 15428–15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira A. H. (2008) Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 7, 97–109. [DOI] [PubMed] [Google Scholar]
- Schapira A. H. and Gegg M. E. (2013) Glucocerebrosidase in the pathogenesis and treatment of Parkinson disease. Proc. Natl Acad. Sci. USA 110, 3214–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira A. H. and Tolosa E. (2010) Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat. Rev. Neurol. 6, 309–317. [DOI] [PubMed] [Google Scholar]
- Schapira A. H., Cooper J. M., Dexter D., Jenner P., Clark J. B. and Marsden C. D. (1989) Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1, 1269. [DOI] [PubMed] [Google Scholar]
- Schapira A. H., Cooper J. M., Dexter D., Clark J. B., Jenner P. and Marsden C. D. (1990) Mitochondrial complex I deficiency in Parkinson's disease. J. Neurochem. 54, 823–827. [DOI] [PubMed] [Google Scholar]
- Setó‐Salvia N., Pagonabarraga J., Houlden H. et al (2012) Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson's disease course. Mov. Disord. 27, 393–399. [DOI] [PubMed] [Google Scholar]
- Sidransky E. (2012) Gaucher disease: insights from a rare Mendelian disorder. Discov. Med. 14, 273–281. [PMC free article] [PubMed] [Google Scholar]
- Sidransky E. and Lopez G. (2012) The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E., Nalls M. A., Aasly J. O. et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med. 361, 1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge J., West C., Westwood B. and Beutler E. (1985) Molecular cloning and nucleotide sequence of human glucocerebrosidase cDNA. Proc. Natl Acad. Sci. USA 82, 7289–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge J., Kuhl W., West C. and Beutler E. (1987a) Complete correction of the enzymatic defect of type I Gaucher disease fibroblasts by retroviral‐mediated gene transfer. Proc. Natl Acad. Sci. USA 84, 906–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge J. A., West C., Kuhl W., Treger L. and Beutler E. (1987b) The human glucocerebrosidase gene has two functional ATG initiator codons. Am. J. Hum. Genet. 41, 1016–1024. [PMC free article] [PubMed] [Google Scholar]
- Sorge J., Gross E., West C. and Beutler E. (1990) High level transcription of the glucocerebrosidase pseudogene in normal subjects and patients with Gaucher disease. J. Clin. Invest. 86, 1137–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz M., Rozenberg R., Pereira Lda. V. and Reis Barbosa E. (2008) Association between Parkinson's disease and glucocerebrosidase mutations in Brazil. Parkinsonism Relat. Disord. 14, 58–62. [DOI] [PubMed] [Google Scholar]
- Steet R. A., Chung S., Wustman B., Powe A., Do H. and Kornfeld S. A. (2006) The iminosugar isofagomine increases the activity of N370S mutant acid beta‐glucosidase in Gaucher fibroblasts by several mechanisms. Proc. Natl Acad. Sci. USA 103, 13813–13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone D. L., Tayebi N., Orvisky E., Stubblefield B., Madike V. and Sidransky E. (2000) Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum. Mutat. 15, 181–188. [DOI] [PubMed] [Google Scholar]
- Sun Q. Y., Guo J. F., Wang L., Yu R. H., Zuo X., Yao L. Y., Pan Q., Xia K. and Tang B. S. (2010a) Glucocerebrosidase gene L444P mutation is a risk factor for Parkinson's disease in Chinese population. Mov. Disord. 25, 1005–1011. [DOI] [PubMed] [Google Scholar]
- Sun Y., Liou B., Ran H. et al (2010b) Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 19, 1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y., Ogawa S. and Sakakibara Y. (2009) Chaperone therapy for neuronopathic lysosomal diseases: competitive inhibitors as chemical chaperones for enhancement of mutant enzyme activities. Perspect. Medicin. Chem. 3, 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan M. and Saunders‐Pullman R. (2013) The association between ß‐glucocerebrosidase mutations and parkinsonism. Curr. Neurol. Neurosci. Rep. 13, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamboli I. Y., Hampel H., Tien N. T., Tolksdorf K., Breiden B., Mathews P. M., Saftig P., Sandhoff K. and Walter J. (2011) Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Abeta generation. J. Neurosci. 31, 1837–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E. K., Tong J., Fook‐Chong S., Yih Y., Wong M. C., Pavanni R. and Zhao Y. (2007) Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch. Neurol. 64, 1056–1058. [DOI] [PubMed] [Google Scholar]
- Tayebi N., Cushner S. and Sidransky E. (1996) Differentiation of the glucocerebrosidase gene from pseudogene by long‐template PCR: implications for Gaucher disease. Am. J. Hum. Genet. 59, 740–741. [PMC free article] [PubMed] [Google Scholar]
- Tayebi N., Callahan M., Madike V., Stubblefield B. K., Orvisky E., Krasnewich D., Fillano J. J. and Sidransky E. (2001) Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol. Genet. Metab. 73, 313–321. [DOI] [PubMed] [Google Scholar]
- Tayebi N., Walker J., Stubblefield B., Orvisky E., Lamarca M. E., Wong K., Rosenbaum H., Schiffmann R., Bembi B. and Sidransky E. (2003) Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 79, 104–109. [DOI] [PubMed] [Google Scholar]
- Toft M., Pielsticker L., Ross O. A., Aasly J. O. and Farrer M. J. (2006) Glucocerebrosidase gene mutations and Parkinson disease in the Norwegian population. Neurology 66, 415–417. [DOI] [PubMed] [Google Scholar]
- Turpin J. C., Dubois G., Brice A., Masson M., Nadaud M. C., Boutry J. M., Schram A. W., Tager J. M. and Baumann N. (1988) Parkinsonian symptomatology in a patient with type I (adult) Gaucher's disease. Lipid Storage Disord. NATO ASI Ser. 150, 103–105. [Google Scholar]
- Vaccaro A. M., Motta M., Tatti M., Scarpa S., Masuelli L., Bhat M., Vanier M. T., Tylki‐Szymanska A. and Salvioli R. (2010) Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum. Mol. Genet. 19, 2987–2997. [DOI] [PubMed] [Google Scholar]
- Varkonyi J., Rosenbaum H., Baumann N., Mackenzie J. J., Simon Z., Aharon‐Peretz J., Walker J. M., Tayebi N. and Sidransky E. (2003) Gaucher disease associated with parkinsonism: four further case reports. Am. J. Med. Genet. A 116A, 348–351. [DOI] [PubMed] [Google Scholar]
- Vila M., Bové J., Dehay B., Rodríguez‐Muela N. and Boya P. (2011) Lysosomal membrane permeabilization in Parkinson disease. Autophagy 7, 98–100. [DOI] [PubMed] [Google Scholar]
- Wang Y., Liu L., Xiong J. et al (2012) Glucocerebrosidase L444P mutation confers genetic risk for Parkinson's disease in central China. Behav. Brain Funct. 1, 8–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbroek W., Gustafson A. M. and Sidransky E. (2011) Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 17, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder‐Rhodes S. E., Evans J. R., Ban M., Mason S. L., Williams‐Gray C. H., Foltynie T., Duran R., Mencacci N. E., Sawcer S. J. and Barker R. A. (2013) Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community‐based incident cohort. Brain 136, 392–399. [DOI] [PubMed] [Google Scholar]
- Wu Y. R., Chen C. M., Chao C. Y., Ro L. S., Lyu R. K., Chang K. H. and Lee‐Chen G. J. (2007) Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J. Neurol. Neurosurg. Psychiatry 78, 977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M., Vogiatzi T., Vekrellis K., Park D. and Stefanis L. (2009) Abberant alpha‐synuclein confers toxicity to neurons in part through inhibition of chaperone‐mediated autophagy. PLoS ONE 4, e5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. H., Xu K., Sun Y. et al (2014) Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 23, 3943–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap T. L., Gruschus J. M., Velayati A., Westbroek W., Goldin E., Moaven N., Sidransky E. and Lee J. C. (2011) Alpha‐synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J. Biol. Chem. 286, 28080–28088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H. (2007) ER stress and diseases. FEBS J. 274, 630–658. [DOI] [PubMed] [Google Scholar]
- Yu Z., Wang T., Xu J. et al (2015) Mutations in the glucocerebrosidase gene are responsible for Chinese patients with Parkinson's disease. J. Hum. Genet. 60, 85–90. [DOI] [PubMed] [Google Scholar]
- Zhang X., Bao Q. Q., Zhuang X. S. et al (2012) Association of common variants in the glucocerebrosidase gene with high susceptibility to Parkinson's disease among Chinese. Chin. J. Physiol. 55, 398–404. [DOI] [PubMed] [Google Scholar]
- Ziegler S. G., Eblan M. J., Gutti U., Hruska K. S., Stubblefield B. K., Goker‐Alpan O., LaMarca M. E. and Sidransky E. (2007) Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol. Genet. Metab. 91, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zokaei N., McNeill A., Proukakis C. et al (2014) Visual short‐term memory deficits associated with GBA mutation and Parkinson's disease. Brain 137(Pt 8), 2303–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]