

Abstract
Prostate cancer is the most common cancer in men in developed countries, and is a target for risk reduction strategies. The effects of alcohol consumption on prostate cancer incidence and survival remain unclear, potentially due to methodological limitations of observational studies. In this study, we investigated the associations of genetic variants in alcohol‐metabolising genes with prostate cancer incidence and survival. We analysed data from 23,868 men with prostate cancer and 23,051 controls from 25 studies within the international PRACTICAL Consortium. Study‐specific associations of 68 single nucleotide polymorphisms (SNPs) in 8 alcohol‐metabolising genes (Alcohol Dehydrogenases (ADHs) and Aldehyde Dehydrogenases (ALDHs)) with prostate cancer diagnosis and prostate cancer‐specific mortality, by grade, were assessed using logistic and Cox regression models, respectively. The data across the 25 studies were meta‐analysed using fixed‐effect and random‐effects models. We found little evidence that variants in alcohol metabolising genes were associated with prostate cancer diagnosis. Four variants in two genes exceeded the multiple testing threshold for associations with prostate cancer mortality in fixed‐effect meta‐analyses. SNPs within ALDH1A2 associated with prostate cancer mortality were rs1441817 (fixed effects hazard ratio, HRfixed = 0.78; 95% confidence interval (95%CI):0.66,0.91; p values = 0.002); rs12910509, HRfixed = 0.76; 95%CI:0.64,0.91; p values = 0.003); and rs8041922 (HRfixed = 0.76; 95%CI:0.64,0.91; p values = 0.002). These SNPs were in linkage disequilibrium with each other. In ALDH1B1, rs10973794 (HRfixed = 1.43; 95%CI:1.14,1.79; p values = 0.002) was associated with prostate cancer mortality in men with low‐grade prostate cancer. These results suggest that alcohol consumption is unlikely to affect prostate cancer incidence, but it may influence disease progression.
Keywords: alcohol, prostate cancer, alcohol metabolising genes, Mendelian randomisation
Short abstract
What's new?
Alcohol may spur prostate cancer progression, though it does not appear to affect incidence, according to new analysis. Variation in genes involved in alcohol metabolism affect how much the body is exposed to carcinogenic metabolites. These authors examined 68 genetic variants in alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) genes, seeking a link with prostate cancer risk. While they found no evidence that these variants alter prostate cancer incidence, they did show that SNPs in the ALDH1A2 gene affect prostate cancer mortality. From a public health standpoint, these results suggest reducing alcohol consumption could slow prostate cancer disease progression.
Abbreviations
- ADH
alcohol dehydrogenase
- ALDH
aldehyde dehydrogenase
- CI
confidence interval
- HR
hazard ratio
- LD
linkage disequilibrium;
- OR
odds ratio
- PRACTICAL
PRostate cancer AssoCiation group to Investigate Cancer‐associated ALterations in the genome
- SNP
single nucleotide polymorphism
Introduction
Prostate cancer is the most common cancer in men in developed countries, with 758,700 new cases diagnosed and 142,000 deaths in 2012.1 With increasing uptake of prostate‐specific antigen (PSA) testing and the ageing population, prostate cancer incidence is increasing.2 The factors influencing prostate cancer incidence and survival after diagnosis are poorly understood, therefore more evidence is needed.3
Alcohol is a carcinogen associated with oropharyngeal, liver, breast, colorectal and oesophageal cancers.4 Functional variation in the genes involved in alcohol metabolism result in altered exposure to the carcinogenic metabolites of ethanol, suggesting a mechanism for genetic sensitivity to alcohol to influence the pathogenesis of cancers.5 For example, populations with an increased prevalence of common genetic variation in the alcohol dehydrogenase gene, that results in reduced enzyme activity, have an increased risk of oesophageal cancer compared with populations with the fully active enzyme.4
At present the role of alcohol use on prostate cancer remains uncertain. The World Cancer Research Fund's extensive report based on systematic reviews described the evidence as limited and inconclusive,6 and the International Association for Research on Cancer did not list this cancer site amongst others more apparently caused by alcohol in their Monograph on alcohol's carcinogenicity.4 There have been conflicting reports of possible associations of alcohol with various stages or histological grades of prostate cancer,7, 8, 9, 10, 11, 12 and meta‐analyses have highlighted the inconsistencies, emphasising the need for further research in this area.13, 14
The majority of evidence about the effects of alcohol on prostate cancer is from observational studies. One potential limitation of traditional observational research is that the findings can potentially be explained by common causes of both exposure and outcome (confounding factors). Other potential sources of bias are reverse causation and recall bias, where having prostate cancer affects drinking behaviour or its reporting, rather than alcohol consumption increasing the risk of prostate cancer. A prospective study design could mitigate both of these problems, but could still be affected by bias in the form of the “sick quitter” effect, where former heavy drinkers reduce their alcohol intake in middle‐age because of comorbidities that may be alcohol‐related.
Mendelian randomisation is an approach that uses genetic variants robustly associated with exposures of interest, or their metabolic effects, as instrumental variables to test the un‐confounded and unbiased causal effects of those exposures and their metabolic effects with cancer.15 Mendelian randomisation analyses rely on two approximate laws of Mendelian genetics,15, 16 that at meiosis alleles segregate without any influence of environmental factors and that the inheritance of one trait is independent of the inheritance of others. This allows genetic variation to be used in epidemiological studies as an un‐confounded proxy for an environmental exposure,15, 16, 17 in this case alcohol consumption, to estimate the influence of cumulative life‐time risk of exposure, to reduce recall bias and the “sick‐quitter” effect and to negate reverse causation. All these features are limitations of previous conventional observational studies.7, 8, 13 Mendelian randomisation has already been used successfully in both cardiovascular18, 19 and cancer epidemiology20, 21, 22, 23, 24 to clarify the causal effects of alcohol on disease.
In this study, we undertook Mendelian randomisation analyses in which we used variants in alcohol metabolising genes influencing metabolism and intake, to test the causal effect of alcohol exposure on prostate cancer risk and progression. The motivation is that if alcohol intake causally increases prostate cancer risk or progression, then genetic variants associated with metabolic effects of alcohol or increased intake will be differentially represented in cases and controls. We stratified the analysis by histological prostate cancer grade, based on Gleason score, as low‐ and high‐grade prostate cancers have differing natural histories which could be influenced by different risk factors.
Material and Methods
Study populations
We used phenotypic and genotypic data from 46,919 men (23,868 cases) in the international Prostate cancer association group to investigate cancer‐associated alterations in the genome (PRACTICAL) consortium. Data were provided by 25 studies within the consortium, based in USA, Australia and European countries. This study population was limited to those of European ethnicity. The studies used a number of methods of recruitment, including screen and clinically detected cases and participants selected due to a family history of prostate cancer. The background characteristics of the participants of each study are shown in Table 1. Gleason scores were used to categorise cancers as low grade (Gleason score ≤6) or high grade (Gleason score ≥7). Further details are available from the consortium website (practical.ccge.medschl.cam.ac.uk). All studies adhered both to national ethical guidelines and to the principles of the Declaration of Helsinki.
Table 1.
Background information on participants contributing to the PRACTICAL Consortium by study
| Age at diagnosis | PSA level at diagnosis (ng/ml) | Family history | Gleason score | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study | Country | Controls | Cases | Mean | SD | Median | Lower quartile | Upper quartile | of disease | 8–10 |
| CAPS | Sweden | 664 | 1,153 | 66.10 | 7.75 | 13.0 | 7.0 | 30.0 | 17.35% | 15.26% |
| CPCS1 | Denmark | 2,771 | 848 | 69.51 | 7.91 | 15.0 | 8.0 | 37.5 | 8.21% | 26.65% |
| CPCS2 | Denmark | 1,009 | 265 | 64.88 | 6.82 | 9.0 | 6.0 | 14.5 | 14.72% | 9.06% |
| EPIC | Europe | 1,079 | 722 | 64.87 | 5.62 | 8.6 | 6.0 | 15.9 | – | 2.22% |
| EPIC‐Norfolk | UK | 917 | 484 | 72.08 | 7.56 | 19.8 | 19.8 | 19.8 | 2.48% | 1.86% |
| ESTHER | Germany | 318 | 313 | 65.52 | 5.09 | 6.9 | 5.0 | 14.0 | 10.54% | 8.63% |
| FHCRC | USA | 730 | 761 | 59.73 | 7.18 | 6.4 | 4.7 | 9.8 | 21.68% | 10.38% |
| IPO‐Porto | Portugal | 66 | 183 | 59.33 | 5.23 | 7.4 | 5.5 | 10.1 | 20.00% | 15.85% |
| MAYO | USA | 488 | 767 | 65.24 | 6.42 | 7.8 | 4.9 | 14.7 | 29.07% | 28.42% |
| MCCS | Australia | 1,170 | 1,698 | 58.45 | 8.46 | 5.4 | 0.0 | 11.4 | 23.45% | 10.31% |
| MEC | USA | 829 | 819 | 69.53 | 7.62 | – | – | – | 13.03% | 34.55% |
| MOFFITT | USA | 100 | 414 | 64.97 | 8.27 | 5.6 | 4.3 | 7.4 | 22.76% | 11.11% |
| PCMUS | Bulgaria | 140 | 151 | 69.27 | 8.71 | 15.8 | 7.4 | 34.0 | 5.30% | 29.80% |
| PPF‐UNIS | UK | 188 | 245 | 68.86 | 7.57 | 8.6 | 6.3 | 14.0 | 25.22% | 9.39% |
| Poland | Poland | 359 | 438 | 67.66 | 7.84 | 11.0 | 6.9 | 26.0 | 10.57% | 11.42% |
| ProMPT | UK | 0 | 166 | 66.33 | 8.64 | 8.8 | 5.7 | 15.3 | 34.62% | 16.87% |
| ProtecT | UK | 1,474 | 1,542 | 62.76 | 5.11 | 5.1 | 3.8 | 8.2 | 7.91% | 5.64% |
| QLD | Australia | 87 | 186 | 61.32 | 6.91 | 5.2 | 2.2 | 7.5 | 36.18% | 3.76% |
| SEARCH | UK | 1,244 | 1,371 | 63.08 | 4.76 | 8.8 | 5.6 | 15.0 | 16.24% | 10.14% |
| STHM1 | Sweden | 2,224 | 2,006 | 66.17 | 6.99 | – | – | – | 20.18% | 7.93% |
| TAMPERE | Finland | 2,413 | 2,754 | 68.18 | 7.96 | 8.6 | 5.6 | 16.3 | – | 13.76% |
| UKGPCS | UK | 4,182 | 4,549 | 63.76 | 7.97 | 9.8 | 5.6 | 24.7 | 23.42% | 14.13% |
| ULM | Germany | 354 | 603 | 63.78 | 6.66 | 9.0 | 6.0 | 15.1 | 44.94% | 12.11% |
| UTAH | USA | 245 | 440 | 62.57 | 8.85 | – | – | – | 51.36% | 15.45% |
| WUGS | USA | 0 | 990 | 60.80 | 7.03 | 5.0 | 4.0 | 7.0 | 42.43% | 7.88% |
Abbreviations: SD, standard deviation; PSA, prostate‐specific antigen.
Genotyping data
The participants were genotyped using a custom Illumina Infinium genotyping array (iCOGS), which was specifically designed for the Collaborative Oncological Gene‐environmental Study (COGS) and recorded 211,155 SNPs (details available from: http://ec.europa.eu/research/health/medical-research/cancer/fp7-projects/cogs_en.html).25, 26 The iCOGS array was designed to investigate SNPs in regions thought to be associated with breast, ovarian and prostate cancer; 68,638 of the SNPs on the array were chosen because of their potential role in prostate cancer aetiology. The other 125,877 SNPs were selected on the basis of potential importance for other cancers and common SNPs known to be associated with any other traits. Individuals with fewer than 95% of genotypes called, or high or low heterozygosity (p < 1 × 10−5) were dropped from our analysis. In total 201,598 SNPs passed quality control. The genotypic data were used to impute SNPs which were not directly genotyped, but were in linkage disequilibrium (LD) with genotyped SNPs. We used the HapMap 2 CEU reference panel and IMPUTE2 software.27
In this study, we searched the iCOGS array database for all alcohol‐metabolising genetic variants (within Alcohol Dehydrogenases (ADHs) or Aldehyde Dehydrogenases (ALDHs) genes), and identified 68 common variants in 5 distinct genomic regions: the ADH cluster on chromosome 4, comprising ADH1A, ADH1B, ADH1C and ADH7; ALDH1A1 and ALDH 1B1 on chromosome 9; ALDH1A2 and ALDH1A3 on chromosome 15. Of these SNPs, 67 were directly genotyped, and one was imputed. An overview of the genes' role in alcohol metabolism and behaviour is shown in Supplementary material Table S1. The characteristics of the SNPs included in this study are shown in Supplementary material Table S2.
Statistical analysis
We converted the genotypic data for each SNP into a count of the number of minor alleles at each locus. We used logistic regression to estimate the associations of the SNPs with prostate cancer risk (cases vs. controls), for all cases and stratified by high‐ vs. low‐grade disease. In a case‐only analysis, we used Cox proportional hazards regression to estimate associations of each SNP with prostate cancer‐specific mortality, stratified by grade of prostate cancer. All regression analyses were adjusted for the first 8 principal components of population stratification, since these genomic regions show marked variation across different populations and so do prostate cancer incidence and survival. When checking the proportional hazards assumption, we found little evidence of violation. These regressions were performed for each study and then meta‐analysed using both fixed‐ and random‐effects models. Studies were excluded from the survival meta‐analysis if there were <5 deaths during the follow up period or <90% completion of follow up data. We investigated between‐study heterogeneity using the Stata metan command to estimate the I 2 statistic assuming a fixed‐effect model; we also report random‐effect models for completeness as such models may be relevant where I 2 values are high (e.g., >75%).28 Using meta‐regression, we investigated whether heterogeneity could be explained by the following a priori defined study‐specific characteristics: mean age at diagnosis, mean PSA at diagnosis, country of study (USA vs. elsewhere), and the percentage of participants with a family history of prostate cancer. Manhattan plots of the associations in the five chromosomal regions were constructed to identify SNPs exceeding the Nyholt corrected p values threshold for association —a multiple testing correction which accounts for LD between the SNPs.29
Sensitivity analyses were conducted by reclassifying low‐ and high‐grade disease as <8 and 8–10 Gleason grade, respectively.
The power of our study was also assessed using reverse power calculations to demonstrate the effect size we would expect to detect given our sample size and α = 0.05 with SNPs of a range of minor allele frequencies.30 The analysis was carried out using Stata v.13.1. The statistical code used to produce these results can be accessed here (https://github.com/nmdavies/practical-alcohol/).
Results
The background characteristics of the participants are summarised in Table 1, by study. Variation between studies reflects their individual recruitment methods (e.g., some studies selected for those men with a positive family history of prostate cancer).
The Manhattan plots with results from both fixed and random effects meta‐analyses testing associations between SNPs in the five genomic regions and prostate cancer risk are presented in Figure 1 (detailed results available in Supplementary material Tables S4–S9, and sensitivity analyses with alternative definitions of low‐ and high‐grade presented in Supplementary material Tables S16–S19). The figure and Supplementary material tables show that no SNP exceeded the Nyholt corrected p values threshold for association with prostate cancer risk.
Figure 1.
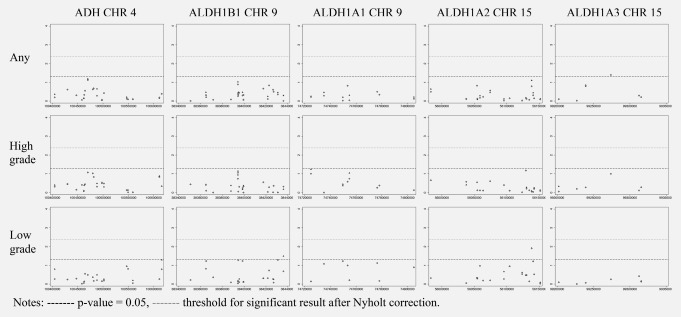
Manhattan plots of association of SNPs, in 5 regions involved in alcohol metabolism, with Prostate Cancer Diagnosis by Prostate Cancer grade.
In case‐only analyses, four SNPs exceeded the Nyholt corrected p values threshold for association with prostate cancer‐specific mortality in the fixed‐effect meta‐analysis (summary Manhattan plots presented in Figure 2, and individual SNP results presented in Supplementary material Tables S10–S15, with results of sensitivity analyses with alternative definitions of low‐ and high‐grade presented in Supplementary material Tables S20–S23). Three SNPs within ALDH1A2 were associated with prostate cancer mortality following diagnosis with any prostate cancer: rs1441817 (fixed effects hazard ratio, HRfixed = 0.78; 95% confidence interval (95%CI):0.66,0.91, p values = 0.002, I 2 = 19.4); rs12910509, HRfixed = 0.76; 95%CI:0.64,0.91, p values = 0.003, I 2 = 23.0); and rs8041922 (HRfixed = 0.76; 95%CI:0.64,0.91, p values = 0.002, I 2 = 25.5). To identify the top independent signal amongst these three, we conducted jointly adjusted analyses. Levels of pairwise LD were too high to attempt study‐specific analyses (pairwise LD rs1441817 and rs12910509 r 2 = 0.89, rs1441817 and rs8041922 r 2 = 0.88, rs12910509 and rs8041922 r 2 = 0.99).31 Pooled analyses were conducted to estimate the joint effects of rs1441817 and rs12910509/rs8041922 (r 2 = 0.99) on prostate cancer survival, with a random effect correction for standard errors. These showed an independent effect of rs1441817, similar in size to that of univariate analyses, but no independent effect of rs12910509/rs8041922 once adjusting for rs1441817. Given the high LD between the three SNPs, they should be taken as representing one underlying genetic signal. Figure 3 presents the forest plot of individual studies contributing to the meta‐analysis of prostate cancer‐specific survival in association with the top independent signal in ALDH1A2. A fourth SNP, rs10973794 in ALDH1B1 (also close to IGFBPL1), was associated with prostate cancer mortality in men with a diagnosis of low‐grade prostate cancer (HRfixed = 1.43; 95%CI:1.14,1.79, p values = 0.002, I 2 = 23.4, Fig. 4). This result was robust to changing the low‐grade definition to <8 (HRfixed = 1.23; 95% CI:1.06,1.41, p values = 0.002, I 2 = 0, Supplementary material Table S22).
Figure 2.
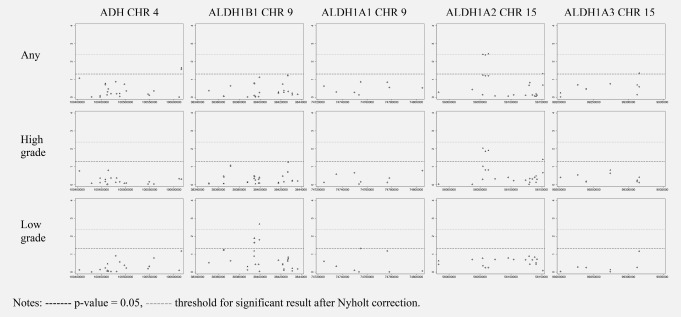
Manhattan plots of association of SNPs, in 5 regions involved in alcohol metabolism, with prostate cancer‐specific survival by prostate Cancer grade.
Figure 3.
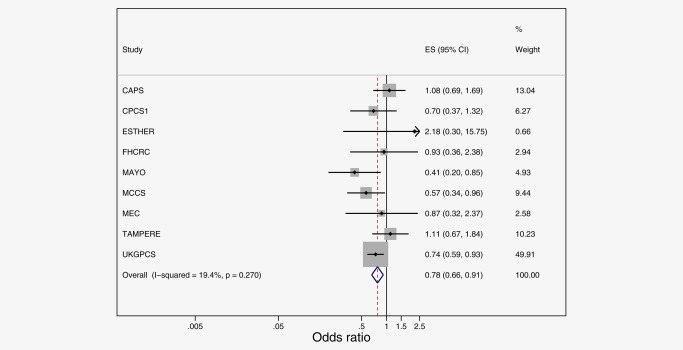
Meta‐analysis of prostate cancer‐specific survival following a diagnosis of any prostate cancer, in association with rs1441817 in ALDH1A2. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 4.
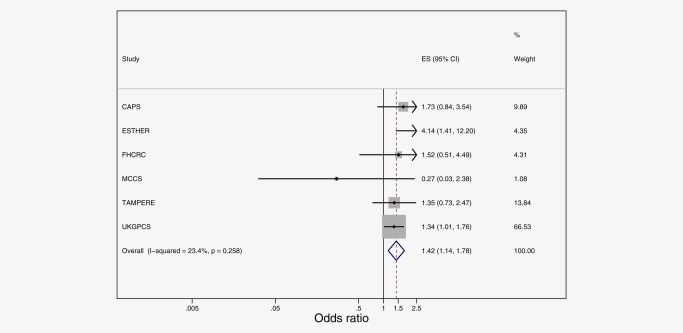
Meta‐analysis of prostate cancer‐specific survival following a diagnosis of low grade prostate cancer, in association with rs10973794 in ALDH1B1. [Color figure can be viewed at wileyonlinelibrary.com]
In general, random effects meta‐analyses yielded weaker evidence of association than fixed‐effect models, as one would expect due to variability across studies producing larger confidence intervals for the former. However, levels of heterogeneity as quantified by the I 2 statistics were low, with point estimates remarkably similar across the two types of meta‐analysis. Meta‐regression analyses found limited evidence that the study‐level characteristics examined had a strong influence on the pooled results (Table 2).
Table 2.
Results of univariate meta‐regressions to test if the association of the two SNPs (representing the two signals observed) is affected by selected study characteristics
| Confidence intervals | |||||
|---|---|---|---|---|---|
| Single nucleotide polymorphism | Study characteristic | Ratio of odds‐ratios | Lower | Upper | p Values |
| rs1441817 | PSA | 1.48 | 0.75 | 2.93 | 0.19 |
| (ALDH1A2) | FHX | 0.63 | 0.35 | 1.14 | 0.10 |
| USA | 0.69 | 0.30 | 1.57 | 0.30 | |
| Age | 1.58 | 1.00 | 2.51 | 0.05 | |
| rs10973794 | PSA | 0.65 | 0.23 | 1.80 | 0.33 |
| (ALDH1B1) | FHX | 0.65 | 0.25 | 1.68 | 0.27 |
| USA | 1.30 | 0.29 | 5.72 | 0.65 | |
| Age | 1.30 | 0.62 | 2.72 | 0.40 | |
PSA, mean PSA at diagnosis.
FHX, percentage family history.
USA, study location in USA vs. rest of world.
Age, mean age at diagnosis.
Discussion
Using data from the PRACTICAL Consortium, we pooled data from 25 studies including a total of 23,868 prostate cancer cases and 23,091 controls, to investigate the association of 68 SNPs within genes thought to be involved with alcohol metabolism with prostate cancer risk and prostate cancer‐specific mortality (amongst men diagnosed with prostate cancer), overall and by Gleason grade. After correcting for multiple testing in the fixed‐effect meta‐analysis, no SNPs exceed the Nyholt threshold for association with a diagnosis of prostate cancer, whereas three SNPs in ALDH1A2 (in strong LD with each other, therefore representing one signal only) exceed the Nyholt threshold for association with prostate cancer‐specific survival. One SNP in ALDH1B1 also exceeds the Nyholt threshold for association with prostate cancer‐specific survival in low‐grade prostate cancer.
Genetic variants in alcohol metabolising genes
There is evidence that genetic variations in ADH and ALDH genes affecting ethanol metabolism32, 33, 34, 35 are associated with altered alcohol intake and risk of alcohol dependence.31, 35, 36, 37 The most extensively studied SNP in the context of alcohol intake is rs1229984 in ADH1B. It has been shown to be associated with increased adverse effects from alcohol intake and reduced consumption.35, 36 On average minor allele carriers drink 17.2% fewer units/week (95%CI:15.6%, 18.9%), are less likely to be in the top third of alcohol drinking volume (odds ratio, OR = 0.7; 95%CI:0.68,0.73) and are less likely to binge drink (OR = 0.78; 95%CI:0.73,0.84).23 It has also been reported to affect cancer risk at various sites.5 In our study, the associations of this SNP with prostate cancer diagnosis and survival were ORfixed = 1.00 (95%CI:0.96,1.03, p values = 0.87), and HRfixed = 1.11 (95%CI:0.95,1.30, p values = 0.17), respectively. Combining the effects of the ADH1B SNP on alcohol intake with the upper confidence intervals from our results implies that a 17% reduction in alcohol consumption is unlikely to reduce prostate cancer risk by >3% and prostate cancer mortality by >5%.
Alcohol is metabolised to acetaldehyde, a known carcinogen, and there is evidence to support the theory that genetic variants in alcohol metabolising genes, which control the production and breakdown of acetaldehyde, contribute to carcinogenesis.4, 5, 20, 24 There is also evidence of a tissue‐specific interaction in the prostate between ethanol and retinoic acid, through modulations of ALDH1A1, ALDH1A2 and ALDH1A3 levels.38 To our knowledge, this is the first comprehensive investigation of the association between ADH and ALDH variants, as genetic proxies for alcohol, and prostate cancer to date. Genetic predisposition to prostate cancer has been examined by GWASs, which shows common genetic variants can explain 33% heritability of prostate cancer but no genome‐wide significant hits are in ADHs or ALDHs39, 40 (however, this lack of evidence from GWASs could be a type 2 error). Similarly, we did not find any evidence of genetic association between ADH/ALDH variants and prostate cancer incidence in this study. Possible reasons for this include type 2 error, especially if the underlying effects of alcohol on prostate cancer incidence are small and limited to the very heavy drinking behaviours and/or to the more aggressive forms of disease, as possibly suggested by the recent literature.7, 12
We have shown that SNPs in ALDH1A2 are associated with altered prostate cancer‐specific mortality in a case‐only analysis. None of these SNPs appear to have regulatory features (www.ensemble.org), so they are unlikely to be causal variants themselves but rather they could be in LD with the causal variants. Recently, ALDH isoforms have been suggested as possible mechanistic mediators of metastasis in prostate cancer in particular41 and other solid tumours in general.42 One study found lack of compelling evidence linking variation in ALDH1 (including ALDH1A1, ALDH1A2, ALDH1A3 and ALDH1B1) with prostate cancer progression,41 but another had reported preliminary evidence for a potential role of ALDH1A2 as a tumour suppressor gene in prostate cancer cell lines43 and decreased expression of ALDH1A2 has been associated with shorter recurrence free survival in patients with prostate cancer.43 In our study, three intronic SNPs in ALDH1A2 were associated with longer survival, none of which were directly or indirectly (through LD) associated with alcohol‐related phenotypes (http://www.ebi.ac.uk/gwas/). One potential explanation for our results may be that these SNPs, or others in LD with them, lead to increased activity in ALDH1A2. We speculate that the observed prostate cancer survival effect could be the result of a net increase in the synthesis of retinoic acid (by ALDH1A2), which is particularly beneficial when the rate of conversion is affected by slower ADH activity in the presence of alcohol consumption (retinol and ethanol both being ADH substrates,44 and ethanol modulating retinoic acid synthesis in the rat prostate38).
Another intronic SNP in ALDH1B1 (also close to IGFBPL1) was found to be associated with increased mortality following a diagnosis of low‐grade prostate cancer. ALDH1B1 is the second most abundant mitochondrial ALDH, after ALDH2, with documented involvement in alcohol metabolism and dependence.45 However, this specific SNP is not known to be in LD with any of the variants associated with alcohol phenotypes to date, therefore we cannot speculate on its specific role in relation to alcohol. Evidence has been previously found linking levels of ALDH1B1 to survival following gastric cancer46 and non‐small‐cell lung cancer,47 but not prostate cancer,41 however the latter was a study in vitro Conversely,ALDH1A1 expression in the prostate has been reported to be a good candidate prognostic biomarker, based on all cause mortality and to a lesser extent prostate cancer‐specific mortality,48 and ALDH1A3 expression is thought to be involved with initiation and progression of several cancers,49 however we did not observe an association with common germ‐line mutations in either of these genes, or did we observe associations with variation in ADH genes. This could be due to different functional effects of variants on metabolic levels (e.g., alcohol and acetaldehyde peak levels and cumulative concentrations), affecting prostate cancer proliferation or survival differently both in terms of effect sizes and pathways.
Differences by tumour grade
In this study, we have found that genetic variants in genes involved in alcohol metabolism were associated with disease‐specific mortality in men with prostate cancer, most of whom had been diagnosed with low‐grade disease. We found a signal specific to low‐grade prostate cancer survival, but none for high‐grade disease. Possible reasons why stronger associations were not seen with high‐grade cancers include: limited power, as there were smaller numbers of high‐ compared with low‐grade cases (this was investigated using a reverse power calculation, to assess the power of this study to detect small associations (Supplementary material Table S3)); patients behaviour may change following a diagnosis, e.g. patients with high‐grade prostate cancer may be too ill to drink and the effect of the SNPs cannot be seen in the absence of drinking; or the findings could reflect a true clinical difference in the way alcohol affects survival for the different grades of prostate cancer.
Strengths and limitations
The strengths of this study include the large sample size and availability of data on both risk and mortality stratified by grade, which is an important predictor of prognosis. Importantly, we used a Mendelian randomisation approach, which minimises the potential for bias due to confounding, information bias (recall bias and sick‐quitter effect) and reverse causation, major limitations of previous studies in this area.7, 8, 13 We were also able to control for confounding by population stratification by adjusting for basic population characteristics. A potential limitation of our study is its power to detect small effects of alcohol on high‐grade disease, with fewer of these cases having been diagnosed and followed‐up.
Data for this study were contributed to the PRACTICAL Consortium from many studies with varying recruitment and inclusion/exclusion criteria, as different screening practices could complicate the interpretation of our results.50 For example, CAPS participants were all diagnosed clinically, whereas ProtecT participants were all screen detected. While the consortium provides a large sample size for investigation, there is inevitably some heterogeneity in the contributing studies. The effect of this was investigated using random effects meta‐analysis and meta‐regression. No one study‐level characteristic had a strong influence on the results, and we were unable to clearly determine the reason for the modest levels of heterogeneity observed, and the consequent variation between the fixed‐effect and random‐effect analyses. Potential explanations include: systematic differences in smaller vs. larger studies, and the former being assigned larger weights in random‐effect models; true variation in the effects of alcohol in the different study populations; effects of study designs that we were not able to investigate, e.g. the different ways cases were ascertained/recruited.
Another possible limitation to consider is the potential influence of pleiotropy. There may be other direct pathways through which the SNPs influence prostate cancer mortality independently of alcohol metabolism and intake. In particular, SNPs in ALDH1A2 could have a role in retinoic acid synthesis, which could affect cancer survival per se and in conjunction with alcohol,38 and we note that the ALDH1B1 SNP is in close proximity to IGFBPL1, which may encode a putative tumour suppressor protein.51 However, there were no other associations of these SNPs, or others in LD with them, reported by the catalogue of published genome‐wide association studies (http://www.ebi.ac.uk/gwas/); therefore the risk of pleiotropy for the genetic variants under study here is likely to be small. Finally, the direction of effect of the SNPs on alcohol intake, apart from rs1229984, is unknown so it is not possible to estimate the effect size of the four SNPs we found to be associated with survival.
Future directions
There are a number of ways in which this work could be taken forward. These include further analysis in larger consortia with longer follow up data availability, or repeating within individual subsets with certain study design characteristics to increase similarity of studies included in analysis (direct replication). Analysis of further genetic variants with known effects on alcohol metabolism or behaviour would also allow further development of this work (indirect replication), as would establishing the magnitude and direction of effect of genetic variants in alcohol metabolising genes on alcohol intake. It would also be interesting to investigate patient behaviour following diagnosis to establish if the varied effect seen between high‐ and low‐grade disease could be due to differences in behaviour.
Conclusion
If confirmed in independent studies or through direct or indirect replication, these findings suggest a role for alcohol in the progression of prostate cancer, whilst also confirming that alcohol is unlikely to have a large impact on prostate cancer carcinogenesis. This has potential public health implications and alcohol intake could be targeted to improve survival from prostate cancer as part of holistic care.
Acknowledgements
The Collaborative Oncological Gene‐environment Study (COGS), within which the PRACTICAL Consortium was assembled, would not have been possible without the contributions of the following: Per Hall (COGS); Douglas F. Easton, Paul Pharoah, Kyriaki Michailidou, Manjeet K. Bolla, Qin Wang (BCAC), Andrew Berchuck (OCAC), Rosalind A. Eeles, Douglas F. Easton, Ali Amin Al Olama, Zsofia Kote‐Jarai, Sara Benlloch (PRACTICAL), Georgia Chenevix‐Trench, Antonis Antoniou, Lesley McGuffog, Fergus Couch and Ken Offit (CIMBA), Joe Dennis, Alison M. Dunning, Andrew Lee, and Ed Dicks, Craig Luccarini and the staff of the Centre for Genetic Epidemiology Laboratory, Javier Benitez, Anna Gonzalez‐Neira and the staff of the CNIO genotyping unit, Jacques Simard and Daniel C. Tessier, Francois Bacot, Daniel Vincent, Sylvie LaBoissière and Frederic Robidoux and the staff of the McGill University and Génome Québec Innovation Centre, Stig E. Bojesen, Sune F. Nielsen, Borge G. Nordestgaard, and the staff of the Copenhagen DNA laboratory, and Julie M. Cunningham, Sharon A. Windebank, Christopher A. Hilker, Jeffrey Meyer and the staff of Mayo Clinic Genotyping Core Facility.
Information on the consortium can be found at http://practical.ccge.medschl.cam.ac.uk/
Additional members from the consortium are Margaret Cook,1 Angela Morgan,2 Artitaya Lophatananon,3,4 Cyril Fisher,2 Daniel Leongamornlert,2 Edward J. Saunders,2 Emma J. Sawyer,2 Koveela Govindasami,2 Malgorzata Tymrakiewicz,2 Michelle Guy, 2 Naomi Livni,2 Rosemary Wilkinson,2 Sara Jugurnauth‐Little,2 Steve Hazel,2 Tokhir Dadaev,2 Melissa C. Southey,5 Liesel M. Fitzgerald,6 John Pedersen,7 John Hopper,8 Ami Karlsson,9 Carin Cavalli‐Bjoerkman,9 Jan‐Erik Johansson,9 Jan Adolfson,9 Markus Aly,9,10 Michael Broms,9 Paer Stattin,9 Brian E. Henderson,11 Fredrick Schumacher,11 Anssi Auvinen,12 Kimmo Taari,13 Liisa Maeaettaenen,14 Paula Kujala,15 Teemu Murtola,16,17 Teuvo L.J. Tammela,17 Tiina Wahlfors,18 Martin Andreas Roder,19 Peter Iversen,19 Peter Klarskov,20 Sune F. Nielsen,21,22 Tim J. Key,23 Hans Wallinder,24 Sven Gustafsson,24 Jenny L. Donovan,25 Freddie Hamdy,26 Angela Cox,27 Anne George,28 Athene Lane,28 Gemma Marsden,26 Michael Davis,25 Paul Brown,25 Paul Pharoah,29 Lisa B. Signorello,30,31 Wei Zheng,32 Shannon K. McDonnell,33 Daniel J. Schaid,33 Liang Wang,33 Lori Tillmans,33 Shaun Riska,33 Thomas Schnoeller,34 Kathleen Herkommer,35 Manuel Luedeke,34 Walther Vogel,36 Dominika Wokozorczyk,37 Jan Lubiski,37 Wojciech Kluzniak,37 Katja Butterbach,38 Christa Stegmaier,39 Bernd Holleczek,39 Babu Zachariah,40 Hui‐Yi Lim,41 Hyun Park,40 James Haley,40 Julio Pow‐Sang,40 Maria Rincon,40 Selina Radlein,40 Thomas Sellers,40 Chavdar Slavov,42 Aleksandrina Vlahova,43 Atanaska Mitkova,44 Darina Kachakova,44 Elenko Popov,42 Svetlana Christova,43 Tihomir Dikov,43 Vanio Mitev,44 Allison Eckert,45 Amanda Spurdle,46 Angus Collins,45 Glenn Wood,45 Greg Malone,45 Judith A. Clements,45 Kris Kerr,45 Megan Turner,45 Pamela Saunders,45 Peter Heathcote,45 Srilakshmi Srinivasan,45 Leire Moya,45 Trina Yeadon,45 Australian Prostate Cancer BioResource,45 Joana Santos,47 Carmen Jerónimo,47 Paula Paulo,47 Pedro Pinto,47 Rui Henrique,47 Sofia Maia,47 Agnieszka Michael,48 Andrzej Kierzek,48 Huihai Wu48 and Robert J. MacInnis6,8
1Centre for Cancer Genetic Epidemiology, Department of Public Health and Primary Care, University of Cambridge, Strangeways Laboratory, Worts Causeway, Cambridge CB1 8RN, United Kingdom, 2The Institute of Cancer Research, Sutton, United Kingdom, 3Institute of Population Health, University of Manchester, Manchester, United Kingdom, 4Warwick Medical School, University of Warwick, Coventry, United Kingdom, 5Genetic Epidemiology Laboratory, Department of Pathology, The University of Melbourne, Grattan Street, Parkville, Vic, Australia, 6Cancer Epidemiology Centre, The Cancer Council Victoria, 615 St Kilda Road, Melbourne, Vic, Australia, 7Tissupath Pty Ltd., Melbourne, Vic Australia, 8Centre for Epidemiology and Biostatistics, Melbourne School of Population and Global Health, The University of Melbourne, Vic, Australia, 9Department of Medical Epidemiology and Biostatistics, Karolinska Institute, Stockholm, Sweden, 10Department of Clinical Sciences at Danderyds Hospital, Stockholm, Sweden, 11Department of Preventive Medicine, Keck School of Medicine, University of Southern California/Norris Comprehensive Cancer Center, Los Angeles, CA, 12Department of Epidemiology, School of Health Sciences, University of Tampere, Tampere, Finland, 13Department of Urology, Helsinki University Central Hospital and University of Helsinki, Helsinki, Finland, 14Finnish Cancer Registry, Helsinki, Finland, 15Fimlab Laboratories, Tampere University Hospital, Tampere, Finland, 16School of Medicine, University of Tampere, Tampere, Finland, 17Department of Urology, Tampere University Hospital and Medical School, University of Tampere, Finland, 18BioMediTech, University of Tampere and FimLab Laboratories, Tampere, Finland, 19Copenhagen Prostate Cancer Center, Department of Urology, Rigshospitalet, Copenhagen University Hospital, Tagensvej 20, 7521, DK‐2200 Copenhagen, Denmark, 20Department of Urology, Herlev Hospital, Copenhagen University Hospital, Herlev Ringvej 75, DK‐230 Herlev, Denmark, 21Department of Clinical Biochemistry, Herlev Hospital, Copenhagen University Hospital, Herlev Ringvej 75, DK‐230 Herlev, Denmark, 22Faculty of Health and Medical Sciences, University of Copenhagen, 23Cancer Epidemiology Unit, Nuffield Department of Clinical Medicine, University of Oxford, Oxford, United Kingdom, 24Department of Epidemiology and Biostatistics, School of Public Health, Imperial College, London, United Kingdom, 25School of Social and Community Medicine, University of Bristol, Canynge Hall, 39 Whatley Road, Bristol, BS8 2PS, United Kingdom, 26Nuffield Department of Surgical Sciences, University of Oxford, Oxford, United Kingdom, Faculty of Medical Science, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom, 27CR‐ United Kingdom /YCR Sheffield Cancer Research Centre, University of Sheffield, Sheffield, United Kingdom, 28University of Cambridge, Department of Oncology, Box 279, Addenbrooke's Hospital, Hills Road Cambridge CB2 0QQ, United Kingdom, 29Centre for Cancer Genetic Epidemiology, Department of Oncology, University of Cambridge, Strangeways Laboratory, Worts Causeway, Cambridge, United Kingdom, 30International Epidemiology Institute, 1555 Research Blvd., Suite 550, Rockville, MD, 31Department of Epidemiology, Harvard School of Public Health, 677 Huntington Avenue, Boston, MA, 32Division of Epidemiology, Department of Medicine, Vanderbilt University Medical Center, 2525 West End Avenue, Suite 800, Nashville, TN, 33Mayo Clinic, Rochester, MN, 34Department of Urology, University Hospital Ulm, Germany, 35Department of Urology, Klinikum rechts der Isar der Technischen Universitaet Muenchen, Munich, Germany, 36Institute of Human Genetics, University Hospital Ulm, Germany, 37International Hereditary Cancer Center, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland, 38Division of Clinical Epidemiology and Aging Research, German Cancer Research Center (DKFZ), 69120 Heidelberg, Germany, 39Saarland Cancer Registry, 66119 Saarbruecken, Germany, 40Department of Cancer Epidemiology, Moffitt Cancer Center, 12902 Magnolia Drive, Tampa, FL, 41Biostatistics Program, Moffitt Cancer Center, 12902 Magnolia Drive, Tampa, FL, 42Department of Urology and Alexandrovska University Hospital, Medical University, Sofia, Bulgaria, 43Department of General and Clinical Pathology, Medical University, Sofia, Bulgaria, 44Department of Medical Chemistry and Biochemistry, Molecular Medicine Center, Medical University, Sofia, 2 Zdrave Str., 1431 Sofia, Bulgaria, 45Australian Prostate Cancer Research Centre‐Qld, Institute of Health and Biomedical Innovation and School of Biomedical Science, Queensland University of Technology, BNE, Australia, 46Molecular Cancer Epidemiology Laboratory, Queensland Institute of Medical Research, BNE, Australia, 47Department of Genetics, Portuguese Oncology Institute, Porto, Portugal, 48The University of Surrey, Guildford, Surrey GU2 7XH, United Kingdom
Conflict of interest
References
- 1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 2. Crawford ED. Understanding the epidemiology, natural history, and key pathways involved in prostate cancer. Urology 2009;73:4–10. [DOI] [PubMed] [Google Scholar]
- 3. Andriole GL. Overview of pivotal studies for prostate cancer risk reduction, past and present. Urology 2009;73:36–43. [DOI] [PubMed] [Google Scholar]
- 4. Baan R, Straif K, Grosse Y, et al. Carcinogenicity of alcoholic beverages. Lancet Oncol 2007;8:292–3. [DOI] [PubMed] [Google Scholar]
- 5. Druesne‐Pecollo N, Tehard B, Mallet Y, et al. Alcohol and genetic polymorphisms: effect on risk of alcohol‐related cancer. Lancet Oncol 2009;10:173–80. [DOI] [PubMed] [Google Scholar]
- 6.Research WCRFaAIfC. Alcoholic beverages. In: Research WCRFaAIfC, ed. Systematic literature review on diet and prostate cancer. Food, nutrition, physical activity, and the prevention of cancer: a global perspective. Second Expert Report ed. Washington DC: AICR, 2007.
- 7. Zuccolo L, Lewis SJ, Donovan JL, et al. Alcohol consumption and PSA‐detected prostate cancer risk—a case–control nested in the ProtecT study. Int J Cancer 2013;132:2176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sawada N, Inoue M, Iwasaki M, et al. Alcohol and smoking and subsequent risk of prostate cancer in Japanese men: the Japan Public Health Center‐based prospective study. Int J Cancer 2014;134:971–8. [DOI] [PubMed] [Google Scholar]
- 9. Watters JL, Park Y, Hollenbeck A, et al. Alcoholic beverages and prostate cancer in a prospective US cohort study. Am J Epidemiol 2010;172:773–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chao C, Haque R, Van Den Eeden SK, et al. Red wine consumption and risk of prostate cancer: the California men's health study. Int J Cancer 2010;126:171–9. [DOI] [PubMed] [Google Scholar]
- 11. Breslow RA, Chen CM, Graubard BI, et al. Prospective study of alcohol consumption quantity and frequency and cancer‐specific mortality in the US population. Am J Epidemiol 2011;174:1044–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gong ZH, Kristal AR, Schenk JM, et al. Alcohol consumption, finasteride, and prostate cancer risk results from the prostate. Cancer 2009;115:3661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Middleton FK, Chikritzhs T, Stockwell T, et al. Alcohol use and prostate cancer: a meta‐analysis. Mol Nutr Food Res 2009;53:240–55. [DOI] [PubMed] [Google Scholar]
- 14. Rota M, Scotti L, Turati F, et al. Alcohol consumption and prostate cancer risk: a meta‐analysis of the dose–risk relation. Eur J Cancer Prev 2012;21:350–9. [DOI] [PubMed] [Google Scholar]
- 15. Davey Smith G, Ebrahim S. ‘Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003;32:1–22. [DOI] [PubMed] [Google Scholar]
- 16. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 2014;23:R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davey Smith G, Lawlor DA, Harbord R, et al. Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med 2007;4:e352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holmes MV, Dale CE, Zuccolo L, et al. Association between alcohol and cardiovascular disease: Mendelian randomisation analysis based on individual participant data. BMJ 2014;349:g4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han HG, Wang HS, Yin ZT, et al. Association of genetic polymorphisms in ADH and ALDH2 with risk of coronary artery disease and myocardial infarction: a meta‐analysis. Gene 2013;526:134–41. [DOI] [PubMed] [Google Scholar]
- 20. Boccia S, Hashibe M, Galli P, et al. Aldehyde dehydrogenase 2 and head and neck cancer: a meta‐analysis implementing a Mendelian randomization approach. Cancer Epidemiol Biomarkers Prev 2009;18:248–54. [DOI] [PubMed] [Google Scholar]
- 21. Hashibe M, McKay JD, Curado MP, et al. Multiple ADH genes are associated with upper aerodigestive cancers. Nat Genet 2008;40:707–9. [DOI] [PubMed] [Google Scholar]
- 22. Guo XF, Wang J, Yu SJ, et al. Meta‐analysis of the ADH1B and ALDH2 polymorphisms and the risk of colorectal cancer in East Asians. Intern Med 2013;52:2693–9. [DOI] [PubMed] [Google Scholar]
- 23. Tsai ST, Wong TY, Ou CY, et al. The interplay between alcohol consumption, oral hygiene, ALDH2 and ADH1B in the risk of head and neck cancer. Int J Cancer 2014;135:2424–36. [DOI] [PubMed] [Google Scholar]
- 24. Hidaka A, Sasazuki S, Matsuo K, et al. Genetic polymorphisms of ADH1B, ADH1C and ALDH2, alcohol consumption, and the risk of gastric cancer: the Japan Public Health Center‐based prospective study. Carcinogenesis 2015;36:223–31. [DOI] [PubMed] [Google Scholar]
- 25. Kote‐Jarai Z, Easton DF, Stanford JL, et al. Multiple novel prostate cancer predisposition loci confirmed by an international study: the PRACTICAL Consortium. Cancer Epidemiol Biomarkers Prev 2008;17:2052–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eeles RA, Al Olama AA, Benlloch S, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet 2013;45:385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Higgins JPT, Thompson SG, Deeks JJ, et al. Measuring inconsistency in meta‐analyses. Br Med J 2003;327:557–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nyholt DR. A simple correction for multiple testing for single‐nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet 2004;74:765–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Purcell S. Genetic Power Calculator, 2008. Available from: http://pngu.mgh.harvard.edu/~purcell/gpc/cc2.html.
- 31. Frank J, Cichon S, Treutlein J, et al. Genome‐wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict Biol 2012;17:171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gyamfi MA, Kocsis MG, He L, et al. The role of retinoid X receptor alpha in regulating alcohol metabolism. J Pharmacol Exp Ther 2006;319:360–8. [DOI] [PubMed] [Google Scholar]
- 33. Birley AJ, James MR, Dickson PA, et al. ADH single nucleotide polymorphism associations with alcohol metabolism in vivo . Hum Mol Genet 2009;18:1533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edenberg HJ. The genetics of alcohol metabolism—role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res Health 2007;30:5–13. [PMC free article] [PubMed] [Google Scholar]
- 35. Linneberg A, Gonzalez‐Quintela A, Vidal C, et al. Genetic determinants of both ethanol and acetaldehyde metabolism influence alcohol hypersensitivity and drinking behaviour among Scandinavians. Clin Exp Allergy 2010;40:123–30. [DOI] [PubMed] [Google Scholar]
- 36. Macgregor S, Lind PA, Bucholz KK, et al. Associations of ADH and ALDH2 gene variation with self report alcohol reactions, consumption and dependence: an integrated analysis. Hum Mol Genet 2009;18:580–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lind PA, Eriksson CJ, Wilhelmsen KC. The role of aldehyde dehydrogenase‐1 (ALDH1A1) polymorphisms in harmful alcohol consumption in a Finnish population. Hum Genomics 2008;3:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fioruci‐Fontanelli BA, Chuffa LG, Mendes LO, et al. Ethanol modulates the synthesis and catabolism of retinoic acid in the rat prostate. Reprod Toxicol 2015;53:1–9. [DOI] [PubMed] [Google Scholar]
- 39. Al Olama AA, Kote‐Jarai Z, Berndt SI, et al. A meta‐analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet 2014;46:1103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eeles R, Goh C, Castro E, et al. The genetic epidemiology of prostate cancer and its clinical implications. Nat Rev Urol 2014;11:18–31. [DOI] [PubMed] [Google Scholar]
- 41. van den Hoogen C, van der Horst G, Cheung H, et al. High aldehyde dehydrogenase activity identifies tumor‐initiating and metastasis‐initiating cells in human prostate cancer. Cancer Res 2010;70:5163–73. [DOI] [PubMed] [Google Scholar]
- 42. Rodriguez‐Torres M, Allan AL. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin Exp Metastasis 2016;33:97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim H, Lapointe J, Kaygusuz G, et al. The retinoic acid synthesis gene ALDH1a2 is a candidate tumor suppressor in prostate cancer. Cancer Res 2005;65:8118–24. [DOI] [PubMed] [Google Scholar]
- 44. Chou CF, Lai CL, Chang YC, et al. Kinetic mechanism of human class IV alcohol dehydrogenase functioning as retinol dehydrogenase. J Biol Chem 2002;277:25209–16. [DOI] [PubMed] [Google Scholar]
- 45. Jackson BC, Holmes RS, Backos DS, et al. Comparative genomics, molecular evolution and computational modeling of ALDH1B1 and ALDH2. Chem Biol Interact 2013;202:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shen JX, Liu J, Li GW, Huang YT, Wu HT. Mining distinct aldehyde dehydrogenase 1 (ALDH1) isoenzymes in gastric cancer. Oncotarget 2013;7:25340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. You Q, Guo H, Xu D. Distinct prognostic values and potential drug targets of ALDH1 isoenzymes in non‐small‐cell lung cancer. Drug Des Dev Ther 2015;9:5087–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao L, Yu N, Guo T, et al. Tissue biomarkers for prognosis of prostate cancer: a systematic review and meta‐analysis. Cancer Epidemiol Biomarkers Prev 2014;23:1047–54. [DOI] [PubMed] [Google Scholar]
- 49. Duan JJ, Cai J, Guo YF, Bian XW, Yu SC. ALDH1A3, a metabolic target for cancer diagnosis and therapy. Int J Cancer 2016;139:965–75. [DOI] [PubMed] [Google Scholar]
- 50. Garcia‐Closas M, de Gonzalez AB. Invited commentary: screening and the elusive etiology of prostate cancer. Am J Epidemiol 2015;182:390–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cai Z, Chen HT, Boyle B, et al. Identification of a novel insulin‐like growth factor binding protein gene homologue with tumor suppressor like properties. Biochem Biophys Res Commun 2005;331:261–6. [DOI] [PubMed] [Google Scholar]