

Significance
Hepatitis C virus is an important human pathogen, and its E2 envelope glycoprotein is the major target of neutralizing antibodies (NAbs) and, hence, a promising vaccine candidate. Many broadly NAbs (bNAbs) to E2 recognize the conserved receptor-binding site, but immunization with soluble E2 antigen rarely elicits a potent bNAb response. Here, we show that soluble E2 is highly stable except for the receptor-binding site and variable loops. Thus, despite high sequence conservation, structural flexibility at the receptor-binding site may distract the immune system from eliciting bNAbs that recognize the conformation required for its function on virions. Stabilization of the E2 CD81 receptor-binding site (CD81bs) by structure-based design may improve its performance as a vaccine candidate.
Keywords: hepatitis C virus, E2, CD81-binding site, conformational flexibility, protein dynamics
Abstract
Hepatitis C virus (HCV) is a major cause of liver disease, affecting over 2% of the world’s population. The HCV envelope glycoproteins E1 and E2 mediate viral entry, with E2 being the main target of neutralizing antibody responses. Structural investigations of E2 have produced templates for vaccine design, including the conserved CD81 receptor-binding site (CD81bs) that is a key target of broadly neutralizing antibodies (bNAbs). Unfortunately, immunization with recombinant E2 and E1E2 rarely elicits sufficient levels of bNAbs for protection. To understand the challenges for eliciting bNAb responses against the CD81bs, we investigated the E2 CD81bs by electron microscopy (EM), hydrogen–deuterium exchange (HDX), molecular dynamics (MD), and calorimetry. By EM, we observed that HCV1, a bNAb recognizing the N-terminal region of the CD81bs, bound a soluble E2 core construct from multiple angles of approach, suggesting components of the CD81bs are flexible. HDX of multiple E2 constructs consistently indicated the entire CD81bs was flexible relative to the rest of the E2 protein, which was further confirmed by MD simulations. However, E2 has a high melting temperature of 84.8 °C, which is more akin to proteins from thermophilic organisms. Thus, recombinant E2 is a highly stable protein overall, but with an exceptionally flexible CD81bs. Such flexibility may promote induction of nonneutralizing antibodies over bNAbs to E2 CD81bs, underscoring the necessity of rigidifying this antigenic region as a target for rational vaccine design.
Hepatitis C virus (HCV) is a leading cause of liver cirrhosis and hepatocellular carcinoma, infecting more than 2% of the world’s population (1). HCV infection is highly prevalent in developing countries and among marginalized populations, such as injection drug users (IDUs) and prisoners in developed countries (2). Effective direct-acting antiviral (DAA) drugs have been developed recently to curb the advance of HCV (3, 4). Nevertheless, new infections are on the rise among young IDUs in developed countries and along drug-trafficking routes (2, 5, 6). Because DAA treatment is prohibitively expensive and does not prevent reinfection (7, 8), an effective vaccine is essential for management of the global HCV epidemic (9–11).
Despite the significant public health burden, no effective prophylactic vaccine against HCV has been developed. High genetic variability of the virus is a major barrier, particularly in the E1 and E2 envelope glycoproteins that are the primary neutralizing antibody (NAb) targets. E2, the receptor-binding protein, mediates cell entry by interacting with the tetraspanin CD81 and several other cell surface molecules (12, 13). The crystal structure of the E2 core domain (E2c) consists of a central β-sandwich flanked by “front” and “back” layers (14) (Fig. 1). The CD81-binding site (CD81bs) has been mapped by mutagenesis and electron microscopy (EM) to various elements: a conserved N-terminal region (residues 412–423), the front layer (residues 424–453), and the adjacent CD81-binding loop (CD81bl; residues 519–535) (14–16) (Fig. 1). The CD81bs is also a conserved antigenic site recognized by multiple broadly neutralizing antibodies (bNAbs) and, therefore, an important target for HCV vaccine design. So far, three distinct clusters of neutralizing epitopes, or antigenic sites, have been identified within the CD81bs. The first antigenic site (AS412) consists of the conserved N-terminal end of the CD81bs spanning residues 412–423. The bNAbs AP33, HCV1, HC33.1, and 3/11 primarily bind to E2 at AS412, and their interactions have been structurally characterized (17–22). The second antigenic site (AS434) consists of a short α-helix in the front layer residues 434–446. The bNAbs HC84-1 and HC84-27 and the weakly neutralizing monoclonal antibody (mAb) 8 primarily recognize this helical region (23, 24). The third cluster of neutralizing epitopes that we call antigenic region 3 (AR3) is a discontinuous surface composed of the entire front layer, including AS434, and the adjacent CD81bl. AR3 is recognized by a family of potent bNAbs consisting of AR3A, AR3B, AR3C, and AR3D (25, 26).
Fig. 1.

Conformational flexibility of a broadly neutralizing epitope on soluble HCV E2c2 revealed by EM. (Left) Crystal structure of E2c is displayed as a ribbon with structural elements indicated and disordered regions shown as dotted lines. The epitopes of mAb AR1B, bNAb HCV1, bNAb AP33, and NAb AR2A are defined; residues that are important for binding, as mapped by alanine scanning experiments, are colored red in the sequences shown. (Right) Three negative-stain EM density maps at ∼30-Å resolution of E2c2 (gray) bound to Fab AR1B (gray), AR2A (gray), and HCV1 (yellow, green, and blue) are shown as transparent surfaces with structural models of the antibodies and E2c superimposed (also Fig. S3). Differences in bNAb HCV1 angles of approach indicated from the three RCT reconstructions are shown.
There is ample evidence that the E2 CD81bs is accessible and immunogenic on the virion and on recombinant E2. First, the CD81bs is recognized by bNAbs with different angles of approach (9) (Fig. S1). Second, although E2 is densely glycosylated, the CD81bs is not obstructed by any of the 11 N-linked glycans (14). Third, antibody responses to the CD81bs are frequently detected in sera from subjects immunized with the candidate vaccine immunogens: soluble E2 or E1E2. However, these serum antibodies are often weak or nonneutralizing despite high anti-E1E2 antibody titers (14, 27–30). Indeed, mAbs have been isolated to the different components of the CD81bs, but only a few (mentioned above) cross-neutralize the highly diverse HCV genotypes (30–33). In a panel of 37 mAbs isolated from mice immunized with E1E2, 20 (54%) and four (11%) bind to the E2 front layer and CD81bl, respectively, but none neutralize HCV (30). The results indicate that the E2 front layer and CD81bl are highly immunogenic but likely contain structural features that act as decoys for the immune system, skewing the response to nonneutralizing epitopes during immunization. Interestingly, in the antibody panel, of seven mAbs that bind subsite AS412, six are neutralizing, suggesting at least that AS412 is presented correctly; however, sufficient levels of NAbs were not produced in the animals. Thus, to understand how to elicit bNAbs to the E2 CD81bs, it is necessary to investigate anti-CD81bs antibodies and the E2 antigens in depth.
Fig. S1.

Angle of approach by antibodies that recognize the HCV E2 receptor-binding site. (A, Left) Crystal structure bNAb HC84-27 (orange) [Protein Data Bank (PDB) ID code 4JZO] is superposed on its peptide epitope in the crystal structure of the HCV E2 core bound to bNAb AR3C (red) (PDB ID code 4MWF) (gray). (A, Right) CD81bs, targeted by both bNAbs, is highlighted, with the general location of the glycosylated region (glycan shield) and HVR1 indicated. (B) Comparison of HIV Env gp120 epitopes at or around the CD4 receptor-binding site, indicating regions protected by the glycan shield and the Env trimer interface. Crystal structures of bNAb VRC01 (red) (PDB ID code 4LST) and non-NAb F105 (wheat) (PDB ID code 3HI1) are shown superposed on the gp120 core antigen (gray). (Right) Common surface recognized by many bnAbs, the CD4-binding site (CD4bs), is highlighted, with the glycan shield shown, as well as the region occluded within the trimeric spike that is not accessible to non-NAbs in the trimer context.
In this study, we first investigated the molecular features of the E2 CD81bs. We used EM to elucidate the interactions between E2 and a bNAb targeting AS412. Next, we mapped the surface accessibility and flexibility of E2 through hydrogen–deuterium exchange mass spectrometry (HDXMS). The HDX results were then corroborated by molecular dynamics (MD) simulations using available crystal structures. To assess the overall stability of E2, we used differential scanning calorimetry (DSC) and determined binding entropies of various E2 ligands using isothermal titration calorimetry (ITC). The combined results suggest recombinant E2 is a highly stable protein with an exceptionally flexible receptor-binding site that can present many nonoptimal conformations/configurations during immunization. Furthermore, we can now use this information to guide engineering efforts to stabilize the E2 CD81bs to advance HCV vaccine design.
Results and Discussion
Flexibility at the E2 CD81bs as Visualized by EM.
The first structural investigations of HCV E2 revealed that AS412 within the E2 CD81bs, when presented as a synthetic peptide, forms a β-hairpin when bound to bNAbs HCV1 and AP33 (17–20). Both HCV1 and AP33 interact with one face of the AS412 β-hairpin, whereas the opposite face contains an N-linked glycan. These initial results suggest that both faces of the AS412 β-hairpin are solvent-accessible and that AS412 might protrude from the E2 protein as a potentially flexible, flap-like structure (18). Interestingly, AS412 is disordered in the published HCV E2c crystal structure (14). Furthermore, recent structures of the AS412 peptide in complex with bNAbs HC33.1 and 3/11 revealed very different, extended conformations for the peptide (21, 22).
To investigate the flexibility of AS412 in the context of E2, we sought to visualize directly any structural heterogeneity in the interaction between bNAb HCV1 and an E2c variant (E2c2; the amino acid sequence of E2c variants are shown in Fig. S2) containing a full variable region 2 (VR2) (Fig. 1 and Fig. S3). Assuming that HCV1 primarily interacts with E2 via AS412, flexibility at AS412 should translate to larger, rigid-body motions for HCV1, as previously observed for anti–HIV-1 bNAb PG9 when bound to HIV-1 envelope glycoprotein gp120 as a monomer (34). Therefore, we formed a complex composed of partially deglycosylated E2c2 bound to the antigen-binding fragments (Fabs) of mAb AR2A, mAb AR1B, and bNAb HCV1, and performed reference-free alignment and classification. Previously, we showed by mutagenesis or negative-stain EM that attachment of AR2A or AR1B to E2 did not significantly alter binding of CD81 or mAb AR3C (14). AR2A and AR1B were incorporated into the complex as fiduciary markers to help orient the E2 structure. Interestingly, substantial heterogeneity was observed at one protrusion in the 2D classes that corresponded to a single Fab. Because of this heterogeneity, we collected random conical tilt (RCT) EM data of the complex and calculated multiple 3D reconstructions (Fig. 1 and Fig. S3). Altogether, the 2D classes were consistent with three separate 3D reconstructions, each containing three protrusions of density corresponding to each of the three Fabs used to construct the complex, with E2c2 at the center. As expected from the 2D classes, there was substantial structural variation for one of the Fabs. To assign identities to each EM density protrusion, we relied on a previous mapping study (25), an alanine scanning experiment (Table S1), the E2c/AR3C Fab complex crystal structure (14), and the E2c-AR2A-CD81 EM structure (14). In this manner, we were able to assign AR2A and AR1B unambiguously to protrusions in the reconstruction that extend in opposite directions from each other. The third EM protrusion was consistent with the location of AS412 on E2 based on the AR1B and AR2A assignments. We therefore identified this more variable protrusion as the HCV1 Fab. Remarkably, the HCV1 Fab exhibited 10–22° variation in the angle of approach between the three 3D reconstructions (Fig. 1). Compared with the nearly 180° variation observed for anti-HIV1 bNAb PG9 when bound to HIV-1 gp120 monomer, the flexibility of HCV1 is modest. However, PG9 recognizes the V1V2 loop, which is 57–80 residues and highly flexible in the context of gp120 monomer, whereas HCV1 recognizes AS412, which is merely 12 residues and only five residues from the first disulfide bond to the core of the E2 protein. Thus, the observed variability of HCV1 is consistent with the originally proposed hypothesis that AS412 is a flap-like structure with structural flexibility, but somewhat constrained by the close proximity to the rest of the E2 protein (18). Of note, the negative-stain EM results are not of sufficient resolution to elucidate whether small, local conformational changes could occur in the E2 front layer and/or CD81bl in the antibody-bound E2.
Fig. S2.

Amino acid sequences of E2 constructs. Sequence alignment of E2 constructs used in this study. The N-linked glycosylation sequons are colored green, and the variable regions are labeled and highlighted in gray. The asterisk indicates glycosylation sequons that were mutated in soluble E2c constructs to facilitate structural characterization.
Fig. S3.

Analysis of negative-stain EM data of antibody/E2 complexes. (A) Two-dimensional class averages of E2/Fab complexes from RCT data. (B) Representative 2D class averages of the E2/Fab complexes demonstrating the different angles between Fabs AR2A and HCV1 that are found relative to the E2 surface. (C) Fourier shell correlation (FSC) curves for the RCT reconstructions of the complexes. The resolution of all three reconstructions is ∼31 Å at an FSC cutoff of 0.5.
Table S1.
Mapping the epitope of NAb AR2A
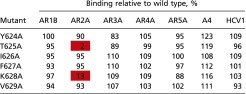 |
The effects of alanine substitution mutagenesis of E2 on binding of a panel of mAbs to discontinuous (AR1B, AR2A, AR3A, AR4A, and AR5A) and continuous (HCV1 and A4) epitopes on E1E2 are shown. The numbers shown indicate the percentage binding level relative to the wild-type E1E2 in an ELISA. Binding levels below 25% of wild type are highlighted in red. Alanine substitution mutagenesis indicates that T625 and K628 of E2 are critical for binding of NAb AR2A.
Mapping the Conformational Flexibility of HCV E2 by HDX.
Next, we sought to extend our investigation on flexibility to the rest of the E2 protein using HDX. HDX tracks the rate of deuterium incorporation into the backbone amides of proteins when exposed to deuterated solvent, and is highly dependent on the conformational flexibility and accessibility of individual residues in the protein (35). Here, we used HDXMS to measure the masses of enzymatically cleaved peptides from E2 after E2 had been exposed to deuterated solvent over different lengths of time and then quenched to stop the exchange (36). In this manner, three complexes were analyzed with HDX: partially deglycosylated E2c2 bound to Fab AR2A (E2c2-AR2A), partially deglycosylated E2c2 bound to Fabs AR2A and AP33 (E2c2-AR2A-AP33), and partially deglycosylated full-length E2 ectodomain (E2ΔTM) bound to Fabs AR2A and AP33 (E2ΔTM-AR2A-AP33). The E2 proteins were enzymatically deglycosylated with endoglycosidase H (EndoH) to maximize the identification of peptides and the overall peptide coverage of the proteins. Fab AR2A, which does not bind to the CD81bs, was required in the deglycosylation step to prevent E2 aggregation and ensure complete protease digestion.
Multiple peptic fragments of the complexes were identified by MS, covering 85% of E2c2 and 60% of E2ΔTM (Figs. S4 and S5). Considering the relatively small size of E2 in relation to its relatively large proportion of prolines that are inaccessible by HDX (25 of 335 E2ΔTM residues are prolines), the variable glycosylation states of the proteins, and the low yield of glycopeptides identifiable in MS, the HDX coverage achieved in this study is on par with a previous HDX study of Ebola virus envelope glycoprotein (37). To facilitate analysis, we focused on high-quality peptides that defined nonoverlapping regions across the protein sequence. Overall, the results indicated that E2c2 in the E2c2/AR2A complex is flexible, with nine of 15 nonoverlapping peptide regions having greater than 50% deuterium exchange by the first time point. E2 peptides with lower deuterium exchange include one peptide within the β-sandwich, which is expected to be relatively stable because it is involved in backbone hydrogen bonds and relatively inaccessible, and five peptide regions within the back layer, which are protected by interaction with Fab AR2A (Fig. 2 A and B and Fig. S5). The most flexible regions are the front layer of the CD81bs and VR2, with over 90% deuterium exchange for all peptides by the first time point (10 s) (Fig. 2A). The variable region 3 (VR3) on E2 is also highly flexible, as expected from its high B-values in the available crystal structures, with 80% deuterium exchange by the first time point compared with 50% deuterium exchange for the peptides covering the β-sandwich. This high global rate of deuterium exchange is comparable to the global deuterium exchange rate of HIV-1 gp120 monomer (38), another viral envelope glycoprotein known to be quite flexible from structural studies (39), and is consistent with the use of flexibility by viral proteins to tolerate mutations (40). To validate our method, we used HDX to map the known bNAb AP33 epitope, which, like the epitope of HCV1, consists of the AS412 with no additional E2 protein components (18, 19) (Fig. S5). As expected, the HDX profile of E2c2-AR2A-AP33 is mostly equivalent to the profile of E2c2-AR2A except with much slower exchange at Asn415-Trp420 within AS412 (Fig. 2B).
Fig. S4.

HDX peptide coverage. E2c2 (Top) and E2ΔTM (Bottom) were incubated in the absence, as well as the presence, of deuteration buffer, and subsequently quenched with a solution containing 6.4 M guanidine HCl and 1.0 M TCEP, followed by fragmentation via pepsin. Analysis by MS was then performed as described in SI Materials and Methods. Daughter peptides identified by MS after HDX experiments are mapped onto the protein sequences. However, the coverage was limited and not complete. The overlapping blue bars represent peptides of a unique sequence and charge state identity (maximum of 10 per cascade of overlapping peptides). Peptides that span more than one row in the figure are represented by black arrows.
Fig. S5.

Conformational flexibility of E2c2 and E2ΔTM as revealed by HDX and MD. The deuteration levels of peptides derived from E2c2 bound to NAb AR2A are aligned beneath the HCV E2 sequence in colored bars representing the following: E2c2 bound to NAb AR2A (Top), E2c2 bound to bNAb AP33 (Middle), and E2ΔTM bound to NAb AR2A and bNAb AP33 (Bottom). Because prolines lack exchangeable amide hydrogens, no colors are indicated. The different colors represent the extent of deuterium exchange, with warmer colors indicating higher deuterium incorporation. Regions on E2 not covered by peptides are shown in white. Horizontal black bars represent individual peptides used to label each region of the sequence, whereas vertical black bars signify the N and C termini of E2 constructs. Antibody-binding sites, variable loop regions, and N-linked glycans (green sticks) are annotated above the sequence, and are colored based on structural components defined in Fig. 1. The average RMSF calculated from a 100-ns MD simulation of unliganded E2c for each residue is aligned below the HDX data.
Fig. 2.

Structural analysis of HDX data. (A, Top) Deuterium incorporation into E2c2 when bound to NAb AR2A after 10 s of exchange is mapped onto the crystal structure of E2c using a color gradient. Disordered regions are represented by dotted lines, and regions not covered by peptic fragments and prolines are shown as thin, white tubes if structured or as gray dots if disordered. (A, Bottom) For clarity, the front layer, the β-sandwich, and the back layer of E2 are displayed individually. (B, Top) Deuterium exchange into E2c2 bound to NAb AR2A after 1,000 s of exchange is displayed as in A. (B, Bottom) Known mAb epitopes are highlighted. (C) HDX of a peptide spanning the N-terminal antigenic region of the CD81bs is plotted for E2c2-AR2A and E2c2-AR2A-AP33. (D) Average HDX of the E2 front layer (cyan), β-sandwich (red), and back layer (green) within the E2c2/AR2A complex is plotted.
Next, we sought to extend our HDX studies to the full-length E2ΔTM, which includes hypervariable region 1 (HVR1) and the C-terminal stalk (residues 384–411 and 646–717, respectively), both of which are truncated in E2c2. A previous EM study with E2ΔTM suggested that HVR1 might pack against a highly conserved hydrophobic surface identified on E2c (14). Thus, HVR1 might aid in stabilizing E2 by covering this hydrophobic surface, which, in turn, might stabilize HVR1 through the interaction. The same study also suggested that the C-terminal stalk region might be packed closely against the E2 back layer, further implying that the additional E2 protein regions on E2ΔTM that are absent in E2c2 could have an overall stabilizing effect. However, the HDX results for E2ΔTM in an E2ΔTM/AR2A/AP33 complex were surprisingly comparable to the results for E2c2 described above (Fig. S5). For example, the peptide regions that overlap between E2ΔTM and E2c2 displayed similar levels of flexibility, suggesting the HVR1 and the C-terminal stalk did not stabilize the core of the E2 protein. Nevertheless, two peptide fragments from the front layer had less deuterium exchange in E2ΔTM compared with E2c2, although this reduced deuterium exchange was modest and only apparent at the earliest time point (10 s) and more subject to experimental variability (Fig. S5). Lower exchange in the front layer is also shown in a previously published HDX study of fully glycosylated E2ΔTM from a different strain (41). Furthermore, over 90% deuterium exchange was observed for all peptides spanning HVR1 of E2ΔTM by the first time point, suggesting HVR1 is at least as flexible as the front layer and VR2 of E2c2. Thus, in addition to being hypervariable in sequence, HVR1 is highly exposed and likely very heterogeneous in conformation, which could serve to distract the immune response. Finally, the C-terminal stalk region also appeared flexible in our study, with four of seven peptide regions having more than 60% deuterium exchange by the first time point. Although less flexible than the front layer or HVR1, the deuterium exchange profile of the C-terminal stalk is comparable to the long CD81bl extending from the β-sandwich (Gly523–Asp535) in E2c2.
Importantly, our HDX studies revealed that the E2 front layer, which constitutes most of the CD81bs, is remarkably flexible. Although displaying slower deuterium exchange than the front layer, the CD81bl is also relatively flexible compared with other E2 regions. The high flexibility was not notably affected by the presence or absence of HVR1 or the C-terminal stalk. The high flexibility of the front layer is surprising because in the crystal structure, 20% of the front layer consists of a helical region, whose backbone amides should be protected through hydrogen bonding, and because the front layer is stapled to the rest of the E2 protein by two disulfide bonds in a manner that partially buries multiple front layer backbone amides. Nonetheless, this finding now provides a possible explanation of recent structural studies of antibodies that target AS434 on the front layer. Specifically, HC84-27 (23) and weakly neutralizing mAb 8 (24) bind to a peptide spanning AS434 from opposite sides (Fig. S6). When superposed on the E2c-AR3C crystal structure, bNAb HC84-27 does not clash with the E2 protein and binds at an angle of approach comparable to bNAb AR3C, the antibody present in the E2c crystal structure. However, mAb 8 would severely clash with E2 if superposed on the E2c structure in the same manner because its epitope is buried by the E2 β-sandwich (24, 42). The ability of mAb 8 to neutralize some viruses, albeit weakly, suggests that major conformational rearrangements at AS434 are possible that would allow mAb 8 to bind without a clash. Such conformational flexibility of AS434 is now supported by the HDX findings. AS434 rearrangement would entail a nearly 180° rotation of the AS434 helix, and would possibly require disulfide bonds that link the front layer with the rest of the E2 core to adopt alternate, extended disulfide conformations. For example, alternating conformations of a disulfide linking the I-EGF1 and I-EGF2 domains of β2 integrin are able to accommodate a remarkable >20-Å hinge motion between the domains (43, 44). Considering the poor neutralizing ability of mAb 8, the alternative AS434 conformation that is recognized by mAb 8 might not be optimal for vaccine design.
Fig. S6.

Opposing modes of recognizing the HCV E2 front layer. (Left) Surface representation of crystal structures of bNAb HC84-27 (Top) (PDB ID code 4JZO) and weakly neutralizing mAb 8 (Bottom) (PDB ID code 4HZL) is shown superposed on their common peptide epitope-spanning residues 430–446 within the E2 front layer, displayed as red (HC84-27 epitope) and blue (mAb 8 epitope) ribbons. (Right) Superposition of the peptide epitopes is shown in detail, indicating nearly equivalent positioning of Trp437, which would point into the protein core in the crystal structure of E2c.
Mapping the Conformational Flexibility of E2 by MD Simulations.
The HDX and EM experiments provided empirical evidence for flexibility at the CD81bs of E2. To assess the flexibility of the CD81bs further, we performed MD analysis on unbound E2c from the published E2c-Fab AR3C crystal structure (14) (Fig. S7). The Cα root mean square fluctuation (RMSF) over a 100-ns simulation was used as a measure of the flexibility. Consistent with the HDX study, MD indicated that the front layer was one of the most flexible regions of E2c (Figs. S5 and S7A). Within the front layer, regions of high flexibility include residues 420–423, 431–434, and 448–452, each with an average 2.7-Å RMSF (Fig. S7C), which is threefold higher than the 0.9-Å RMSF observed for the β-sandwich. Overall, the unliganded E2 front layer appeared to prefer conformations that drift away from the central β-sandwich of the E2 protein, with a 5- to 6-Å RMSD relative to the antibody-bound conformation (Fig. 3C). Interestingly, the most populated cluster of conformations adopted by the front layer is characterized by an extended conformation of the AS434 helical region, in which loops extending from the helix deviate 7–8 Å from the bNAb-bound conformation in the crystal structure (Fig. S7). This conformational difference is surprising because the Cys452–Cys620 disulfide bond staples the nearby C terminus of the front layer to the more stable back layer. However, the MD results revealed that this disulfide bond appears to accommodate the conformational differences by potentially adopting alternate orientations (Fig. S7E), as previously observed for the integrin subunits discussed above (43, 44). Together with the HDX data, the MD findings further support the conclusion that the front layer is conformationally flexible.
Fig. S7.

MD simulation of E2. (A) Conformational flexibility of HCV E2c based on cluster analysis of MD simulation. The conformations sampled by unliganded E2c during a 100-ns MD simulation were clustered using the backbone rmsd-based hierarchical average linkage clustering, with a merging distance cutoff of 2 Å over the entire trajectory. Based on this cutoff, 10 clusters were generated, with representatives from each cluster shown as superposed cartoons. Individual structural components are shown separately, as indicated by the circled areas in the figure. The cartoons are rainbow-colored based on sequence distance from the N terminus. The initial AR3C antibody-bound conformation from the crystal structure is also displayed as a gray cartoon. (B) Conformational flexibility of the HCV E2c front layer based on a second cluster analysis of the MD simulation. The conformations sampled by unliganded E2c during a 100-ns MD simulation were subjected to the all-atom rmsd-based hierarchical average linkage, clustering with a merging distance cutoff of 2 Å (details are provided in SI Materials and Methods). Based on this cutoff, 37 clusters were generated, with representatives from each cluster shown as superposed cartoons. The cartoons are rainbow-colored as in A. The structures of the E2c front layer from the top three most populated clusters are shown below, together with the initial AR3C antibody-bound conformation (gray) for comparison. (C) RMSF of each residue of unliganded E2c after a 100-ns MD simulation is mapped onto the crystal structure of E2c by a color gradient and presented as in Fig. S5. (D) Average RMSF of the front layer, β-sandwich, and back layer are plotted. (E) Most populated conformation of the E2c front layer after 100 ns of MD (red ribbon) is displayed, with the front layer bNAb-bound conformation (gray ribbon) from the crystal structure. The two structures are presented after being superposed at the back sheet of the β-sandwich, which is at the core of the protein. The C termini of the two conformations of the front layers are displayed below to highlight the alternate orientations adopted by a disulfide bond. This disulfide bond staples the front layer to the more stable back layer of E2c.
Fig. 3.

Calorimetry studies of HCV E2ΔTM. (A) DSC denaturation curves for E2ΔTM, E2c, E2c3, and E2cΔFL are displayed. The Tm and ΔT1/2 values are indicated next to the curves. The raw data are shown in black, whereas the fitted curves, from which are derived the Tm and ΔT1/2 values, are shown as red dots. (B, Left) ITC analysis of E2ΔTM binding to AR1B, AR2A, AR3C, CD81, AP33, and HCV1 is presented in a graph. (B, Right) Binding Kd of each interaction is shown. Values for −TΔS, ΔH, ΔG, and Kd are the averages of results from three ITC experiments. Representative isotherms are illustrated in Fig. S8.
Thermal Stability of HCV E2.
The observed high conformational flexibility of recombinant E2 suggests that E2 might be a relatively unstable protein, which could be detrimental for vaccine development. To assess the global thermal stability of E2, we first analyzed E2ΔTM using DSC (Fig. 3A). Unexpectedly, the DSC profile of E2ΔTM displayed a single unfolding peak with a high thermal denaturation midpoint (Tm) of 84.8 °C, comparable to the DSC profiles of proteins from thermophilic organisms (45), which generally display Tm values above 80 °C. Furthermore, the E2ΔTM Tm is substantially higher than other viral envelope proteins, such as HIV-1 gp140 (68.1 °C) (46) and influenza hemagglutinin (67.9 °C) (47). However, the DSC unfolding peak of E2ΔTM is relatively broad, with a transition width (ΔT1/2) of 10.6 °C, which is nearly twice the ΔT1/2 of HIV-1 gp140 (46, 48). A broad ΔT1/2 may be indicative of a multistep, noncooperative unfolding transition, suggesting the protein may contain different domains that unfold independently (49, 50). Next, to determine the influence of the C-terminal stalk region and the N-terminal HVR1 on the stability of E2, we analyzed the E2c construct that lacks these components. As suggested by the HDX analysis above, lack of the stalk and HVR1 had only a modest effect on the stability of E2c, which has a Tm of 81.5 °C. However, the unfolding peak of E2c remained broad, with a ΔT1/2 of 10.0 °C, indicating that this construct still displayed a noncooperative unfolding behavior.
We next investigated how the flexible, surface-accessible regions that are poorly defined in the electron density in the E2c crystal structure might affect the melting profile of E2. Thus, we modified the E2c construct to E2c3 by truncating loops at residues 569–581 and 585–597, which overlap VR3 and the following disordered region. The removal of these loops from E2c did not affect protein expression and purification, although E2c3 had a lower Tm of 75.7 °C, nearly twice the difference in Tm between E2c and E2ΔTM, but with a similar ΔT1/2 (11.2 °C). This finding indicated that truncation of a flexible and variable region between the β-sandwich and back layer had a destabilizing effect on E2, but did not result in a more cooperative melting curve.
Lastly, to study the contribution of the front layer to the overall thermal stability of E2, we generated an E2c construct lacking the front layer (E2cΔFL). The E2cΔFL had a Tm of 77.9 °C, suggesting it is less stable than E2c, but surprisingly more stable than E2c3. The result implies that E2 thermal stability is more tolerant of a front layer deletion than truncation of the flexible VR3 region defined previously (14). A recent crystal structure of E2cΔFL from a genotype 2a HCV further supports a relatively minimal role of the E2 front layer in overall E2 stability (41). Thus, despite the flanking disulfide bridges to the β-sandwich and back layer, the front layer might be more akin to a flexible loop than a rigid structural element stably integrated with the rest of the E2 protein.
Thermodynamic Parameters of HCV E2 Interactions with Ligands.
The change in entropy during protein binding, −TΔS, can be an indication of structural ordering induced by ligand binding. For example, with interactions between HIV-1 gp120 monomer and CD4 (and similar ligands), high and unfavorable −TΔS correlates with the induction of structural rearrangement (51). Here, we used ITC to measure the −TΔS of binding to HCV E2ΔTM by the CD81bs-targeting ligands bNAbs AR3C, AP33, and HCV1 and by the large external loop of CD81 (Fig. 3B and Fig. S8). For comparison, we also measured binding to E2ΔTM by a non-CD81bs–directed ligand mAb, AR1B.
Fig. S8.

ITC thermograms. Representative ITC thermograms with curves fitting the data are displayed for the interactions between E2ΔTM and various antibodies. Note that the thermogram for the addition of CD81 to a preformed complex of E2ΔTM and AR2A is displayed for E2+AR2A+CD81.
The ITC results indicated that all ligands tested bound tightly to E2ΔTM. The −TΔS of the CD81bs-targeting AR3C (2.79 kcal/mol) and CD81 (−0.44 kcal/mol) was modest. However, the −TΔS of AR3C and CD81 did not notably differ from the −TΔS of the non–CD81bs-directed antibodies AR2A (1.25 kcal/mol) and AR1B (3.43 kcal/mol). By contrast, the −TΔS of AS412 targeting bNAbs AP33 (6.26 kcal/mol) and HCV1 (5.44 kcal/mol) was nearly twofold higher than for AR3C, suggesting more stabilization on binding to the 12-residue AS412 than the other CD81bs components. In comparison to the high −TΔS observed for CD4 binding to gp120 (55 kcal/mol) (51), it appears that CD81bs recognition does not entail large, global conformational changes in E2.
Conclusion
Our investigations of E2 antigens by EM, HDX, and MD have shown that the CD81bs exhibits high structural flexibility. Our results extend and confirm previous observations and speculations made from crystal structures showing that the AS412 region is disordered when unbound (14, 41) and adopts a hairpin or open-loop conformation when complexed with different NAbs (17–22). Such alternate conformations appear to be critical for virus neutralization. A recent study by Sandomenico et al. (52) showed that antibodies raised against cyclized AS412 peptides that adopt an alternative conformation were not neutralizing. From the present study, the CD81bs front layer resembles a highly flexible loop despite being held by two disulfide bonds and despite high sequence conservation, which is generally not associated with flexible loops in viral proteins. High flexibility is consistent with the observation that soluble E2 and E1E2 can elicit antibodies that can bind the CD81bs with high affinity but, nonetheless, cannot neutralize the virus (27–31). Some of these non-NAbs or weak NAbs could be similar to mAb 8, which recognizes a conformation likely to be poorly accessible on the virus (Fig. S6). By mapping E2 conformational dynamics, the data here provide a molecular explanation for why it is difficult to elicit CD81bs NAbs to the virus using recombinant E2. In contrast to HIV, which has a recessed CD4 receptor-binding site, the CD81bs on HCV is fully accessible (Fig. S1), and yet NAbs to CD81bs are difficult to elicit because of conformational flexibility. Interestingly, the DSC findings indicated that although E2 is flexible, it is also thermally stable, at least in its core region, presumably due to its high density of disulfide bonds. It appears that in E2, HCV has evolved a highly stable β-sandwich scaffold stabilized by conserved disulfide bonds to accommodate both sequence variation and conformational flexibility. Of note, the observations made with the soluble E2 antigens may differ in the context of E2 on the viral surface. Nonetheless, next-generation soluble E2 antigens with a more stabilized CD81bs that is in a functional conformation recognized by NAbs may now be engineered based on these E2 flexibility results and published structures. Indeed, antigen stabilization as a rational vaccine design strategy to enhance NAb production has been demonstrated for respiratory syncytial virus (53), and is being actively pursued for HIV (54, 55). Further work in determining E2 CD81bs flexibility on the E1E2 complex and the virion is warranted to improve immunogen design targeting this important conserved functional site on HCV.
Materials and Methods
Antibodies.
Fabs were either generated from papain (Sigma) digestion of the corresponding IgGs or expressed in Escherichia coli (BL21 strain; Novagen) as described previously (14).
E2 Proteins.
The four soluble E2 constructs in this study were E2ΔTM, E2c2, E2c3, and E2cΔFL (Fig. S2). The constructs were produced in 293F cells in the presence of kifunesine.
HDX.
Protein complexes analyzed by HDX were introduced to an Orbitrap Elite Mass Spectrometer (ThermoFisher Scientific) for electrospray ionization and accurate mass measurements. DXMS Explorer (Sierra Analytics) was used for the calculation of the deuteration level of all of the peptides as described previously (56, 57).
ITC.
ITC binding experiments were performed using a MicroCal Auto-iTC200 instrument (GE Healthcare).
DSC.
Thermal melting curves of fully glycosylated HCV E2 glycoproteins were obtained with a MicroCal VP-Capillary calorimeter (Malvern).
EM.
Size exclusion chromatography-purified, EndoH-treated E2/Fab complexes were imaged on an FEI Tecnai F20 instrument at 200 kV transmission electron microscope (TEM) equipped with a Gatan Ultrascan 4k × 4k CCD. Data were collected using Leginon Multi-Scale Imaging and processed with TiltPicker (58), Xmipp (59), SPIDER (60), and EMAN1 (61).
MD Simulations.
MD simulations were performed using the Desmond MD package with the Maestro–Desmond interoperability tool, version 2.0 (62).
Further details of materials and methods used in this study are provided in SI Materials and Methods.
SI Materials and Methods
Expression and Purification of Antibodies.
Fabs HCV1, AP33, and AR2A were generated from papain (Sigma) digestion of the corresponding IgGs. Fabs were purified using Protein A agarose (General Electric), followed by size exclusion chromatography (SEC) using a Superdex 200 instrument (GE Healthcare) (14, 17, 18). Fabs AR3C and AR1B were expressed in Escherichia coli (BL21 strain; Novagen) as described previously (14). In short, bacteria transformed with the Fab expression plasmids were inoculated into superbroth growth media supplemented with ampicillin and grown to the log phase at 37 °C. The bacteria were induced with isopropyl β-d-1-thiogalactopyranoside to a final concentration of 200 μM and incubated at 16 °C overnight with shaking. On the next day, the bacterial culture was harvested by centrifugation, and the cell pellets were resuspended in PBS and treated with lysozyme (Amresco, Inc.) and protease inhibitor mixture (Sigma). Soluble Fabs were extracted from the cells by sonication, and clarified cell lysates were purified over a Protein G affinity column (63). The antibody Fab fragments were eluted with 0.2 M glycine (pH 2.2), buffer-exchanged to PBS, and purified further by SEC.
Generation of Soluble E2 Constructs for Protein Expression and Purification.
The four soluble E2 constructs in this study were E2ΔTM, E2c2, E2c3, and E2cΔFL (Fig. S2). E2ΔTM contains the E2 sequence for residues 384–717. E2c2 is an E2 core construct lacking the N-terminal HVR1 (residues 384–411) and C-terminal stalk region (residues 646–746), with the fourth and ninth glycosylation sites mutated out as in E2c (14). E2c3 is an E2c variant with the VR3 loop (residues 569–581) and is a predicted disordered region (residues 585–597) replaced by one or two glycine residues. E2cΔFL (E2 residues 492–645) is another E2c variant with the front layer and VR2 truncated. The constructs were cloned into the pCMV-Tag4-tpa mammalian expression vector and cotransfected with pAdvantage (Promega) in 293F cells in the presence of 10 μM kifunensine. Supernatant was purified over an antibody affinity column consisting of AR3A or AR1B IgG chemically conjugated to Protein A agarose beads. Bound E2 proteins were eluted with 0.2 M glycine (pH 2.2), buffer-exchanged to PBS, and purified further by SEC. Purified proteins were evaluated by SDS/PAGE and ELISA using an in-house anti-HCV antibody panel (AR1A, AR1B, AR2A, and AR3A) and a large external loop of CD81 (CD81-LEL).
Protein Complex Formation and Deglycosylation.
E2 proteins were deglycosylated before analysis by HDX and EM. To minimize E2 aggregation during deglycosylation, E2 proteins were complexed with Fab AR2A or AR1B at a 1:1.2 molar ratio. The complex was then deglycosylated with EndoH in 200 mM NaCl and 50 mM sodium citrate (pH 5.5) at 37 °C for 1 h following the manufacturer’s protocol (New England Biolabs) before final purification by SEC. EndoH cleaves between the first and second GlcNAc moieties next to the Asn.
HDX Sample Preparation.
For HDX, three complexes were assembled: EndoH-treated E2c2 bound to Fab AR2A (E2c2-AR2A), EndoH-treated E2c2 bound to Fabs AR2A and AP33 (E2c2-AR2A-AP33), and EndoH-treated full-length E2ΔTM bound to Fabs AR2A and AP33 (E2ΔTM-AR2A-AP33). Fab AR2A was added to E2 first at a 1.2:1 molar excess before the complex was deglycosylated using EndoH as previously described (14). This binary complex was first purified by SEC before addition of the other Fabs. The complete complexes were exchanged into 1× sodium PBS at 0.5–1.0 mg/mL before analysis by HDX.
HDX.
Before the deuteration studies, disulfide reduction and quench conditions that produced optimal sequence coverage were established as previously described (37, 64, 65). For E2c2-Fab AR2A (0.6 mg/mL), Fab AP33 (2 mg/mL), and E2c2-Fab AR2A-Fab AP33 (0.57 mg/mL), functional deuteration of proteins was performed by mixing 2 μL of stock solution with 4 μL of D2O buffer [8.3 mM Tris, 150 mM NaCl in D2O (pDread 7.2)] at 0 °C. At 10 s, 100 s, and 1,000 s, 9 μL of ice-cold quench buffer [6.4 M GluCl, 1 M Tris(2-carboxyethyl)phosphine (TCEP) (pH 2.85)] was added to stop the exchange reaction. Quenched samples were incubated on ice for 5 min; then, 5 μL of quench diluent [0.8% formic acid, 16.6% (vol/vol) glycerol] was added, and samples were frozen on dry ice. The nondeuterated and equilibrium-deuterated control samples were prepared by diluting 2 μL of stock proteins into 4 μL of H2O buffer [8.3 mM Tris, 150 mM NaCl (pH 7.2)] with 1 min of incubation on ice or 4 μL of 0.8% formic acid in 99.9% D2O with incubation at room temperature for 24 h, and quenched as above for the functional deuterated samples. The HDX of E2ΔTM-Fab AR2A-Fab AP33 was performed separately, but in the same manner except for an additional time point at 30 s. All samples were subsequently thawed at 4 °C and passed over a pepsin column (16-μL bed volume) at a flow rate of 20 μL⋅min−1. Proteolytic peptide fragments were collected on a C18 trap (Michrom Magic C18AQ; 0.2 × 2 μm) and separated using a C18 reverse phase analytical column (Michrom Magic C18AQ; 0.2 × 50 3 μm, 200 Å) running a linear gradient of 6.4% (vol/vol) to 38.4% (vol/vol) acetonitrile in 0.05% trifluoroacetic acid over 30 min. The peptide-containing eluent was introduced to an Orbitrap Elite Mass Spectrometer (ThermoFisher Scientific) for electrospray ionization (ESI) and accurate mass measurements. The instrument was operated in the positive ESI mode, and mass spectra were recorded in both MS1 and tandem MS mode from m/z 150–2,000 at a mass-resolving power of 60,000 (at m/z 400). Proteome Discoverer software (ThermoFisher) was used to identify the sequence of the peptide ions. DXMS Explorer (Sierra Analytics, Inc.) was used for calculation of the deuteration level of all of the peptides as described previously (56, 57).
ITC.
ITC binding experiments were performed using a MicroCal Auto-iTC200 instrument (GE Healthcare). All proteins were extensively dialyzed against a buffer containing 20 mM Tris and 150 mM NaCl (pH 7.4) before conducting the titrations. Subsequently, protein concentrations were adjusted and confirmed using calculated extinction coefficients and absorbance at 280 nm. In the syringe, the ligands were Fab HCV1, Fab AP33, Fab AR3C, Fab AR2A, Fab AR1B, and CD81 at concentrations ranging between 45 and 88.4 μM. Fully glycosylated E2ΔTM was in the cell at concentrations ranging between 4.5 and 10 μM. Soluble CD81-LEL naturally forms dimers, and the dimer can bind to two E2 proteins, which could potentially complicate the binding analysis. Therefore, E2ΔTM was first complexed to mAb AR2A, which is known to prevent the CD81 dimer from binding to a second E2ΔTM (14). The protein-binding experiments were performed with the following parameters: cell at 25 °C, 16 injections of 2.5 μL each, injection interval of 180 s, injection duration of 5 s, and reference power of 5 μcal. To perform sequential binding experiments to characterize CD81 binding by ITC, the mixed sample from the first titration (E2ΔTM + Fab AR2A) was left in the cell and the concentration of the HCV component was recalculated based on the dilution from the first experiment (∼88% of the initial concentration). Subsequently, CD81 was added in a second titration. To calculate the affinity constants (Kd), the molar reaction enthalpy, and the stoichiometry of binding, Origin 7.0 software was used by fitting the integrated titration peaks using a single-site binding model.
DSC.
Thermal melting curves of fully glycosylated HCV E2 glycoproteins were obtained with a MicroCal VP-Capillary calorimeter (Malvern). The purified E2 glycoproteins produced from 293S cells were buffer exchanged into 1× PBS and concentrated to 0.5–1 μM before analysis by the instrument. Melting was probed at a scan rate of 90 °C⋅h−1. Data processing, including buffer correction, normalization, and baseline subtraction, was conducted using the standardized protocol from the Origin 7.0 software.
EM Sample Preparation and Imaging.
SEC-purified EndoH-treated E2 proteins bound to Fab AR2A, Fab AR1B, and Fab HCV1 were diluted to 9.1 μg/mL, applied to a freshly glow-discharged C-flat grid prepared for RCT, and negatively stained with 2% uranyl formate. For RCT, we used carbon-coated, 400-copper mesh grids with 2-μm holes at 2-μm regular spacing. Over the carbon was a layer of nitrocellulose with another thin layer of carbon. The complexes were imaged at a magnification of 100,000× on an FEI Tecnai F20 instrument at 200 kV TEM equipped with a Gatan Ultrascan 4k × 4k CCD, resulting in a pixel size of 1.09 Å per pixel (calibrated using catalase crystal diffraction) at a dose of ∼25 electron (e)/Å2. Data were collected using Leginon Multi-Scale Imaging for RCT (MSI-RCT raster 3.1) software at angles of 0° and −55° (66).
Data Processing, 3D Volume Reconstruction, and Model Fitting.
Tiltpicker was used to select tilt pairs automatically from raw EM micrographs that were then binned by 2, resulting in 2.17 Å per pixel and placed into 160 × 160 pixel boxes (58). Particles were aligned with Xmipp Maximum likelihood alignment, resulting in a final stack of 6,364 raw particles (59). Three RCT models were created using Spider from class averages representing one of three positions of Fab HCV1 (60). Model refinement was conducted in EMAN1, resulting in ∼30-Å resolution reconstructions based on a 0.5 Fourier shell correlation cutoff value (61) (Fig. S3).
SI MD Simulations
E2c Structure Preparation for MD Simulation.
The starting coordinates for E2c were obtained from our E2c-AR3C crystal structure (Protein Data Bank ID code 4MWF) (14). The structure was prepared by assigning bond orders and adding hydrogen atoms using the default protocol from the protein preparation wizard interface with Maestro 8.5 in the Schrödinger biologics suite (https://www.schrodinger.com). Force-field parameters for standard amino acid residues were obtained from the optimized potentials for a liquid simulations-all atom (OPLS-AA) force field (67). The prepared structure was subsequently minimized in MacroModel 9.7 (https://www.schrodinger.com) using the steepest descent method with a maximum of 500 iterations, with a gradient convergence threshold of 0.05 and constant dielectric with a dielectric constant of 1. The OPLS-AA force field with extended cutoff values [van der Waals (VDW) = 8.0 Å, electrostatic = 20.0 Å, H-bond = 4.0 Å] was used for the minimization. We note that the MD simulations are carried out on the known structure of the E2c and differences could occur for MD simulations of intact E2 and in E1/E2 on the viral surface. However, these structures are not yet known and, hence, we cannot simulate them.
System Preparation for MD Simulation.
MD simulations were performed using the Desmond MD package with the Maestro–Desmond interoperability tool, version 2.0 (62). The systems were set up for explicit solvent simulations. The solute was placed in an orthorhombic box containing simple point charge (SPC) water at 1.5 nm from solute boundaries. A physiological salt concentration of 0.15 M was used.
Simulation Setup.
An MD simulation for a period of 100 ns was performed using the following three-step protocol: (i) minimization, (ii) equilibration, and (iii) production run.
Minimization.
The setup system was minimized to remove close contacts between solute/solvent molecules using the Limited Memory Broyden–Fletcher–Goldfarb–Shanno method with maximum iterations of 200 cycles and a convergence threshold of 1.0 kcal⋅mol−1⋅Å−1. The step size was 0.005 ps, and the switch criterion was 25.0 kcal⋅mol−1⋅Å−1. The short-range interactions used a cutoff radius of 9.0 Å, whereas the long-range coulombic interactions were taken into account using smooth particle mesh Ewald (PME) with an Ewald tolerance of 1 e−09/Å.
Equilibration protocol.
During equilibration, 2,000 steps of steepest descent minimization of water molecules and ions were performed to allow water molecules to assume a lower energy geometry, whereas the solute was restrained with a force constant of 50 kcal⋅mol−1⋅Å2. The resulting system was then subjected to 2,000 steps of minimization with no restraints, reaching a root mean square gradient of 0.1 to assure relaxation of the structures, followed by 12 ps of heating from 10 to 300 K in a constant volume ensemble with restraints on the solute heavy atoms (50 kcal⋅mol−1⋅Å2). Annealing was followed by 12 ps of constant-pressure unrestrained simulation at 300 K, where convergence of energies, temperature, pressure, and density of the systems was monitored.
Productions run protocol.
The output coordinates and velocities from the equilibrated run were subsequently used in a production run over a period of 100 ns at 300 K using an isothermal/isobaric (NPT, where N= number of particles, P= constant pressure, T= constant temperature) ensemble. Hydrogen bonds were constrained with a variant of the M-SHAKE algorithm (68). To maintain 1 atm at 300 K in the NPT ensemble, the system was coupled to a Martyna–Tobias–Klein barostat (69) with a relaxation time of 2 ps with the isotropic coupling style, and a Nose–Hoover thermostat with a relaxation time of 1 ps (70). Long-range electrostatic interactions were modeled using a PME method (71). The VDW interactions and real-space contributions to the electrostatics were truncated at 10 Å, and the long-range VDW contributions to the energy and the pressure were estimated by assuming a homogeneous distribution of VDW spheres with a dispersion coefficient 69.5 kcal⋅mol−1⋅Å. A reversible reference system propagator algorithms (RESPA) integrator (72) with steps of 2 fs was used for bonded and short-range nonbonded interactions, and it was used at 6 fs for long-range electrostatics. During the course of simulation, a quality check of the system was performed by monitoring the total energy profile and the Cα rmsd of structures in the MD trajectory.
Cluster Analysis of the E2c MD Trajectory.
Cluster analysis of the E2c MD simulation trajectory was performed using Maestro (https://www.schrodinger.com), version 8.5. Two independent clustering approaches were used to understand the flexibility of E2c better. First, a trajectory was subjected to the rmsd-based hierarchical average linkage clustering, with a merging distance cutoff of 2 Å. Backbone atom-based clustering led to 10 clusters, which, in agreement with the HDX and visual analysis of the trajectory, confirmed that the β-sandwich region is stable, whereas the front layer is exceptionally flexible (Fig. S7A). Second, to study the conformational preferences of the front layer, all-atom–based clustering was performed. For this clustering, the residues from the most stable elements of the β-sandwich and the back layer (residues 64–75, 80–86, 117–131,174–199, and 207–211), as well as the front layer (residues 9–41), were used. In this way, the clustering was driven by the conformational flexibility of the front layer. The calculation resulted in 37 clusters (Fig. S7B); three of the most populated clusters were selected for further analysis.
Acknowledgments
R.U.K. thanks the Swiss National Science Foundation for a postdoctoral fellowship. This work is supported by NIH Grants AI079031 and AI123861 (to M.L.); NIH Grants AI106005 and AI123365 (to M.L. and I.A.W.); NIH Grant GM094586 (to I.A.W.); and NIH Grants AI117905, GM020501, and AI101436 (to S.L.), as well as the Skaggs Institute (I.A.W.). This article is manuscript 29353 from The Scripps Research Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The electron microscopy reconstruction of the complex consisting of hepatitis C virus (HCV) E2 core domain bound to antigen-binding fragments antigenic region 1B (AR1B), AR2A, and HCV1 has been deposited with the Electron Microscopy Data Bank under accession codes EMD-8338, EMD-8339, and EMD-9340.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1609780113/-/DCSupplemental.
References
- 1.Messina JP, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015;61(1):77–87. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thursz M, Fontanet A. HCV transmission in industrialized countries and resource-constrained areas. Nat Rev Gastroenterol Hepatol. 2014;11(1):28–35. doi: 10.1038/nrgastro.2013.179. [DOI] [PubMed] [Google Scholar]
- 3.Feeney ER, Chung RT. Antiviral treatment of hepatitis C. BMJ. 2014;348:g3308. doi: 10.1136/bmj.g3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pawlotsky JM, Feld JJ, Zeuzem S, Hoofnagle JH. From non-A, non-B hepatitis to hepatitis C virus cure. J Hepatol. 2015;62(1) Suppl:S87–S99. doi: 10.1016/j.jhep.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, et al. Hepatitis C virus genotype diversity among intravenous drug users in Yunnan Province, Southwestern China. PLoS One. 2013;8(12):e82598. doi: 10.1371/journal.pone.0082598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ly KN, et al. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann Intern Med. 2012;156(4):271–278. doi: 10.7326/0003-4819-156-4-201202210-00004. [DOI] [PubMed] [Google Scholar]
- 7.Marco A, et al. Hepatitis C virus reinfection among prisoners with sustained virological response after treatment for chronic hepatitis C. J Hepatol. 2013;59(1):45–51. doi: 10.1016/j.jhep.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Midgard H, et al. Hepatitis C reinfection after sustained virological response. J Hepatol. 2016;64(5):1020–1026. doi: 10.1016/j.jhep.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Kong L, Jackson KN, Wilson IA, Law M. Capitalizing on knowledge of hepatitis C virus neutralizing epitopes for rational vaccine design. Curr Opin Virol. 2015;11:148–157. doi: 10.1016/j.coviro.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker CM, Grakoui A. Hepatitis C virus: Why do we need a vaccine to prevent a curable persistent infection? Curr Opin Immunol. 2015;35:137–143. doi: 10.1016/j.coi.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox AL. MEDICINE. Global control of hepatitis C virus. Science. 2015;349(6250):790–791. doi: 10.1126/science.aad1302. [DOI] [PubMed] [Google Scholar]
- 12.Zona L, et al. CD81-receptor associations--impact for hepatitis C virus entry and antiviral therapies. Viruses. 2014;6(2):875–892. doi: 10.3390/v6020875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Douam F, Lavillette D, Cosset FL. The mechanism of HCV entry into host cells. Prog Mol Biol Transl Sci. 2015;129:63–107. doi: 10.1016/bs.pmbts.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Kong L, et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science. 2013;342(6162):1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owsianka AM, et al. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J Virol. 2006;80(17):8695–8704. doi: 10.1128/JVI.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drummer HE, Wilson KA, Poumbourios P. Identification of the hepatitis C virus E2 glycoprotein binding site on the large extracellular loop of CD81. J Virol. 2002;76(21):11143–11147. doi: 10.1128/JVI.76.21.11143-11147.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong L, et al. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J Virol. 2012;86(23):13085–13088. doi: 10.1128/JVI.01939-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong L, et al. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci USA. 2012;109(24):9499–9504. doi: 10.1073/pnas.1202924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Potter JA, et al. Toward a hepatitis C virus vaccine: The structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J Virol. 2012;86(23):12923–12932. doi: 10.1128/JVI.02052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pantua H, et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J Mol Biol. 2013;425(11):1899–1914. doi: 10.1016/j.jmb.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 21.Meola A, et al. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J Virol. 2015;89(4):2170–2181. doi: 10.1128/JVI.02190-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, et al. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J Biol Chem. 2015;290(16):10117–10125. doi: 10.1074/jbc.M115.643528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krey T, et al. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 2013;9(5):e1003364. doi: 10.1371/journal.ppat.1003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng L, et al. Structural evidence for a bifurcated mode of action in the antibody-mediated neutralization of hepatitis C virus. Proc Natl Acad Sci USA. 2013;110(18):7418–7422. doi: 10.1073/pnas.1305306110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Law M, et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med. 2008;14(1):25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 26.Giang E, et al. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci USA. 2012;109(16):6205–6210. doi: 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ray R, et al. Characterization of antibodies induced by vaccination with hepatitis C virus envelope glycoproteins. J Infect Dis. 2010;202(6):862–866. doi: 10.1086/655902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer K, Banerjee A, Frey SE, Belshe RB, Ray R. A weak neutralizing antibody response to hepatitis C virus envelope glycoprotein enhances virus infection. PLoS One. 2011;6(8):e23699. doi: 10.1371/journal.pone.0023699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang P, et al. Hepatitis C virus epitope-specific neutralizing antibodies in Igs prepared from human plasma. Proc Natl Acad Sci USA. 2007;104(20):8449–8454. doi: 10.1073/pnas.0703039104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruwona TB, Giang E, Nieusma T, Law M. Fine mapping of murine antibody responses to immunization with a novel soluble form of hepatitis C virus envelope glycoprotein complex. J Virol. 2014;88(18):10459–10471. doi: 10.1128/JVI.01584-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ball JK, Tarr AW, McKeating JA. The past, present and future of neutralizing antibodies for hepatitis C virus. Antiviral Res. 2014;105:100–111. doi: 10.1016/j.antiviral.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellnitz S, et al. Binding of hepatitis C virus-like particles derived from infectious clone H77C to defined human cell lines. J Virol. 2002;76(3):1181–1193. doi: 10.1128/JVI.76.3.1181-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Triyatni M, et al. Structural features of envelope proteins on hepatitis C virus-like particles as determined by anti-envelope monoclonal antibodies and CD81 binding. Virology. 2002;298(1):124–132. doi: 10.1006/viro.2002.1463. [DOI] [PubMed] [Google Scholar]
- 34.McLellan JS, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480(7377):336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom. 2006;17(11):1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40(3):1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 37.Bale S, et al. Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Negl Trop Dis. 2011;5(11):e1395. doi: 10.1371/journal.pntd.0001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong L, et al. Local conformational stability of HIV-1 gp120 in unliganded and CD4-bound states as defined by amide hydrogen/deuterium exchange. J Virol. 2010;84(19):10311–10321. doi: 10.1128/JVI.00688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, et al. Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science. 2009;326(5956):1123–1127. doi: 10.1126/science.1175868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, Tawfik DS. Do viral proteins possess unique biophysical features? Trends Biochem Sci. 2009;34(2):53–59. doi: 10.1016/j.tibs.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 41.Khan AG, et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature. 2014;509(7500):381–384. doi: 10.1038/nature13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng L, et al. Discrete conformations of epitope II on the hepatitis C virus E2 protein for antibody-mediated neutralization and nonneutralization. Proc Natl Acad Sci USA. 2014;111(29):10690–10695. doi: 10.1073/pnas.1411317111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi M, et al. A structural hypothesis for the transition between bent and extended conformations of the leukocyte β2 integrins. J Biol Chem. 2007;282(41):30198–30206. doi: 10.1074/jbc.M701670200. [DOI] [PubMed] [Google Scholar]
- 44.Smagghe BJ, Huang PS, Ban YE, Baker D, Springer TA. Modulation of integrin activation by an entropic spring in the β-knee. J Biol Chem. 2010;285(43):32954–32966. doi: 10.1074/jbc.M110.145177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Razvi A, Scholtz JM. Lessons in stability from thermophilic proteins. Protein Sci. 2006;15(7):1569–1578. doi: 10.1110/ps.062130306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanders RW, et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013;9(9):e1003618. doi: 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Epand RM, Epand RF. Thermal denaturation of influenza virus and its relationship to membrane fusion. Biochem J. 2002;365(Pt 3):841–848. doi: 10.1042/BJ20020290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brower ET, Schön A, Freire E. Naturally occurring variability in the envelope glycoprotein of HIV-1 and development of cell entry inhibitors. Biochemistry. 2010;49(11):2359–2367. doi: 10.1021/bi1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morar-Mitrica S, Nesta D, Crotts G. Differential scanning calorimetry (DSC) for biopharmaceutical development: Old concepts, new applications. Biopharma Asia. 2013;2(4):44–55. [Google Scholar]
- 50.Deller MC, Kong L, Rupp B. Protein stability: A crystallographer’s perspective. Acta Crystallogr F Struct Biol Commun. 2016;72(Pt 2):72–95. doi: 10.1107/S2053230X15024619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Myszka DG, et al. Energetics of the HIV gp120-CD4 binding reaction. Proc Natl Acad Sci USA. 2000;97(16):9026–9031. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandomenico A, et al. Generation and characterization of monoclonal antibodies against a cyclic variant of hepatitis C virus E2 epitope 412-422. J Virol. 2016;90(7):3745–3759. doi: 10.1128/JVI.02397-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McLellan JS, et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science. 2013;342(6158):592–598. doi: 10.1126/science.1243283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guenaga J, et al. Well-ordered trimeric HIV-1 subtype B and C soluble spike mimetics generated by negative selection display native-like properties. PLoS Pathog. 2015;11(1):e1004570. doi: 10.1371/journal.ppat.1004570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kong L, et al. Uncleaved prefusion-optimized gp140 trimers derived from analysis of HIV-1 envelope metastability. Nat Commun. 2016;7:12040. doi: 10.1038/ncomms12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mendillo ML, et al. A conserved MutS homolog connector domain interface interacts with MutL homologs. Proc Natl Acad Sci USA. 2009;106(52):22223–22228. doi: 10.1073/pnas.0912250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen H, et al. Allosteric inhibition of complement function by a staphylococcal immune evasion protein. Proc Natl Acad Sci USA. 2010;107(41):17621–17626. doi: 10.1073/pnas.1003750107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: Software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166(2):205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheres SH, et al. Maximum-likelihood multi-reference refinement for electron microscopy images. J Mol Biol. 2005;348(1):139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 60.Frank J, et al. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116(1):190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 61.Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128(1):82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 62.Bowers KJ, et al. 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (SC06) (ACM Press, New York)
- 63.Erntell M, Myhre EB, Sjöbring U, Björck L. Streptococcal protein G has affinity for both Fab- and Fc-fragments of human IgG. Mol Immunol. 1988;25(2):121–126. doi: 10.1016/0161-5890(88)90059-4. [DOI] [PubMed] [Google Scholar]
- 64.Hastie KM, et al. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc Natl Acad Sci USA. 2011;108(48):19365–19370. doi: 10.1073/pnas.1108515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marsh JJ, et al. Structural insights into fibrinogen dynamics using amide hydrogen/deuterium exchange mass spectrometry. Biochemistry. 2013;52(32):5491–5502. doi: 10.1021/bi4007995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suloway C, et al. Automated molecular microscopy: The new Leginon system. J Struct Biol. 2005;151(1):41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 67.Jorgensen WL, Maxwell DS, TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 68.Krautler V, van Gunsteren WF, Hunenberger PH. A fast SHAKE: Algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem. 2001;22:501–508. [Google Scholar]
- 69.Martyna GJ, Tobias DJ, Klein ML. Constant-pressure molecular-dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 70.Hoover WG. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A Gen Phys. 1985;31(3):1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 71.Darden TA, York DM, Pedersen LG. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 72.Tuckerman M, Berne BJ, Martyna GJ. Reversible multiple time scale molecular-dynamics. J Chem Phys. 1992;97:1990–2001. [Google Scholar]