

Abstract
The basis of the extraordinary sensitivity and frequency selectivity of the cochlea is a chloride‐sensitive protein called prestin which can produce an electromechanical response and which resides in the basolateral plasma membrane of outer hair cells (OHCs). The compound 9‐anthracenecarboxylic acid (9‐AC), an inhibitor of chloride channels, has been found to reduce the electromechanical response of the cochlea and the OHC mechanical impedance. To elucidate these 9‐AC effects, the functional electromechanical status of prestin was assayed by measuring the nonlinear capacitance of OHCs from the guinea‐pig cochlea and of prestin‐transfected human embryonic kidney 293 (HEK 293) cells. Extracellular application of 9‐AC caused reversible, dose‐dependent and chloride‐sensitive reduction in OHC nonlinear charge transfer, Q max. Prestin‐transfected cells also showed reversible reduction in Q max. For OHCs, intracellular 9‐AC application as well as reduced intracellular pH had no detectable effect on the reduction in Q max by extracellularly applied 9‐AC. In the prestin‐transfected cells, cytosolic application of 9‐AC approximately halved the blocking efficacy of extracellularly applied 9‐AC. OHC inside‐out patches presented the whole‐cell blocking characteristics. Disruption of the cytoskeleton by preventing actin polymerization with latrunculin A or by decoupling of spectrin from actin with diamide did not affect the 9‐AC‐evoked reduction in Q max. We conclude that 9‐AC acts on the electromechanical transducer principally by interaction with prestin rather than acting via the cytoskeleton, chloride channels or pH. The 9‐AC block presents characteristics in common with salicylate, but is almost an order of magnitude faster. 9‐AC provides a new tool for elucidating the molecular dynamics of prestin function.
Keywords: auditory system, channelopathies, cochlea, electromechanical transduction, sensory hair cells
Introduction
The chloride‐channel inhibitor 9‐anthracenecarboxylic acid (9‐AC) (Jentsch et al., 2002) specifically blocks the skeletal muscle‐type chloride channel ClC‐1 (Estévez et al., 2003). Malfunction of this channel, essential for the stability of the resting membrane potential in skeletal muscle (Steinmeyer et al., 1991), causes myotonia (Rüdel & Lehmann‐Horn, 1985), a pathological condition also induced by 9‐AC (Moffett & Tang, 1968). ClC‐1 channels are also present in outer hair cells (OHCs) of the cochlea (Kawasaki et al., 1999), where chloride ions are essential for normal hearing (Santos‐Sacchi et al., 2006). Patients suffering from myotonic dystrophy also have a concurrent high risk of sensorineural hearing loss (Wright et al., 1988).
The cellular basis of the extraordinary frequency selectivity and sensitivity of the mammalian cochlea is the OHC (Ashmore, 2008; Dallos, 2008). In response to a change in its transmembrane potential (Brownell et al., 1985; Ashmore, 1987; Dallos & Evans, 1995) the OHC produces electromechanical force (Frank et al., 1999) to amplify the vibration response of the cochlea. The molecular basis of this electromechanical transducer is the motor protein prestin (Zheng et al., 2000), located in the OHC basolateral plasma membrane (Kalinec et al., 1992; Huang & Santos‐Sacchi, 1994; Cimerman et al., 2013). Prestin is essential for cochlear amplification and, therefore, normal hearing (Liberman et al., 2002; Dallos, 2008).
The electromechanical action of prestin requires intracellular chloride ions that must be translocated to an interaction site within prestin (Oliver et al., 2001; Rybalchenko & Santos‐Sacchi, 2003; Schaechinger et al., 2011; Gorbunov et al., 2014). There is evidence that a cAMP‐activated chloride channel, the cystic fibrosis transmembrane conductance regulator (CFTR), can physically interact with prestin to enhance voltage‐dependent charge displacement associated with motor activity (Homma et al., 2010). Evidence that chloride ions influence the electromechanical force derives from vibration measurements of acoustically induced motion of the basilar membrane in vivo (Santos‐Sacchi et al., 2006) and of electrically induced motion of the reticular lamina in vitro (Scherer & Gummer, 2004; Nowotny & Gummer, 2006), structures which are located at the basal and apical aspects of the OHC, respectively. 9‐AC, the chloride channel blocker used in the reticular lamina experiments, has also been shown to reduce the magnitude of the imaginary part of the OHC axial mechanical impedance (Eckrich et al., 2008). Although the mechanical impedance data suggest that the axial stiffness of the cell was reduced by 9‐AC, the mechanisms of 9‐AC block are unknown.
Given the importance of the chloride sensitivity of prestin and of putative links between hearing loss and chloride‐associated channelopathies, such as in myotonic dystrophy, the aim of the present study was to elucidate mechanisms of 9‐AC block of OHC function. We enquired whether (i) 9‐AC interacts with prestin, (ii) 9‐AC interacts with the cytoskeleton, and/or (iii) 9‐AC's blocking effects are mediated by chloride channels and/or change in intracellular pH. This aim was achieved by measuring the nonlinear capacitance (NLC)–the electrical component of the electromechanical transducer– of isolated OHCs and of human embryonic kidney (HEK) 293 cells transfected with prestin.
Materials and methods
Ethical approval
The study was approved by the Animal Protection, Veterinary Service and Veterinary Medicine Department of the University of Tübingen and by the Regional Council Tübingen (Reference numbers: 09.01.2012, 17.12.2009 and 10.01.2008), complying with legal requirements of the European Communities Council Directive of 24 November 1986 (86/609/EEC) for the protection of animals used for experimental purposes.
Preparation of OHCs
OHCs were isolated from the third and fourth cochlear turns of pigmented guinea pigs (n = 84, weight 300–900 g). Cell lengths ranged from 55 to 80 μm. Animals were bred in the Animal Facilities of the University of Tübingen (Einrichtung für Tierschutz, Tierärztlichen Dienst und Labortierkunde, directed by Dr Franz Iglauer). Animals were anesthetized by intraperitoneal injection of a mixture of 100 mg/kg ketamine and 4 mg/kg xylazine, and were killed by cervical dislocation. Temporal bones were dissected and placed in ice‐chilled Hanks' balanced salt solution (HBSS; Biochrom KG, Berlin, Germany), containing (in mm): NaCl, 137; KCl, 5.4; CaCl2, 1.25; NaHCO3, 4.2; MgSO47H2O, 0.81; KH2PO4, 0.44; Na2HPO42H2O, 0.34; glucose, 5.0; and HEPES, 10; with osmolarity 310 mOsm/L adjusted with d‐(+)‐glucose and pH 7.25. HBSS was used as a basic extracellular fluid throughout the preparation steps and experiments, unless otherwise stated. All chemicals were from Sigma–Aldrich (Taufkirchen, Germany) unless otherwise stated. HEPES was from MERCK (Darmstadt, Germany).
The bulla was opened and most of its wall was removed with a bone rongeur and placed in fresh HBSS. The majority of the cochlear wall was removed with a scalpel and the modiolus cut at its base to remove it from the cochlea. Stria vascularis was removed with forceps, while the organ of Corti was separated from the modiolus with a sharpened stainless‐steel fine needle. Pieces of organ of Corti originating from the upper third of the cochlea were placed in the experimental chamber containing 200 μL HBSS. OHCs were dissociated by gentle reflux using a 100‐μL Eppendorf pipette. After the OHCs settled down on a coverslip pre‐coated with poly‐l‐lysine (0.01%), requiring ~ 10 min, the chamber was filled with 2 mL HBSS. This volume eliminated significant hydrostatic pressure changes that might have been otherwise produced by fluid evaporation. OHCs were used within 2 h post mortem. All experiments were conducted at a controlled room temperature of 21.5 ± 0.5 °C.
Transfection of HEK 293 cells
Cells were transiently transfected with vector DNA rat prestin labelled with GFP (Halet, 2005) as previously described (Heidrych et al., 2008). Briefly, HEK 293 cells (German Collection of Microorganisms and Cell Cultures GmbH; DSMZ No. ACC‐305) were cultured and transfected in 1 mL Dulbecco's modified Eagle's medium (DMEM; Biochrom KG) containing 10% fetal calf serum (Gibco®) at 37 °C and 5% CO2 in 95% humidity. Culturing was performed in 12 culture‐dish wells containing sterile glass coverslips pre‐coated with poly‐d‐lysine (0.01%). After overnight incubation, cells were transfected with the vector DNA using the transfection reagent Lipofectamine™ 2000 (Invitrogen™ Life Technologies GmbH, Darmstadt, Germany) according to the manufacturer's instruction. Each well contained ~ 2.5 × 105 cells and received 2.4 μg DNA and 4 μL Lipofectamine™ 2000. Medium was changed 6 h after transfection. Experiments were performed after an incubation time of 48–72 h in extracellular solution containing (in mm): NaCl, 90; TEA‐Cl, 20; CsCl, 20; CoCl2, 2; MgCl2, 1.48; CaCl2, 2; and HEPES, 10 (pH 7.2, osmolarity 320 mOsm/L). This solution is known to minimize endogenous current amplitudes in these cells (Santos‐Sacchi & Navarrete, 2002).
Electrophysiology and drug application
Patch‐clamp recordings were performed to measure the NLC of OHCs and HEK 293 cells. For this purpose, a Zeiss Axioskop2 FS mot microscope (Zeiss, Heidelberg, Germany) was equipped with an EPC‐9 patch‐clamp amplifier (Heka Elektronik, Lambrecht/Pfalz, Germany). Patch pipettes were fabricated with a P‐87 Flaming/Brown type micropipette puller (Sutter Instrument Company, Novato, USA), using soda‐glass capillaries (Hildenberg GmbH, Malsfeld, Germany) for OHCs and borosilicate‐glass capillaries (GC150F‐10, Harvard Apparatus Ltd., Edenbridge, United Kingdom) for HEK 293 cells. Pipettes were filled with (intracellular) solution containing (in mm): CsCl, 135; HEPES, 10; MgCl2, 2; CaCl2, 0.1; and EGTA, 11 (pH 7.2, osmolarity 320 mOsm/L). Pipette osmolarity was chosen 10 mOsm/L above extracellular osmolarity to maintain a constant OHC intracellular pressure in equilibrium with atmospheric pressure.
The pipette resistance, measured in the (extracellular) bath solution, was 1.5–2 MΩ for the soda‐glass and 4–5 MΩ for the borosilicate‐glass pipettes. In the case of current–voltage (I‐V) relationship measurement (in HEK 293 cells), the in situ series resistance was 20–42 MΩ before online compensation of the capacitive transient; the series resistance was reduced by at least 60% by compensation. In the case of membrane‐capacitance measurements, the series resistance was compensated offline for each measurement point, based on the series‐resistance values provided by the patch‐clamp software. The series resistance was 1.6–6 MΩ for OHCs and 6–21 MΩ for HEK 293 cells.
Drugs were applied locally extracellular to the cell, and in some experiments added to the patch‐pipette solution. Extracellular drug application was performed through a Y‐shaped perfusion capillary system with tip positioned ~ 450 μm from the cell, in front of the patch capillary, and had a tip diameter of ~ 350 μm. A perfusion rate of 14 μL/min was set using a four‐channel Ismatec perfusion pump (IDEX Health & Science GmbH, Wertheim, Germany). In the following, ‘drug application’ means that the channel of the perfusion system was switched from the control solution to the extracellular solution that contained the drug; ‘washout’ means that the channel was switched back to the control solution. For extracellular drug application, a minimum drug pre‐incubation interval of 20 s was chosen to ensure that the extracellular solution had been completely replaced. For intracellular drug application, experiments were never started earlier than 1 min after patching the cell; this time interval ensured that the intracellular solution was replaced by the patch‐pipette solution, as ascertained in preliminary experiments using the calcium indicator fluorescence dye fluo‐3.
Stock solutions of 9‐AC and 9‐anthracene‐methanol (9‐AM) were prepared in dimethyl sulfoxide (DMSO) in concentrations of (in mm) 0.05, 0.5, 5, 50, 500 and 2500, and diluted 1 : 1000 or in the case of 2.5‐M stock solution 1 : 500, in fresh HBSS on the day of the experiment. The highest concentrations of 9‐AC stock solution were extensively sonicated (minimum of 10 min) in an ultrasound bath (Transsonic T460; Allpax, Papenburg, Germany) before the final dilutions were made. All 9‐AC‐containing HBSS were sonicated before use. The final DMSO concentration did not exceed 0.2%. Osmolarity and pH of the 9‐AC and 9‐AM solutions were controlled and corrected with d‐(+)‐glucose and NaOH application, respectively.
Two different methods were used to reduce the intracellular chloride concentration: either 115 mm CsCl was substituted with 40 mm Cs2SO4 in the patch solution (labelled as ‘Cs2SO4’ in Fig. 4) or 130 mm CsCl was substituted with 130 mm potassium gluconate in the intracellular solution and with 130 mm sodium gluconate in the extracellular solution (labelled as ‘redCl’ in Fig. 4).
Figure 4.
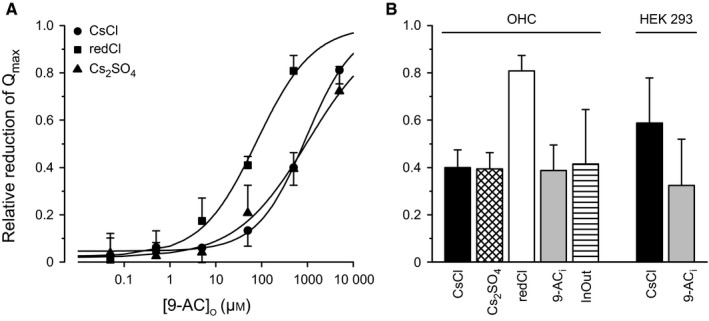
The 9‐AC block is chloride‐sensitive and 9‐AC can access from both sides of the plasma membrane. (A) Dose dependency of the extracellularly applied 9‐AC on the maximum amount of motor charge moved, Q max, measured with extracellular 9‐AC concentrations of 0.05, 0.5, 5, 50, 500 and 5000 μm. The effect on Q max is presented as relative reduction. Circles labelled ‘CsCl’, the control condition with intracellular solution containing 135 mm CsCl. Triangles labelled ‘Cs2 SO 4’, intracellular chloride concentration reduced by substituting 115 mm chloride with sulfate in the patch pipette. Squares labelled ‘redCl’, both intracellular and extracellular chloride concentrations reduced by substituting 130 mm CsCl with gluconate in both the patch‐pipette and bath solutions. This latter condition significantly shifted the dose dependency to the left. Lines indicate logistic‐function fits (Eqn (3)) with estimates of the IC 50 and the slope parameter, respectively, of 928 ± 66, 1012 ± 192, 79 ± 14 μm and 0.82 ± 0.04, 0.56 ± 0.06, 0.71 ± 0.08 for the three conditions. (B) The relative reduction in Q max in response to extracellular application of 500 μm 9‐AC, using the indicated intracellular solutions, patch configuration, drugs and cell types. Labelling on the abscissae has the following meaning. ‘CsCl’ (n = 11 for OHCs, n = 11 for HEK 293 cells), ‘Cs2 SO 4’ (n = 5), and ‘redCl’ (n = 4), as in panel A. ‘9‐AC i’, 500 μm 9‐AC in the patch solution (n = 5 for OHCs, n = 5 for HEK 293 cells). ‘InOut’, inside‐out excised‐patch configuration (n = 3); all other measurements were made in the whole‐cell configuration. Data for the bar labelled ‘OHC/CsCl’ are the same as those for the bar labelled ‘Control’ in Fig. 2D. Notice for OHCs, only intracellular and extracellular chloride substitution with gluconate (redCl; n = 4) increased the relative reduction in Q max compared with CsCl control. That is, the blocking efficacy of extracellular 9‐AC was increased by lowering the OHC [Cl−] gradient by reducing the intra‐ and extracellular [Cl−]. Notice for HEK 293 cells, the addition of 9‐AC to the cytosol (9‐AC i; n = 5) decreased the blocking efficacy of extracellularly applied 9‐AC.
To investigate a possible involvement of the cytoskeleton in the blocking action of 9‐AC, the compounds diamide (Adachi & Iwasa, 1997; Song & Santos‐Sacchi, 2013) and latrunculin A (Matsumoto et al., 2010; Song & Santos‐Sacchi, 2013) were used to disrupt the cytoskeleton. Stock solution of diamide was prepared in distilled water. OHCs were pre‐incubated with 2 mm diamide for 30 min; the extracellular fluid was replaced with drug‐free HBSS shortly before recordings started. Stock solution of latrunculin A was prepared in DMSO. Latrunculin A was applied by local perfusion of the extracellular solution and was also contained in the patch‐capillary solution; intra‐ and extracellular concentrations were both 10 nm. For four of the five diamide‐treated cells, the hydrostatic pressure in the patch pipette needed to be increased (0.1–0.2 kPa relative to atmospheric pressure) to avoid cytoplasm leakage into the patch pipette and, therefore, to prevent the cells from collapsing. This correction was not required for the latrunculin‐treated cells.
Confocal microscopy
To select transfected HEK 293 cells, the coverslips with cell cultures were mounted in a plastic chamber and placed on the stage of a Zeiss Axioskop2 FS mot microscope (Zeiss, Heidelberg, Germany) equipped with a Zeiss LSM 510 confocal system (Kaneko et al., 2006). The GFP protein expression was visualized with a FITC filter set (BP 450‐490, FT 510, LP 515) and cells possessing a typical fluorescence‐signal pattern in the plasma membrane were selected. At the end of the capacitance measurements, images were recorded in 1‐μm vertical steps and 1024 × 1024 pixel² resolution; the voxel size was 0.3 × 0.3 × 1.6 μm3. Fluorescence images presented in this manuscript are for single confocal planes. The GFP labelling was excited with an argon laser (λex = 488 nm) and the emitted light was recorded above 505 nm. The pinhole diameter was set as 1 au. A Zeiss 40× IR‐Achroplan water‐immersion objective with NA 0.8, and zeiss lsm 510 software was used.
Capacitance measurement and parameter estimation
The Lindau–Neher algorithm was used to measure capacitance. The stimulus voltage was a sinusoid of frequency 1 kHz and amplitude 20 mV (peak‐to‐peak), superimposed on a constant holding potential, which was increased from −200 mV to +100 mV in 20‐mV steps. Each data point represents the averaged steady‐state response to 80 stimulus cycles. The initial holding potential was −60 mV. The whole‐cell current was sampled at 10 kHz and low‐pass filtered with a four‐pole Bessel filter with cut‐off frequency of 3 kHz. The Sine+DC mode of the LockIn extension was used in the pulse v8.66 software (Heka Elektronik, Lambrecht/Pfalz, Germany) to measure the membrane capacitance (Lindau & Neher, 1988; Gillis, 2000).
The voltage dependence of the capacitance was fitted with the first derivative of a two‐state Boltzmann function representing motor function plus a two‐state Boltzmann function representing voltage‐driven changes in membrane dimensions and/or dielectric constant (Santos‐Sacchi & Navarrete, 2002):
| (1) |
where
| (2) |
in which V is the membrane potential, C 0 is the voltage‐independent (linear) component of the membrane capacitance, Q max is the maximum motor charge moved, V h is the voltage at peak capacitance, z is the valence, e is the electron charge, k is Boltzmann's constant, T is absolute temperature and ΔC sa is the maximum increase in capacitance when all motors switch from the fully contracted state to the fully expanded state at asymptotically large hyperpolarizing potentials. Within the realms of the model, C 0 is the membrane capacitance when all motors are in the fully contracted state at asymptotically large depolarizing potentials.
The parameters Q max, V h, α, C 0 and ΔC sa were fitted with the Levenberg–Marquardt algorithm in Origin7 (OriginLab Corporation, MA, USA.). For five fit parameters and the 16 data points for capacitance as function of membrane voltage, each parameter estimate has 11 degrees of freedom.
Dose–response data were normalized to pre‐application values, averaged across cells and fitted with a logistic function of the form:
| (3) |
where x is the 9‐AC concentration and the fit parameters are: A 1 and A 2, the estimates of y at asymptotically small and large concentrations, respectively; IC 50, the estimate of the half‐maximal inhibitory concentration; n, the estimate of the slope parameter. A 2 was set to unity because the block asymptoted to 100% at the highest concentrations.
Detection of intracellular pH change
For measuring intracellular pH changes in response to extracellular 9‐AC application, isolated OHCs were incubated with the membrane‐permeant acetoxymethyl (AM) ester derivative of the fluorescence dye 2′,7′‐bis‐(2‐carboxyethyl)‐5‐(and‐6)‐carboxyfluorescein (BCECF‐AM, 1.5 μm; Molecular Probes, Leiden, The Netherlands) for 15 min. Although this dye probably does not provide information about intracellular pH in close proximity to prestin (Mistrik et al., 2012), for present purposes it is suitable for indicating cytosolic pH changes. The dye loading process was terminated by replacing the extracellular fluid with dye‐free HBSS using a perfusion pump (Ismatec SA, Glattbrugg, Switzerland). The confocal microscope was used to measure the intracellular BCECF‐fluorescence intensity. The probe was excited close to its isosbestic point of λiso = 439 nm with the λex = 458 nm argon laser beam. Emitted light above 505 nm was detected using a long‐pass filter. Because both the excitation and the emission intensities of the dye decrease when the dye environment becomes more acidic, the observed decreasing signal intensities are equivalent to changes in pH toward more acidic values. The dye‐emitted fluorescence signal was measured in the centre of the cell with a region of interest (ROI) of approximate size 5 × 15 μm2; the confocal plane was located in the middle of the cell. The relative time course of the fluorescence signal intensities was independent of the vertical position and the size of the ROIs; the time course was quantified by fitting with a single exponential equation. The power of the excitation laser was kept as low as possible to minimize dye bleaching. For experiments with 9‐AC without DMSO, the drug was diluted in HBBS with extensive treatment (minimum of 10 min) in an ultrasound bath.
Statistical analysis
Fitted parameters and collated data are presented as sample mean and SD. Statistically significant difference between sample means was evaluated by the Student's t‐test without assuming equality of variances (Guttman et al., 1971) and was defined as statistically significant at the 95% level of confidence (P < 0.05). The test value is given as t ν, where the subscript ν denotes the number of degrees of freedom. The P‐value for the result occurring at chance under the null hypothesis is denoted as either P 1 or P 2 to distinguish, respectively, between a one‐sided (directional) test and a two‐sided (non‐directional) test.
Results
9‐AC reversibly reduces the NLC of OHCs
The effect of extracellular 500 μm 9‐AC on the voltage dependence of the OHC capacitance is illustrated in Fig. 1A. The OHC was clamped from −200 mV to +100 mV in 20‐mV steps from an initial holding potential of −60 mV. The patch solution contained 135 mm CsCl. The 9‐AC reversibly reduced the nonlinear motor component of the capacitance. Data were fitted using Eqn (1) (Fig. 1A, lines). Fit parameters are given in Table 1. The maximum motor‐charge transfer, Q max, decreased by 46% and the voltage at peak capacitance, V h, shifted by 25 mV in the positive direction. There were small but significant increases in ΔC sa (0.9 pF; t 22 = 2.55, P 1 = 0.0091) and α (2.0 mV; t 22 = 2.53, P 1 = 0.0095). There was no significant change in C 0 (t 22 = 0.85, P 2 = 0.41). All OHCs possessed similar 9‐AC sensitivity; on average, extracellular application of 500 μm 9‐AC reduced Q max by 40 ± 8% (n = 11; Fig. 2D, Control), shifted V h in the positive direction (for all cells) by 13 ± 8 mV (n = 11), and did not significantly affect the parameters α, ΔC sa and C 0. These results suggest that 9‐AC alters prestin‐associated charge movement.
Figure 1.
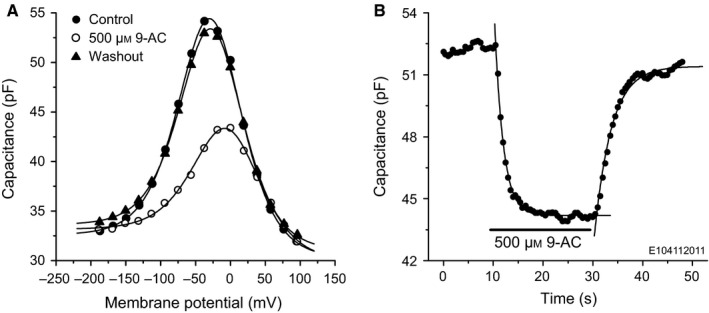
9‐AC reversibly reduces the NLC of OHCs. (A) Capacitance of a control OHC (filled circles), voltage‐clamped with intracellular solution containing 135 mm CsCl. Extracellular application of 500 μm 9‐AC (open circles) reduces the NLC. The block is reversible (closed triangles). Lines show the results of the fits using Eqn (1); fit parameters are given in Table 1. (B) Time course of the 9‐AC block of NLC measured at −20 mV. Lines are single exponential fits with time constants of 1.7 ± 0.1 s and 3.4 ± 0.2 s for the on‐ and off‐drug phases, respectively. E104112011, acronym identifying the cell; the same convention is used in the following figures.
Table 1.
| Control | 500 μm 9‐AC | Washout | |
|---|---|---|---|
| C 0 (pF) | 30.2 ± 0.3 | 29.9 ± 0.4 | 31.0 ± 0.2 |
| α (mV) | 30.6 ± 0.5 | 32.6 ± 1.0 | 30.3 ± 0.3 |
| V h (mV) | −28.6 ± 0.6 | −3.8 ± 1.3 | −26.9 ± 0.4 |
| Q max (pC) | 2.81 ± 0.06 | 1.53 ± 0.07 | 2.55 ± 0.03 |
| ΔC sa (pF) | 2.4 ± 0.3 | 3.3 ± 0.4 | 2.6 ± 0.2 |
Figure 2.
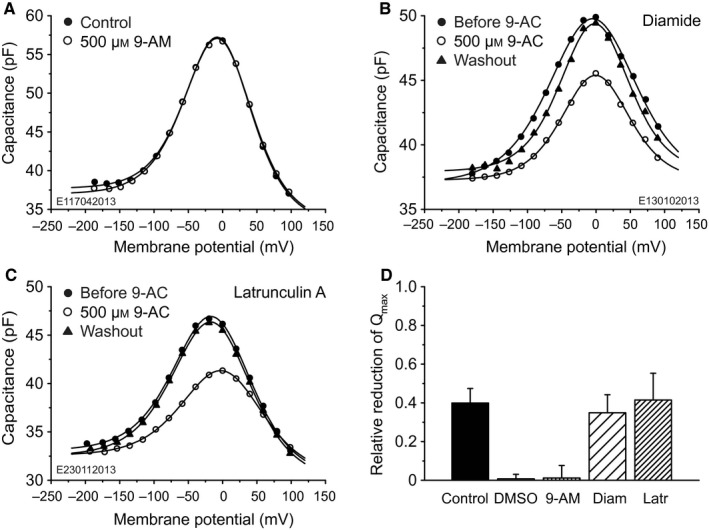
9‐AC block requires negative charge and appears independent of the integrity of the cytoskeleton. (A–C) Capacitances of three voltage‐clamped OHCs. Labelling conventions are as in Fig. 1A. (A) Extracellular application of 500 μm 9‐AM, an electrically neutral analog of 9‐AC. There is no detectable effect on cell capacitance, meaning that the negative charge of 9‐AC is mandatory for 9‐AC block. (B) Application of 500 μm 9‐AC to an OHC treated extracellularly with diamide. NLC is reversibly reduced; fit parameters are given in Table 3. (C) Application of 500 μm 9‐AC to an OHC treated intracellularly and extracellularly with latrunculin A. NLC is reversibly reduced; fit parameters are given in Table 4. (D) The relative reduction in Q max in response to extracellular application of 500 μm 9‐AC, DMSO or 500 μm 9‐AM. Labelling on the abscissa has the following meaning. Control, extracellular application of 500 μm 9‐AC to control OHCs (n = 11). DMSO, extracellular application of DMSO to control OHCs (n = 6). 9‐AM, extracellular application of 500 μm 9‐AM to control OHCs (n = 6). Diam, diamide‐treated OHCs (n = 5) with extracellular application of 500 μm 9‐AC. Latr, latrunculin‐treated OHCs (n = 7) with extracellular application of 500 μm 9‐AC. There is no significant difference for relative Q max reductions for cells treated with diamide or latrunculin A compared with untreated cells, indicating that the cytoskeleton is not a primary target of 9‐AC. Relative reductions for DMSO and 9‐AM applications are not significantly different from zero and there was also no effect on the other capacitance‐defining parameters; that is, neither DMSO nor 9‐AM had a detectable effect on NLC.
Control experiments, in which DMSO instead of 9‐AC was locally applied, showed that DMSO had no observable effect on NLC: Q max reduction was 0.75 ± 2.3% (n = 6; Fig. 2D, DMSO), which is not significantly different from 0% (t 5 = 0.77, P 2 = 0.47). Likewise, there was no effect of DMSO on the other NLC parameters.
The speed of the 9‐AC block was quantified by monitoring the capacitance every 0.5 s at the membrane potential of −20 mV (Fig. 1B). The time course of both the blocking and recovery phases can each be quantified by a single exponential function. For the OHC illustrated in Fig. 1B, the time constants for the block and recovery phases were, respectively, 1.7 ± 0.1 s and 3.4 ± 0.2 s. On average, the time constants for block and recovery were 2.8 ± 1.5 s (n = 4) and 7.1 ± 4.5 s (n = 3), respectively. These values are 1–2 orders of magnitude smaller than for 9‐AC block of oocyte‐expressed ClC‐1 channels, where the binding site of the drug is located on the intracellular side of the channel pore (Estévez et al., 2003). The relatively fast block for OHCs suggests that the binding site and dynamics for the drug might be different from those for ClC‐1 channels, the site possibly being inside the lipid bilayer or even at the extracellular plasma membrane.
The negative charge of 9‐AC is crucial for NLC reduction
Experiments were also conducted to test the effect of 500 μm 9‐AM, an electro‐neutral analog of 9‐AC, on the NLC of OHCs. 9‐AM had no effect on capacitance; an example is given in Fig. 2A. On average, the Q max reduction was 1.1 ± 6.5% (n = 6; Fig. 2D, 9‐AM), which is not significantly different from 0% (t 5 = 0.44, P 2 = 0.68). This result demonstrates that the anionic site on 9‐AC is mandatory for reducing NLC.
There is no evidence for a cytoskeletal involvement in NLC reduction
A possible involvement of the cytoskeleton on the 9‐AC‐induced reduction in NLC was investigated by disrupting the cytoskeleton with the actin–spectrin decoupler, diamide (Adachi & Iwasa, 1997; Song & Santos‐Sacchi, 2013), and also applying a blocker of actin polymerization, latrunculin A (Matsumoto et al., 2010; Song & Santos‐Sacchi, 2013).
In the case of diamide‐treated cells but not the latrunculin‐treated cells, after breakthrough of the plasma membrane to establish the whole‐cell patch configuration, there appeared to be an obvious net motion of cytoplasmic material in the general direction of the patch electrode (four of five cells), as one might expect for extensive disruption of the cytoskeleton and probably other cytoplasmic structures. Therefore, the patch‐pipette pressure was increased (0.1–0.2 kPa relative to atmospheric pressure) to arrest cytoplasm leakage.
Single‐cell examples of the NLC for diamide‐ and latrunculin‐treated cells are given, respectively, in Fig. 2B and C; they are denoted by ‘Before 9‐AC’. Also shown is the effect of 500 μm 9‐AC applied extracellularly. The capacitance fit parameters for the cells in Figs. 2B and C are given, respectively, in Tables 3 and 4. The population means and SDs of the capacitance fit parameters before application of 9‐AC are presented in Table 2 for the untreated (Control) and treated (Diamide, Latrunculin A) cells.
Table 3.
| Before 9‐AC | 500 μm 9‐AC | Washout | |
|---|---|---|---|
| C 0 (pF) | 37.0 ± 0.8 | 37.3 ± 0.3 | 37.8 ± 0.6 |
| α (mV) | 42.4 ± 1.7 | 33.1 ± 0.8 | 33.8 ± 1.4 |
| V h (mV) | −4.7 ± 2.2 | −0.6 ± 1.1 | −1.76 ± 1.9 |
| Q max (pC) | 2.16 ± 0.16 | 1.07 ± 0.04 | 1.56 ± 0.10 |
| ΔC sa (pF) | 0.0 ± 0.7 | 0.0 ± 0.3 | 0.1 ± 0.6 |
Table 4.
| Before 9‐AC | 500 μm 9‐AC | Washout | |
|---|---|---|---|
| C 0 (pF) | 30.3 ± 0.4 | 30.4 ± 0.2 | 29.9 ± 0.3 |
| α (mV) | 38.1 ± 0.8 | 39.7 ± 0.6 | 38.3 ± 0.7 |
| V h (mV) | −13.5 ± 0.9 | 1.58 ± 0.78 | −14.2 ± 0.8 |
| Q max (pC) | 2.33 ± 0.07 | 1.60 ± 0.04 | 2.31 ± 0.06 |
| ΔC sa (pF) | 2.8 ± 0.3 | 2.2 ± 0.2 | 2.6 ± 0.3 |
Table 2.
Mean and SD of the fit‐parameter populations for capacitance data from control, diamide‐treated and latrunculin‐treated OHCs before application of 9‐AC
| Control | Diamide | Latrunculin A | |
|---|---|---|---|
| (n = 11) | (n = 5) | (n = 7) | |
| C 0 (pF) | 31.0 ± 4.8 | 33.7 ± 5.7 | 27.4 ± 3.6 |
| α (mV) | 32.7 ± 5.1 | 41.7 ± 5.8 | 33.6 ± 3.5 |
| V h (mV) | −20.9 ± 13.7 | −5.6 ± 15.7 | −16.1 ± 9.1 |
| Q max (pC) | 2.30 ± 0.35 | 1.81 ± 0.31 | 2.00 ± 0.41 |
| ΔC sa (pF) | 3.5 ± 0.8 | 1.4 ± 2.3 | 2.8 ± 0.6 |
Referring first to the data before application of 9‐AC, the treated cells exhibit the bell‐shaped NLC function found for untreated OHCs. Although the fit parameters for the latrunculin‐treated cells are similar to those for the untreated cells, this is not the case for the diamide‐treated cells for α, V h and ΔC sa. On average, α is ~ 30% larger than for untreated cells (t 22 = 1.65, P 1 = 0.057; Table 2). In other words, where the effective charge transfer, z, was 0.78 ± 0.12 for untreated cells, it was only 0.60 ± 0.09 for the diamide‐treated cells [Eqn (2) with kT/e = 25.4 mV]. Thus, as hypothesized by Adachi & Iwasa (1997), based on the voltage dependence of motility, the diamide might be acting not only on the cytoskeleton but also directly on the motor. The V h for the diamide‐treated cells tended to be slightly more positive (15 mV) than for the control cells (t 7 = 1.98, P 1 = 0.044). Finally, unlike the situation for the untreated cells, ΔC sa for diamide‐treated cells appears to be not significantly different from 0 pF (t 4 = 1.38, P 2 = 0.24, for the average data in Table 2; t 11 = 0.00, P 2 = 1.00 for the cell in Fig. 2B and Table 3). However, this is most probably a statistical rather than a physical feature of the diamide data because α and V h are much larger than for untreated cells, so that ΔC sa could not be fitted with statistical confidence when the maximum membrane potential is limited to 100 mV. In other words, the data do not permit conclusions about ΔC sa for diamide‐treated cells.
Application of 9‐AC to these two types of treated cells caused reversible reduction of the NLC, in a similar way to that found for untreated cells. There was a reversible shift of V h in the positive direction for latrunculin‐treated but no significant shift for diamide‐treated cells. On average, the Q max reduction was 35 ± 9% for diamide‐treated cells (n = 5; Fig. 2D, Diam) and 42 ± 14% for latrunculin‐treated cells (n = 7; Fig. 2D, Latr); these values are not significantly different from those for untreated OHCs (t 7 = 1.12, P 2 = 0.30, and t 9 = 0.39, P 2 = 0.71, respectively). On average, the positive shift in V h for the latrunculin‐treated cells was 24 ± 10 mV (n = 7), which is significantly larger than the shift of 13 ± 8 mV (n = 11) for the untreated OHCs (t 11 = 2.58, P 1 = 0.013). On average, there was no significant change in V h for the diamide‐treated cells (−3 ± 16 mV, n = 5, t 4 = 0.42, P 2 = 0.70).
The parameter C 0 was not affected by 9‐AC treatment but the parameter α was reversibly changed by a small but significant amount, increasing for latrunculin‐treated OHCs by 2.2 ± 1.6 mV (P 1 = 0.0095) but decreasing for diamide‐treated OHCs by 5.0 ± 4.0 mV (P 1 = 0.024). An ~ 2‐mV increase of α for latrunculin‐treated OHCs (e.g. 1.6 ± 1.0 mV; Fig. 2C, Table 4) is similar to that found for some control cells (e.g. 2.0 ± 1.1 mV; Fig. 2A, Table 1), although on average there was no change of α for control OHCs. For the latrunculin‐treated OHCs, there was a reversible reduction in ΔC sa by 1.09 ± 1.06 pF, which is significantly less than 0 pF (t 6 = 2.72, P 1 = 0.017), and amounts to ~ 40% of the value of ΔC sa before 9‐AC application (Table 2). However, these changes inΔC sa and α are on the boarder of statistical detectability and, as for the case of the control OHCs, are if anything considered as being second‐order effects.
In general, the similarity in form and magnitude of the effect of 9‐AC on the treated and untreated cells suggests that the cytoskeleton is not the primary target of 9‐AC.
9‐AC reduces NLC of prestin‐transfected HEK 293 cells
Although the bell‐shaped form of the voltage dependence of the capacitance reflects the presence of functional prestin in the plasma membrane, the 9‐AC sensitivity of the NLC is not conclusive evidence of an interaction between the drug and prestin. For example, ClC‐1 channels, which are also known to be expressed in OHCs (Kawasaki et al., 1999), might also be the target of 9‐AC, in spite of the relative speed of the block compared with that for oocyte‐expressed ClC‐1 channels. The interaction between prestin and 9‐AC was, therefore, sought by investigating voltage dependence of the membrane capacitance for prestin‐transfected HEK 293 cells. GFP labelling was used to visualize prestin expression in the HEK 293 cells; Fig. 3A shows the typical plasma‐membrane staining. The membrane capacitance was measured for cells clamped from −200 mV to +100 mV in 20‐mV steps from a holding potential of −60 mV (Fig. 3B). Lines indicate fits using Eqn (1); fit parameters are given in Table 5. The principal effect of 9‐AC was to reversibly reduce the nonlinear motor component of the capacitance. On average, 500 μm 9‐AC reduced Q max by 59 ± 19% (n = 11; Fig. 4B, HEK 293/CsCl), significantly larger (t 13 = 3.06, P 1 = 0.0046) than the reduction obtained for OHCs.
Figure 3.
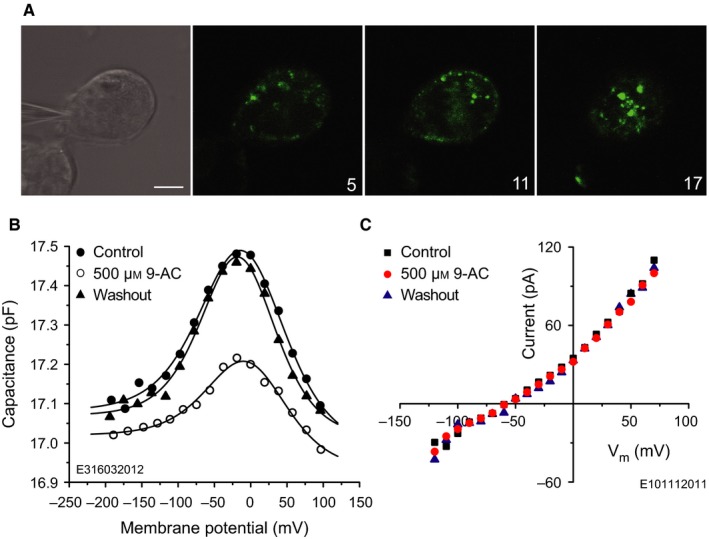
9‐AC reversibly reduces the NLC of prestin‐transfected HEK 293 cells. (A) Prestin‐transfected HEK 293 cell possessing the typical dotted fluorescence in the plasma membrane. Cells were voltage‐clamped using 135 mm CsCl intracellular solution (upper left/DIC image). GFP labelling was recorded with confocal microscopy using an argon laser with excitation wavelength of 488 nm. The emitted light was detected above 505 nm. Numbers in the bottom right corners of the fluorescence images indicate the level of the confocal section in microns. (B) Capacitance of the prestin‐transfected HEK 293 cell in A. Extracellular application of 500 μm 9‐AC reversibly reduces the NLC. Lines indicate fits using Eqn (1) with parameters given in Table 5. (C) I‐V characteristics of a non‐transfected HEK 293 cell, indicating that 9‐AC does not influence voltage‐gated chloride conductances in this cell line. Scale bar, 10 μm.
Table 5.
Fit parameters using Eqn (1) for capacitance data from the prestin‐transfected HEK 293 cell in Fig. 3B
| Control | 500 μm 9‐AC | Washout | |
|---|---|---|---|
| C 0 (pF) | 16.99 ± 0.03 | 16.93 ± 0.03 | 17.02 ± 0.02 |
| α (mV) | 39.6 ± 2.1 | 36.9 ± 3.0 | 33.7 ± 1.5 |
| V h (mV) | −10.2 ± 2.5 | −3.2 ± 3.5 | −14.9 ± 1.8 |
| Q max (fC) | 71 ± 6 | 34 ± 4 | 56 ± 4 |
| ΔC sa (fF) | 97 ± 28 | 87 ± 22 | 75 ± 17 |
V h tended to change upon application of 9‐AC but the change was not consistently in any one direction, the average being 13 ± 19 mV (n = 11). There was no significant change in α or ΔC sa. However, after application of 9‐AC there was a relatively small (compared with the Q max effect) but significant change in C 0 detectable in nine of the 11 HEK 293 cells, which was, with one exception, a decrease; on average (n = 11), the relative change was −1.1 ± 1.1% (t 10 = 3.25, P 1 = 0.0044). [Relative rather than absolute changes in C 0 are presented for the population of HEK 293 cells because of the relatively large spread of C 0 values across cells (8.8–50.3 pF). These values are correlated with cell size, as ascertained by estimating the surface area of the cell as a sphere of diameter equal to the arithmetic mean of the minor and major axes of the confocal image of the largest cell cross‐section: linear regression of C 0 with surface area yielded a slope of 1.26 ± 0.07 μF/cm² (n = 8; squared correlation coefficient = 0.74). This slope is an estimate of the specific capacitance of the HEK 293 cells.] Three cells showed recovery of C 0 after washout; one is shown in Fig. 3B. On average (n = 8), the washout value of the relative change in C 0 was −1.26 ± 1.39%, which is not significantly different from the pre‐washout value (t 12 = 0.43, P 2 = 0.67), meaning that further change in C 0 was not detectable in these eight cells. As the reduction in C 0 was usually irreversible, it is not possible to conclude whether it is directly dependent on 9‐AC. The relative changes are small, being no more than an order of magnitude larger than the voltage‐dependent capacitance observed in native HEK 293 cells (Farrell et al., 2006).
To check whether the action of 9‐AC on prestin‐transfected HEK 293 cells might have been conveyed by endogenous chloride channels, the effect of 9‐AC on the I‐V characteristic of native (non‐transfected) HEK 293 cells was tested. The same intracellular and extracellular solutions were used as in the NLC measurements on prestin‐transfected HEK 293 cells. There was no significant effect of 9‐AC on the I‐V characteristic (n = 5); an example is given in Fig. 3C. Therefore, because 9‐AC reversibly reduced the motor component of the NLC of the prestin‐transfected HEK 293 cells and had no effect on their endogenous conductances, we conclude that the effect of 9‐AC on prestin in these cells is not conveyed by endogenous chloride channels.
These results with transfected HEK 293 cells, together with those for the cytoskeleton disruption experiments, support the hypothesis of an interaction between prestin and 9‐AC in OHCs.
9‐AC block of NLC is chloride‐dependent
As chloride ions underpin prestin function (Oliver et al., 2001; Rybalchenko & Santos‐Sacchi, 2003), we investigated whether 9‐AC might compete with Cl− to block the activity of the motor protein. To this end, the intracellular chloride concentration, [Cl−]i, was reduced and evidence for a facilitator effect on the action of 9‐AC examined. First, the dependence of NLC on 9‐AC concentration was measured in the range 0.05 μm–5 mm for the usual [CsCl] in the patch electrode. The dependence was parameterized using the relative reduction in Q max and is plotted as a logarithmic function of 9‐AC concentration (Fig. 4A, circles labelled CsCl). This dose–response data were fitted with a logistic function [Eqn (3); solid line in Fig. 4A]; IC 50 and the slope parameter were 928 ± 66 μm and 0.82 ± 0.04, respectively. Second, [Cl−]i was reduced by patching OHCs with an intracellular solution in which 115 mm Cl− was substituted with 40 mm (Fig. 4A, triangle labelled Cs2SO4). There was no statistically significant shift (n = 5; Fig. 4B, OHC/Cs2SO4). The IC 50 and the slope parameter were 1012 ± 192 μm and 0.56 ± 0.06, respectively.
Since the reduced [Cl−]i did not appear to influence the efficacy of the extracellularly applied 9‐AC, we suggest that: (i) Cl− and 9‐AC might act at different sites on prestin, or (ii) Cl− and 9‐AC might interact at different sites of the motor complex, or (iii) this method of reducing the [Cl−]i was inadequate. With respect to the third possibility, the relatively large [Cl−] gradient formed by 145 mm extracellular and 24 mm intracellular in the preceding experiment could have caused Cl− influx through, for example, a Cl−‐permeable stretch‐sensitive conductance in the basolateral membrane (Rybalchenko & Santos‐Sacchi, 2003). Therefore, to examine whether Cl− influx might have influenced the preceding [Cl−]i‐reduction results, [Cl−] was reduced not only intracellularly but also extracellularly by substituting 130 mm Cl− with 130 mm gluconate in both solutions. That is, compared with the preceding experiment, the 9‐AC dose‐dependency is now tested for not only a lower [Cl−]i = 9 mm but also a lower gradient by using a lower extracellular concentration, [Cl−]o = 15 mm. For this condition, there is a significant leftward shift of the dose–response curve (Fig. 4A, squares labelled redCl), the logistic‐function fit yielding an IC 50 of 79 ± 14 μm. The slope parameter was 0.71 ± 0.08. On average, at the 9‐AC concentration of 500 μm, the relative Q max reduction was 81 ± 7% (n = 4; Fig. 4B, OHC/redCl), which is double the value of 40 ± 8% found for the 135 mm CsCl intracellular solution (n = 11; Fig. 4B, OHC/CsCl). In conclusion, these latter experiments demonstrate that the 9‐AC block is indeed chloride‐sensitive and suggest that 9‐AC might compete for chloride binding sites on prestin.
9‐AC can block NLC intracellularly
Previous studies with ClC‐1 channels showed that 9‐AC binds to a hydrophobic pocket close to the Cl− binding site of the channel that is accessible from the cytoplasm (Estévez et al., 2003). To investigate whether in OHCs the 9‐AC can act by intracellular binding, 500 μm 9‐AC was added to the intracellular solution and the blocking performance of extracellularly applied 500 μm 9‐AC ascertained. The presence of intracellular 500 μm 9‐AC had no detectable effect (t 6 = 0.30, P 2 = 0.78) on the blocking efficacy of extracellularly applied 500 μm 9‐AC; on average, the relative Q max reduction was 39 ± 11% (n = 5; Fig. 4B, OHC/9‐ACi).
One possible explanation for this result is that 9‐AC has relatively little access to the intracellular surface of the plasma membrane when approached from the cytosol, perhaps due to the presence of the subsurface cisternae. To examine this possibility, membrane patches were excised from the basolateral plasma membrane and capacitance measured in the inside‐out patch configuration to allow direct access of 9‐AC to the cytosolic surface of the plasma membrane. Perfusion of the experimental chamber with 500 μm 9‐AC reduced Q max by 42 ± 23% (n = 3; Fig. 4B, OHC/InOut), which is not significantly different from the reduction for the control situation for the intact cell (t 2 = 0.26, P 2 = 0.82). These results indicate that the intracellular surface of the plasma membrane is sensitive to 9‐AC and suggest that in the whole‐cell condition the subsurface cisternae hinder access of the drug from the cytosol.
Since HEK 293 cells do not possess subsurface cisternae, intracellular 9‐AC‐application experiments were also conducted on this cell type. In the presence of 500 μm 9‐AC in the patch pipette the extracellularly applied 500 μm 9‐AC reduced Q max by 32 ± 20% (n = 5; Fig. 4B, HEK 293/9‐ACi). This reduction is about half the value found without 9‐AC in the patch pipette (59 ± 19%, n = 11; Fig. 4B, HEK 293/CsCl); the difference is statistically significant (t 8 = 2.60, P 1 = 0.032). Taken together with the results of the inside‐out patch experiments on excised OHC plasma membrane, these data show that 9‐AC can access the binding site from both extracellular and intracellular surfaces of the plasma membrane and, therefore, that 9‐AC can act by intracellular binding.
9‐AC can penetrate the plasma membrane
Having established that 9‐AC can access the binding site by both extracellular and intracellular routes, we enquired whether 9‐AC can penetrate the plasma membrane from the extracellular side. That is, we sought further evidence for intracellular or intramembranous binding. As 9‐AC is an acid, it might be expected to reduce the intracellular pH if it were to diffuse through the plasma membrane. Intracellular pH changes were demonstrated using the fluorescence pH indicator BCECF (Fig. 5). Extracellular application of 500 μm 9‐AC reversibly reduced the intracellular fluorescence signal intensity with a time constant of 2.0 ± 0.8 s (n = 8) for the usual case where the drug was diluted in HBSS containing DMSO. The time constant was 5.7 ± 2.7 s (n = 7) when the drug was diluted in HBSS alone. That is, the time constant is significantly longer (t 7 = 3.49, P 1 = 0.0051) in the absence of DMSO. These data indicate that 9‐AC is able to penetrate the plasma membrane and that DMSO can facilitate that diffusion.
Figure 5.
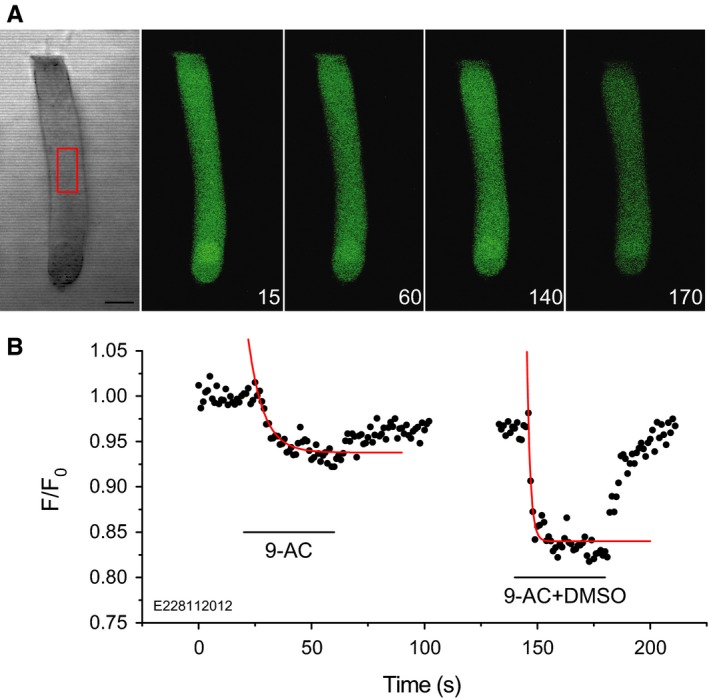
Extracellular 9‐AC reduces intracellular pH. (A) Fluorescence monitoring of the time course of intracellular pH, using the fluorescence dye BCECF, in response to extracellular 500 μm 9‐AC. Numbers in the bottom right corners of the fluorescence images indicate time in seconds. The region of interest indicated by the red rectangle in the middle of the isolated OHC was chosen to calculate fluorescence intensity values, F, normalized to the averaged initial value F0 recorded at the beginning of the experiment. (B) Time course of the relative fluorescence intensity for the cell in A. Including DMSO in the perfusion fluid resulted in a larger and faster fluorescence signal (right panel) than when 9‐AC alone was present (left panel). Red lines indicate single exponential fits with time constants of 6.7 ± 1.1 s and 1.5 ± 0.3 s, respectively, in the absence and presence of DMSO. Scale bar, 10 μm.
For non‐transfected HEK 293 cells, the onset time‐constant of the fluorescence signal for extracellular application of 500 μm 9‐AC was 2.9 ± 0.9 s (n = 7; data not illustrated). Although slightly longer than the OHC value (2.0 ± 0.8 s, n = 8), the difference is only just statistically significant (t 12 = 2.03, P 1 = 0.033). Therefore, it is concluded that the speed of diffusion through the OHC lateral wall is prestin‐independent.
9‐AC‐induced reduction in NLC is not due to intracellular acidification
These pH experiments beg the question as to whether the 9‐AC‐induced reduction in NLC might be a pH effect. Indeed, the pH time constant for the standard condition of 9‐AC diluted in HBSS containing DMSO (2.0 ± 0.8 s, n = 8) was not significantly different (t 4 = 1.00, P 2 = 0.37) from the time constant for the 9‐AC induced decrease for NLC (2.8 ± 1.5 s, n = 4). Addressing this point, application of DMSO in the absence of 9‐AC reduced intracellular pH (data not shown) with a time constant of 1.8 ± 0.4 s (n = 6). This value is not significantly different to the pH time constant in the presence of 9‐AC (t 9 = 0.61, P 2 = 0.56). However, as shown earlier (Fig. 2D), application of DMSO alone has negligible effect on NLC, as does application of the electrically neutral analog of 9‐AC, 9‐AM. These results imply that the 9‐AC‐induced reduction of NLC is not due to intracellular acidification.
Discussion
Electromechanical properties of the voltage‐dependent chloride‐channel blocker 9‐AC, described in the current paper, are similar in several aspects to electromechanical properties of the anionic amphipathic sodium salicylate. Applied to isolated OHCs, sodium salicylate causes reversible reduction in electromotility (Shehata et al., 1991; Russell & Schauz, 1995; Tunstall et al., 1995; Kakehata & Santos‐Sacchi, 1996; Homma & Dallos, 2011), NLC (Tunstall et al., 1995; Kakehata & Santos‐Sacchi, 1996; Oliver et al., 2001; Santos‐Sacchi & Navarrete, 2002; Homma & Dallos, 2011; Santos‐Sacchi & Song, 2014), electromechanical force (Hallworth, 1997), axial stiffness (Russell & Schauz, 1995) and lateral‐wall stiffness parameters (Lue & Brownell, 1999; Zhou & Raphael, 2005). Sodium salicylate has been reported not to change viscoelastic properties of the plasma membrane (Ermilov et al., 2005) or the strength of interaction between the cytoskeleton and the plasma membrane (Morimoto et al., 2002). For prestin‐transfected kidney or ovary cells, sodium salicylate causes reduction in NLC (Zheng et al., 2000) and electromechanical force (Ludwig et al., 2001), as well as electromotility provided the cell is distorted into a non‐cylindrical shape to allow change in surface area without changing cytoplasmic volume (Zheng et al., 2000). Application of sodium salicylate to the organ of Corti reduces the vibration amplitude of the basilar membrane in response to acoustic (Murugasu & Russell, 1995; Santos‐Sacchi et al., 2006) and electrical (Mammano & Ashmore, 1993; Brownell et al., 2011) stimulation. After rapidly permeating the OHC plasma membrane (Tunstall et al., 1995; Kakehata & Santos‐Sacchi, 1996), presumably in its uncharged form, sodium salicylate dissociates and the anion then competes with chloride for binding on prestin (Oliver et al., 2001), reducing the NLC and the electromechanical force available for mechanical feedback into the organ of Corti.
The current study demonstrates that the voltage‐dependent chloride channel blocker 9‐AC also reduces the NLC of OHCs and prestin‐transfected HEK 293 cells. However, unlike salicylate, ΔC sa was not affected by 9‐AC except at a very high concentration (5 mm), where it was reduced by ~ 1 pF (data not illustrated). Salicylate causes a dose‐dependent increase in ΔC sa of 1–2 pF (Santos‐Sacchi & Navarrete, 2002; Homma & Dallos, 2011; Santos‐Sacchi & Song, 2014).
The present data indicate that 9‐AC, similarly to salicylate, reduces NLC principally by interacting with prestin rather than by interacting with the cytoskeleton or with chloride channels, or by elevating intracellular [H+].
Chloride sensitivity of the 9‐AC block
The 9‐AC block of NLC is chloride‐sensitive, as demonstrated by substituting 130 mm Cl− with gluconate both intracellularly and extracellularly (Fig. 4A, redCl). These data suggest that 9‐AC and Cl− might compete for the same putative anion‐binding site on prestin or that the binding sites for 9‐AC and Cl− are overlapping. Similar competitive‐antagonist behaviour was found between Cl− and salicylate when added to the cytoplasmic side of the plasma membrane (Oliver et al., 2001). The 9‐AC dose‐dependency of the Q max reduction is similar to salicylate dose‐dependency: for the 135 mm extracellular and 140 mm intracellular Cl− concentrations, the IC 50 and the slope parameter estimated from the logistic‐function fit were, respectively, 928 ± 66 μm and 0.82 ± 0.04 for extracellular 9‐AC (Fig. 4A) and 964 μm and 0.86 for extracellular salicylate (Santos‐Sacchi et al., 2006; their Fig. 2). The absence of an effect of 9‐AM, an electrically neutral form of 9‐AC, on the NLC strengthens the possibility that the anionic charged 9‐AC prefers to interact with the prestin binding site of the chloride ion, and hints at the binding site being intracellular.
Access route of 9‐AC to its binding site
The 9‐AC has a high affinity for ClC‐1 channels (Estévez et al., 2003), channels which are also expressed in OHCs (Kawasaki et al., 1999). As 9‐AC is very hydrophobic, when applied extracellularly it can reach the intracellularly located inhibitory site of the ClC‐1 channel by diffusion through the lipid bilayer (Pusch et al., 2002). According to experiments on excised patches of Xenopus oocytes (Estévez et al., 2003), complete block of the ClC‐1 channel requires hundreds of seconds (time constant τ = 142 ± 29 s) when 100 μm 9‐AC is applied extracellularly and requires a shorter time (τ = 38.3 ± 3.4 s) when applied intracellularly; washout was reported to be practically impossible. These time constants are one to two orders of magnitude longer than those found for extracellular application to the OHC (Fig. 1B) and, therefore, indicative of different membrane penetrating and/or blocking mechanisms from those for ClC‐1 channels.
The speed of intracellular pH change (τ = 2.0 ± 0.8 s) in the present study indicates that 9‐AC can rapidly penetrate plasma membrane. Therefore, the relatively large time constants in the ClC‐1 study (Estévez et al., 2003) most probably originate from the dynamics of (intracellular) block on the channel protein, rather than being limited by transmembrane diffusion. The speed of 9‐AC‐induced block of charge movement (τ = 2.8 ± 1.5 s) and of recovery from block (τ = 7.1 ± 4.5 s) observed for OHCs could indicate either that the 9‐AC acts extracellularly or that it can permeate the plasma membrane sufficiently fast to enable rapid intracellular block (or intra‐membrane block via an intracellular route). In other words, because 9‐AC is membrane‐permeable, one cannot decide from these observations whether 9‐AC acts predominantly intracellularly or extracellularly.
Although absolute pH change was not measured, the relative change in the fluorescence signal (Fig. 5B) suggests that there was sufficient anionized 9‐AC in the cytosol to allow for intracellular block. This assertion is based on the following. Assuming that 9‐AC diffuses through the membrane in its non‐dissociated (uncharged) state and then is deprotonated in the cytosol (Greger, 1990), we can estimate the intracellular concentration of anionic 9‐AC, denoted by , by applying the Henderson–Hasselbalch equation intracellularly and extracellularly under the assumption that the intracellular and extracellular concentrations of non‐dissociated 9‐AC are equal, as was done by Kakehata & Santos‐Sacchi (1996) for sodium salicylate. Designating intracellular and extracellular parameters by the subscripts i and o, respectively, the result is . Based on the calibration by Mistrik et al. (2012) for BCECF‐AM in OHCs (change of 0.31 pH units for 10% change in fluorescence) and the data in Fig. 5B, we estimate that without DMSO and with DMSO, which for = 500 μm and gives the estimates = 330 μm without DMSO and = 204 μm with DMSO. Although ‘rough’ estimates, they indicate that the concentration of intracellular (anionized) 9‐AC was within an order of magnitude of the extracellular concentration.
The fact that the 9‐AC block of NLC is reversible (Fig. 1A) and the recovery time constant (Fig. 1B) small for OHCs compared with ClC‐1 channels, together with the finding that the presence of intracellular 9‐AC does not influence the efficacy of the extracellular block in the whole‐cell configuration (Fig. 4B, OHC/9‐ACi), potentially support the hypothesis that the target for 9‐AC block is located extracellularly. However, the subsurface cisternae, composed of many membranous layers in the intact OHC of the guinea pig (Holley, 1996), might have acted as a trap for the intracellular 9‐AC, preventing the drug from accessing the plasma membrane from the intracellular side, at least on the time scale of our experiments. The results of two further experiments support this interpretation of inaccessibility due to subsurface cisternae. First, the excised plasma‐membrane patches do not possess subsurface cisternae and, therefore, allow direct access of 9‐AC to the intracellular surface when applied in the inside‐out patch configuration. The NLC was blocked in this patch configuration, the amount being similar to that for extracellular application (Fig. 4B, OHC/InOut). Second, the experiments with prestin‐transfected HEK 293 cells (cells not possessing subsurface cisternae), where the intracellular presence of 9‐AC significantly reduced the efficacy of the extracellularly applied drug (Fig. 4B, HEK 293/9‐ACi), also revealed that 9‐AC can modify prestin function not only from the extracellular side but also from the cytosolic side of the plasma membrane. Thus, in spite of the speed of the response to extracellularly applied 9‐AC, the results of the intracellular pH experiments with OHCs, the excised‐patch experiments with OHCs and the intracellular 9‐AC experiments with prestin‐transfected HEK 293 cells strongly support the hypothesis that the blocking site is intracellular, or within the membrane via an intracellular route.
Does 9‐AC interact with prestin?
To elucidate possible interaction mechanisms between 9‐AC and prestin, 9‐AC was applied to prestin‐transfected HEK 293 cells because these cells do not possess an organized cytoskeletal network. This approach is based, of course, on the success of earlier experiments, where the effect of salicylate on the electromechanical properties of prestin‐transfected kidney or ovary cells was examined (Zheng et al., 2000; Ludwig et al., 2001). 9‐AC reversibly reduced the NLC of the prestin‐transfected cells (Fig. 2A; Fig. 4B, HEK 293/CsCl), similar but larger than the reduction found for OHCs. This result suggests that 9‐AC interacts with prestin. However, the results do not exclude the possibilities that interaction is indirect via endogenous chloride channels, the OHC cytoskeleton or elevation of intracellular [H+]. Therefore, three further sets of experiments were performed.
Chloride experiments
Although five different endogenous chloride conductances have been found in a HEK 293 cell line (Zhu et al., 1998), there is no agreement as to whether these chloride channels are expressed in all types of HEK 293 cell lines (Pusch et al., 1994) and it is not known whether all five are voltage‐sensitive (Zhu et al., 1998) and whether any of them show sensitivity to 9‐AC. However, we were able to exclude the possibility of an effect of 9‐AC on endogenous chloride channels in our cell line because 9‐AC did not affect the I‐V characteristic of native (non‐transfected) HEK 293 cells (Fig. 3C). The cystic fibrosis transmembrane conductance regulator (CFTR) channel is known to be expressed in OHCs (Homma et al., 2010). The possibility that in OHCs this channel conveys the interaction between 9‐AC and prestin can also be excluded because this cAMP‐activated chloride channel requires activation with cAMP, forskolin (Rommens et al., 1991; Ai et al., 2004) or a cocktail of cAMP/ATP/IBMX (Ai et al., 2004; Homma et al., 2010), situations that did not exist in our experiments. Moreover, one can safely exclude the possibility that the 9‐AC block was mediated by ClC‐1 channels, known to be present in OHCs (Kawasaki et al., 1999), because the block of motor charge was at least two orders of magnitude faster than the block of ClC‐1 channels (Estévez et al., 2003).
Cytoskeleton experiments
In earlier cytoskeleton experiments investigating the electromechanical action of OHCs, diamide has been used (Adachi & Iwasa, 1997; Song & Santos‐Sacchi, 2013) because it decouples spectrin from actin (Becker et al., 1986), and latrunculin A has been used (Matsumoto et al., 2010; Song & Santos‐Sacchi, 2013) because it prevents actin polymerization (Morton et al., 2000). The NLC data for the diamide‐treated cells, before application of the 9‐AC, presented a remarkable difference to that for the untreated cells and the latrunculin‐treated cells: α was ~ 30% larger (Table 2). The diamide‐treated cells were more difficult to patch, requiring more precise pressure control for breakthrough of the plasma membrane to establish whole‐cell configuration. In most cases, after opening the cell the pressure in the pipette had to be increased by 0.1–0.2 kPa to avoid cytosol leaking into the patch pipette. As membrane tension can act directly on the motor complex (Kakehata & Santos‐Sacchi, 1995), it is likely that turgor pressure is also one reason for the larger values of α. In addition, it is possible that diamide can also act directly on the motor, as suggested by Adachi & Iwasa (1997) based on their finding of reduced voltage dependence of motility due to diamide, the effective charge transfer, z, decreasing from 0.76 to 0.69 (their table 2). Nevertheless, irrespective of the treatment (diamide or latrunculin A) the principal effect of extracellular 9‐AC application was unambiguous and similar, namely, always a reduction in Q max and of similar magnitude to that for untreated cells (Fig. 2; Tables 3 and 4).
As for the untreated cells, the latrunculin‐treated cells also presented a shift in V h to more positive membrane potentials. However, the shift was, on average, 11 mV larger than for untreated cells, possibly because disruption of the cytoskeleton might have caused an increase in membrane tension which directly affected the motor, as postulated by others from the results of trypsin (Kakehata & Santos‐Sacchi, 1995) as well as diamide and latrunculin (Song & Santos‐Sacchi, 2013) experiments.
As for the untreated cells, there was no detectable 9‐AC‐induced change in C 0. On average, there was a relatively small 2‐mV increase of α for the latrunculin‐treated cells. This increase was on the border of statistical detectability and, as such, is similar to the situation for untreated cells where, on average, an effect of 9‐AC on α was not detected. However, ΔC sa for the latrunculin‐treated cells was significantly affected by 9‐AC; when detectable (6/7 cells), it was reduced. The direction of ΔC sa change was opposite to that known for sodium salicylate applied to untreated OHCs (Homma & Dallos, 2011; Santos‐Sacchi & Navarrete (2002); Santos‐Sacchi & Song, 2014), hinting at a basic difference between the mechanisms of salicylate and 9‐AC blocks.
Taken together, it is concluded that the most probable target of 9‐AC is not the cytoskeleton but is within the plasma membrane, the HEK 293 cell experiments suggesting prestin as the site.
Intracellular pH experiments
Salicylate, known to act on the inner side of the plasma membrane, readily permeates the plasma membrane of OHCs (presumably) in its uncharged form (Tunstall et al., 1995; Kakehata & Santos‐Sacchi, 1996; Zhi et al., 1996). However, the time constant for salicylate‐induced decrease in intracellular pH is about an order of magnitude longer than that for 9‐AC (based on fig. 5A in Tunstall et al. (1995), τ ≈ 15 s at an extracellular salicylate concentration of 5 mm, which is slightly above the IC 50 for NLC; similar time constants are given in Kakehata & Santos‐Sacchi (1996), where τ ≈ 19 and 11 s, respectively, for 1 and 10 mm salicylate). The recovery time‐constant for salicylate‐induced pH change is also, not unexpectedly, much longer than that for 9‐AC (τ ≈ 23 s based on fig. 5A in Tunstall et al., 1995). As in the case of 9‐AC, the time course for salicylate‐induced pH decrease is similar to that for NLC decrease, the time constant for NLC decrease being 10–20 s depending on concentration and application route, extracellular (Tunstall et al., 1995) or intracellular (Kakehata & Santos‐Sacchi, 1996). Importantly, although the pH and NLC time courses in response to 9‐AC are much shorter than those for salicylate, the pH and NLC changes are interdependent for both substances. This interdependence provides evidence that the elevation of [H+] is not the primary cause of the reduction in NLC, as was also originally concluded by Tunstall et al. (1995) for salicylate.
Further evidence that intracellular acidification is an unlikely explanation for the 9‐AC block of NLC derives from the DMSO experiments. Application of DMSO did not change NLC (Fig. 2D) but it did reduce intracellular pH when applied to the 9‐AC solution (Fig. 5B). Moreover, application of 9‐AM, which was also diluted in DMSO, had no significant effect on NLC (Fig. 2D), although intracellular pH was probably changed by DMSO. In this property 9‐AC also shows similarity to salicylate, because it also reduces NLC independently of changes in intracellular pH (Tunstall et al., 1995).
Therefore, again it is concluded that the most probable reason for the 9‐AC‐evoked reduction in charge movement for prestin‐transfected HEK 293 cells and, therefore, for OHCs is interaction between 9‐AC and prestin.
Future experiments
The possibility of a chloride‐binding motif in prestin has been proposed by Bai et al. (2009), based on its homology with a known chloride‐binding site in a bacterial chloride channel (Estévez & Jentsch, 2002) and its position on the intracellular surface of prestin (Navaratnam et al., 2005). However, based on molecular simulations and cysteine substitution experiments, it has been suggested that the chloride (anion) binding site is located centrally within prestin (Gorbunov et al., 2014). According to their simulations, this anion‐binding site is occluded from the extracellular side. Recently, an additional binding site for Cl− and other NLC‐inducing anions was identified computationally within a deep cavity of the C‐terminus of mammalian prestin (Lolli et al., 2016). The cavity remains unchanged upon anion binding. The authors therefore suggested that the site might accommodate a reservoir of anions that can be rapidly released, without conformational rearrangement, for translocation to the anion pathway identified in the transmembrane domain by Gorbunov et al. (2014). These findings suggest the possibility of multiple interaction sites of 9‐AC and salicylate with prestin. Indeed, given the rapid action on the electromechanical transducer by 9‐AC compared with salicylate, the interaction sites for the two compounds might differ. To answer the questions whether the interaction between the negatively charged 9‐AC and prestin is direct and whether chloride‐binding domains might also be involved will require site‐directed mutation experiments and molecular modelling.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG), grant Gu 194/9‐1,2. We thank Dr J. Dettling and Dr A. Breß for providing the prestin‐transfected HEK 293 cells.
Abbreviations
- 9‐AC
9‐anthracenecarboxylic acid
- 9‐AM
9‐anthracene‐methanol
- DMSO
dimethyl sulfoxide
- HBBS
Hanks' balanced salt solution
- HEK
human embryonic kidney
- IC50
half‐maximal inhibitory concentration
- I‐V
current–voltage
- NLC
nonlinear capacitance
- OHC
outer hair cell
[The copyright line was updated on 28 June 2016, after first online publication.]
References
- Adachi, M. & Iwasa, K.H. (1997) Effect of diamide on force generation and axial stiffness of the cochlear outer hair cell. Biophys. J ., 73, 2809–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai, T. , Bompadre, S.G. , Sohma, Y. , Wang, X. , Li, M. & Hwang, T.C. (2004) Direct effects of 9‐anthracene compounds on cystic fibrosis transmembrane conductance regulator gating. Pflug. Arch., 449, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore, J.F. (1987) A fast motile response in guinea‐pig outer hair cells: the cellular basis of the cochlear amplifier. J. Physiol., 388, 323–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore, J. (2008) Cochlear outer hair cell motility. Physiol. Rev., 88, 173–210. [DOI] [PubMed] [Google Scholar]
- Bai, J.P. , Surguchev, A. , Montoya, S. , Aronson, P.S. , Santos‐Sacchi, J. & Navaratnam, D. (2009) Prestin's anion transport and voltage‐sensing capabilities are independent. Biophys. J., 96, 3179–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, P.S. , Cohen, C.M. & Lux, S.E. (1986) The effect of mild diamide oxidation on the structure and function of human erythrocyte spectrin. J. Biol. Chem., 261, 4620–4628. [PubMed] [Google Scholar]
- Brownell, W.E. , Bader, C.R. , Bertrand, D. & de Ribaupierre, Y. (1985) Evoked mechanical responses of isolated cochlear outer hair cells. Science, 227, 194–196. [DOI] [PubMed] [Google Scholar]
- Brownell, W.E. , Jacob, S. , Hakizimana, P. , Ulfendahl, M. & Fridberger, A. (2011) Membrane cholesterol modulates cochlear electromechanics. Eur. J. Physiol., 461, 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimerman, J. , Waldhaus, J. , Harasztosi, C. , Duncker, S.V. , Dettling, J. , Heidrych, P. , Bress, A. , Gampe‐Braig, C. , Frank, G. , Gummer, A.W. , Oliver, D. , Knipper, M. & Zimmermann, U. (2013) Generation of somatic electromechanical force by outer hair cells may be influenced by prestin‐CASK interaction at the basal junction with the Deiter's cell. Histochem. Cell Biol., 140, 119–135. [DOI] [PubMed] [Google Scholar]
- Dallos, P. (2008) Cochlear amplification, outer hair cells and prestin. Curr. Opin. Neurobiol., 18, 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos, P. & Evans, B.N. (1995) High‐frequency motility of outer hair cells and the cochlear amplifier. Science, 267, 2006–2009. [DOI] [PubMed] [Google Scholar]
- Eckrich, T. , Nowotny, M. , Harasztosi, C. , Scherer, M. & Gummer, A.W. (2008) Impedance measurements of isolated outer hair cells. ARO Midwinter Meeting, Abstract 517.
- Ermilov, S.A. , Murdock, D.R. , El‐Daye, D. , Brownell, W.E. & Anvari, B. (2005) Effects of salicylate on plasma membrane mechanics. J. Neurophysiol., 94, 2105–2110. [DOI] [PubMed] [Google Scholar]
- Estévez, R. & Jentsch, T.J. (2002) CLC chloride channels: correlating structure with function. Curr. Opin. Struc. Biol., 12, 531–539. [DOI] [PubMed] [Google Scholar]
- Estévez, R. , Schroeder, B.C. , Accardi, A. , Jentsch, T.J. & Pusch, M. (2003) Conservation of chloride channel structure revealed by an inhibitor binding site in ClC‐1. Neuron, 38, 47–59. [DOI] [PubMed] [Google Scholar]
- Farrell, B. , Shope, C.D. & Brownell, W.W. (2006) Voltage‐dependent capacitance of human embryonic kidney cells. Phys. Rev. E, 73, 041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, G. , Hemmert, W. & Gummer, A.W. (1999) Limiting dynamics of high‐frequency electromechanical transduction of outer hair cells. Proc. Natl. Acad. Sci. USA, 96, 4420–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis, K.D. (2000) Admittance‐based measurement of membrane capacitance using the EPC‐9 patch‐clamp amplifier. Pflug. Arch., 439, 655–664. [DOI] [PubMed] [Google Scholar]
- Gorbunov, D. , Sturlese, M. , Nies, F. , Kluge, M. , Bellanda, M. , Battistuta, R. & Oliver, D. (2014) Molecular architecture and the structural basis for anion interaction in prestin and SLC26 transporters. Nat. Commun., 5, 3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger, R. (1990) Choride channel blockers In Fleischer S. & Fleischer B. (Eds), Biomembranes. Part V. Cellular and Subcelluar Transport: Epithelial Cells. Academic Press, Inc., San Diego, pp. 793–810. [Google Scholar]
- Guttman, I. , Wilks, S.S. & Hunter, J.S. (1971) Introductory Engineering Statistics. Wiley, New York, pp. 173–174. [Google Scholar]
- Halet, G. (2005) Imaging phosphoinositide dynamics using GFP‐tagged protein domains. Biol. Cell, 97, 501–518. [DOI] [PubMed] [Google Scholar]
- Hallworth, R. (1997) Modulation of outer hair cell compliance and force by agents that affect hearing. Hearing Res., 114, 204–212. [DOI] [PubMed] [Google Scholar]
- Heidrych, P. , Zimmermann, U. , Bress, A. , Pusch, C.M. , Ruth, P. , Pfister, M. , Knipper, M. & Blin, N. (2008) Rab8b GTPase, a protein transport regulator, is an interacting partner of otoferlin, defective in a human autosomal recessive deafness form. Hum. Mol. Genet., 17, 3814–3821. [DOI] [PubMed] [Google Scholar]
- Holley, M.C. (1996) Outer hair cell motility In Dallos P., Popper A.N. & Fay R.R. (Eds), The Cochlea. Springer‐Verlag, New York Inc, pp. 386–434. [Google Scholar]
- Homma, K. & Dallos, P. (2011) Evidence that prestin has at least two voltage‐dependent steps. J. Biol. Chem., 286, 2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma, K. , Miller, K.K. , Anderson, C.T. , Sengupta, S. , Du, G.G. , Aguinaga, S. , Cheatham, M. , Dallos, P. & Zheng, J. (2010) Interaction between CFTR and prestin (SLC26A5). Biochim. Biophys. Acta, 1798, 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, G. & Santos‐Sacchi, J. (1994) Motility voltage sensor of the outer hair cell resides within the lateral plasma membrane. Proc. Natl. Acad. Sci. USA, 91, 12268–12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch, T.J. , Stein, V. , Weinreich, F. & Zdebik, A.A. (2002) Molecular structure and physiological function of chloride channels. Physiol. Rev., 82, 503–568. [DOI] [PubMed] [Google Scholar]
- Kakehata, S. & Santos‐Sacchi, J. (1995) Membrane tension directly shifts voltage dependence of outer hair cell mitility and associated gating charge. Biophys. J., 68, 2190–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakehata, S. & Santos‐Sacchi, J. (1996) Effects of salicylate and lanthanides on outer hair cell motility and associated gating charge. J. Neurosci., 16, 4881–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinec, F. , Holley, M.C. , Iwasa, K.H. , Lim, D.J. & Kachar, B. (1992) A membrane‐based force generation mechanism in auditory sensory cells. Proc. Natl. Acad. Sci. USA, 89, 8671–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko, T. , Harasztosi, C. , Mack, A.F. & Gummer, A.W. (2006) Membrane traffic in outer hair cells of the adult mammalian cochlea. Eur. J. Neurosci., 23, 2712–2722. [DOI] [PubMed] [Google Scholar]
- Kawasaki, E. , Hattori, N. , Miyamoto, E. , Yamashita, T. & Inagaki, C. (1999) Single‐cell RT‐PCR demonstrates expression of voltage‐dependent chloride channels (ClC‐1, ClC‐2 and ClC‐3) in outer hair cells of rat cochlea. Brain Res., 838, 166–170. [DOI] [PubMed] [Google Scholar]
- Liberman, M.C. , Gao, J. , He, D.Z. , Wu, X. , Jia, S. & Zuo, J. (2002) Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature, 419, 300–304. [DOI] [PubMed] [Google Scholar]
- Lindau, M. & Neher, E. (1988) Patch‐clamp techniques for time‐resolved capacitance measurements in single cells. Pflug. Arch., 411, 137–146. [DOI] [PubMed] [Google Scholar]
- Lolli, G. , Pasqualetto, E. , Costanzi, E. , Bonetto, G. & Battistutta, R. (2016) The STAS domain of mammalian SLC26A5 prestin harbours an anion‐binding site. Biochem. J., 473, 365–370. [DOI] [PubMed] [Google Scholar]
- Ludwig, J. , Oliver, D. , Frank, G. , Klöcker, N. , Gummer, A.W. & Fakler, B. (2001) Reciprocal electromechanical properties of rat prestin: the motor molecule from rat outer hair cells. Proc. Natl. Acad. Sci. USA, 98, 4178–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue, A.J. & Brownell, W.E. (1999) Salicylate induced changes in outer hair cell lateral wall stiffness. Hearing Res., 135, 163–168. [DOI] [PubMed] [Google Scholar]
- Mammano, F. & Ashmore, J.F. (1993) Reverse transduction measured in the isolated cochlea by laser Michelson interferometry. Nature, 365, 838–841. [DOI] [PubMed] [Google Scholar]
- Matsumoto, N. , Kitani, R. , Maricle, A. , Mueller, M. & Kalinec, F. (2010) Pivotal role of actin depolymerization in the regulation of cochlear outer hair cell motility. Biophys. J., 99, 2067–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistrik, P. , Daudet, N. , Morandell, K. & Ashmore, J.F. (2012) Mammalian prestin is a weak Cl‐/HCO3 ‐ electrogenic antiporter. J. Physiol., 590, 5597–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett, R.B. & Tang, A.H. (1968) Skeletal muscle stimulants substituted benzoic acids. J. Med. Chem., 11, 1020–1022. [DOI] [PubMed] [Google Scholar]
- Morimoto, N. , Raphael, R.M. , Nygren, A. & Brownell, W.E. (2002) Excess plasma membrane and effects of ionic amphipaths on mechanics of outer hair cell lateral wall. Am. J. Physiol‐Cell Ph., 282, C1076–C1086. [DOI] [PubMed] [Google Scholar]
- Morton, W.M. , Ayscough, K.R. & McLaughlin, P.J. (2000) Latrunculin Alters the actin‐monomer subunit interface to prevent polymerization. Nat. Cell Biol., 2, 376–378. [DOI] [PubMed] [Google Scholar]
- Murugasu, E. & Russell, I.J. (1995) Salicylate ototoxicity: effects on basilar membrane displacement, cochlear microphonics, and neural responses in the basal turn of the guinea pig cochlea. Audit. Neurosci., 1, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaratnam, D. , Bai, J.P. , Samaranayake, H. & Santos‐Sacchi, J. (2005) N‐terminal‐mediated homomultimerization of prestin, the outer hair cell motor protein. Biophys. J., 89, 3345–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny, M. & Gummer, A.W. (2006) Nanomechanics of the subtectorial space caused by electromechanics of cochlear outer hair cells. Proc. Natl. Acad. Sci. USA, 103, 2120–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver, D. , He, D.Z. , Klocker, N. , Ludwig, J. , Schulte, U. , Waldegger, S. , Ruppersberg, J.P. , Dallos, P. & Fakler, B. (2001) Intracellular anions as the voltage sensor of prestin, the outer hair cell motor protein. Science, 292, 2340–2343. [DOI] [PubMed] [Google Scholar]
- Pusch, M. , Steinmeyer, K. & Jentsch, T.J. (1994) Low single channel conductance of the major skeletal muscle chloride channel, ClC‐1. Biophys. J., 66, 149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch, M. , Accardi, A. , Liantonio, A. , Guida, P. , Traverso, S. , Camerino, D.C. & Conti, F. (2002) Mechanisms of block of muscle type CLC chloride channels (Review). Mol. Membr. Biol., 19, 285–292. [DOI] [PubMed] [Google Scholar]
- Rommens, J.M. , Dho, S. , Bear, C.E. , Kartner, N. , Kennedy, D. , Riordan, J.R. , Tsui, L.C. & Foskett, J.K. (1991) cAMP‐inducible chloride conductance in mouse fibroblast lines stably expressing the human cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA, 88, 7500–7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüdel, R. & Lehmann‐Horn, F. (1985) Membrane changes in cells from myotonia patients. Physiol. Rev., 65, 310–356. [DOI] [PubMed] [Google Scholar]
- Russell, I.J. & Schauz, C. (1995) Salicylate ototoxicity: effects on the stiffness and electromotility of outer hair cells isolated from the guinea pig cochlea. Audit. Neurosci., 1, 309–319. [Google Scholar]
- Rybalchenko, V. & Santos‐Sacchi, J. (2003) Cl− flux through a non‐selective, stretch‐sensitive conductance influences the outer hair cell motor of the guinea‐pig. J. Physiol., 547, 873–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Sacchi, J. & Navarrete, E. (2002) Voltage‐dependent changes in specific membrane capacitance caused by prestin, the outer hair cell lateral membrane motor. Pflug. Arch., 444, 99–106. [DOI] [PubMed] [Google Scholar]
- Santos‐Sacchi, J. & Song, L. (2014) Chloride and salicylate influence prestin‐dependent specific membrane capacitance: support for the area motor model. J. Biol. Chem., 289, 10823–10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Sacchi, J. , Song, L. , Zheng, J. & Nuttall, A.L. (2006) Control of mammalian cochlear amplification by chloride anions. J. Neurosci., 26, 3992–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaechinger, T.J. , Gorbunov, D. , Halaszovich, C.R. , Moser, T. , Kügler, S. , Fakler, B. & Oliver, D. (2011) A synthetic prestin reveals protein domains and molecular operation of outer hair cell piezoelectricity. EMBO J., 30, 2793–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer, M.P. & Gummer, A.W. (2004) Vibration pattern of the organ of Corti up to 50 kHz: evidence for resonant electromechanical force. Proc. Natl. Acad. Sci. USA, 101, 17652–17657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata, W.E. , Brownell, W.E. & Dieler, R. (1991) Effects of salicylate on shape, electromotility and membrane characteristics of isolated outer hair cells from guinea pig cochlea. Acta Oto‐laryngol., 111, 707–718. [DOI] [PubMed] [Google Scholar]
- Song, L. & Santos‐Sacchi, J. (2013) Disparities in voltage‐sensor charge and electromotility imply slow chloride‐driven state transitions in the solute carrier SLC26a5. Proc. Natl. Acad. Sci. USA, 110, 3883–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer, K. , Ortland, C. & Jentsch, T.J. (1991) Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature, 354, 301–304. [DOI] [PubMed] [Google Scholar]
- Tunstall, M.J. , Gale, J.E. & Ashmore, J.F. (1995) Action of salicylate on membrane capacitance of outer hair cells from the guinea‐pig cochlea. J. Physiol., 485, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, R.B. , Glantz, R.H. & Butcher, J. (1988) Hearing loss in myotonic dystrophy. Ann. Neurol., 23, 202–203. [DOI] [PubMed] [Google Scholar]
- Zheng, J. , Shen, W. , He, D.Z. , Long, K.B. , Madison, L.D. & Dallos, P. (2000) Prestin is the motor protein of cochlear outer hair cells. Nature, 405, 149–155. [DOI] [PubMed] [Google Scholar]
- Zhi, M. , Phillips, B. & Brownell, W.E. (1996) Alkaline cytoplasm and pH gradients in outer hair cell: evidence for a proton pump. Assoc. Res. Otolaryngol., 19, 538. [Google Scholar]
- Zhou, Y. & Raphael, R.M. (2005) Effect of salicylate on the elasticity, bending stiffness, and strength of SOPC membranes. Biophys. J., 89, 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, G. , Zhang, Y. , Xu, H. & Jiang, C. (1998) Identification of endogenous outward currents in the human embryonic kidney (HEK 293) cell line. J. Neurosci. Meth., 81, 73–83. [DOI] [PubMed] [Google Scholar]