

Abstract
Objective
While large fetal copy number aberrations can generally be detected through sequencing of DNA in maternal blood, the reliability of tests depends on the fraction of DNA that originates from the fetus. Existing methods to determine this fetal fraction require additional work or are limited to male fetuses. We aimed to create a sex‐independent approach without additional work.
Methods
DNA fragments used for noninvasive prenatal testing are cut only by natural processes; thus, influences on cutting by the packaging of DNA in nucleosomes will be preserved in sequencing. As cuts are expected to be made preferentially in linker regions, the shorter fetal fragments should be enriched for reads starting in nucleosome covered positions.
Results
We generated genome‐wide nucleosome profiles based on single end sequencing of cell‐free DNA. We found a difference between DNA digestion of fetal cell‐free DNA and maternal cell‐free DNA and used this to calculate the fraction of fetal DNA in maternal plasma for both male and female fetuses.
Conclusion
Our method facilitates cost‐effective noninvasive prenatal testing, as the fetal DNA fraction can be estimated without the need for expensive paired‐end sequencing or additional tests.
The methodology is implemented as a tool, which we called SANEFALCON (Single reAds Nucleosome‐basEd FetAL fraCtiON). It is available for academic and non‐profit purposes under Creative Commons Attribution‐NonCommercial‐ShareAlike 4.0 International Public License. github.com/rstraver/sanefalcon. © 2016 The Authors. Prenatal Diagnosis published by John Wiley & Sons, Ltd.
Short abstract
What's Already Known About This Topic?
Fetal DNA is found in small and varying amounts in maternal blood, enough to detect fetal aberrations such as Down syndrome through next generation sequencing methods.
Fetal DNA is generally shorter, and this is believed to be influenced by nucleosomes.
What Does This Study Add?
We obtained nucleosome positions from cell‐free DNA over a combination of low‐coverage samples.
Our method shows how fetal fragments are influenced by nucleosomes compared with maternal fragments.
We deduced the fetal fraction using the distribution of reads starting around nucleosome positions, independent of the fetal sex.
Introduction
Low‐coverage next generation sequencing (NGS) data are used for noninvasive prenatal testing (NIPT), avoiding the 2–3 : 1001, 2, 3 chance for miscarriage introduced by invasive sampling. Although previous work has shown this is a reliable method for screening,4, 5, 6, 7, 8 its reliability depends heavily on the assumption that there is enough fetal DNA in the sample tested.9, 10 The determination of the fetal fraction can either be done with a separate test, such as a quantitative PCR, or directly from the sequencing data. The latter method is preferred, as it directly reflects the fetal fraction in the data that are also used to determine fetal aneuploidy. In contrast, a separate test will not detect loss of fetal fraction during stages of laboratory work‐up for NGS (such as library preparation). In case of a male fetus, fetal fraction determination can be performed based on Y‐chromosomal sequences. Several algorithms have been described,11, 12 including the freely available DEFRAG algorithm (https://github.com/rstraver/wisecondor/blob/master/defrag.py). Several approaches to determine fetal fraction for all pregnancies (male and female fetuses) have been described. One method uses differences in C‐methylation between maternal and fetal DNA,13 but the additional laboratory work required makes this option rather unattractive for routine diagnostic purposes. Additionally, such approaches require splitting the sample in two, one part for the actual NGS analysis and another for determining the C‐methylation, thus introducing a possible difference in fetal fraction between the two subsamples at the end of the analysis. Another option is to use differences in single nucleotide polymorphisms (SNPs) to distinguish between fetal and maternal DNA. This can be performed if the method for trisomy detection is based on SNPs.14 The fetal fraction can also be determined from the distribution of reads containing SNPs,15 but the requirement of an extremely high‐coverage per sample makes this approach too expensive for daily diagnostic routines.
Another promising way to obtain an indication of the fetal fraction is to use paired‐end data. Fetal DNA consists of shorter fragments than maternal DNA.16 Yu et al. recently showed a correlation between insert sizes and fetal fraction when using paired‐end sequencing.17 Although this appears to be a useful solution, the increased cost and turn‐around time of paired‐end sequencing limit its clinical implementation as a routine procedure. The correlation with the fetal fraction was suspected to be caused by nucleosomes as the insert sizes of maternal DNA appear to concentrate around the length of a nucleosome with some linker DNA still attached (167 bp), while fetal DNA seems to be biased toward lengths smaller than and up to the length of nucleosomes (147 bp18). However, no laboratory experiments were performed to prove this. As these approaches have not been compared with the same set of samples, it is currently not possible to define which method works best.
We hypothesized that if the differences between maternal and fetal DNA fragment sizes are caused by differential nucleosomal packaging of the DNA during apoptosis, or by differences in the strength of nucleosome binding, we should be able to find a correlation between the fetal fraction and the positioning of read fragments around the nucleosomes. To confirm this hypothesis, we developed a method that takes this principle into account by determining the distribution of reads with respect to nucleosome positions.
Cell‐free DNA (cfDNA) in maternal plasma is already digested into small fragments by natural processes. Because of the size of these fragments, no additional shearing is required before sequencing. Thus, sequencing reads obtained from these fragments all start at positions where they were cut by natural processes. Enzymes that cut the DNA into smaller fragments can easily make cuts in between nucleosomes, in the areas described as linker DNA (Figure 1a). The DNA that is wound around a nucleosome is harder to reach and left uncut. Therefore, DNA fragments with nucleosomes still attached are protected from these cutting processes until they are unwound (Figure 1b). Hence, longer DNA fragments are expected to start further upstream from nucleosomes (Figure 1c). As a result, the distribution in read start positions will change if the relative amount of large read fragments changes with respect to the amount of short fragments (Figure 1d). To detect nucleosome positions, we combine several low coverage samples into a single high‐coverage dataset (Figure 1e). By looking for high and low concentrations of reads in small areas, we can determine where nucleosomes are positioned in our samples. For a single sample, we aligned all nucleosomes on top of each other to obtain a single artificial nucleosome with high coverage (Figure 1f). This artificial nucleosome then provides us insight in the distribution of DNA fragment degradation (Figure 1g) and can be used to determine the fraction of fetal DNA and maternal DNA in cell‐free maternal plasma.
Figure 1.
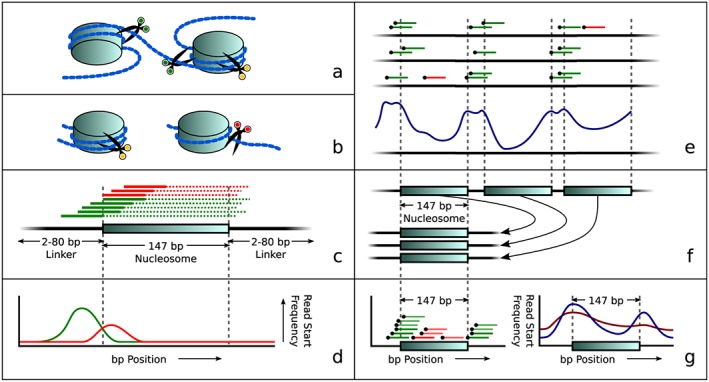
Overview of the principle of our approach: nucleosome‐dependent differences in degradation of maternal and fetal DNA lead to different start sites of sequence reads. (a) Simplified visualization of DNA wound around nucleosomes with three examples of cut positions (green scissors means able to cut in this view while yellow scissors indicate that cutting is protected by the histone complex). (b) Later during degradation, the fragment may either still be protected by a nucleosome (left, yellow scissors), or it may be unwound, allowing it to be cut at the previously inaccessible position (now indicated by red scissors. (c) Theoretical distribution of reads around a nucleosome. Colors correspond to scissor colors in panels (a) and (b). The solid lines depict reads while dotted lines represent the unsequenced parts of these DNA fragments. (d) Idealized read start frequency distribution for reads upstream of the nucleosome center, overstating the difference in positions where cuts were made. (e) Combining several low coverage samples (top) reveals a genome‐wide repetitive pattern (bottom) that matches in size and frequency to nucleosome and linker DNA positions. (f) Aligning all nucleosome positions to a single aligned nucleosome. (g) Read start positions in the aligned nucleosome (left) show the aligned nucleosome profile (right), which can then be used to determine differences in read distributions related to fetal fractions: Blue shows a sample with a low fetal fraction; red shows a sample with a higher fetal fraction
Methods
Data
DNA was isolated using a Qiasymphony (Qiagen, Hilden, Germany) from a total of 398 blood plasma samples taken from pregnant women, most of them with a gestational age of 12–15 weeks. With 5500 SOLiD™ Fragment Library kit (LifeTech PN 4464413, Carlsbad, CA, USA) and TruSeq adapters (Illumina FC‐121‐4001 and FC‐121‐4002, San Diego, CA, USA), NGS libraries were made using a Biomek FX robot (Beckman Coulter, Pasadena, CA, USA). Eight NGS libraries were pooled per rapid flow cell lane of the HiSeq2500 (Illumina). Samples were split over several runs obtained over a period of 12 months. This way we captured variations that occur in daily practice. We obtained 8–16 million 51 bp single end reads per sample. Sequence data were demultiplexed allowing one mismatch in the sequence tag, then mapped to Hg19 using bwa, allowing zero mismatches and removing any read that had multiple mappable positions. Pregnancies of 153 male fetuses were used to determine a fetal fraction reference using the fraction of reads mapped to chromosome Y.
Generating nucleosome start position profiles
To obtain nucleosome positioning for our data type, we combined (RETRO filtered, WISECONDOR19 in the Supporting Information) reads from all samples into a single meta‐sample. For all reads, we only took the start position into account. Reverse‐mapped reads had their start position adjusted for their length by adding their length minus one base pair to their first position on the genome. This was carried out as the BAM file format reports the first base pair position of every read on the reference genome rather than the first position of the read sequence, creating an offset in positions if not accounted for. Every base pair in the reference sequence received a score describing the likeliness of this base pair position being the center of a nucleosome. This score is calculated as the average amount of read start positions in 20 bp left and right of the center 147 bp area around the targeted base pair, divided by the average read start count for the 147 bp center area. Formally,
where s(x) is the nucleosome center score for base pair position x and RP(y) the number of reads starting at position y.
The algorithm saves the maximum value from the set of scored base pair positions (across the whole genome) and ignores all scores 147 bp left and right of this position further on to exclude possibly overlapping nucleosome calls. Picking the maximum‐scored base pair is repeatedly carried out until no position scores higher than 1 remain. Pseudocode for these steps is shown in Algorithm S1. For further analysis, we removed any nucleosome call that had a lower average count of reads in either 20 bp side regions than the center 147 bp region. This ensures we only take calls into account that were indeed likely nucleosome centers rather than slopes or randomly occurring peaks.
Aligned nucleosome profile per sample
Detected nucleosome positions were aligned onto each other at their centers. We determined the number of reads that started at any position within 147 base pairs upstream and downstream in respect to nucleosome positions. Pseudocode for this process is shown in Algorithm S2.
Leave set out training
To remove overfitting on the training data (Figure S1), we built an artificial nucleosome per training sample based on a nucleosome position reference track that was built without that specific training sample. For example, to obtain an aligned nucleosome profile for training sample A, we could use all training samples except for sample A itself to obtain a nucleosome track, then use this track to obtain a nucleosome profile for sample A. The nucleosome profile per sample can then be used to determine the correlations per base pair with the chromosome Y‐based fetal fraction that is used when calculating the fetal fraction per sample. For the test samples, the reference nucleosome track is built from all training samples together.
To make sure we do not provide any run specific kind of information to our nucleosome track during training, we also removed all samples from the run the training sample originated from when creating the reference nucleosome track. To reduce sample size variation per run, we combined actual sequencing runs into 12 run sets with close to 25 samples each. This resulted in creating 12 nucleosome tracks, one per run set, where every run set gets a reference nucleosome track (based on 11 run sets) for which its own samples were not used to determine nucleosome positions. An overview of this procedure is shown in Figure S2, and the pseudocode for this process is shown in Algorithm S3. Next, the nucleosome tracks per training sample are used to calculate the correlation per base pair position with the fetal fraction determined on the Y chromosome. This is outlined in Algorithm S4. The fetal fraction for a test sample is then determined by adding the read start frequencies per base pair after weighting them by their respective correlation score. Pseudocode for this is shown in Algorithm S5.
Ethical background
This study describes the development of a new software tool to predict fetal fraction. Although NIPT data were used retrospectively to develop and validate the tool, no individual reports were issued. Furthermore, the outcome of the tool (prediction of fetal fraction) has no relation at all with clinical outcome of the mother or the fetus; it will only support the reliability of the outcome of the NIPT analysis. For that reason, no approval of a medical ethical committee was required.
Results
Extracted nucleosome positions match nucleosome characteristics
We combined a training set of 298 low‐coverage single end (51 bp) read samples (Illumina HiSeq2500) of cfDNA extracted from pregnant women (Table 1) into a single set of reads (see Section on Methods). The smoothed read start signal (blue) shows small regions where reads are more or less concentrated compared with adjacent regions. The peak pattern closely matches the sizes of nucleosomes and linker DNA, which respectively are 147 and 2–80 bp, with a mean of 20 bp (Figure S3). By applying a nucleosome filter over read start positions, we inferred the most likely nucleosome positioning over the entire genome in this dataset (see Section on Methods). Figure 2a shows the results of our approach on a 2000 bp region (nucleosome score per bp shown in red). Local maxima are considered to be the nucleosome centers (black lines in Figure 2a). The total number of nucleosomes found was 13 521 603, of which 13 429 874 were left after filtering. We also calculated dinucleotide patterns in the sequence of the observed nucleosome positions aligned on their centers, shown in Figure 2b. These dinucleotide frequencies in both the nucleosome core and the linker regions are in concordance with what is known from earlier studies.20, 21
Table 1.
Overview of the number of samples used, split by fetal sex and analysis group
| XX | XY | Total | |
|---|---|---|---|
| Training | 147 | 151 | 298 |
| Test | 39 | 59 | 98 |
| Paired‐end | 11 | 10 | 21 |
Figure 2.
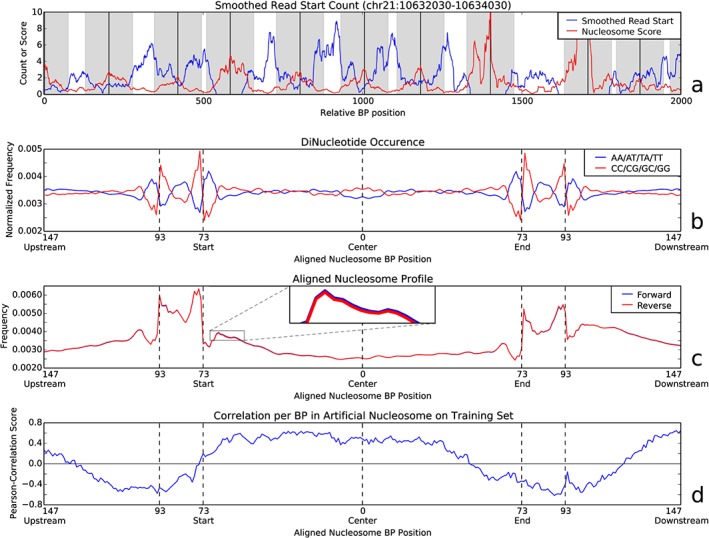
(a) The red line shows the nucleosome positioning scores as determined by the nucleosome filter for a 2000 bp area on chromosome 21. The blue line shows the smoothed read start position count. Black vertical lines show likely nucleosome centers with their respective covered regions (147 bp) in gray. Note that the distance between any two nucleosome centers matches that of the combined nucleosome size (147 bp) and linker DNA length (2–80 bp). (b) Occurrences of dinucleotides found in the reference genome around detected nucleosome positions. Values are relative to their expected values as obtained by counting occurrences over the whole genome. This shows clear preferences for certain dinucleotides at the start and end of the nucleosomes, as well as in the center area. (c) Read start profile across aligned nucleosome centers for all training samples combined. All autosomal chromosome nucleosome positions have been combined to create a single nucleosome profile per strand. The reverse strand (red) was mirrored to show the similarity in read start behavior between the two strands. A cutout shows a zoomed‐in region to visualize the very small differences between the two strands. The linker DNA is clearly identifiable by the increased amount of read starts in the regions just over 73 bp upstream and downstream of the nucleosome center. (d) Read start correlation profile using all male pregnancy training samples, showing the correlation between the frequency of reads starting at any position in the aligned nucleosome profile and the chromosome Y‐based reference values
Aligned nucleosome profile reflects DNA‐degradation process
For every sample, reads over all autosomal chromosomes were combined into an aligned nucleosome profile by aligning their start positions to the nearest downstream nucleosome center position and summing together all relative position frequencies over all nucleosomes (Figure 2c). As discussed before, cfDNA was not sheared before sequencing; therefore, the read start distribution profile reflects the degradation process of cfDNA. As shown, there is a high concentration of read starts in linker DNA regions compared with the nucleosome covered center. Furthermore, the pattern of the linker DNA on both ends is almost the same; there is a decrease in coverage of read starts in the middle of the linker DNA, and read starts in the nucleosome covered region decrease in coverage from its start toward the center.
Nucleosome‐based fetal fraction correlated with reference‐based fetal fraction
We determined the fetal DNA fraction for every male pregnancy sample (153 out of 298) in our training set by dividing the amount of reads mapped to chromosome Y by the total amount of reads mapped to autosomal chromosomes. By relating the Y‐based reference fetal fraction and aligned nucleosome profile per sample, we can deduce the correlation between the frequency of reads starting at any base pair position relative to the nucleosome center and the corresponding fetal fraction for that sample (Figure 2d). This shows that the frequency of reads starting within the nucleosome is positively correlated to the fetal fraction. Reads starting outside of the nucleosome covered regions are negatively correlated to the fetal fraction and thus considered mostly maternal. Hence, fetal DNA start positions are enriched in nucleosome covered areas (any position within the region marked by 73 bp upstream and downstream of the nucleosome center), while maternal DNA is enriched in the expected linker region and beyond (any position outside the 73 bp upstream and downstream region in Figure 2d). The increased correlation near 147 bp upstream and downstream (Figure 2d) was expected, as these regions overlap strongly with neighboring nucleosomes (linker DNA is known to vary between 2 and 80 bp, but averages at about 20 bp in length).
Predicting fetal fraction
To predict the fetal fraction, we calculated a weighted sum of read start positions over the aligned nucleosome profile per sample using the correlations scores as weights (see Section on Methods). This prediction correlates strongly (Pearson correlation: 0.636, P‐value 1.61e–18, 151 training samples) with the fraction of reads on chromosome Y, as shown in Figure 3a. When the weighted sum is applied to the independent test set, this correlation stays nearly unchanged (Pearson correlation: 0.654, P‐value 1.86e–08, 59 test samples).
Figure 3.
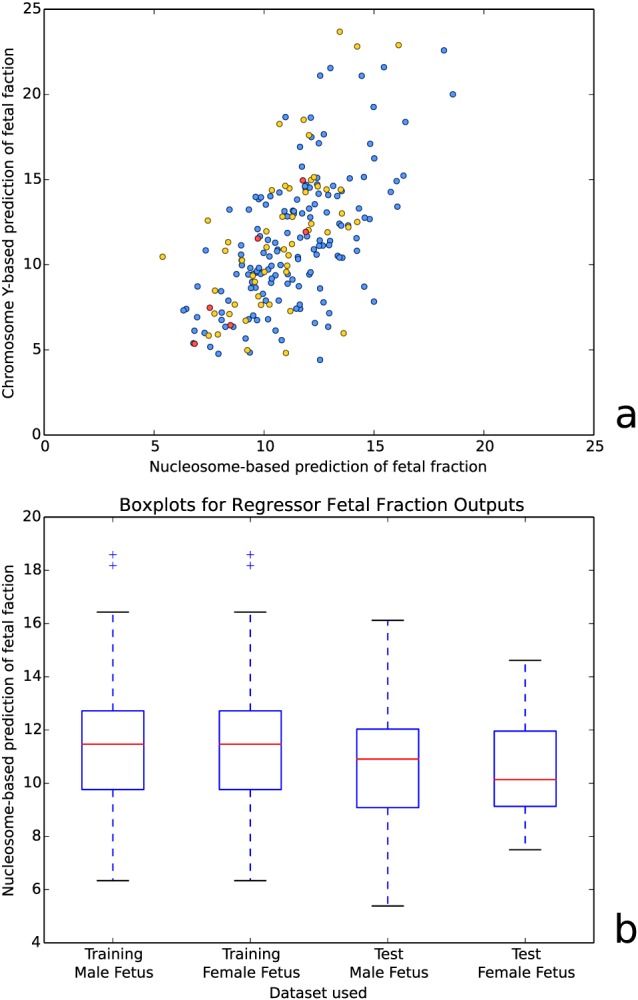
(a) Nucleosome‐based prediction of fetal fraction versus chromosome Y‐based prediction of fetal fraction for both training (blue) and test (red and yellow) samples of XY pregnancies. Red dots mark test samples from a single run. (b) Boxplots of the nucleosome‐based fetal fraction prediction for both training and test data, separated per fetal sex. Additional information on the accuracy of our method can be found in Table S1
The correlation between predicted fetal fractions (weighted sum) and fractions based on chromosome Y was negatively influenced by inter‐run variations. Runs with sufficient male pregnancies to determine a correlation per run had a higher correlation between these approaches (e.g. a correlation of 0.851 and P‐value 3.16e–02 over six samples from a single run, as marked red in Figure 3a).
To see if male fetus samples behave differently from female fetus samples in our method, we tested for changes in the distribution of the predicted fetal fraction. We used 147 female pregnancy samples in our training set and 39 in our test set. While male pregnancy training samples were used for both the nucleosome detection and determining the weights of the predictor, female pregnancy training samples were only used for nucleosome detection. As shown in Figure 3b, both groups have nearly the same estimated fetal fraction distribution as their male pregnancy counterparts. These nonsignificant variations are more likely caused by variable fetal fractions than by a systematic error in our model.
Fragment start position correlates strongly with fragment sizes
To compare our method with the fragment size‐based method using paired‐end sequencing by Yu et al.17, we tested for correlations between the read start frequency per base pair in the aligned nucleosome profile and the fragment size frequencies in an additional 21 samples subjected to paired‐end sequencing (Table 1). As shown in Figure 4, reads starting near the end of a nucleosome up to the start of linker DNA (147–93 bp upstream or 50–93 bp downstream) are strongly correlated to read pairs with fragment sizes over 175 bp (denoted by A in Figure 4), while fragment sizes between 30 and 160 bp are strongly correlated with reads starting in the first half of nucleosomes (73–0 bp upstream, denoted by B in Figure 4).
Figure 4.
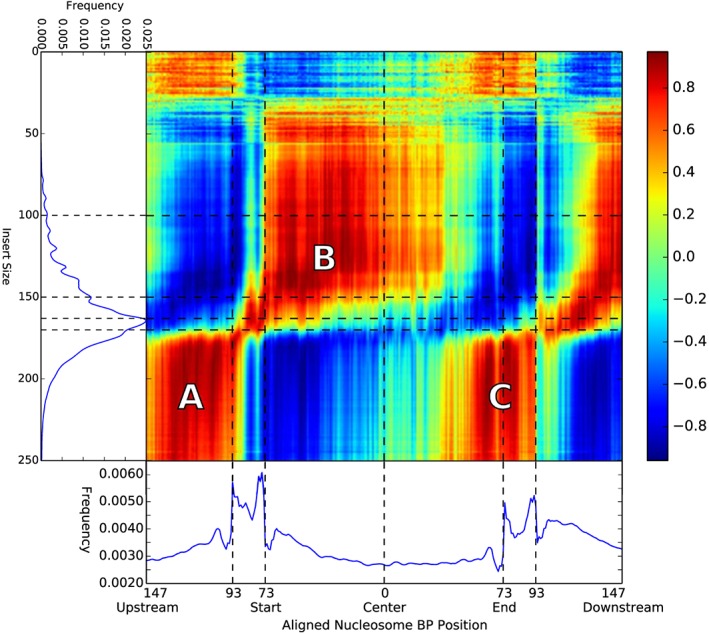
Correlation per base pair of the aligned nucleosome profile with different fragment sizes in paired‐end data. As clearly visible, larger insert sizes correlate to the regions around the nucleosome positions (marked A), while the first half of the nucleosome covered area (73 upstream to 0 center, marked B) correlates strongly with fragment sizes up to 150 bp. The positive correlation between fragment sizes over 175 and nucleosome position 50–90 bp downstream (marked C) match expectations of cutting behavior when a DNA fragment contains two nucleosomes
This finding further suggests that maternal DNA consists of larger fragments that span over linker DNA while fetal fragments are digested downstream, past the start of nucleosome covered positions. When we applied our (trained) nucleosome correlation profile to estimate the fetal fraction per (paired‐end) sample, this resulted in a correlation of 0.891 (P‐value 6.00e–08) with the fragment size method as described by Yu et al.17
Discussion
We have demonstrated that a putative genome‐wide nucleosome profile can be deduced from the read‐start positions when sequencing cfDNA. Based on these profiles, we have been able to infer an estimation of the fetal fraction per sample using low coverage single end read data. At the heart of our approach lies the idea that cfDNA is cut in small pieces and that these cut positions are preferably located in DNA that is not tightly bound to nucleosomes. If fetal DNA is degraded more extensively than maternal DNA, it is expected to have less nucleosomes still attached. Consequently, more cuts in DNA loci normally bound by nucleosomes will be observed in fetal cfDNA compared with maternal cfDNA.
The success of our method depends on an accurate in silico detection of nucleosome positions along the genome. We built such a nucleosome track from cfDNA sequencing data, making use of read start positions acquired over many different samples. This results in a recognizable genome‐wide nucleosome track, as shown by dinucleotide distributions in Figure 2b. It should be noted that no golden standard is available for such a nucleosome track, so formally, we cannot prove that our track represents actual nucleosomes, despite fulfilling many criteria such as periodicity, linker length and dinucleotide distribution. Existing work on determining nucleosome positioning is often based on a high‐coverage single sample of a specific cell type after additional lab work to obtain nucleosome‐focussed fragments.20, 21, 22, 23 Alternatively, nucleosome tracks are predictions from a model that is based on other species such as yeast and then applied to the human reference genome.24 These models do show strong overlap with true nucleosome positioning but have shown not to provide useful information for our approach as the obtained correlations are too weak for our model (Figure S4). Consequently, it is important to extract the nucleosome track from cfDNA and probably under the same sequencing conditions, as we already observed run‐dependent effects (Figure 3a).
Considering the distributions of the aligned nucleosomes, we hypothesized that fetal DNA fragments are either further digested or have a different nucleosome positioning. Reasons for the different digestion can be either different processes working on the fetal DNA or the same digestion process advanced to a later state before a blood sample is taken for analysis (Figures 1a and b). An important reason to believe the differences we found are caused by the more advanced state of DNA degradation can be derived from the correlation plots shown in Figures 2d and 4. Close observation of Figure 2 shows that there is already a negative correlation with the chromosome Y‐based fetal fraction from roughly 50 bp downstream onwards, which indicates enrichment of maternal DNA. This is counter intuitive because of the starting positions of these fragments are still inside the (protected) nucleosome. Assuming that the downstream fragment ends that are not sequenced because of the single‐end sequencing are cut in linker DNA, short fragments would start between 49 bp downstream and the end of the nucleosome. However, reads from fragments smaller than 51 bp include partial adapter sequences and thus become unmappable (we did not trim adapters from the sequenced reads). Inspecting Figure 4 shows that this region is positively correlated with large fragment sizes, see area denoted by C. Consequently, reads that start at this position actually belong to large fragments covering the next nucleosome and thus are preferentially maternal, causing correlation with maternal DNA instead of fetal DNA, explaining the observed negative correlation in Figure 2. This supports the theory of a relatively more advanced digestion process for fetal DNA compared with maternal DNA.
Although our method does require a large amount of data for the training phase (we used 298 samples), any additional sample to be tested can be sequenced at relatively low costs compared with other methods as no additional laboratory tests are needed: The algorithm works using low‐coverage data and was tested on single end 51 bp reads. As the method is independent of read length, shorter (or longer) reads can be used as long as reliable mapping is assured. However, although 298 samples is a lot for setting up these tests, we deem this number rather small for our approach. Considering the coverage of the combined samples is still close to zero when only considering read start positions, the nucleosome position detection reliability could be drastically improved by increasing the amount of training data. Additionally, the correlation found between the read frequency anywhere in the nucleosome profile and the fetal fraction as obtained from chromosome Y could be improved by adding more data with their respective fetal fractions. Currently, there is no golden standard for determining the fetal fraction during NIPT analysis. We compared our method to methods based on the presence of chromosome Y in male pregnancies and to the size‐based method using paired‐end sequencing. Our results demonstrate that our new method is as reliable as the other two methods, but has the benefit that it works independent of fetal sex, and that no expensive paired‐end sequencing is needed.
Conclusion
We have developed a reliable method to create a genome‐wide nucleosome profile from cfDNA and a method to determine the fetal fraction for any sample without requiring a change in workflow or cost per sample using current NIPT procedures.
What's Already Known About This Topic?
Fetal DNA is found in small and varying amounts in maternal blood, enough to detect fetal aberrations such as Down syndrome through next generation sequencing methods.
Fetal DNA is generally shorter, and this is believed to be influenced by nucleosomes.
What Does This Study Add?
We obtained nucleosome positions from cell‐free DNA over a combination of low‐coverage samples.
Our method shows how fetal fragments are influenced by nucleosomes compared with maternal fragments.
We deduced the fetal fraction using the distribution of reads starting around nucleosome positions, independent of the fetal sex.
Supporting information
Supporting info item
Straver, R. , Oudejans, C. B. M. , Sistermans, E. A. , and Reinders, M. J. T. (2016) Calculating the fetal fraction for noninvasive prenatal testing based on genome‐wide nucleosome profiles. Prenat Diagn, 36: 614–621. doi: 10.1002/pd.4816.
Funding sources: None
Conflicts of interest: None declared
References
- 1. Tabor A, Philip J, Madsen M, et al. Randomised controlled trial of genetic amniocentesis in 4606 low‐risk women. Lancet 1986;327(8493):1287–93. [DOI] [PubMed] [Google Scholar]
- 2. Harris RA, Washington AE, Nease RF, Kuppermann M. Cost utility of prenatal diagnosis and the risk‐based threshold. Lancet 2004;363(9405):276–82. [DOI] [PubMed] [Google Scholar]
- 3. Tabor A, Vestergaard CHF, Lidegaard Ø. Fetal loss rate after chorionic villus sampling and amniocentesis: an 11‐year national registry study. Ultrasound Obstet Gynecol 2009;34(1):19–24. [DOI] [PubMed] [Google Scholar]
- 4. Chiu RWK, Chan KCA, Gao Y, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci 2008;105(51):20458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lo YMD, Chan KCA, Sun H, et al. Maternal plasma DNA sequencing reveals the genome‐wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2(61):61ra91. [DOI] [PubMed] [Google Scholar]
- 6. Chiu RW, Sun H, Akolekar R, et al. Maternal plasma DNA analysis with massively parallel sequencing by ligation for noninvasive prenatal diagnosis of trisomy 21. Clin Chem 2009;56(3):459–63. [DOI] [PubMed] [Google Scholar]
- 7. Chiu RWK, Akolekar R, Zheng YWL, et al. Non‐invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ 2011;342(jan11 1):c7401–c7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen EZ, Chiu RWK, Sun H, et al. Noninvasive of fetal trisomy 18 and trisomy 13 by maternal plasma DNA sequencing. PLoS One 2011;6(7):e21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ehrich M, Deciu C, Zwiefelhofer T, et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol 2011;204(3):205.e1-11. [DOI] [PubMed] [Google Scholar]
- 10. Palomaki GE, Kloza EM, Lambert‐Messerlian GM, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med 2011;13(11):913–20. [DOI] [PubMed] [Google Scholar]
- 11. Fan HC, Blumenfeld YJ, Chitkara U, et al. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci 2008;105(42):16266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song Y, Huang S, Zhou X, et al. Non‐invasive prenatal testing for fetal aneuploidies in the first trimester of pregnancy. Ultrasound Obstet Gynecol 2015;45(1):55–60. [DOI] [PubMed] [Google Scholar]
- 13. Poon LL, Leung TN, Lau TK, et al. Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin Chem 2002;48(1):35–41. [PubMed] [Google Scholar]
- 14. Dar P, Curnow KJ, Gross SJ, et al. Clinical experience and follow‐up with large scale single‐nucleotide polymorphism–based noninvasive prenatal aneuploidy testing. Am J Obstet Gynecol 2014;211(5):527–e1. [DOI] [PubMed] [Google Scholar]
- 15. Jiang P, Chan KCA, Liao GJW, et al. FetalQuant: deducing fractional fetal DNA concentration from massively parallel sequencing of DNA in maternal plasma. Bioinformatics 2012;28(22):2883–90. [DOI] [PubMed] [Google Scholar]
- 16. Chan KA. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem 2004;50(1):88–92. [DOI] [PubMed] [Google Scholar]
- 17. Yu SCY, Chan KCA, Zheng YWL, et al. Size‐based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc Natl Acad Sci 2014;111(23):8583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cutter AR, Hayes JJ. A brief review of nucleosome structure. FEBS Lett 2015;589(20 Pt A):2914–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Straver R, Sistermans EA, Holstege H, et al. WISECONDOR: detection of fetal aberrations from shallow sequencing maternal plasma based on a within‐sample comparison scheme. Nucleic Acids Res 2013;42(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaffney DJ, Mcvicker G, Pai AA, et al. Controls of nucleosome positioning in the human genome. PLoS Genet 2012;8(11):e1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pedersen JS, Valen E, Velazquez AMV, et al. Genome‐wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res 2013;24(3):454–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schones DE, Cui K, Cuddapah S, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008;132(5):887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valouev A, Johnson SM, Boyd SD, et al. Determinants of nucleosome organization in primary human cells. Nature 2011;474(7352):516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaplan N, Moore IK, Fondufe‐Mittendorf Y, et al. The DNA‐encoded nucleosome organization of a eukaryotic genome. Nature 2008;458(7236):362–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item