

Abstract
Eukaryotic translation initiation factor 4E (eIF4E) is considered as the corner stone in the cap-dependent translation initiation machinery. Its role is to recruit mRNA to the ribosome through recognition of the 5′-terminal mRNA cap structure (m7GpppN, where G is guanosine, N is any nucleotide). eIF4E is implicated in cell transformation, tumourigenesis, and angiogenesis by facilitating translation of oncogenic mRNAs; it is thus regarded as an attractive anticancer drug target. We have used two approaches to design cap-binding inhibitors of eIF4E by modifying the N7-substituent of m7GMP and replacing the phosphate group with isosteres such as squaramides, sulfonamides, and tetrazoles, as well as by structure-based virtual screening aimed at identifying non-nucleotide cap-binding antagonists. Phosphomimetic nucleotide derivatives and highly ranking virtual hits were evaluated in a series of in vitro and cell-based assays to identify the first non-nucleotide eIF4E cap-binding inhibitor with activities in cell-based assays, N-[(5,6-dihydro-6-oxo-1,3-dioxolo[4,5-g]quinolin-7-yl)methyl]-N′-(2-methyl-propyl)-N-(phenyl-methyl)thiourea (14), including down-regulation of oncogenic proteins and suppression of RNA incorporation into polysomes. Although we did not observe cellular activity with any of our modified m7GMP phosphate isostere compounds, we obtained X-ray crystallography structures of three such compounds in complex with eIF4E, 5′-deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)amino-N7-methyl-guanosine (4a), N7-3-chlorobenzyl-5′-deoxy-5′-(1,2-dioxo-3-hydroxy-cyclobut-3-en-4-yl)amino-guanosine (4f), and N7-benzyl-5′-deoxy-5′-(trifluoromethyl-sulfamoyl)guanosine (7a). Collectively, the data we present on structure-based design of eIF4E cap-binding inhibitors should facilitate the optimisation of such compounds as potential anticancer agents.
Keywords: Cancer, eIF4E, Protein synthesis, mRNA translation, Cap-binding inhibitor
Abbreviations: eIF, eukaryotic translation initiation factor; 4E-BP, eF4E-binding protein; FP, fluorescence polarisation; m7G, N7-methyl guanosine; RRL, rabbit reticulocyte lysate
Graphical abstract

1. Introduction
In eukaryotes, most mRNAs are translated using a cap-dependent mechanism, which consists of three stages: initiation, elongation, and termination. The initiation step requires a number of eukaryotic translation initiation factors (eIFs) [1]. Initially, the 40S ribosomal subunit associates with Met-tRNAi and a group of initiation factors, including eIF1, eIF1A, eIF2, eIF3, and eIF5 to form the 43S pre-initiation complex [2]. This complex is then recruited to the 5′-end of capped mRNA by another complex of factors (known as eIF4F) consisting of eIF4E, eIF4G, and eIF4A. eIF4E is an indispensable element for cap-dependent translation initiation and plays a major role in recognition of the mRNA cap structure (m7GpppN, where N is any nucleotide). It binds to eIF4G, which serves as a scaffolding protein to gather eIF4A (ATP-dependent RNA helicase), eIF4B, eIF3, and poly-A-binding protein to form the 48S complex [3], [4]. This complex then scans the mRNA for an initiation codon and, once this is located, the 60S ribosomal subunit joins to form the elongation-competent fully functional 80S.
Under typical circumstances, eIF4E is the least abundant of the cellular translation initiation components [5] and it represents the rate-limiting element for the initiation process [6], [7]. Under resting conditions, eIF4E is kept in an inactive form bound to 4E-binding proteins (4E-BPs); once the latter have been phosphorylated through the PI3K-AKT-mTOR signalling pathway, eIF4E is released and can associate with eIF4G to launch translation.
eIF4E is overexpressed in numerous human tumours, and it contributes to transformation, tumourigenesis, and progression of cancers [8], [9]. eIF4E overexpression facilitates translation of weak and highly structured mRNAs that typically encode proteins involved in cancer pathology, such as proto-oncoproteins (e.g. cyclin D1, ornithine decarboxylase), angiogenesis factors (e.g. FGF-2, VEGF), and factors related to tumour invasiveness, such as MMP-9 [10], due to the ability of eIF4E to stimulate eIF4A [11]. For these reasons, eIF4E represents an attractive cancer drug target [12]. Moreover, a recent study suggests that targeting eIF4E for cancer treatment has minimal effects on growth of –and protein synthesis in– healthy cells [13].
Strategies to design eIF4E–mRNA cap-binding antagonists have been based on nucleoside (purine N7-substitutions) [14], [15], [16], [17] and nucleotide (altered phosphate groups) [17], [18], [19] modifications of 7-methylguanosine (m7G) nucleotides (Fig. 1) [12], including recent work affording for the first time m7G monophosphate (m7GMP) nucleotide mimetic compounds with high affinity for eIF4E [20]. However, to date no cell-permeable small-molecule eIF4E cap-binding antagonists have been reported. As with antiviral agents derived from nucleotides, prodrugs that mask the ionic nature of phosphate groups, such as phosphate esters of phosphoramidates, may offer an avenue for the design of cell-permeable eIF4E inhibitors, but this strategy has not progressed very far as yet [21].
Fig. 1.

(a) mRNA cap recognition (represented by m7GTP shown as yellow CPK sticks) and binding of eIF4G or 4E-BPs (represented by a peptide derived from 4E-BP1 shown as a cyan cartoon) occurs on opposite faces of eIF4E (green cartoon). Apart from direct cap-binding antagonists, allosteric inhibitors (binding pose of 4-EGI1 [22] shown as magenta sticks) and inhibitors derived from eIF4G and 4E-BPs [23] are being developed. Whereas m7GTP-binding is dominated by polar interactions between the cationic N-methylpurine system and eIF4E residues W56, W102, and E103 (cation–π interaction and H-bonds), as well as the phosphate groups with residues R157, K159, and K162, (b) GMP derivatives with N7-substituents other than methyl, such as 4-fluorobenzyl [24] (green) or (4-chlorophenoxy)ethyl [20] (cyan), also make hydrophobic contacts with two concave lipophilic pockets (surface representations) behind the W56—W102 stack. Figure constructed from PDB entries 2V8W, 2V8Y, 4DT6, and 4TPW. 3D-Structure illustrations in this and subsequent Figures were prepared using MacPyMOL (The PyMOL Molecular Graphics System, Version 1.2, Schrödinger, LLC). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Here we report on new methods to design cap antagonists and we show a group of nucleotide mimetic compounds with phosphate group isosteres and various purine N7-substituents. The biological activities of these compounds were assessed using a range of techniques and the eIF4E-binding modes of three compounds were determined experimentally using X-ray crystallography. However, despite phosphate group modifications, these nucleotide mimetics still exhibit poor cellular bioactivity. Therefore, a computer-aided drug design method was exploited to design a set of non-nucleotide compounds to provide the first small-molecule eIF4E inhibitor possessing cellular activity consistent with blocking of eIF4E-mediated initiation of translation.
2. Results and discussion
2.1. Design and synthesis of nucleotide monophosphate mimetic eIF4E inhibitors
One of the main challenges in the design of inhibitors of eIF4E cap binding has been to achieve membrane permeability and thus cellular bioavailability [20]. This is due to the fact that the affinity of eIF4E for the RNA m7GpppN cap structure derives in large part from polar interactions between the ligand triphosphate group and the receptor protein (Fig. 1a) [15], [25]. In order to address the permeability problem, as well as the intrinsic hydrolytic and enzymatic lability of nucleotides, we aimed to design nucleoside monophosphate mimetics [25], [26]. Several phosphate group replacements were investigated, such as squaramides, sulfonamides, and tetrazoles. Since e.g. m7GMP has much lower affinity for eIF4E than m7GTP [25], we expected to compensate the loss in affinity with nucleoside monophosphate mimetics through optimal purine N7-substituent modifications, based on previous studies demonstrating that replacing the N7-methyl group with bulkier groups contributed significantly to ligand affinity (Fig. 1b) [27], [28].
Squaric acid possesses similar charge distribution, polarity, and acidic properties as phosphoric acid, hence the squaramide group could represent an isostere for the phosphate group [29], [30]. Similarly, sulfonamide derivatives have been reported as phosphate mimics in the design of tyrosine phosphatase inhibitors [31]. The third candidate we considered for phosphate group replacement was a tetrazole [32], as this system mimics the acidic features of the phosphate group, provided the proton attached to one of the tetrazole ring nitrogens is not replaced [33].
The key synthetic precursor for the preparation of our nucleotide cap mimetics was the protected guanosine derivative 1e (Scheme 1) [34], which was prepared by first blocking the 2′,3′-vicinal glycol of guanosine as the acetonide 1a [35], followed by dimethoxytritylation of the exocyclic amine group (1b) [36], and transformation of the 5′-hydroxyl group to the amine via successive conversion to the iodide 1c and azide 1d, and reduction of the latter [37]. The amine 1e was condensed either with dimethyl squarate [29] to afford the squaramide derivative 2 or with polar sulfonyl chlorides to furnish the sulfonamide derivatives 5. These intermediates were N7-alkylated using a range of alkyl or aryl bromides [14] to provide the methyl squaramides 3 and sulfonamides 6. The methyl squaramide groups of the former were first demethylated with sodium iodide in acetone [38], followed by treatment with aqueous formic acid to afford the squaramides 4. The acetonide group of precursors 6 was removed similarly with formic acid to provide the sulfonamides 7 [39]. The tetrazole derivatives 11 were also elaborated from 1e, by Boc-protection of the 5′-amino group (8), followed by N7-alkylation (9), global deprotection (10), and acylation of the 5′-amino group with 1H-tetrazole-5-acetic acid.
Scheme 1.

Preparation of nucleotide mimetics 4a–4i, 7a–7c, and 11a–11ba. a Conditions: (i) DMTr-Cl, Me3SiCl; (ii) (PhO)3P+MeI−, THF, −78 °C; (iii) NaN3, DMF, 85 °C; (iv) Ph3P, pyridine, NH4OH; (v) dimethyl squarate, iPr2NEt, MeOH; (vi) R-X, DMF; (vii) NaI, Me2CO, Δ, then 80% aq HCOOH, 24 h; (viii) RSO2Cl, Et3N, CH2Cl2; (ix) 80% aq HCOOH, 12 h; (x) Boc2O, Et3N, CH2Cl2, 24 h; (xi) 5-tetrazole acetic acid, DIC, Et3N, DMF.
2.2. Design of non-nucleotide eIF4E inhibitors
As an alternative to nucleoside monophosphate mimetics we considered structure-based design of eIF4E cap-binding antagonists. For this purpose we docked multi-conformer libraries of lead-like molecules from the ZINC [40] database to a model of the Bn7GMP-binding site of eIF4E derived from a complex crystal structure we reported earlier (PDB entry 2V8Y) [15] using shape-based fast rigid exhaustive docking (FRED) [41], [42]. We applied a combination of scoring functions, including functions based on Poisson-Boltzmann electrostatic potentials, in order to ensure that interactions of virtual hits mimicking the cation–π interaction [43], [44] of the N7-methylguanine system in the mRNA cap with the eIF4E W56—W102 indole side chains (Fig. 1a) would be rewarded. The virtual screening protocol was validated by docking and scoring a set of 500 randomly-chosen diverse decoy molecules from the ZINC database spiked with a set of N7-alkylated cap-derived nucleotides known to bind eIF4E, as well their non-alkylated counterparts, which are known not to interact with eIF4E [25]. This analysis revealed that the N7-alkylated cap-derived nucleotides populated the top 12% of the hit list from the combined scoring protocol. Based on this result we proceeded to screen the entire lead-like subset of the ZINC database. We acquired several hundred of the top-ranking virtual hits and submitted these to the mass spectrometry-based assay described earlier [45], as well as the fluorescence polarisation (FP) assay described below. The chemical structure and predicted properties of the most highly scored virtual hits are shown in Table 1.
Table 1.
Chemical structures and predicted properties of virtual hits 12–16 from structure-based design.
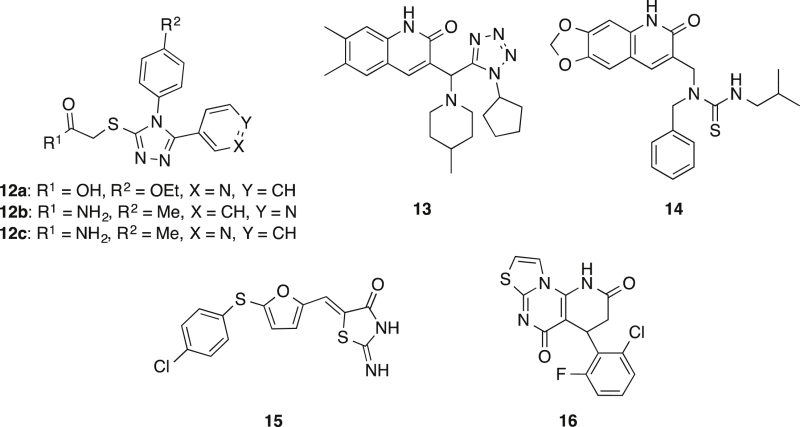 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | Source | CASa | Mr | Predicted propertiesb |
||||||
| logP | logDc | TPSAd (Å2) | nRote | logSif | Drug-likeg | Lead-likeh | ||||
| 12a | Asinex BAS04026402 |
557064-00-3 | 356.4 | 0.9 | −1.1 | 90 | 7 | −4.7 | yes | yes |
| 12b | Asinex BAS02054521 |
338425-77-7 | 325.4 | 1.2 | 1.8 | 87 | 5 | −4.9 | yes | yes |
| 12c | Asinex BAS02563297 |
333412-47-8 | 325.4 | 1.2 | 1.8 | 87 | 5 | −4.9 | yes | yes |
| 13 | Asinex ASN03777735 |
459445-48-8 | 420.6 | 4.3 | 4.3 | 76 | 4 | −5.9 | yes | no |
| 14 | Asinex ASN03775814 |
461002-66-4 | 423.5 | 4.0 | 4.0 | 63 | 6 | −7.1 | yes | no |
| 15 | ChemBridge 6373744 |
426248-94-4 | 336.8 | 3.8 | 3.8 | 66 | 3 | −6.9 | yes | yes |
| 16 | ChemBridge 8893829 |
899375-48-5 | 349.8 | 2.3 | 2.3 | 62 | 1 | −4.5 | yes | yes |
Chemical Abstracts Registry Number.
Using ChemAxon InstantJChem version 15.6.8.0.
Predicted logD at pH = 7.4.
Topological polar surface area.
Number of non-terminal rotatable bonds.
Log10 of predicted molar intrinsic aqueous solubility.
Criteria: nRot < 10 and TPSA < 140 Å2[46].
Criteria: Mr ≤ 450, −4 ≤ logD7.4 ≤ 4, number of rings ≤ 4, nRot ≤ 10, number of H-bond donors ≤ 5, number of H-bond acceptors ≤ 8 [47].
2.3. Biological activity of nucleotide monophosphate mimetic and non-nucleotide eIF4E inhibitors
In order to assess our m7GMP-derived compounds and virtual screening hits as mRNA cap-binding antagonists, we developed an FP assay (Fig. 2) based on displacement of a fluorescently labelled m7GDP derivative from eIF4E by cap-binding inhibitors. Assay optimisation showed that a tracer concentration of 10 nM and an eIF4E concentration of 90 nM was optimal. All test compounds were screened initially at 100 μM and actives were titrated to calculate Kd values, summarised in Table 2.
Fig. 2.

(a) Recombinant eIF4E or bovine serum albumin (BSA) was titrated against the FP probe ATTO-465-labelled 2′/3′-O-(2-aminoethylcarbamoyl)-7-methyl-guanosine-5′-diphosphate (EDA-m7GDP-ATTO-465, structure shown inset; 10 nM) and a Kd value with respect to eIF4E binding was calculated. (b) The FP assay is specific for the unique structural feature of the mRNA cap and can distinguish between m7GTP and GTP (eIF4E concentration: 90 nM).
Table 2.
Summary of biological activities of compounds 4–16.a
| Cmpd | FP assay |
RNA cross-linking assay |
Reticulocyte lysate assay |
35S-Met incorporation assay |
|||
|---|---|---|---|---|---|---|---|
| Inhibition (%)b | Kd (μM) | Inhibition (%)b | IC50 (μM) | IC50 (μM) | IC50 (HeLa) | IC50 (MCF-7) | |
| m7GTP | 97.3 ± 2.4c | 0.37 ± 0.03 | 92.8 ± 1.6c | 0.72 ± 0.14 | 8.6 ± 1.1 | Inactive | Inactive |
| m7GMP | 86.3 ± 5.0 | 3.9 ± 0.5 | 97.5 ± 0.8 | 1.7 ± 0.1 | 46.9 ± 3.4 | Inactive | Inactive |
| 4a | 14.4 ± 3.2 | n.d.d | Inactive | n.d. | n.d. | n.d. | n.d. |
| 4b | 9.1 ± 5.5 | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 4c | 15.2 ± 4.7 | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 4d | 2.0 ± 6.3 | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 4e | Inactive | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 4f | 13.2 ± 4.2 | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 4g | 3.9 ± 5.2 | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 4h | 1.8 ± 7.7 | n.d. | 6.5 ± 7.1 | n.d. | n.d. | n.d. | n.d. |
| 4i | Inactive | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 7a | 89.0 ± 1.7 | 56.5 ± 6.9 | 42.9 ± 4.8 | 12.6 ± 0.5 | 65.9 ± 13.6 | Inactive | Inactive |
| 7b | 49.4 ± 6.3 | 133 ± 28 | 48.6 ± 1.3 | 18.7 ± 5.0 | 165 ± 32 | Inactive | Inactive |
| 7c | Inactive | n.d. | 22.2 ± 1.8 | n.d. | n.d. | n.d. | n.d. |
| 11a | 9.7 ± 2.9 | n.d. | 25.8 ± 2.9 | n.d. | n.d. | n.d. | n.d. |
| 11b | 30.2 ± 2.8 | n.d. | 15.3 ± 4.8 | n.d. | n.d. | n.d. | n.d. |
| 12a | 13.6 ± 2.2 | n.d. | 10.2 ± 3.3 | n.d. | n.d. | n.d. | n.d. |
| 12b | 25.8 ± 5.9 | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 12c | 8.5 ± 2.8 | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 13 | Inactive | n.d. | Inactive | n.d. | n.d. | n.d. | n.d. |
| 14 | 68.1 ± 0.8 | 57.8 ± 13.5 | 47.5 ± 6.7 | 38.6 ± 10.6 | 65.1 ± 12.5 | 35.2 ± 4.0 | 37.7 ± 10.4 |
| 15 | Inactive | n.d. | 3.5 ± 3.2 | n.d. | n.d. | n.d. | n.d. |
| 16 | Inactive | n.d. | 33.8 ± 8.1 | n.d. | n.d. | n.d. | n.d. |
All experiments were carried out on three independent occasions.
At 100 μM concentration unless otherwise indicated.
At 10 μM concentration.
Not determined.
As expected, m7GTP had almost 10-fold higher affinity for eIF4E in our FP assay than m7GMP. Although the majority of the squaramide (4), sulfonamide (7), and tetrazole (11) phosphate-mimetic analogues inhibited tracer binding in the FP assay at the prescreen concentration of 100 μM to some extent, only the sulfonamides 7a and 7b showed levels of inhibition that permitted construction of a sufficiently wide dose–response titration for accurate Kd determination. Of the squaramides, all derivatives except 4a, 4c, and 4f showed insignificant inhibition (<10%) in the prescreen. Surprisingly, in the context of the squaramide phosphomimetic group, the expected enhancement of affinity upon replacing the N7-methyl group (4a) with a benzyl group (4b) was not observed. Furthermore, substitution of the phenyl group with small and electronically neutral groups (3-Me, 4c; 3-Cl, 4f) retained the modest activity of 4a and 4b, whereas introduction of larger and more polar substituents reduced or abolished activity. The trifluoromethyl- and the para-carboxyphenyl sulfonamides 7a and 7b were the only nucleoside monophosphate mimetic compounds for which Kd values could be obtained, and these are significantly higher than that of m7GMP. The tetrazole derivatives 11 had very modest activity. Here the N7-benzyl derivative 11b was somewhat more active than the N7-methyl compound 11a.
Amongst the virtual screening hits, a cluster of 4,5-bisaryl-2-carboxymethylthio-1,2,3-triazoles 12a–c was identified but these showed low activity in the FP prescreen and Kd values could not be determined. Of the somewhat structurally related quinolinones 13 and 14, the former was essentially without activity in the FP assay, whereas the latter was clearly active, at a level similar to the best of the nucleotide derivatives (7b). The thiazolone 15 and the pyridothiazolopyrimidine 16 showed no activity in the FP assay.
Further to confirm inhibition of eIF4E cap binding, a radiometric competition binding assay was developed based on an in vitro UV cross-linking technique widely used to study protein–RNA interactions [48]. In this assay, a radiolabelled and capped RNA was incubated with eIF4E in the presence or absence of putative inhibitors 4–16. The RNA–eIF4E complexes formed were cross-linked, separated by SDS-PAGE, and quantitated by autoradiography. The assay was validated with positive controls (m7GMP and m7GTP), test compounds were pre-screened at 100 μM (Table 2), and for sufficiently active compounds half-maximal inhibitory concentration (IC50) values were determined (Fig. 3).
Fig. 3.

Dose–response curves for m7GMP, m7GTP, as well as compounds 7a, 7b, and 14 in the radiolabelled and capped RNA (5 nM) – eIF4E (1.5 μM) UV cross-linking assay. Inhibitor concentrations are plotted against autoradiography band intensities following separation of the cross-linked RNA–eIF4E complexes.
As expected, m7GMP and m7GTP displayed similar activity levels in the UV cross-linking assay as in the FP assays. The squaramides 4 were inactive with the exception of the derivative 4h. All three sulfonamides 7 showed activity in the UV cross-linking assay and for 7a and 7b IC50 values could be determine (Fig. 3). For the inhibitors from the virtual screen (11–16), the UV cross-linking assay results mirrored those from the FP assay but an IC50 value could only be determined for quinolinone 14.
For the nucleoside monophosphate mimetic compounds, which we expected to be devoid of cellular bioactivity, we also sought to obtain a functional read-out of eIF4E inhibition. For this purpose we employed a cell-free rabbit reticulocyte lysate (RRL) system to observe the translational consequences of eIF4E inhibition [49]. Test compounds m7GMP, m7GTP, as well as the sulfonamides 7a and 7b were incubated in the RRL system, which was primed with luciferase RNA, and expression of luciferase was then quantitated. From the results (Table 2) it can be observed that all four test compounds inhibited luciferase synthesis. For m7GMP and m7GTP the IC50 values in the RRL assay were significantly higher than those determined for competitive binding, perhaps due to limited stability of these nucleotides in RRL, whereas the drop-off in activity was much less pronounced for sulfonamides 7a and 7b. RRLs were then primed with bicistronic mRNAs where the initiation of translation of the upstream Renilla cistron occurs in a cap-dependent manner, while the translation of the downstream firefly luciferase cistron is driven by the HCV internal ribosome entry segment and is therefore independent of the cap structure at the 5′ end of the mRNA (Fig. 4). This bicistronic RNA allowed the comparison of the effect of quinonlinone 14 and the para-carboxyphenyl sulfonamides 7a and 7b on cap-dependent and cap-independent translation simultaneously. The data show that while, as expected, there was inhibition of synthesis from the cap-dependent Renillia luciferase mRNA, there was no effect on the translation initiation of the firefly luciferase mediated by the HCV-IRES, suggesting that sulfonamides 7a and 7b and quinolinone 14 inhibit cap-dependent translation (Fig. 4).
Fig. 4.

Compounds 7a, 7b, and 14 inhibit cap-dependent translation, but not cap-independent translation driven by the HCV IRES: (a) Schematic of the construct containing the HCV IRES. Cap-dependent translation drives expression from the Renilla luciferase transcript, whereas cap-independent translation drives protein synthesis from the Firefly luciferase transcript. (b) Rabbit reticulocyte lysates were primed with pRHCVF RNA and treated with compound 14 (i) or 7a or 7b (ii) at the doses shown. The compounds were dissolved in DMSO, which is known to inhibit the reticulocyte lysate system when compared to the control without DMSO. Therefore a 1% solution of DMSO was used as an additional control and the degree of inhibition measured relative to this value. Experiments were performed on three independent occasions, significance (***P < 0.001) was calculated using Student's t-test (error bars S.D.). Black bars denote Renilla luciferase and grey bars Firefly luciferase.
In order to investigate the effect of the eIF4E inhibitors on overall protein synthesis in cells, we employed 35S-Met labelling [50] to measure the amount of newly synthesised total protein in HeLa cells after incubation for 24 h with test compounds (the protein synthesis inhibitor puromycin was used as a positive control). None of the nucleoside phosphates (m7GMP and m7GTP) or phosphate mimetics (7a and 7b) showed measurable inhibition of cellular protein synthesis, indicating that membrane permeability of the nucleoside phosphate mimetic compounds, including the sulfonamides 7a and 7b, is still sub-optimal. Quinolinone 14, on the other hand, which was the only virtual screening hit with activity in the UV cross-linking assay, also inhibited protein synthesis in both HeLa and MCF-7 cells with IC50 values similar to the activity levels observed in cell-free assays, suggesting that this compound is cell-permeable. Based on the predicted physicochemical properties of this compound (Table 2), this finding was not unexpected.
For quinolinone 14, which was observed to inhibit cellular protein synthesis, we investigated if this effect was indeed due to suppression of translation, as would be expected of an eIF4E cap-binding inhibitor. For this purpose we used polysome analysis [51]. HeLa cells were treated for 2 h with test compound 14 or puromycin as a positive control, harvested, treated with cycloheximide in order to trap RNA–ribosome complexes, and cell lysates were fractionated (Fig. 5a, i–iv). For cells treated with quinolinone 14, there was a decrease of the amount of RNA incorporated in multiple ribosomes (polysomes) compared to untreated cells and a concomitant increase in the sub-polysomal material, consistent with a block in initiation (Fig. 5a and b), supporting our data which shows 14 to suppress cap-dependent translation in cells.
Fig. 5.

(ai and aii) Cells, either control (grey line) or treated with 25 mM compound 14 (pink line) were lysed and post-nuclear extracts were applied to 10–50% sucrose gradients to allow the separation of actively translating ribosomes (polysomes) from subpolysomal material (e.g. 40S and 60S ribosomal subunits). (aiii) Analysis of the areas under the curves attributable to the polysomes and subpolysomes in the control (ai) and treated (aii) samples showed a decrease in the amount of polysomes in treated cells and a concomitant increase in the subpolysomal fractions. (aiv) To examine whether there was a decrease in the amount of c-myc mRNA associated with the polysomes following treatment with compound 14, RT-qPCR was performed on individual fractions. The data show that there is a shift of c-myc mRNA to fractions that contain fewer polysomes indicative of translational repression of this mRNA. (b) To examine the effect of higher doses of compound 14 on polysome formation, cells were treated with 50 μM (pink line) and 100 μM (red line) and gradients repeated. The data show a greater reduction in the amount of polysomal material under these conditions. (c) Western blot analyses of HeLa lysates treated for 2 h with vehicle only (DMSO) or with compound 14 (20, 50 and 100 μM). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To examine whether there was an effect on the expression of proteins which are known to be eIF4E dependent and are associated with tumorigenesis, lysates of HeLa cells treated for 2 h with quinolinone 14 were probed by western blotting for c-myc and cyclin D1 protein, with actin used as a loading control. The data show that at 50 and 100 μM treatment concentrations of 14 there were decreases in levels of both proteins (Fig. 5c). To confirm that the decrease in protein levels corresponded with altered polysomal association, mRNAs were isolated from polysomal and sub-polysomal regions of the gradients and RT-PCR was used to assess the relative amount of c-myc mRNA before and after treatment. The data show that following treatment with quinolinone 14 there was a small shift of the c-myc mRNA into the sub-polysomes, consistent with a decrease in its translation (Fig. 5 aiv).
2.4. X-ray crystallography
eIF4E was co-crystallised with either an eIF4G- or a 4E-BP-derived peptide (designated 4G and 4EBP, respectively) and one of three of the m7GMP-derived analogues (squaramides 4a and 4f, sulfonamide 7a). All complex structures showed the characteristic cupped hand structure of eIF4E [52] with the ligands occupying the cap-binding site.
The initial eIF4E:4G:7a complex structure (Fig. 6b and c) was solved to a resolution of 1.71 Å (Table 3) with the ligand 7a residing in the cap-binding site, accompanied by a single sulfate ion (from the crystallisation buffer). The guanine of 7a makes three H-bonds, two with the side chain of E103 and one with the backbone carbonyl of W102. The sulfate ion interacts directly with eIF4E through R157 and makes additional water-bridged interactions with R112, N155, R157, and K162, as well as to the sulfonamide group of 7a. The ligand 7a itself interacts with four neighbouring or bridging waters. The unusual binding mode observed was unpredicted as it was expected that the guanine would make the same or similar cation-π stacking interaction as seen with all other cap-based analogues to date (compare Fig. 6a) [12], [15], [20]. However, the presence of the sulfate ion allows 7a to ‘fold′ back on itself and the positively charged guanine forms a stabilising intramolecular electrostatic interaction with the electronegative sulfonamide group. In order to accommodate this ligand conformation, W102 is reoriented out of the binding pocket from its normal cation-π interaction position. W56 remains in the usual position, providing a stacking interaction with the guanine. In comparison to other complex structures with cap derivatives (Fig. 6a) the sulfate ion occupies the same position as the α-phosphate. Presumably the unusual bound conformation of 7a observed was favoured due to high sulfate concentration in the crystallisation buffer.
Fig. 6.

(a) eIF4E binding pose of Bn7GMP (PDB code 2V8X). (b) Binding pose of sulfonamide 7a, accompanied by a sulfate ion in eIF4E (crystallographic water molecules are indicated by red spheres). (c) Electron density (red mesh, 0.53 electrons/Å3) contoured at 1.5 Å around ligand 7a and sulfate ion. (d) Binding pose of 7a following back-soaking (see text). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 3.
Crystallographic data.
| eIF4E:4G:7a (PDB 5EI3) | eIF4E:4G:7a back-soak (PDB 5EIR) | eIF4E:4EBP:4a (PDB 5EKV) | eIF4E:4G:4f PDB 5EHC) | |
|---|---|---|---|---|
| Data collection | ||||
| Resolution (Å) | 1.71 | 2.69 | 3.61 | 2.40 |
| Space group | P21221 | P21221 | P212121 | P21221 |
| Unit cell (Å) | ||||
| a | 38.6 | 38.3 | 38.5 | 38.5 |
| b | 52.1 | 52.2 | 100.8 | 52.2 |
| c | 125.2 | 123.7 | 136.5 | 125.7 |
| Unique reflectionsa | 28165 (4039) | 6814 (854) | 6413 (889) | 10425 (1466) |
| Completeness (%)a | 100 (100) | 93.8 (85.8) | 97.8 (95.5) | 99.7 (99.7) |
| Rmerge (%)a | 9.4 (34.2) | 16.0 (35.9) | 28.2 (42.3) | 19.2 (33.9) |
| I/σa | 15.7 (5.3) | 5.5 (3.0) | 4.6 (3.1) | 8.9 (5.3) |
| Refinement | ||||
| R (%) | 16.6 | 24.8 | 26.8 | 21.8 |
| Rfree (%) | 18.7 | 27.2 | 34.1 | 26.4 |
| Mean temperature factor B (Å2)b | ||||
| eIF4E | 14.1 | 24.7 | 17.1/20.8e | 19.4 |
| 4G/4EBP | 14.0 | 28.1 | 17.5/18.8e | 19.2 |
| Ligand | 22.1 | 38.5 (36.9)d | 18.4/24.1e | 42.4 |
| SO4 | 29.5 | (46.1)d | – | – |
| Occupancy (%)b | ||||
| Ligand | 1.0 | 0.52 (0.48)d | 0.88/0.74e | 1.0 |
| SO4 | 1.0 | 0.48 | – | – |
| All atoms used in refinement | 2103 | 1790 | 3265 | 1846 |
| Water | 355 | 32 | 0 | 139 |
| Validationc (% of all residues) | ||||
| Favoured | 98.0 | 99.0 | 97.8 | 97.9 |
| Allowed | 100.0 | 100.0 | 100.0 | 100.0 |
| Disallowed | 0.0 | 0.0 | 0.0 | 0.0 |
Values in parentheses refer to the highest-resolution shell.
B-factor and occupancy refinement were performed using Phenix.
Validation was performed using MolProbity.
Values in parenthesis refer to the lowest occupancy model.
Values for both copies in the asymmetric unit.
In order to test this hypothesis, a crystal from these conditions was transferred to an identical crystallisation drop but lacking ammonium sulfate. The structure from this crystal was solved to 2.69 Å (Table 3) and shows that there is mixed occupancy for the ligand. The occupancies for both conformations are almost equal (52:48) with the higher occupancy conformation being one where the trifluoromethylsulfonamide group occupies the α-phosphate binding position (Fig. 6d), previously occupied by sulfate. As expected, the displacement of the sulfate was accompanied by repositioning of W102 to form the usual cation-π ‘sandwich′ interaction with the positively charged guanine moiety and W56.
The squaramide complex structures eIF4E:4EBP:4a and eIF4E:4f were solved to 3.61 Å and 2.40 Å, respectively (Table 3). Despite low overall resolution, the eIF4E co-crystal structure with ligand 4a clearly shows that the squaramide moiety acts as a phosphate mimic by making a similar interaction with R157 despite an altered conformation (Fig. 7a). The positively charged N7-methyl guanine system forms the characteristic cation-π interaction as expected. Fig. 7b shows compound 4f bound to the cap-binding site of eIF4E. The guanine group is located in its normal binding position, stacking with the side-chain of W56. As predicted, the squaramide occupies the α-phosphate position (Fig. 7c), making interactions with the side-chain of R157. The chloro substituent of the N7-benzyl group causes a rotation of approximately 90° in relation to other N7-benzyl substituted cap analogues (Fig. 7c). In turn, to accommodate the extra space required by the chloro group, the side chain of W102 is re-oriented away from making the usual cation-π stacking interaction to a position similar to that seen with 7a in the presence of sulfate (compare Fig. 6, Fig. 7b).
Fig. 7.

X-ray crystallographic eIF4E-binding poses of compound 4a (a) and 4f (b). (a) Alignment of experimental binding poses of Bn7GMP (PDB code 2V8X; green), 4a (magenta), and 4f (cyan). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We were unable to obtain complex crystal structures with unambiguous ligand density of the eIF4E inhibitors 12–16 from the virtual screen. However, it is interesting to compare the predicted binding modes of these compounds with the experimental eIF4E complexes of the m7GMP-derived derivatives (Fig. 8). Re-docking of Bn7GMP to the receptor structure of the Bn7GMP complex using the protocol used for virtual screening showed (Fig. 8a) virtually identical positioning of the N7-benzylguanine portion of the ligand, whereas the phosphate group in the re-docked pose bound more deeply into the cavity behind W56 and making polar interactions with the side chains of R157, W56, and D90, compared to the experimental binding pose, in which the phosphate group only interacts with R157. As a result of the different positioning of the phosphate group, the ribose systems in the experimental and modelled poses are also positioned somewhat differently.
Fig. 8.

Predicted eIF4E binding poses of compounds 12–16. (a) eIF4E binding pose of Bn7GMP as determined crystallographically (PDB code 2V8X; cyan), compared with re-docked pose of Bn7GMP (green) using the same docking protocol as in the virtual screen. Docked poses of virtual hits (b) 12a (green), 12b (grey), and 12c (magenta); (c) 13; (d) 14; (e) 15); (f) 16. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The top-ranking docking solutions for the 4,5-bisaryl-2-carboxymethylthio-1,2,3-triazoles 12a–c all showed similar poses, with the substituted phenyl groups stacking between W56 and W102, the unsubstituted aryl group inserting into the cavity behind W102, and the carboxymethyl substituents occupying the polar pocket behind W56 (Fig. 8b). Quinolinones 13 and 14 are predicted to stack between W56 and W102 in a similar manner, disposing the remaining substituents into the subpockets behind the W residues as shown (Fig. 8c and d). Thiazolone 15 and the pyridothiazolopyrimidine 16 again show stacked aromatic systems involving W56 and W102 but these ligands do not show good interactions with the other subpockets of the cap-binding site.
3. Conclusions
In the present study we report a new set of nucleotide-based cap analogues with phosphate modifications. Crystal structures of sulfonamide 7a and squaramides 4a and 4f in complex with eIF4E were obtained and show similar binding modes to cap derivatives co-crystallised with eIF4E previously. However, our structures highlight the flexibility of the m7G-binding pocket, especially residue W102 that forms part of the cation-π interaction characteristic of cap-recognition by eIF4E. This finding suggests that cap-binding inhibitors of eIF4E devoid of a permanent cationic structure may be possible. All of the confirmed eIF4E inhibitors (12a, 14, 16) from our alternative design strategy based on virtual screening of lead-like non-nucleotide compounds support this suggestion. We present likely eIF4E-binding modes for these compounds but the possibility that these induce conformational changes in the cap-binding pocket of eIF4E cannot be excluded.
As has been found elsewhere [20], [25], imparting sufficient membrane permeability in eIF4E cap-binding inhibitors for such compounds effectively to block cellular eIF4E function is difficult. Despite modulation of the ionic and polar nature of the phosphate group in m7GMP derivatives through replacement with phosphate isosteres, none of our nucleoside monophosphate mimetic compounds showed cellular activity. The situation was different with the non-nucleotidic eIF4E inhibitors identified through virtual screening. These compounds possess physicochemical properties (Table 1) consistent with good permeability and we show that one of these compounds, the quinolinone 14, not only inhibited eIF4E cap-binding in a biochemical and cell-free functional assay, but also acted as a protein synthesis inhibitor at the level of RNA translation in cellular assays. Although compound 14 is not particularly lead-like due to excessive ring count and lipophilicity, as well as low intrinsic solubility (Table 1), our results confirm for the first time that cellular eIF4E can be targeted with non-nucleotidic compounds and inherently drug-like compounds.
4. Materials and methods
4.1. Chemistry
Flash chromatography was carried out using silica gel cartridges (Sorbsil® and ISOLUTE®). TLC was performed using Merck silica gel 60 F254 plates and visualisation by UV irradiation at 254 nm or by staining with KMnO4 or ninhydrin solutions, as appropriate. 1H- 13C-, 19F-, and 31P NMR spectra were obtained at room temperature using a Bruker 400 Ultrashield spectrophotometer at 400.13 MHz, 100.61 MHz, 376.50 MHz, and 161.97 MHz, respectively. Samples were prepared in the deuterated solvents Me2SO-d6 (unless otherwise indicated). Chemical shifts (δ) are expressed in ppm relative to SiMe4. Mass spectra were obtained using a Waters 2759 spectrometer using both positive and negative electrospray ionisation. Infrared spectra were recorded on a Nicolet IR-200 FT-IR spectrophotometer using the KBr disc technique. Analytical RP-HPLC was performed with a Shimadzu UFLCXR system coupled to a mass spectrometer (Applied Biosystems API2000). Phenomenex columns thermostated at 40 °C were used: C8-110A (method 1); Luna 3u (PFP2) 110A, (method 2), and C18 (method 3); all with dimensions 50 × 2 mm. The flow rate was 0.5 mL/min and UV detection was at 220 and 254 nm. Gradient elution: pre-equilibration for 1 min with 10% solvent B in solvent A, 10–98% solvent B over 2 min, 98% B for 2 min, 98 to 10% B over 0.5 min, and finally 10% B for 1 min (solvent A: 0.1% HCOOH in H2O, solvent B: 0.1% HCOOH in MeCN). Preparative RP-HPLC was carried out using a Waters automated LC system, with Waters 2525 pump, Waters 2767 sample manager, and Waters 2487 dual wavelength absorbance detector system or a Dionex ICS-3000 HPLC system with an Ultimate-3000 detector, using Phenomenex Luna PFP or HILIC (5 μM) columns (250 mm × 10 mm). The purity of all compounds screened in the biological assays was examined by LC−MS analysis and was found to be ≥ 95%.
4.2. N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethylidene)guanosine (1b)
2′,3′-O-(1-Methyl-ethylidene)guanosine 1a (mp 202–204 °C, lit [53] > 200 °C; 5 g, 15.5 mmol) was co-evaporated with pyridine (3 × 30 mL) and dissolved in dry pyridine (100 mL) under N2. Me3SiCl (9.8 mL, 77.4 mmol) was added over 5 min by syringe. The solution was stirred for 3 h, when DMTrtCl (6.3 g, 18.5 mmol) was added. Stirring (under N2) was continued for 21 h. The reaction was quenched with NH4OH (30 mL of a 35% solution in H2O). The mixture was stirred for 2 h and evaporated. The residue was dissolved in EtOAc (100 mL) and the solution was washed with H2O, brine, and aq NaHCO3 (50 mL each). The organic layer was evaporated and the residue was chromatographed (SiO2; CH2Cl2—MeOH, 9:1) to afford the title compound as a white solid (76%). Mp 179–181 °C. 1H NMR: δ 10.66 (bs,1H, NH), 7.82 (s, 1H), 7.58 (s, 1H) 7.17–7.29 (m, 9H), 6.84–6.86 (m, 4H), 5.38 (d, J = 3.9 Hz, 1H), 4.76 (dd, J1 = 6.2 Hz, J2 = 3.9 Hz, 1H), 4.39 (dd, J1 = 6.2 Hz, J2 = 2.4 Hz, 1H), 3.96 (td, J1 = 6 Hz, J2 = 2.4 Hz, 1H), 3.71 (s, 6H), 3.27–3.35 (m, 2H), 1.35 (s, 3H), 1.20 (s, 3H). 13C NMR: δ 157.67, 156.39, 150.94, 149.29, 145.10, 136.93, 136.85, 136.61, 129.72, 129.61, 128.24, 127.61, 126.47, 117.39, 112.92, 88.86, 85.14, 81.78, 80.94, 69.48, 61.39, 54.94, 27.09, 25.45. IR (KBr): ν 3342, 1685, 1607, 1568, 1508 cm−1. MS (ESI+): m/z [M+H]+ calcd for C34H36N5O7, 626.3; found: 626.1.
4.3. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-iodo-2′,3′-O-(1-methylethylidene)guanosine (1c)
A stirred solution of 1b (1 g, 1.6 mmol) in anhydrous THF (100 mL) was cooled to −78 °C. (PhO)3P+MeI− (1.45 g, 3.2 mmol) was added and the solution was allowed to reach r.t. over 30 min. THF was evaporated and the residue was diluted with EtOAc (50 mL). This solution was washed with Na2S2O3 (3 × 30 mL of a 25% solution in H2O). The organic phase was treated with MgSO4, filtered, and evaporated. The residue was dissolved in EtOAc (15 mL) and precipitated with hexane (100 mL). The precipitate was chromatographed (SiO2; CH2Cl2—MeOH, 9.2:0.8). The product was again precipitated from EtOAc–hexane to afford the title compound as a white solid (70%). Mp 148–150 °C. 1H NMR: δ 10.76 (s, 1H, NH), 7.81 (s, 1H), 7.57 (s, 1H) 7.11–7.35 (m, 9H), 6.87–6.89 (d, J = 10 Hz, 4H), 5.53 (d, J = 4.2 Hz, 1H), 4.66 (dd, J1 = 4.2 Hz, J2 = 6.2 Hz, 1H), 4.18 (dd, J1 = 6.2 Hz, J2 = 2.4 Hz, 1H), 4.04 (td, J1 = 6.2 Hz, J2 = 2.4 Hz, 1H), 3.75 (s, 6H), 2.77–2.91 (m, 2H), 1.40 (s, 3H), 1.21 (s, 3H). 13C NMR: δ 157.81, 156.34, 151.09, 149.06, 144.95, 137.49, 136.85, 136.64, 129.59, 129.51, 128.19, 127.76, 126.70, 118.00, 113.44, 113.05, 113.03, 89.49, 83.72, 82.78, 80.45, 69.41, 55.00, 27.09, 25.52, 5.75. IR (KBr): ν 3344, 1688, 1606, 1567, 1508 cm−1. MS (ESI+): m/z [M+H]+ calcd for C34H35IN5O6, 736.16; found, 736.14.
4.4. 5′-Azido-5′-deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethylidene)guanosine (1d)
To a stirred solution of 1c (1.95 g, 2.65 mmol) in dry DMF (50 mL) NaN3 (490 mg, 7.5 mmol) was added under N2 and the reaction was heated to 85 °C for 24 h [CAUTION! Use of inorganic azide: reaction carried out behind a safety shield]. After cooling, the solution was evaporated and the residue was treated with H2O (40 mL). The mixture was stirred for 30 min. The precipitate was filtered and washed with H2O (2 × 30 mL) and Et2O (2 × 30 mL) and was then dried to afford the title compound as a white solid (77%). Mp 136–138 °C. 1H NMR: δ 10.76 (bs, 1H, NH), 7.81 (s, 1H), 7.57 (s, 1H), 7.13–7.35 (m, 9H), 6.86–6.90 (m, 4H), 5.53 (d, J = 3.9 Hz, 1H), 4.72 (dd, J1 = 6.2 Hz, J2 = 3.9 Hz, 1H), 4.17 (dd, J1 = 6.2 Hz, J2 = 2.4 Hz, 1H), 4.04 (m, 1H), 3.75 (s, 6H), 3.00–3.08 (m, 2H), 1.38 (s, 3H), 1.21 (s, 3H). 13C NMR: δ 157.80, 156.36, 151.12, 149.08, 144.97, 137.42, 136.84, 136.78, 129.58, 129.52, 128.20, 127.77, 126.65, 117.92, 113.34, 113.04, 89.04, 82.63, 81.14, 80.59, 69.40, 54.98, 51.36, 27.09, 25.53. IR (KBr): ν 3353, 2102, 1686, 1606, 1568, 1508 cm−1. MS (EMI+): m/z [M+H]+ calcd for C34H35N8O6, 651.27; found, 651.24.
4.5. 5′-Amino-5′-deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethylidene)guanosine (1e)
To a stirred solution of 1d (1 g, 1.5 mmol) in dry pyridine (15 mL) Ph3P (840 mg, 3.1 mmol) was added under N2 and the reaction was stirred for 3 h. The mixture was cooled to 0 °C and concentrated. NH4OH (2.8 mL of 35% solution in H2O) was added. Stirring was continued for 20 h, when the mixture was evaporated. The residue was chromatographed (Si2O; CHCl3—MeOH—Et3N, 9:1:0.1) to afford the title compound as a white solid (48%). Mp 203–205 °C. 1H NMR: δ 10.76 (bs, 1H, NH), 7.81 (s, 1H), 7.57 (s, 1H) 7.14–7.30 (m, 9H), 6.86 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz, 4H), 5.45 (d, J = 3.9 Hz, 1H), 4.76 (dd, J1 = 6.4 Hz, J2 = 3.9 Hz, 1H), 4.23 (dd, J1 = 6.4 Hz, J2 = 3.0 Hz, 1H), 3.87–3.95 (m, 1H), 3.75 (s, 6H), 2.59–2.67 (m, 2H), 1.37 (s, 3H), 1.20 (s, 3H). 13C NMR: δ 157.71, 156.35, 151.15, 149.17, 144.99, 137.19, 137.02, 129.62, 128.23, 127.68, 126.55, 117.66, 113.40, 112.97, 88.51, 83.40, 81.17, 81.10, 69.46, 55.00, 42.30, 27.11, 25.62. IR (KBr): ν 3374, 1683, 1606, 1570, 1508 cm−1. MS (ESI+): m/z [M+H]+ calcd for C34H37N6O6, 625.28; found, 625.26.
4.6. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-2′, 3′-O-(1-methylethylidene)guanosine (2)
To a stirred solution of 1e (300 mg, 0.48 mmol) in MeOH (25 mL) iPr2NEt (40 μL, 1.92 mmol) was added. After 5 min, dimethyl squarate (68 mg, 0.48 mmol) was added to the reaction mixture, which was stirred for 4 h. It was then evaporated and the residue was chromatographed (Si2O; CHCl3—MeOH, 9:1) to afford the title compound as a white solid (61%). Mp 170–172 °C. 1H NMR: δ 10.77 (s, 1H), 8.5–8.7 (m, 1H), 7.74 (s, 1H), 7.60 (s, 1H) 7.11–7.33 (m, 9H), 6.88 (d, J = 8.4 Hz, 4H), 5.49 (dd, J1 = 14.4 Hz, J2 = 4 Hz, 1H), 4.61 (m, J = 3.6 Hz, 1H), 4.21–4.28 (m, 4H), 3.94 (m, J = 2.4 Hz, 1H), 3.68 (s, 6H), 2.80–3.26 (m, 1H), 1.38 (s, 3H), 1.21 (s, 3H). 1H NMR (75 °C): δ 10.61 (s, 1H), 8.41 (s, 1H), 7.68 (s, 1H), 7.42 (s, 1H), 7.16–7.31 (m, 9H), 6.83–6.88 (m, 4H), 5.46 (d, 1H, J = 3.6 Hz), 4.8 (dd, 1H, J1 = 6.3 Hz, J2 = 3.6 Hz), 4.33 (dd, 1H, J1 = 6.3 Hz, J2 = 3.1 Hz), 4.24 (s, 3H), 4.00 (dd, 1H, J1 = 6.3 Hz, J2 = 3.1 Hz), 3.72 (s, 6H), 3.03 (s, 2H), 1.38 (s, 3H), 1.21 (s, 3H). 13C NMR: δ 189.25, 189.04, 182.53, 177.62, 177.16, 172.29, 171.93, 157.78, 156.37, 151.04, 149.00, 145.01, 137.83, 136.95, 129.63, 129.50, 128.19, 127.82, 126.64, 118.07, 113.21, 113.06, 89.11, 88.95, 82.97, 82.58, 81.09, 80.93, 80.36, 80.19, 69.34, 60.10, 59.96, 54,97, 45.80, 45.14,27.15, 25.66. IR (KBr): ν 3412, 2935, 1702, 1608, 1508 cm−1. MS (ESI+): m/z [M+H]+ calcd for C39H39N6O9, 735.28; found, 735.24.
4.7. General method 1: N7-Alkylation of 5′-amino-5′-deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethylidene)guanosine derivatives 2, 5, and 8
To a solution of compound 2, 5a, 5b, or 8 (1 eq) in DMF, the appropriate alkyl or aryl halide (10 eq) was added and the mixture was stirred for 24–48 h, with monitoring by TLC (CHCl3—MeOH, 17:3) and LC-MS. After completion of reaction, the mixture was evaporated in vacuo and the residue was triturated with Et2O with the aid of sonication. The precipitate was filtered and chromatographed (SiO2; CHCl3—MeOH, 9:1) to afford the pure title compound.
4.8. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-N7-methyl-2′,3′-O-(1-methylethylidene)guanosine (3a)
Using general method 1 from 2 and methyl bromide. White solid (57%). Mp 183–185 °C. 1H NMR: δ 9.88 (s, 1H), 8.78 (m, 1H), 8.29 (m, 1H), 7.66–7.88 (m, 10H), 7.27–7.34 (m, 4H), 6.1 (m, 1H), 5.53 (m, 1H), 5.1 (m, 1H), 4.82 (m, 4H), 4.58 (s, 3H), 4.19–4.22 (m, 2H), 4.20 (s, 6H), 1.89 (s, 3H), 1.64 (s, 3H). 13C NMR: δ 188.88, 188.58, 182,81, 177.31, 172.32, 171.82, 157.94, 157,85, 153.04, 147.17, 144.30, 138.09, 136.16, 129.69, 129.60, 128.22, 127.92, 126.90, 116.95, 113.16, 108.60, 91.65, 86.12, 85.77, 85.05, 80.94, 69.80, 60.27, 55.04, 45,64, 35.69, 26.74, 25.36. MS (ESI+): m/z [M]+ calcd for C40H41N6O9, 749.29; found, 749.26.
4.9. N7-Benzyl-5′-deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-2′,3′-O-(1-methylethylidene)guanosine (3b)
Using general method 1 from 2 and benzyl bromide. White solid (47%). Mp 182–185 °C. 1H NMR: δ 11.60 (s, 1H), 9.47 (s, 1H), 8.55–8.80 (m, 1H, NH), 8.14 (s, 1H), 7.14–7.42 (m, 14H), 6.82–6.85 (m, 4H), 5.6 (bs, 1H), 5.53 (m, 2H), 4.76–4.9 (m, 1H), 4.12–4.44 (m, 5H), 3.68 (s, 6H), 3.07 (m, 2H), 1.38 (s, 3H), 1.15 (s, 3H). 13C NMR: δ 189.29, 189.11, 182.69, 182.36, 177.91, 177.37, 172.26, 171.78, 157.80, 154.06, 147.70, 144.52, 137.37, 136.24, 134.10, 129.76, 129.67, 128.73, 128.68, 128.31, 127.79, 126.73, 116.91, 113.03, 107.80, 92.15, 86.32, 85.39, 81.04, 69.68, 60.25, 60.10, 54.97, 51.42, 45.40, 44.97, 26.79, 25.42. MS (ESI+): m/z [M]+ calcd for C46H45N6O9, 825.32; found, 825.29.
4.10. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-N7-(3-methylbenzyl)-2′,3′-O-(1-methylethylidene)guanosine (3c)
Using general method 1 from 2 and 3-methylbenzyl bromide. Off-white solid (67%). 1H NMR: δ 9.44 (s, 1H), 8.54–8.84 (m, 1H, NH), 7.10–7.33 (m, 14H), 6.79–6.88 (m, 4H), 5.64 (m, 1H), 5.31–5.48 (m, 2H), 4.79–4.94 (m, 1H), 4.12–4.41 (m, 5H), 3.66 (s, 6H), 2.93–3.28 (m, 2H), 2.26 (s, 3H), 1.38 (s, 3H), 1.15 (s, 3H). 13C NMR: δ 189.29, 189.08, 182.63, 182.32, 177.91, 177.33, 172.24, 171.77, 157.76, 154.96, 154.69, 147.72, 144.62, 137.95, 137.23, 136.40, 133.97, 129.80, 129.72, 129.41, 128.63, 127.75, 126.66, 125.83, 112.99, 107.74, 92.18, 86.29, 85.29, 81.73, 69.69, 60.52, 60.10, 54.96, 51.30, 45.29, 44.95, 26.78, 25.46, 20.94. MS (ESI+): m/z [M]+ calcd for C47H47N6O9, 839.3; found, 839.3.
4.11. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-N7-(3-methoxybenzyl)-2′,3′-O-(1-methylethylidene)guanosine (3d)
Using general method 1 from 2 and 3-methoxybenzyl bromide. Off-white solid (54%). 1H NMR: δ 9.40 (s, 1H), 8.54–8.82 (m, 1H, NH), 6.79–7.32 (m, 18H), 5.6 (m, 1H), 5.39–5.49 (m, 2H), 4.72–4.94 (m, 1H), 4.09–4.31 (m, 5H), 3.72 (s, 3H), 3.67 (s, 6H), 2.90–3.25 (m, 2H), 1.38 (s, 3H), 1.15 (s, 3H). 13C NMR: δ 189.39, 189.05, 182.60, 182.28, 177.89, 177.38, 172.15, 171.85, 159.34, 157.72, 147.65, 144.70, 137.09, 136.50, 135.48, 129.90, 129.73, 128.30, 127.71, 126.62, 120.74, 118.43, 114.70, 113.9, 112.95, 107.85, 92.14, 86.73, 81.37, 79.16, 69.52, 60.19, 60.05, 55.08, 54.92, 51.16, 45.27, 44.97 26.74, 25.40. MS (ESI+): m/z [M]+ calcd for C47H47N6O10, 855.33; found, 855.32.
4.12. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-2′, 3′-O-(1-methylethylidene)-N7-(3-(trifluoromethyl)benzyl)guanosine (3e)
Using general method 1 from 2 and 3-trifluoromethylbenzyl bromide. White solid (76%). 1H NMR: δ 9.43 (s, 1H), 8.52–8.82 (m, 1H, NH), 7.82–7.92 (m, 18H), 7.66–7.77 (m, 2H), 7.65–7.75 (m, 1H) 7.08–7.35 (m, 9H) 6.76–6.90 (m, 4H), 5.42–5.69 (m, 3H), 4.81–4.99 (m, 1H), 4.15–4.45 (m, 5H), 3.67 (s, 6H), 2.89–3.26 (m, 2H), 1.38 (s, 3H), 1.13 (s, 3H). 13C NMR: δ 189.32, 189.06, 182.61, 182.29, 178.75, 177.33, 172.30, 172.22, 157.71, 147.82, 144.75, 136.51, 135.42, 133.04, 129.81, 129.50, 128.73, 127.69, 126.59, 125.76, 125.56, 125.32, 124.82, 122.62, 118.12, 112.93, 107.75, 92.31, 86.66, 85.46 81.09, 69.57, 60.08, 54.93, 50.70, 44.92, 26.72, 25.40. 19F NMR (DMSO-d6): δ −60.98. IR (KBr): ν 3422, 2934, 1804, 1708, 1608, 1508 cm−1. MS (ESI+): m/z [M]+ calcd for C47H44F9N6O9, 893.3; found, 893.2.
4.13. N7-(3-Chlorobenzyl)-5′-deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxy-cyclo-but-3-en-4-yl)amino-2′,3′-O-(1-methylethylidene)guanosine (3f)
Using general method 1 from 2 and 3-chlorobenzyl bromide. Off-white solid (63%). Mp 202–204 °C. 1H NMR: δ 9.39 (s, 1H), 8.50–8.89 (m, 1H, NH), 7.10–7.62 (m, 14H), 6.74–6.93 (m, 4H), 5.63 (s, 1 H), 5.38–5.56 (m, 2H), 4.60–5.00 (m, 1H), 4.08–4.55 (m, 5H), 3.68 (s, 6H), 2.93–3.28 (m, 2H), 2.26 (s, 3H), 1.39 (s, 3H), 1.15 (s, 3H). 13C NMR: δ 189.32, 189.08, 182.64, 182.29, 177.94, 172.23, 171.81, 157.62, 147.87, 136.72, 136.45, 133.14, 130.49, 129.80, 129.75, 128.75, 128.46, 127.60, 126.46, 112.84, 107.65, 92.27, 90.43, 86,53 81.07, 69.48, 60.11, 54.92, 50.38, 45.65, 26.78, 25.46. IR (KBr): ν 3424, 2933, 1803, 1709, 1606, 1508 cm−1. MS (ESI+): m/z [M]+ calcd for C46H44ClN6O9, 859.3; found, 859.2.
4.14. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-N7-(4-fluorobenzyl)-2′,3′-O-(1-methylethylidene)guanosine (3g)
Using general method 1 from 2 and 4-fluorobenzyl bromide. White solid (47%). Mp: 200–202 °C. 1H NMR: δ 11.87 (s, 1H), 9.37 (s, 1H), 8.44–8.85 (m, 1H, NH), 7.46–7.55 (m, 3H) 7.08–7.36 (m, 11H) 6.75–6.95 (m, 4H), 5.61 (s, 1H), 5.48 (m, 2H), 4.73–4.90 (m, 1H) 4.12–4.45 (m, 5H), 3.68 (s, 6H), 2.90–3.26 (m, 2H), 1.38 (s, 3H), 1.15 (s, 3H). 13C NMR: δ 189.47, 189.10, 182.58, 182.31, 178.49, 172.03, 161.01, 157.76, 154.56, 147.72, 144.90, 138.19, 137.38, 136.47, 131.14, 130.37, 129.73, 129.65, 128.29, 127.76,126.66, 115.66, 115.46, 113.00, 107.75, 92.13, 86.55, 81.01, 69.60, 60.10, 54.94, 50.81, 45.43, 26.76, 25.40. 19F NMR: δ −113.08. MS (ESI+): m/z [M]+ calcd for C46H44FN6O9, 843.3; found, 843.2.
4.15. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-N7-(4-isopropylbenzyl)-2′,3′-O-(1-methylethylidene)guanosine 3h
Using general method 1 from 2 and 4-isopropylbenzyl bromide. White solid (46%). Mp 184–186 °C. 1H NMR: δ 9.41 (s, 1H), 8.54–8.79 (m, 1H, NH), 7.11–7.38 (m, 14H), 6.75–6.91 (m, 4H), 5.63 (s, 1H), 5.32–5.52 (m, 2H), 4.73–4.91 (m, 1H), 4.09–4.41 (m, 5H), 3.67 (s, 6H), 2.86 (sp, J = 6.8 Hz, 1H), 2.26 (s, 3H), 1.38 (s, 3H), 1.1–1.0 (m, 10H). 13C NMR: δ 189.32, 189.13, 182.85, 177.96, 77.32, 172.24, 171.83, 157.78, 154.28, 149.09, 147.71, 147.70, 145.17, 136.62, 136.25, 131.57, 129.75, 128.82, 128.31, 127.78, 126.64, 113.01, 107.79, 92.15, 90.6, 86.25, 81.44, 81.08, 69.59, 59.75, 54.98, 51.12, 45.29, 33.16, 26.78, 25.43, 23.73. MS (ESI+): m/z [M]+ calcd for C49H51N6O9, 867.4; found, 867.3.
4.16. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-5′-(1,2-dioxo-3-methoxycyclobut-3-en-4-yl)amino-N7-(3,5-(di-tert-butyl)benzyl)-2′,3′-O-(1-methylethylidene)guanosine (3i)
Using general method 1 from 2 to 3,5-(di-tert-butyl)benzyl bromide. White solid (48%). Mp 210–212 °C (dec). 1H NMR: δ 9.51 (s, 1H), 8.56–8.82 (m, 1H, NH), 8.33 (m, 1H), 7.11–7.45 (m, 13H), 6.75–6.90 (m, 4H), 5.58 (m, 1H), 5.37–5.49 (m, 2H), 4.76–4.92 (m, 1H), 4.02–4.52 (m, 5H), 3.66 (s, 6H), 2.87–3.27 (m, 2H), 1.38 (s, 3H), 1.24 (s, 18H), 1.10 (s, 3H). 13C NMR: δ 189.08, 182.59, 182.34, 177.92, 177.34, 172.24, 171.82, 157.73, 150.94, 147.74, 144.69, 136.39, 133.41, 129.84, 129.74, 128.40, 127.68, 126.63, 123.58, 122.58, 112.91, 107.91, 92.51, 86.82, 86.00, 81.65, 81.07, 69.67, 60.13, 54.91, 51.77, 45.23, 44.73, 34.55, 31.12, 26.70, 25.32. IR (KBr): ν 3434, 2958, 1802, 1708, 1607, 1508 cm−1. MS (ESI+): m/z [M]+ calcd for C54H61N6O9, 937.4; found, 937.4.
4.17. General method 2: deprotection of 3a–3i
To a stirred solution of 3 (1 eq) in Me2CO, NaI (2 eq) was added and the mixture was heated under reflux for 48 h. After completion of the reaction, the solvent was evaporated and the residue was treated with HCOOH (1 mL/0.1 mmol of 80% aq soln) for 12 h. After cooling, the mixture was evaporated and the residue was triturated with Me2CO with the aid of sonication. The precipitate was filtered, washed successively with Me2CO and hexane, and was dried.
4.18. 5′-Deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)amino-N7-methyl-guanosine (4a)
Using general method 2 from 3a. White solid (38%). Mp 192–194 °C. 1H NMR: δ 9.48 (s, 1H), 8.11 (br s, 1H), 7.14–7.19 (m, 1H), 5.73 (d, J = 6.3 Hz, 1H), 5.51 (br s, 2H), 4.76 (t, J = 6.3 Hz, 1H), 4.13–4.22 (m, 2H), 4.02 (s, 3H), 3.98 (dd, J1 = 4.8 Hz, J2 = 2.6 Hz, 1H), 3.46–3.52 (m, 1H). 13C NMR: δ 197.95, 188.93, 180.62, 157.19, 154.06, 148.95, 137.79, 108.39, 90.36, 85.76, 71.59, 70.56, 44.67, 35.60. IR (KBr): ν 3367, 1794, 1709, 1674, 1619, 1600, 1549, 1519 cm−1. Anal. RP-HPLC: tR 0.77 min (method 1, >95%). MS (ESI+): m/z [M]+ calcd for C15H17N6O7, 393.12; found, 393.11.
4.19. N7-Benzyl-5′-deoxy-5′-(1,2-dioxo-3-hydroxy-cyclobut-3-en-4-yl)amino-guanosine (4b)
Using general method 2 from 3b. White solid (35%). Mp 186–188 °C. 1H NMR: δ 11.70 (s, 1H), 9.75 (s, 1H), 7.52–7.57 (m, 2H), 7.36–7.43 (m, 3H), 7.11–7.18 (m, 1H), 5.76 (d, J = 6.0 Hz, 1H), 5.63 (s, 1H), 4.74 (d, J1 = 10.8 Hz, J2 = 5.8 Hz, 1H), 4.12–4.26 (m, 2H), 3.98 (dd, J1 = 10.8 Hz, J2 = 4.2 Hz, 1H), 3.48–3.53 (m, 1H). 13C NMR: δ 197.88, 189.00, 180.63, 156.74, 154.46, 149.28, 137.58, 134.48, 128.76, 128.63, 128.54, 107.58, 90.65, 85.66, 71.73, 70.60, 51.43, 44.72. IR (KBr): ν 3394, 1791, 1706, 1632, 1606, 1532 cm−1. Anal. RP-HPLC: tR 2.33 min (method 1, >95%), tR 1.51 min (method 2, >94%). MS (ESI+): m/z [M]+ calcd for C21H21N6O7, 469.15; found, 469.14.
4.20. 5′-Deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)amino-N7-3-methylbenzyl-guano-sine (4c)
Using general method 2 from 3c. White solid (37%). Mp 187–190 °C. 1H NMR: δ 11.75 (br s, 1H), 9.80 (s, 1H), 7.28–7.35 (m, 2H), 7.15–7.22 (m, 1H) 7.06–7.15 (m, 1H), 5.77 (d, J = 5.9 Hz, 1H), 5.57 (s, 2H), 4.73 (t, J = 5.9 Hz, 1H), 4.13–4.22 (m, 4H), 3.98 (m, 1H), 3.47–3.58 (m, 1H), 2.30 (s, 3H). 13C NMR: δ 197.70, 188.90, 180.55, 155.79, 153.21, 149.17, 138.21, 137.96, 134.22, 129.44, 129.06, 128.84, 125.62, 107.59, 90.71, 85.61, 71.79, 70.58, 51.49, 44.74, 20.97. Anal. RP-HPLC: tR: 2.52 (method 1, >95%), tR: 1.98 min (method 2, >95%). MS (ESI+): m/z [M]+ calcd for C22H23N6O7, 483.16; found, 483.15.
4.21. 5′-Deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)amino-N7-3-methoxybenzyl-guano-sine (4d)
Using general method 2 from 3d. White solid (36%). Mp 177–179 °C. 1H NMR: δ 11.69 (s, 1H), 9.78 (s, 1H), 7.32 (m, 1H), 7.07–7.14 (m, 3H), 6.95 (m, 1H), 5.77 (d, J = 6.0 Hz, 1H), 5.58 (s, 2H), 5.51 (br s, 2H), 4.73 (t, J = 6.0 Hz, 1H), 4.11–4.21 (m, 2H), 3.98 (m 1H), 3.75 (s, 3H), 3.49–3.55 (m, 1H). 13C NMR: δ 197.50, 188.86, 180.38, 159.49, 155.74, 153.17, 149.17, 138.03, 135.64, 130.13, 120.60, 114.34, 114.18, 107.59, 90.71, 85.64, 71.75, 70.57, 55.17, 51.45, 44.74. Anal. RP-HPLC: tR 2.42 min (method 1, >95%), tR 1.92 min (method 2, >95%). MS (ESI+): m/z [M]+ calcd for C22H23N6O8, 499.16; found, 499.15.
4.22. 5′-Deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)amino-N7-3-(trifluoromethyl)benz-yl-guanosine (4e)
Using general method 2 from 3e. The crude product was purified by preparative HPLC using a Luna HILIC column tR 8.0 min; gradient from 0% A to 100% A over 12.5 min, where A is 5:4:1 MeCN—H2O—50 mM aq NH4HCOO pH 3.2, and B is 9:1 MeCN–50 mM aq NH4HCOO pH 3.2, to afford 4e as a white solid (14.2%). Mp 221–224 °C. 1H NMR: δ 8.21 (s, 1H), 7.97 (s, 1H) 7.49–7.59 (m, 4H), 5.60–5.78 (m, 3H) 4.71 (dd, J1 = 10.4 Hz, J2 = 4.8 Hz, 1H), 4.08–4.27 (m, 2H), 3.98 (dd, J1 = 4.7 Hz, J2 = 3.2 Hz, 1H), 3.48–3.55 (m, 1H). 13C NMR: δ 197.79, 189.28, 181.22, 155.78, 153.24, 149.24, 138.20, 138.08, 135.47, 132.91, 130.07, 129.27, 125.60, 125.38, 107.58, 90.76, 85.54, 71.87, 70.52, 50.93, 44.69. 19F NMR: δ −61.02. Anal. RP-HPLC: tR: 2.67 min (method 1, >95%), tR 2.15 min (method 2, >95%). MS (ESI+): m/z [M]+ calcd for C22H20F3N6O7, 537.13; found, 537.13.
4.23. N7-3-Chlorobenzyl-5′-deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)aminoguanosine (4f)
Using general method 2 from 3f. White solid (44%). Mp 193–195 °C. 1H NMR: δ 11.73 (s, 1H), 9.80 (s, 1H), 7.42–7.66 (m, 4H), 7.13–7.18 (m, 1H) 5.77 (d, J = 6.0 Hz, 1H), 5.61 (s, 2H), 4.71 (t, J = 5.7 Hz, 1H), 4.11–4.21 (m, 2H), 3.98 (m, 1H), 3.52 (s, 3H), 3.51–3.56 (m, 1H). 13C NMR: δ 197.30, 188.80, 180.34, 155.76, 153.16, 149.25, 138.17, 136.54, 133.34, 130.83, 128.78, 128.45, 127.37, 107.55, 90.74, 85.49, 71.94, 70.55, 50.85, 44.76. Anal. RP-HPLC: tR 2.54 min (method 1, >95%), tR 2.02 min (method 2, >94%). MS (ESI+): m/z [M]+ calcd for C21H20ClN6O7, 503.11; found, 503.10.
4.24. 5′-Deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)amino-N7-4-flurobenzyl-guanosine (4g)
Using general method 2 from 3g. White solid (31%). 1H NMR: δ 9.69 (s, 1H), 7.64–7.70 (m, 2H), 7.23–7.26 (m, 4H), 5.74 (d, J = 5.9 Hz, 1H), 5.62 (s, 2H), 4.74 (dd, J1 = 5.9 Hz, J2 = 4.8 Hz, 1H), 4.12–4.25 (m, 2H), 3.98 (dd, J1 = 7.1 Hz, J2 = 3.2 Hz, 1H), 3.75 (s, 3H), 3.46–3.52 (m, 1H). 13C NMR: δ 197.94, 188.97, 180.73, 158.03, 156.09, 149.33, 136.87, 131.22, 131.14, 130.85, 115.83, 115.62, 107.50, 90.54, 85.54, 71.74, 70.61, 50.53, 44.76. 19F NMR: δ −113.16. Anal. RP-HPLC: tR 2.44 min (method 1, >95%), tR 1.90 min (method 2, >95%). MS (ESI+): m/z [M]+ calcd for C21H20FN6O7, 487.14; found, 487.14.
4.25. 5′-Deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)amino-N7-4-isopropylbenzyl-guanosine (4h)
Using general method 2 from 3h. White solid (11%). Mp 167–169 °C. 1H NMR: δ 9.64 (s, 1H), 8.23 (s, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 3H), 5.73 (d, J = 6.3 Hz, 1H), 5.30–5.60 (m, 4H), 4.79 (t, J = 6.3 Hz, 1H), 4.09–4.30 (m, 2H), 3.96 (m, 1H), 3.45–3.51 (m, 1H), 2.87 (sp, J = 6.8 Hz, 1H), 1.17 (d, J = 6.8 Hz, 6H). 13C NMR: δ 197.78, 188.98, 180.52, 155.77, 153.16, 149.10, 147.89, 137.85, 135.17, 128.69, 127.50, 126.83, 126.48, 107.50, 90.71, 85.62, 71.79, 70.55, 51.27, 44.68, 33.17, 33.09, 23.79, 23.73. Anal. RP-HPLC: tR: 2.73 min (method 1, >95%), tR 2.21 min (method 2, >95%). MS (ESI+): m/z [M]+ calcd for C24H27N6O7, 511.19; found, 511.19.
4.26. N7-3,5-(di-tert-butyl)benzyl-5′-deoxy-5′-(1,2-dioxo-3-hydroxycyclobut-3-en-4-yl)amino-guanosine (4i)
Using general method 2 from 3i. White solid (15%). 1H NMR: δ 9.53 (s, 1H), 8.40 (s, 2H), 8.28 (s, 1H), 7.45 (d, J = 1.65 Hz, 2H), 7.37 (m, 1H), 5.73 (d, J = 6.9 Hz, 1H), 5.62 (s, 2H), 4.89 (dd, J1 = 6.9 Hz, J2 = 4.8 Hz, 1H), 4.21 (td, J1 = 8.1 Hz, J2 = 3.2 Hz, 1H), 4.14 (dd, J1 = 4.8 Hz, J2 = 3.2 Hz, 1H), 3.96 (dd, J1 = 4.8 Hz, J2 = 1.8 Hz, 1H), 3.40 (m, 1H), 1.26 (s, 18H). 13C NMR: δ 198.14, 188.89, 180.75, 165.84, 150.93, 150.61, 149.23, 135.27, 134.31, 123.01, 122.20, 107.92, 90.46, 86.06, 70.93, 70.83, 51.30, 44.88, 31.19. Anal. RP-HPLC: tR 3.07 min (method 1, >86%), tR 2.54 min (method 2, >95%). MS (ESI+): m/z [M]+ calcd for C29H37N6O7, 581.27; found, 581.27.
4.27. 5′-Deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethylidene)-5′-(trifluoromethyl-sulfamoyl)guanosine (5a)
To a stirred solution of 1e (200 mg, 0.32 mmol) in dry CH2Cl2 (10 mL), Et3N (128 μL, 1.28 mmol) was added. The mixture was cooled to 0 °C and CF3SO2Cl (33 μL, 0.32 mmol) was added drop-wise via syringe. After stirring for 48 h, the mixture was evaporated and the residue was chromatographed (Si2O; CHCl3—MeOH, 9:1) to afford the title compound as a yellow oil (49%). 1H NMR: δ 10.78 (s, 1H), 9.70 (s, 1H), 7.82 (s, 1H), 7.63 (s, 1H), 7.13–7.34 (m, 9H), 6.88 (d, J = 7.5 Hz, 4H), 5.54 (d, J = 4.0 Hz, 1H), 4.72 (dd, J1 = 6.2 Hz, J2 = 4.0 Hz, 1H), 4.19 (dd, J1 = 6.2 Hz, J2 = 3.1 Hz, 1H), 3.87–3.93 (m, 1H), 3.72 (s, 6H), 2.93–3.09 (m, 2H), 1.39 (s, 3H), 1.21 (s, 3H). 13C NMR: δ 157.87, 156.47, 151.20, 149.22, 145.06, 137.49, 137.08, 136.89, 129.65, 129.60, 128.30, 127.78, 126.66, (q, J = 324 Hz, 124 Hz, 121 Hz, 118 Hz, 115 Hz, CF3), 124.35, 121.15, 117.94, 114.74, 117.89, 113.83, 113.08, 88.58, 82.91, 80.83, 80.81, 69.52, 54.98, 45.11, 27.16, 25.71. 19F NMR: δ −77.46. MS (ESI+): m/z [M+H]+ calcd for C35H36F3N6O8S, 757.2; found, 757.2.
4.28. 5′-(4-Carboxyphenyl)sulfamoyl-5′-deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methyl-ethylidene)guanosine (5b)
To a stirred solution of 6e (300 mg, 0.48 mmol) in CH2Cl2 (25 mL) Et3N was added (254 μL, 1.92 mmol). After cooling to 0 °C, 4-carboxybenzenesulfonyl chloride (105 mg, 0.48 mmol) was added and the mixture was allowed to reach r.t. and was stirred for 24 h. It was evaporated and the residue was chromatographed (Si2O; CH2Cl2—MeOH, 8.5:1.5) to afford the title compound as a white solid (73%). 1H NMR: δ 10.92 (bs, 1 H), 8.08 (dd, J1 = 6.8 Hz, J2 = 1.7 Hz, 2 H), 7.79 (dd, J1 = 6.8 Hz, J2 = 1.7 Hz, 2 H), 7.77 (s, 1 H), 7.76 (s, 1 H) 7.08–7.24 (m, 9 H), 6.78–6.92 (m, 4H), 5.39 (d, J = 4.1 Hz, 1 H), 4.74 (dd, J1 = 6.4 Hz, J2 = 4.1 Hz, 1H), 4.14 (dd, J1 = 6.4 Hz, J2 = 3.1 Hz, 1 H), 3.83 (td, J1 = 6.4 Hz, J2 = 3.1 Hz, 1 H), 3.68 (s, 6H), 2.67–2.74 (m, 2 H), 1.33 (s, 3 H), 1.17 (s, 3 H). 13C NMR: δ 173.09, 157.66, 151.19, 149.11, 144.97, 140.37, 137.16, 136.26, 129.85, 129.53, 128.17, 127.60, 126.25, 124.71, 117.71, 113.59, 112.92, 88.39, 82.69, 80.97, 69.44, 59.82, 44.42, 27.87, 25.58. IR (KBr): ν 3314, 2985, 2674, 2488, 1685, 1605, 1570, 1508 cm−1. MS (ESI+): m/z [M+H]+ calcd for C41H41N6O10 S 809.26 found: 809.23.
4.29. N7-Benzyl-5′-deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethylidene)-5′-(trifluoro-methylsulfamoyl)guanosine (6a)
Using general method 1 from 5a and benzyl bromide. Light yellow solid (53%). 1H NMR: δ 11.42 (br s, 1H), 10.11 (s, 1H), 8.18 (s, 1 H), 7.45–7.53 (m, 2H), 7.15–7.41 (m, 13H), 6.74–6.93 (m, 4H), 5.34–5.51 (m, 3H), 4.99 (dd, J1 = 5.8 Hz, J2 = 1.3 Hz, 1H), 4.65 (d, J = 5.7 Hz, 1H), 4.30–4.39 (m, 1H), 3.70 (s, 6H), 3.03–3.10 (m, 2H), 1.37 (s, 3H), 1.11 (s, 3H). 13C NMR: δ 157.71, 152.92, 147.71, 144.44, 137.76, 136.24, 136.08, 134.33, 129.89, 129.73, 128.88, 128.62, 128.35, 127.60, 126.58, 112.90, 112.84, 111.86, 106.78, 92.42, 89.39, 83.96, 81.86, 69.69, 54.90, 51.53, 47.55, 43.65, 26.66, 24.97. 19F NMR: δ −76.09. IR (KBr): ν 3430, 2933, 1706, 1607, 1509. MS (ESI+): m/z [M]+ calcd for C42H42F3N6O8S, 847.27; found, 847.25.
4.30. 5′-(4-Carboxyphenyl)sulfamoyl-5′-deoxy-N2-(4,4′-dimethoxytrityl)-N7-methyl-2′,3′-O-(1-methylethylidene)guanosine (6b)
Using general method 1 from 5b and methyl iodide. The crude product was purified by SiO2 column chromatography (CHCl3—MeOH, 7.5:2) to furnish pure title compound as a white solid (61%). 1H NMR (MeOH-d4): δ 8.13 (d, J = 8.4 Hz, 2H) 7.85 (d, J = 8.4 Hz, 2 H), 7.16–7.38 (m, 9 H), 6.77–6.87 (m, 4H), 5.60 (d, J = 1.2 Hz,1 H), 5.09 (dd, J1 = 6 Hz, J2 = 2 Hz, 1 H), 4.33 (dd, J1 = 6 Hz, J2 = 1.7 Hz, 1 H), 4.26 (td, J1 = 6 Hz, J2 = 1.7 Hz, 1 H), 4.07 (s, 3 H) 3.74 (s, 6 H), 2.76–2.90 (m, 2H), 1.41 (s, 3 H), 1.11 (s, 3 H). 13C NMR (MeOH-d4): δ 160.08, 153.04, 149.21, 143.94, 139.37, 137.58, 131.27, 131.22, 129.88, 129.33, 128.99, 128.18, 127.78, 114.87, 114.23, 113.99, 113.02, 95.08, 88.21, 84.35, 83.41, 72.10, 65.31, 55.74, 36.65, 27.13, 25.82. MS (ESI+): m/z [M+2H]+ calcd for C42H45N6O10S, 823.3; found, 823.2.
4.31. N7-Benzyl-5′-(4-carboxyphenyl)sulfamoyl-5′-deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethylidene)guanosine (6c)
Using general method 1 from 5c and benzyl bromide. The crude product was purified by SiO2 column chromatography (CHCl3—MeOH, 8:2) to furnish pure title compound as a white solid (58%). 1H NMR (MeOH-d4): δ 9.16 (s,1H), 8.14 (d, J = 8.5 Hz, 2 H), 7.80 (d, J = 8.5 Hz, 2H), 7.49–7.54 (m, 2 H), 7.35–7.39 (m, 3 H), 7.16–7.36 (m, 9 H), 6.74–6.83 (m, 4H), 5.54–5.68 (m, 3H), 5.04 (dd, J1 = 6.1 Hz, J2 = 1.7 Hz, 1 H), 4.27 (dd, J1 = 6.1 Hz, J2 = 2.0 Hz, 1H), 4.17–4.23 (m, 1 H), 3.73 (s, 6 H), 2.75–2.86 (m, 2H), 1.39 (s, 3H), 1.10 (s, 3H). 13C NMR (MeOH-d4): δ 160.08, 154.91, 149.52, 145.95, 143.85, 137.61, 135.27, 131.22, 131.19, 129.87, 129.51, 129.01, 128.20, 127.78, 114.98, 114.25, 114.17, 109.76, 95.04, 88.06, 84.17, 82.94, 72.10, 55.73, 53.74, 43.81, 27.14, 25.87. MS (ESI+): m/z [M]+ calcd for C48H47N6O10S, 899.3; found, 899.2.
4.32. General method 3: deprotection of 6a–6c and 9a–9b
A solution of 6 or 9 in HCOOH (1 mL/0.1 mmol of 80% aq solution) was stirred for 12 h. The mixture was evaporated and the residue was triturated with Me2CO with the aid of sonication. The precipitate was filtered, washed successively with Me2CO and hexane, and was dried.
4.33. N7-Benzyl-5′-deoxy-5′-(trifluoromethylsulfamoyl)guanosine (7a)
Using general method 3 from 6a. The crude product was purified by preparative HPLC using a Luna PFP column (tR 4.9 min; isocratic elution at 35% B in A, where A is 0.1% CF3COOH in H2O, and B is 0.1% CF3COOH in MeCN) to afford 7a as a white solid (26%). 1H NMR: δ 9.27 (s, 1H), 7.49–7.56 (m, 2H), 7.36–7.44 (m, 3H), 5.94 (d, J = 4.0 Hz, 1H), 5.66 (dd, J1 = 21.0 Hz, J2 = 14.4 Hz, 2H), 4.19 (dd, J1 = 5.2 Hz, J2 = 4.0 Hz, 1H), 4.29 (t, J = 5.2 Hz, 1H), 4.13–4.17 (m, 1H), 3.57–3.63 (m, 2H). 13C NMR: δ 157.48, 154.98, 151.33, 134.99, 130.38, 130.27, 129.81, 122.94, 119.75, 109.10, 92.73, 85.28, 74.84, 72.12, 53.63, 46.45. 19F NMR: δ −76.39. Anal. RP-HPLC: tR 2.63 min (method 1, >95%), tR 2.23 min (method 2, >63%). MS (ESI+): m/z [M]+ calcd for C18H20F3N6O6S, 505.11; found, 505.11.
4.34. 5′-(4-Carboxyphenyl)sulfamoyl-5′-deoxy-N7-methylguanosine (7b)
Using general method 3 from 6b. The crude product was purified by preparative HPLC using a LUNA PFP column (tR 4.0 min, gradient of 5%–50% B in A over 15 min, where A is 0.1% CF3COOH in H2O, and B is 0.1% CF3COOH in MeCN) to afford 7b as a white solid (44%). 1H NMR (DMSO-d6 and D2O): δ 9.26 (s, 1H), 8.28 (s, 1H), 8.11 (s, 1H), 8.00 (d, J = 7.7 Hz, 2H), 7.76 (d, J = 7.7 Hz, 2H), 7.58 (br s, 2H), 5.75 (d, J = 4.8 Hz, 1H), 4.50 (t, J = 4.8 Hz, 1H), 4.06 (t, J = 4.8 Hz, 1H), 4.01 (s, 3H), 3.97 (dd, J1 = 9.6 Hz, J2 = 4.8 Hz), 3.10 (m, 2H, H5′). 13C NMR (DMSO-d6 and D2O): δ 173.36, 171.03, 149.41, 140.95, 140.23, 129.31, 127.48, 126.57, 125.85, 109.03, 90.32, 83.60, 72.82, 70.51, 43.70, 35.74. Anal. RP-HPLC: tR 1.09 min (method 1, >95%), tR 0.77 min (method 2, >95%). MS (ESI+): m/z [M]+ calcd for C41H40N6O10S, 481.11; found, 481.11.
4.35. N7-Benzyl-5′-(4-carboxyphenyl)sulfamoyl-5′-deoxyguanosine (7c)
Using general method 3 from 6c. The crude product was purified by RP-HPLC with a LUNA PFP column (tR 19.8 min, gradient of 10%–30% B in A over 20 min, where A is 0.1% CF3COOH in H2O, and B is 0.1% CF3COOH in MeCN) to afford 7c as a white solid (38%). 1H NMR: δ 9.56 (s, 1H), 8.11 (s, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.87 (d, J = 8.4 Hz, 2H), 7.37–7.51 (m, 7H), 5.79 (d, J = 4.4 Hz, 1H), 5.59 (s, 2H), 4.50 (t, J = 4.8 Hz, 1H), 4.09 (t, J = 4.8 Hz, 1H), 3.89–4.02 (m, 1H), 3.09–3.24 (m, 2H). 13C NMR: δ 166.17, 155.77, 153.16, 149.62, 144.32, 136.48, 134.30, 130.00, 128.84, 128.20, 126.70, 106.79, 89.14, 83.71, 73.45, 70.58, 51.61, 49.29. Anal. RP-HPLC: tR 2.49 min (method 1, >95%), tR 2.09 min (method 2, >95%). MS (ESI+): m/z [M]+ calcd for C24H25N6O8S, 577.14; found, 577.14.
4.36. 5′-(tert-Butyloxycarbamoyl)-5′-deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethyli-dene)guanosine (8)
To a stirred solution of 1e (0.5 g, 0.8 mmol) in CH2Cl2 (30 mL) was added Et3N (220 μL, 1.6 mmol). The mixture was cooled to 0 °C and Boc2O (170 mg, 0.8 mmol) was added. Stirring was continued for 3 h, when the mixture was evaporated. The residue was purified by SiO2 column chromatography (CH2Cl2—MeOH, 9:1) to afford pure title compound as a white solid (375 mg, 65%). 1H NMR: δ 10.66 (s, 1H, NH), 7.76 (s, 1H), 7.58 (s, 1H) 7.10–7.33 (m, 9H), 6.83–6.89 (d, J = 8.5 Hz, 4H), 5.36 (d, J = 3.9 Hz, 1H), 4.76 (dd, J1 = 6.0 Hz, J2 = 3.9 Hz, 1H), 4.27 (m, 1H), 3.82 (m, 1H), 3.70 (s, 6H), 2.61–2.87 (m, 2H), 1.38 (s, 9H), 1.35 (s, 3H), 1.18 (s, 3H). 13C NMR: δ 157.73, 156.38, 155.55, 150.95, 149.23, 145.08, 137.22, 136.97, 136.74, 129.67, 128.24, 127.72, 126.54, 117.58, 113.26, 113.03, 112.99, 88.68, 82.41, 81.33, 80.38, 77.91, 69.39, 54.96, 41.85, 28.17, 27.16, 25.57. MS (ESI+): m/z [M+H]+ calcd for C39H45N6O8, 725.33; found, 725.32.
4.37. 5′-(tert-Butyloxycarbamoyl)-5′-deoxy-N2-(4,4′-dimethoxytrityl)-N7-methyl-2′,3′-O-(1-methylethylidene)guanosine (9a)
Using general method 1 from 8 and methyl iodide. Yellow powder (98%). Mp 106–107 °C. 1H NMR: δ 11.48 (s, 1H, NH), 9.20 (s, 1H), 8.07 (s, 1H) 7.15–7.34 (m, 9H), 6.89 (m, 1H), 6.86 (d, J = 8.9 Hz, 4H), 5.45 (d, J = 2.1 Hz, 1H), 4.82 (dd, J1 = 6.1 Hz, J2 = 2.1 Hz, 1H), 4.39 (dd, J1 = 6.1 Hz, J2 = 1.4 Hz, 1H), 4.08 (m, 1H), 3.93 (m, 3H), 3.71 (s, 6H), 2.72–3.03 (m, 2H), 1.38 (s, 9H), 1.35 (s, 12H), 1.10 (s, 3H). 13C NMR: δ 157.89, 155.63, 152.80, 147.19, 144.32, 137.82, 136.10, 136.03, 129.74, 129.66, 128.26, 127.81, 126.81, 113.07, 112.75, 108.54, 92.19, 86.13, 82.00, 81.33, 78.18, 69.81, 55.02, 41.63, 35.62, 28.11, 26.61, 25.14. MS (ESI+): m/z [M]+ calcd for C40H47N6O8, 739.34; found, 739.34.
4.38. N7-Benzyl-5′-(tert-butyloxycarbamoyl)-5′-deoxy-N2-(4,4′-dimethoxytrityl)-2′,3′-O-(1-methylethylidene)guanosine (9b)
Using general method 1 from 8 and benzyl bromide. The crude product was purified by SiO2 column chromatography (CHCl3—MeOH, 9:1) to afford the title compound as a white powder (73%). 1H NMR ((CD3)2CO): δ 10.11 (s, 1H), 7.02–7.67 (m, 15H), 6.66–6.94 (m, 4H), 6.45–6.65 (t, J = 5.9 Hz, 1H), 5.42–5.77 (m, 3H), 5.12 (m, 1H), 4.72 (m, 1H), 4.22–4.44 (m, 1H), 3.67 (s, 6H), 3.14–3.48 (m, 2H), 1.43 (s, 3H), 1.40 (s, 9H), 1.17 (s, 3H). 13C NMR ((CD3)2CO): δ 158.99, 157.32, 157.10, 149.16, 146.44, 138.26, 138.18, 136.48, 135.60, 131.44, 131.37, 130.02, 129.57, 129.46, 128.16, 127.08, 114.08, 113.42, 108.70, 93.82, 89.03, 84.51, 82.73, 79.29, 71.32, 55.37, 52.32, 43.47, 34.40, 28.63, 27.53, 26.03. MS (ESI+): m/z [M]+ calcd for C46H51N6O8, 815.4; found, 815.3.
4.39. 5′-Amino-5′-deoxy-N7-methylguanosine (10a)
Using general method 3 from 9a. After completion of the reaction the solvent was evaporated and the residue was suspended in H2O and extracted with CHCl3. The combined extracts were dried (MgSO4) and evaporated. The residue was purified by preparative HPLC using a Luna HILIC column (tR 8.0 min; gradient from 0% A to 100% A over 12.5 min, where A is 5:4:1 MeCN—H2O–50 mM aq NH4HCOO pH 3.2, and B is 9:1 MeCN–50mM aq NH4HCOO pH 3.2) to afford the title compound as a white solid (51%). 1H NMR: δ 9.32 (s, 1H), 8.26 (s, 2H), 6.99 (br s, 2H), 5.82 (dd, J = 4.7 Hz, 1H), 4.47 (t, J = 4.7, 1H), 4.18 (t, J = 4.7 Hz, 1H), 4.18 (t, J = 4.7 Hz, 1H), 4.04 (m, 1H), 4.00 (s, 3H), 3.41 (m, 2H). 13C NMR: δ 158.68, 156.83, 149.26, 135.39, 107.69, 89.51, 82.52, 73.43, 70.57, 40.94, 35.46. MS (ESI+): m/z [M]+ calcd for C11H17N6O4, 297.13; found, 297.13.
4.40. 5′-Amino-N7-benzyl-5′-deoxyguanosine (10b)
Using general method 3 from 9b. The crude product was triturated with Me2CO and the colourless precipitate was filtered and dried (44%). 1H NMR: δ 8.50 (s, 1H), 7.51–7.61 (m, 2H), 7.32–7.43 (m, 3H), 5.98 (d, J = 4.4 Hz, 2H), 5.70 (s, 2H), 4.87 (t, J = 4.4 Hz, 1H), 4.38 (m, 1H), 4.32 (m, 1H), 3.36–3.64 (m, 2H). 13C NMR: δ 157.18, 154.77, 149.61, 136.66, 134.65, 128.90, 128.77, 128.38, 106.99, 90.31, 81.50, 73.10, 70.85, 51.40, 40.80. Anal. RP-HPLC: tR 3.51 min (method 1, >95%). MS (ESI+): m/z [M]+ calcd for C17H21N6O4, 373.2; found, 373.2.
4.41. 5′-Deoxy-N7-methyl-5′-((1H-tetrazol-5-yl)methylcarbamoyl)guanosine (11a)
To a stirred solution of 10a (15 mg, 0.05 mmol) in DMF (1 mL) were added Et3N (8 μL, 0.05 mmol), N,N′-diisopropylcarbodiimide (8 μL, 0.05 mmol), and 1H-tetrazole-5-acetic acid (7 mg, 0.05 mmol). The mixture was stirred for 24 h at r.t. and diluted with Me2CO (10 mL). The precipitate was collected, dried, and purified by preparative HPLC using a Luna HILIC column (gradient from 0% A to 100% A over 12.5 min, where A is 5:4:1 MeCN—H2O–50 mM aq NH4HCOO pH 3.2, and B is 9:1 MeCN–50mM aq NH4HCOO pH 3.2) to afford the title compound as a white solid (3 mg, 14%). 1H NMR (D2O): δ 8.26 (s, 1H), 5.77 (d, J = 4.3 Hz, 1H), 4.72 (t, J = 4.3 Hz, 1H), 4.51 (m, 2H), 4.09 (m, 2H), 3.88 (s, 3H), 3.76 (s, 2H), 3.47 (m, 2H). 13C NMR (D2O): δ 171.20, 163.14, 162.86, 155.40, 155.08, 149.44, 108.68, 90.23, 83.33, 73.67, 70.69, 40.39, 35.79, 31.89. Anal. RP-HPLC: tR 0.67 min (method 1: >95%), tR 0.33 min, (method 2, >95%.) MS (ESI+): m/z [M+] calcd for C14H19N10O5, 407.15; found, 407.16.
4.42. N7-Benzyl-5′-deoxy-5′-((1H-tetrazol-5-yl)methylcarbamoyl)guanosine (11b)
This compound was obtained from 10b analogously to preparation and purification of 11a (34%). 1H NMR: δ 10.19 (s, 1H), 8.38 (t, J = 5.6 Hz, 1H), 8.31 (s, 2H), 7.74 (s, 1H), 7.49–7.58 (m, 2H), 7.21–7.41 (m, 3H), 5.85 (d, J = 5.3 Hz, 1H), 5.75 (dd, J1 = 20.0 Hz, J2 = 15.0 Hz, 1H), 4.40 (t, J = 5.2 Hz, 1H), 4.06 (m, 2H), 3.34–3.67 (m, 4H). 13C NMR: δ 169.95, 165.46, 158.39, 155.95, 150.63, 136.32, 135.42, 129.19, 129.00, 128.92, 107.00, 88.05, 84.92, 74.52, 70.80, 51.77, 40.90, 34.05. Anal. RP-HPLC: tR 1.40 min (method 1, >95%), tR 0.77 min (method 2, >95%). MS (ESI+): m/z [M+] calcd for C20H23N10O5, 483.18; found, 483.15.
4.43. Virtual screening
An eIF4E–Bn7GMP complex crystal structure (PDB code 2V8X) [15] was used for docking purposes, and docking was based on the lead-like subset of compounds in the ZINC database [40]. Receptor preparation was performed using the Biopolymer function in SYBYL 8.0 (Tripos International, 1699 South Hanley Rd., St. Louis, MO, 63144, USA). The entire binding site occupied by Bn7GMP in the eIF4E template structure was defined as the docking site. Ligand 3D co-ordinates were generated using CONCORD (R. S. Pearlman, “Concord,” distributed by Tripos International, St. Louis, MO, 63144, USA). Conformer (up to 200 per compound) generation (OMEGA version 2.4.6) [54], [55], as well as docking and scoring (FRED module of the OEDocking version 3.0.0) [41], [42], were carried out using OpenEye Scientific Software (Santa Fe, NM, USA; http://www.eyesopen.com). Scoring of docked poses was performed using a combination of the OEDocking scoring functions Chemgauss3 and Zapbind (using Poisson-Boltzman electrostatics approximation), together with a semi-empirical method using the LocalSCFv2.0 application (Fujitsu FQS Poland Sp. z o. o., Krakow, Poland) [56]. Virtual hit compounds were acquired from vendors (Table 1) and were characterised by LC-MS. Two different batches of compound 14 with slightly different appearance and aqueous solubility were obtained. However, these were indistinguishable by LC-MS and NMR, and spectral data was in accord with the structure shown.
4.44. Western blotting
SDS-PAGE and electroblotting were carried out as usual. For Western blotting, the following antibodies were used: rabbit anti-BCL, Cell Signalling #2872; mouse anti c-Myc (9E10), SantaCruz #sc-040; rabbit anti-GAPDH, Cell Signalling #2118; rabbit anti-eIF4E-BP1, Cell Signalling #9644; rabbit anti-phospho-eIF4E-BP1, Cell Signalling #9459; rabbit anti-eIF4E, Cell Signalling #9742; rabbit anti-eIF2A (D7D2) XP, Cell Signalling #5324; mouse anti-eIF2A, Cell Signalling #2103; rabbit anti-cyclin D1, SantaCruz #753; mouse anti-β-actin, Sigma #A3854; (primary); anti-mouse-HRP conjugate, 1:5,000, Dako P0447; anti-rabbit-HRP conjugate, 1:10,000, GE Healthcare NA9340V.
4.45. Recombinant eIF4E
Expression and purification of eIF4E followed a modified procedure of that already published [15]. Briefly, the procedure was modified to allow for rapid, scale-up production of eIF4E and will be published separately (manuscript in preparation). Following purification, the protein concentration was adjusted to 1 mg mL−1 (Vivaspin concentrator, 5 kDa cut-off) and the solution was flash-frozen in 200-μL aliquots, which were stored at −80 °C.
4.46. Fluorescence polarisation assay
A fluorescent probe (EDA-m7GDP–ATTO-465; NU-827-465) was obtained from Jena Biosciences; 96-well black plates (Optiplate®) were from Perkin Elmer. Recombinant eIF4E and the probe were diluted (20 mM HEPES, pH 7.4, 100 mM KCl, 1 mM DTT) to obtain the required concentration. m7GTP (Sigma-Aldrich) and m7GMP were used as positive controls. The final volume in each well was 100 μL. Compound screening was performed using a final DMSO concentration of 1%. Fluorescence polarisation measurements were recorded on a Perkin-Elmer Envision 2104 plate reader. For Kd calculation, logarithmic serial dilutions of test compounds were prepared. Background-corrected readings were plotted against test compound concentration in GaphPad Prism (Version 5.0) in order to obtain Kd values.
4.47. Crystallisation of ligand-bound eIF4E
A 3.2-mM solution of 4G-I (eIF4G-derived peptide, KKRYDREFLLGFQF) or 4EBP-I (4E-BP-derived peptide, RIIYDRKFLMECRN) in crystallisation buffer (50 mM Tris, pH 8.0, 150 mM KCl, 1 mM DTT) was added to an equal volume of a solution of recombinant human eIF4E (8 mg mL−1). A ligand solution (50 or 100 mM) in DMSO was added to a final DMSO concentration of 2.5%. Thin needle-like crystals grew over a period of 3–7 d by the hanging drop method (1:1 protein–mother liquor) over 21–31% PEG-8,000, 1–3% (NH4)2SO4, 100 mM HEPES, pH 7.5. Drops containing crystals were added to 50 μL mother liquor vortex-mixed for 3 min. This was used as a seed stock (at 1:1 and 1:10 dilution) for seeding back into the same conditions. Needle-like crystals grew in 1–7 d and were frozen directly in liquid N2. For back-soaking experiments with ligand 7a complex crystals, crystals were transferred to a drop containing a 1:1 ratio of crystallisation buffer and 100 mM HEPES, pH 7.5, 21% PEG-8,000, supplemented with 7a. The drop was equilibrated over 100 mM HEPES, pH 7.5, 21% PEG-8000 for 5 min before flash-freezing the crystals in liquid N3.
X-ray crystallography data collection and structure refinement. Data were collected at Diamond Light Source, UK, from single crystals using beamlines I02, I04, and I04-1, at 100 K, and were processed using either iMOSFLM [57] or XDS [58] using scaling with SCALA [59]. The structures were solved with the AutoMR command in the PHENIX suite [60], which uses the programme PHASER [61]. A single eIF4E and 4E-BP chain from the unit cell of PDB entry 2V8Y (ligand removed) was used as the search model and all structures were subjected to several rounds of refinement in PHENIX and manual refinement in COOT [62], including ligand fitting and a rebuilding of the 4G-I peptide chain (where necessary).
4.48. Rabbit reticulocyte lysate in vitro translation assays
PCR (hot KOD polymerase kit, Novagen®; Qiaquick PCR purification kit, QIAGEN) was used to amplify the luciferase DNA sequence from a pGl3 vector with the primers GCGGCCGGCCGCCCCGACTCTAGAA and GGGGTAATACGACTCACTATAGGGTCC-ACCTCGAATCACTAGTCAGCTG. Alternatively, the plasmid pRHCVF, which contains Renilla luciferase as the upstream cistron and firefly luciferase as the downstream cistron preceded by the HCV IRES, was digested with HpaI and use to prime in vitro transcription reactions. In vitro RNA synthesis was performed on the resulting DNA using a mMessage mMachine® T7 kit (Ambion). The resulting capped RNAs were purified using microSpin G-50 columns (GE Healthcare), analysed by agarose gel electrophoresis, and were stored in aliquots at −80 °C. An in-vitro translation Rabbit Reticulocyte Lysate System kit (Promega) was used as per the manufacturer's instructions. Briefly, 20 ng capped RNA was incubated with rabbit reticulocyte lysate in a final reaction volume of 20 μL. The mixture was incubated at 30 °C for 90 min and then placed on ice for immediate analysis. Diluent (H2O) was replaced by test compound solutions in the course of compound screening and the final DMSO concentration was limited to 1%. Assays were performed with m7GTP as a positive control and a DMSO negative control (which was assigned a 100% value).
4.49. Ultraviolet cross-linking assay
In a way similar to RNA luciferase synthesis, a 392-base coding region of the pGL3 vector was amplified and the corresponding RNA was generated using the T7 RiboMax large-scale production system (Promega) according to the manufacturer's instructions. The resulting transcript was then cap-radiolabelled with 32P-[α-GTP] (specific activity 400 Ci mmol−1, Hartmann Analytic GmbH) using a vaccinia virus capping system (New England Biolabs) and purified using a microSpin G-50 column. A 1-μL aliquot of the labelled RNA was transferred to a scintillation vial with 2 mL scintillation liquid (Ecoscint A, National Diagnostics), and was read in a scintillation counter (Wallac Winspectral 1414 liquid scintillation counter). The disintegration per minute readings were converted to Ci units, and the concentration of radiolabelled capped RNA was then calculated based the original 32P-GTP stock concentration after correction based on decay time. Assay procedures: to work out suitable concentrations of RNA and eIF4E to be used in the assay, a titration experiment of eIF4E against a constant concentration of labelled RNA was carried out. The assay was performed using 96-well flat-bottom plates (Corning, Costar®), in a volume of 20 μL for each reaction (in a ratio of 10:5:5 for buffer, RNA, eIF4E, respectively). Plates were kept on ice and were exposed to a UV radiation source of 275 J m2 at a distance of 3 cm for a period of 30 min. The plate wells were then treated with a mixture of RNase (0.1 mg mL−1 RNase A, 0.1 mg mL−1 RNase T1, 0.1 mg mL−1 RNase V1) at 37 °C for 30 min. Samples were then separated by SDS-PAGE and the gels were dried and exposed to autoradiography film for 24 h, before being read using a Typhoon 9500 FLA scanner. Band intensities were quantified using image analysis software (Image J, Version 1.46) relative to a DMSO controls.
4.50. 35S-methionine incorporation assay
HeLa cells were seeded into 12-well plates at 1 × 105 cells per well for 2-h and at 6 × 104 cells per well for 24-h treatment experiments. Cells were incubated for 24 h until they reached 70% confluency and were then treated with test compounds. Cells were methionine-starved for 30 min by changing to a Met-free DMEM medium, with renewed test compound addition. A 1.5-μL aliquot of 35S-methionine (30 μCi mL−1, Hartmann Analytic) was added to each well of the plates, which were incubated for 30 min. The plates were placed on ice and medium was removed. The cells were washed with warm phosphate-buffered saline (PBS) twice and lysed by storing at −80 °C for 30 min in lysis buffer (Promega). For quantification, protein was precipitated with trichloroacetic acid, collected, dried, transferred to scintillation liquid, and read in a scintillation counter.
4.51. Polysome profiling assay
HeLa cells were seeded in 15-cm diameter dishes and were left for 24 h to reach 70% confluency. Cells were treated with test compound for 2 h and cycloheximide (0.1 mg mL−1) was added. Cells were harvested on ice and then lysed using lysis buffer 300 mM NaCl, 15 mM MgCl2, 15 mM Tris (pH 7.5) containing 1 mg mL−1 heparin sulfate and 0.1 mg mL−1 cycloheximide, supplemented with 0.1% (v/v) Triton X-100. Cell lysates were loaded on sucrose gradients (10–60% sucrose in 12-mL Sorvall centrifuge tubes) made with the same buffer, and were ultracentrifuged using a SW40Ti (Beckman Coulter) centrifuge at 38,000 r.p.m. for 2 h. The gradients were then separated into fractions of 1 mL with continuous UV absorbance recording.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
PDB ID codes
5EI3, 5EIR, 5EKV, 5EHC.
Acknowledgements
The work presented here was funded by the Association for International Cancer Research (AICR; grant reference 06-063), the Medical Research Council (MRC), and Cancer Research UK (CRUK; grant reference C18648/A10174) and was supported by the Wellcome Trust Multi-User Equipment Grants 081287/Z/06/Z and 101527/Z/13/Z. FS was supported by a Scholarship from the University of Damascus, Syria.
References
- 1.Kapp L.D., Lorsch J.R. The molecular mechanics of eukaryotic translation. Annu. Rev. Biochem. 2004;73:657–704. doi: 10.1146/annurev.biochem.73.030403.080419. [DOI] [PubMed] [Google Scholar]
- 2.Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borman A.M., Michel Y.M., Malnou C.E., Kean K.M. Free poly(A) stimulates capped mRNA translation in vitro through the eIF4G-poly(A)-binding protein interaction. J. Biol. Chem. 2002;277:36818–36824. doi: 10.1074/jbc.M205065200. [DOI] [PubMed] [Google Scholar]
- 4.von der Haar T., Gross J.D., Wagner G., McCarthy J.E. The mRNA cap-binding protein eIF4E in post-transcriptional gene expression. Nat. Struct. Mol. Biol. 2004;11:503–511. doi: 10.1038/nsmb779. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh B., Benyumov A.O., Ghosh P., Jia Y., Avdulov S., Dahlberg P.S., Peterson M., Smith K., Polunovsky V.A., Bitterman P.B., Wagner C.R. Nontoxic chemical interdiction of the epithelial-to-mesenchymal transition by targeting cap-dependent translation. ACS Chem. Biol. 2009;4:367–377. doi: 10.1021/cb9000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gingras A.C., Raught B., Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 7.Goodfellow I.G., Roberts L.O. Eukaryotic initiation factor 4E. Int. J. Biochem. Cell. Biol. 2008;40:2675–2680. doi: 10.1016/j.biocel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Benedetti A., Graff J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–3199. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 9.Graff J.R., Zimmer S.G. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin. Exp. Metastasis. 2003;20:265–273. doi: 10.1023/a:1022943419011. [DOI] [PubMed] [Google Scholar]
- 10.Zimmer S.G., DeBenedetti A., Graff J.R. Translational control of malignancy: the mRNA cap-binding protein, eIF-4E, as a central regulator of tumor formation, growth, invasion and metastasis. Anticancer Res. 2000;20:1343–1351. [PubMed] [Google Scholar]
- 11.Feoktistova K., Tuvshintogs E., Do A., Fraser C.S. Human eIF4E promotes mRNA restructuring by stimulating eIF4A helicase activity. Proc. Natl. Acad. Sci. U. S. A. 2013;110:13339–13344. doi: 10.1073/pnas.1303781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia Y., Polunovsky V., Bitterman P.B., Wagner C.R. Cap-dependent translation initiation factor eIF4E: an emerging anticancer drug target. Med. Res. Rev. 2012;32:786–814. doi: 10.1002/med.21260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graff J.R., Konicek B.W., Vincent T.M., Lynch R.L., Monteith D., Weir S.N., Schwier P., Capen A., Goode R.L., Dowless M.S., Chen Y., Zhang H., Sissons S., Cox K., McNulty A.M., Parsons S.H., Wang T., Sams L., Geeganage S., Douglass L.E., Neubauer B.L., Dean N.M., Blanchard K., Shou J., Stancato L.F., Carter J.H., Marcusson E.G. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Invest. 2007;117:2638–2648. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darzynkiewicz E., Stepinski J., Ekiel I., Goyer C., Sonenberg N., Temeriusz A., Jin Y., Sijuwade T., Haber D., Tahara S.M. Inhibition of eukaryotic translation by nucleoside 5′-monophosphate analogs of mRNA 5′-cap: changes in N7 substituent affect analog activity. Biochemistry. 1989;28:4771–4778. doi: 10.1021/bi00437a038. [DOI] [PubMed] [Google Scholar]
- 15.Brown C.J., McNae I., Fischer P.M., Walkinshaw M.D. Crystallographic and mass spectrometric characterisation of eIF4E with N7-alkylated cap derivatives. J. Mol. Biol. 2007;372:7–15. doi: 10.1016/j.jmb.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 16.Jia Y., Chiu T.L., Amin E.A., Polunovsky V., Bitterman P.B., Wagner C.R. Design, synthesis and evaluation of analogs of initiation factor 4E (eIF4E) cap-binding antagonist Bn-7-GMP. Eur. J. Med. Chem. 2010;45:1304–1313. doi: 10.1016/j.ejmech.2009.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh P., Park C., Peterson M.S., Bitterman P.B., Polunovsky V.A., Wagner C.R. Synthesis and evaluation of potential inhibitors of eIF4E cap binding to 7-methyl GTP. Bioorg. Med. Chem. Lett. 2005;15:2177–2180. doi: 10.1016/j.bmcl.2005.01.080. [DOI] [PubMed] [Google Scholar]
- 18.Darzynkiewicz E., Ekiel I., Lassota P. Tahara. Inhibition of eukaryotic translation by analogs of messenger RNA 5′-cap: chemical and biological consequences of 5′-phosphate modifications of 7-methylguanosine 5′-monophosphate. Biochemistry. 1987;26:4372–4380. doi: 10.1021/bi00388a028. [DOI] [PubMed] [Google Scholar]
- 19.Kowalska J., Lukaszewicz M., Zuberek J., Ziemniak M., Darzynkiewicz E., Jemielity J. Phosphorothioate analogs of m7GTP are enzymatically stable inhibitors of cap-dependent translation. Bioorg. Med. Chem. Lett. 2009;19:1921–1925. doi: 10.1016/j.bmcl.2009.02.053. [DOI] [PubMed] [Google Scholar]
- 20.Chen X., Kopecky D.J., Mihalic J., Jeffries S., Min X., Heath J., Deignan J., Lai S., Fu Z., Guimaraes C., Shen S., Li S., Johnstone S., Thibault S., Xu H., Cardozo M., Shen W., Walker N., Kayser F., Wang Z. Structure-guided design, synthesis, and evaluation of guanine-derived inhibitors of the eIF4E mRNA-cap interaction. J. Med. Chem. 2012;55:3837–3851. doi: 10.1021/jm300037x. [DOI] [PubMed] [Google Scholar]
- 21.Li S., Jia Y., Jacobson B., McCauley J., Kratzke R., Bitterman P.B., Wagner C.R. Treatment of breast and lung cancer cells with a N-7 benzyl guanosine monophosphate tryptamine phosphoramidate pronucleotide (4Ei-1) results in chemosensitization to gemcitabine and induced elF4E proteasomal degradation. Mol. Pharm. 2013;10:523–531. doi: 10.1021/mp300699d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papadopoulos E., Jenni S., Kabha E., Takrouri K.J., Yi T., Salvi N., Luna R.E., Gavathiotis E., Mahalingam P., Arthanari H., Rodriguez-Mias R., Yefidoff-Freedman R., Aktas B.H., Chorev M., Halperin J.A., Wagner G. Structure of the eukaryotic translation initiation factor eIF4E in complex with 4EGI-1 reveals an allosteric mechanism for dissociating eIF4G. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E3187–E3195. doi: 10.1073/pnas.1410250111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lama D., Quah S.T., Verma C.S., Lakshminarayanan R., Beuerman R.W., Lane D.P., Brown C.J. Rational optimization of conformational effects induced by hydrocarbon staples in peptides and their binding interfaces. Sci. Rep. 2013;3:3451. doi: 10.1038/srep03451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown C.J., Dastidar S.G., See H.Y., Coomber D.W., Ortiz-Lombardia M., Verma C., Lane D.P. Rational design and biophysical characterization of thioredoxin-based aptamers: insights into peptide grafting. J. Mol. Biol. 2010;395:871–883. doi: 10.1016/j.jmb.2009.10.069. [DOI] [PubMed] [Google Scholar]
- 25.Fischer P.M. Cap in hand: targeting eIF4E. Cell Cycle. 2009;8:2535–2541. doi: 10.4161/cc.8.16.9301. [DOI] [PubMed] [Google Scholar]
- 26.Beaulieu P.L., Cameron D.R., Ferland J.M., Gauthier J., Ghiro E., Gillard J., Gorys V., Poirier M., Rancourt J., Wernic D., Llinas-Brunet M., Betageri R., Cardozo M., Hickey E.R., Ingraham R., Jakes S., Kabcenell A., Kirrane T., Lukas S., Patel U., Proudfoot J., Sharma R., Tong L., Moss N. Ligands for the tyrosine kinase p56lck SH2 domain: discovery of potent dipeptide derivatives with monocharged, nonhydrolyzable phosphate replacements. J. Med. Chem. 1999;42:1757–1766. doi: 10.1021/jm980676t. [DOI] [PubMed] [Google Scholar]
- 27.Darzynkiewicz E., Stepinski J., Ekiel I., Goyer C., Sonenberg N., Temeriusz A., Jin Y., Sijuwade T., Haber D., Tahara S.M. Inhibition of eukaryotic translation by nucleoside 5′-monophosphate analogues of mRNA 5′-cap: changes in N7 substituent affect analogue activity. Biochemistry. 1989;28:4771–4778. doi: 10.1021/bi00437a038. [DOI] [PubMed] [Google Scholar]
- 28.Jia Y., Chiu T.L., Amin E.A., Polunovsky V., Bitterman P.B., Wagner C.R. Design, synthesis and evaluation of analogs of initiation factor 4E (eIF4E) cap-binding antagonist Bn7-GMP. Eur. J. Med. Chem. 2010;45:1304–1313. doi: 10.1016/j.ejmech.2009.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seio K., Miyashita T., Sato K., Sekine M. Synthesis and properties of new nucleotide analogues possessing squaramide moieties as new phosphate isosters. Eur. J. Org. Chem. 2005:5163–5170. [Google Scholar]
- 30.Sato K., Seio K., Sekine M. Squaryl group as a new mimic of phosphate group in modified oligodeoxynucleotides: synthesis and properties of new oligodeoxynucleotide analogues containing an internucleotidic squaryldiamide linkage. J. Am. Chem. Soc. 2002;124:12715–12724. doi: 10.1021/ja027131f. [DOI] [PubMed] [Google Scholar]
- 31.Huang P., Ramphal J., Wei J., Liang C., Jallal B., McMahon G., Tang C. Structure-based design and discovery of novel inhibitors of protein tyrosine phosphatases. Bioorg. Med. Chem. 2003;11:1835–1849. doi: 10.1016/s0968-0896(03)00039-7. [DOI] [PubMed] [Google Scholar]
- 32.Zingle C., Kuntz L., Tritsch D., Grosdemange-Billiard C., Rohmer M. Isoprenoid biosynthesis via the methylerythritol phosphate pathway: structural variations around phosphonate anchor and spacer of fosmidomycin, a potent inhibitor of deoxyxylulose phosphate reductoisomerase. J. Org. Chem. 2010;75:3203–3207. doi: 10.1021/jo9024732. [DOI] [PubMed] [Google Scholar]
- 33.Herr R.J. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods. Bioorg. Med. Chem. 2002;10:3379–3393. doi: 10.1016/s0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 34.Sekine M., Seio Y., Miyashita T., Sato K. Japan Science and Technology Agency; Japan: 2005. Preparation of Nucleoside Analogs Having Squaric Acid or Deltic Acid-derived Moiety. Jpn. Kokai Tokkyo Koho JP2005162631. [Google Scholar]
- 35.Zhang B.L., Cui Z.Y., Sun L.L. Synthesis of 5 ′-deoxy-5 '-thioguanosine-5′-monophosphorothioate and its incorporation into RNA 5 '-termini. Org. Lett. 2001;3:275–278. doi: 10.1021/ol006916s. [DOI] [PubMed] [Google Scholar]
- 36.McGall G.H., Barone A.D., Diggelmann M., Fodor S.P.A., Gentalen E., Ngo N. The efficiency of light-directed synthesis of DNA arrays on glass substrates. J. Am. Chem. Soc. 1997;119:5081–5090. [Google Scholar]
- 37.Dean D.K. An improved synthesis of 5′-amino-5′-deoxyguanosine. Synth. Commun. 2002;32:1517–1521. [Google Scholar]
- 38.Delfino J.M., Stankovic C.J., Schreiber S.L., Richards F.M. Synthesis of a bipolar phosphatidylethanolamine: a model compound for a membrane-spanning probe. Tetrahedron Lett. 1987;28:2323–2326. [Google Scholar]
- 39.Moriguchi T., Asai N., Okada K., Seio K., Sasaki T., Sekine M. First synthesis and anticancer activity of phosmidosine and its related compounds. J. Org. Chem. 2002;67:3290–3300. doi: 10.1021/jo016176g. [DOI] [PubMed] [Google Scholar]
- 40.Irwin J.J., Sterling T., Mysinger M.M., Bolstad E.S., Coleman R.G. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGann M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011;51:578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 42.McGann M. FRED and HYBRID docking performance on standardized datasets. J. Comp. Aided Mol. Des. 2012;26:897–906. doi: 10.1007/s10822-012-9584-8. [DOI] [PubMed] [Google Scholar]
- 43.Dougherty D.A. Cation-m interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 44.Ma J.C., Dougherty D.A. The cation-pi interaction. Chem. Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 45.Patwardhan G., Meullenet F., Nowicki M.W., Oldham N.J., Willis A.E., Walkinshaw M.D., Fischer P.M. The 238th ACS National Meeting, Washington, DC, USA. 2009. Molecular modeling leading the way: toward nonnucleotide analogs targeting the eIF4E-mRNA cap interaction as novel anticancer agents. p Abs. MEDI 461. [Google Scholar]
- 46.Veber D.F., Johnson S.R., Cheng H.-Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 47.Oprea T.I., Davis A.M., Teague S.J., Leeson P.D. Is there a difference between leads and drugs? A historical perspective. J. Chem. Inf. Comp. Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- 48.Lindqvist L., Imataka H., Pelletier J. Cap-dependent eukaryotic initiation factor-mRNA interactions probed by cross-linking. RNA. 2008;14:960–969. doi: 10.1261/rna.971208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pelham H.R., Jackson R.J. An efficient mRNA-dependent translation system from reticulocyte lysates. Eur. J. Biochem. 1976;67:247–256. doi: 10.1111/j.1432-1033.1976.tb10656.x. [DOI] [PubMed] [Google Scholar]
- 50.Chan J., Khan S.N., Harvey I., Merrick W., Pelletier J. Eukaryotic protein synthesis inhibitors identified by comparison of cytotoxicity profiles. RNA. 2004;10:528–543. doi: 10.1261/rna.5200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He S.L., Green R. Polysome analysis of mammalian cells. Methods Enzymol. 2013;530:183–192. doi: 10.1016/B978-0-12-420037-1.00010-5. [DOI] [PubMed] [Google Scholar]
- 52.Marcotrigiano J., Gingras A.-C., Sonenberg N., Burley S.K. Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell. 1997;89:951–961. doi: 10.1016/s0092-8674(00)80280-9. [DOI] [PubMed] [Google Scholar]
- 53.Schmitz K., Hahn F., Schepers U. Modified GSMP synthesis greatly improves the disulfide crosslink of T7 run-off siRNAs with cell penetrating peptides. Synlett. 2010:2959–2963. [Google Scholar]
- 54.Hawkins P.C.D., Skillman A.G., Warren G.L., Ellingson B.A., Stahl M.T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and cambridge structural database. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hawkins P.C.D., Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012;52:2919–2936. doi: 10.1021/ci300314k. [DOI] [PubMed] [Google Scholar]
- 56.Anikin N.A., Anisimov V.M., Bugaenko V.L., Bobrikov V.V., Andreyev A.M. LocalSCF method for semiempirical quantum-chemical calculation of ultralarge biomolecules. J. Chem. Phys. 2004;121:1266–1270. doi: 10.1063/1.1764496. [DOI] [PubMed] [Google Scholar]
- 57.Battye T.G.G., Kontogiannis L., Johnson O., Powell H.R., Leslie A.G.W. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kabsch W. XDS. Acta Crystallogr. D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., McNicholas S.J., Murshudov G.N., Pannu N.S., Potterton E.A., Powell H.R., Read R.J., Vagin A., Wilson K.S. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]