

Abstract
Capillary electrophoresis (CE) has been identified as a useful platform for detecting, quantifying and screening for modulators of protein-protein interactions (PPIs). In this method, one protein binding partner is labeled with a fluorophore, the protein binding partners are mixed, and then the complex separated from free protein allowing direct determination of bound to free ratios. Although possessing many advantages for PPI studies, the method is limited by the need to have separation conditions that both prevent protein adsorption to capillary and maintain protein interactions during the separation. In this work, we use protein cross-linking capillary electrophoresis (PXCE) to overcome this limitation. In PXCE, the proteins are cross-linked under binding conditions and then separated. This approach eliminates the need to maintain non-covalent interactions during electrophoresis and facilitates method development. We report PXCE methods for an antibody-antigen interaction and heterodimer and homodimer heat shock protein complexes. Complexes are cross-linked by short treatments with formaldehyde after reaching binding equilibrium. Cross-linked complexes are separated by electrophoretic mobility using free solution CE or by size using sieving electrophoresis of SDS-complexes. The method gives good quantitative results, e.g., a lysozyme-antibody interaction was found to have Kd = 24 ± 3 nM by PXCE and Kd = 17 ± 2 nM using isothermal calorimetry (ITC). Heat shock protein 70 (Hsp70) in complex with bcl2 associated athanogene 3 (Bag3) was found to have Kd = 25 ± 5 nM by PXCE which agrees with Kd values reported without cross-linking. Hsp70-Bag3 binding site mutants and small molecule inhibitors of Hsp70-Bag3 were characterized by PXCE with good agreement to inhibitory constants and IC50 values obtained by a bead-based flow cytometry protein interaction assay (FCPIA). PXCE allows rapid method development for quantitative analysis of PPIs.
Graphical Abstract
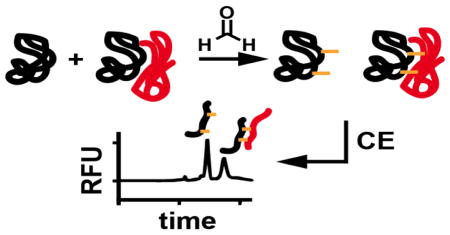
Introduction
Protein-protein interactions (PPIs) control many cellular functions. As a result, it is important to be able to quantify these interactions. It is also of interest to identify small molecule modulators of PPI for use as probes for chemical biology and as possible drugs. The diversity and transient nature of PPI can make them challenging to study. Several techniques have been developed for PPI analysis including flow cytometry protein interaction assays (FCPIA), isothermal titration calorimetry (ITC), fluorescence polarization, in silico methods, nuclear magnetic resonance (NMR), surface plasmon resonance (SPR), fluorescence resonance energy transfer (FRET) and AlphaLisa. Each of these techniques has strengths and weaknesses and can be chosen for different applications. For example, for screening chemical libraries to identify potential modulators of PPI, many of these techniques are impractical because of quantification, throughput, or sample consumption considerations. In this work we explore the use of protein cross-linking CE (PXCE) for detecting and quantifying PPIs.
PXCE is a variant of affinity probe capillary electrophoresis (APCE). In APCE, an equilibrated mixture of binding partners is electrophoresed to allow for detection of non-covalent interactions.1–3 APCE has been used to investigate many biomolecular interactions such as protein-protein4–7, antibody-antigen8–13, protein-DNA10,14–17, protein-peptide10,18 and protein-aptamer19–21. Typically, one binding partner is fluorescently labeled enabling sensitive detection by laser induced fluorescence (LIF). APCE offers advantages of low sample volume requirements, high throughput, and highly sensitive direct detection of free protein and protein complex. These advantages make APCE a potentially powerful approach for characterizing PPI and other non-covalent biomolecular interactions. The utility of this approach for screening for modulators of PPI was demonstrated in a study of the heat shock protein 70 (Hsp70) and Bcl-2 associated athanogene 3 (Bag3) interaction.4 The CE assay was found to be more selective than an FCPIA screen based on the minimal perturbation of the proteins for the assay and the ability to discern fluorescent test compounds and protein aggregation in the CE data. These features eliminated many false positives.
Although possessing many advantages for detecting, quantifying, and screening PPIs, APCE is limited by the need to have separation conditions that both maintain protein interactions over the course of the separation and also prevent protein adsorption to the capillary. Strategies to minimize protein-wall interactions include capillary derivatization,4,15,16,19 extreme pH,22,23 surfactant additives24 and high ionic strength buffers.25,26 Techniques to minimize protein adsorption to the capillary are often not compatible with maintaining non-covalent protein interactions or require optimization for each protein binding partner of interest. As a result, it is often difficult and slow to develop CE methods for PPI, greatly limiting the use of this technique.
In this work, we examine protein cross-linking prior to CE separation for detecting and quantifying PPI. This process allows complexes to be formed under binding conditions and then separated under non-native or denaturing conditions, facilitating method development. Previously protein cross-linking prior to CE analysis has been used to check the success of cross-linking for different carbodiimide cross-linkers.27 In another study, cross-linking prior to CE was used to screen for dimer formation in therapeutic antibody samples.28 Good agreement was found for results by CE and size exclusion chromatography without cross-linking suggesting the potential for more in-depth, quantitative assays.
Here, the utility of PXCE was investigated for determining Kd of three protein-protein complexes: the antibody-antigen complex of lysozyme-anti-lysozyme, Hsp70-Bag3 heterodimer and heat shock protein 90 (Hsp90) homodimer. PXCE was also applied to quantify inhibition of PPIs with Hsp70-Bag3 binding site mutants and small molecule inhibitors. Formaldehyde was chosen as the cross-linking reagent because of its short reaction time and reactivity toward many amino acid residues.29 The complexes chosen present different challenges and opportunities. Lysozyme and Bag3 have been identified as a difficult proteins to analyze using CE because they strongly adsorb to the inner wall of fused silica capillaries resulting in missing peaks4 or poor peak shape22. Heat shock proteins including Hsp70 and Hsp90 and their co-chaperone interactions have been identified as potential drug targets.30,31
Experimental Section
Chemicals and Materials
Unless otherwise specified reagents were purchased from Sigma Aldrich (St. Louis, MO). Lysozyme, Alexa Fluor 488 5-SDP ester and Alexa Fluorophore 488 NHS ester were purchased from ThermoFisher Scientific (Waltham, MA). All separation and assay buffers were made using water deionized to 18 MΩ using a Series 1090 E-pure system (Barnstead Thermolyne Cooperation; Dubuque, IA).
Protein Purification and Labeling
Hsp70 and Bag3 were expressed and purified as previously reported.4,32–34 Hsp90 was subcloned into pET28 vector to incorporate N-terminal 6x-His tag. Plasmid was transformed into BL21(DE3) One Shot start cells (Invitrogen; Carlsbad, CA) and purified on a Nickel-NTA column followed by size exclusion chromatography (SEC) on a HiLoad 16/600 Superdex 200 PG column (GE Healthcare; Piscataway, NJ). The concentrated SEC fraction was labeled with Alexa Fluor 488 NHS Ester according to manufacturer instructions and dialyzed into phosphate buffered saline, pH 7.4. Hsp70 and Hsp90 were labeled with Alexa Fluor 488 5-SDP ester. Lysozyme was incubated with a final concentration of 100 μg/mL fluorescein isothiocyanate (FITC) for 1 h at room temperature and dialyzed into phosphate buffer, pH 7.5.
Protein Cross-linking Capillary Electrophoresis
Protein samples were allowed to equilibrate in 25 mM HEPES, 10 mM KCl, 5 mM MgCl2 and 0.3% (w/v) Tween-20, pH 7.5. Small molecules were dissolved in DMSO and spiked into protein samples to a final concentration of 1% DMSO for all dose response samples, including positive and negative controls. Samples were incubated for at least 15 min prior to cross-linking. Proteins were cross-linked at room temperature by addition of formaldehyde, prepared from paraformaldehyde, to a final concentration of 1% (w/v) formaldehyde for 10 min unless otherwise stated. Cross-linking reactions were quenched by adding Tris to a total concentration of 20 mM and, for gel electrophoretic analysis, 0.2% (w/v) SDS.
All CE experiments were carried out using a Beckman Coulter P/ACE MDQ (Fullerton, CA) equipped with a Sapphire laser (Coherent; Santa Clara, CA) with 488/520 nm λexcitation/λemission filters for LIF. Data were collected by 32 Karat software and analyzed using Cutter 7.0.35 Binding data and IC50 curves were fit by non-linear regression using Prism 6.0 (GraphPad Software; San Diego, CA). All separations were carried out in 360 μm outer diameter fused silica capillary with 50 μm internal diameter for free solution electrophoresis and 40 μm internal diameter for gel electrophoresis separations (Polymicro Technologies; Phoenix, AZ). The total capillary length was 30 cm with 10 cm to detection window. Free solution electrophoresis of FITC labeled lysozyme (FITC-lysozyme) and anti-lysozyme was carried out in 10 mM sodium tetraborate, pH 10, electrophoresis buffer. Samples were injected by pressure at 0.5 psi for 5 s and electrophoresed with an applied field of 500 V/cm.
For gel electrophoresis separations of Hsp90 Alexa Fluor 488 (Hsp90-488), Hsp70 Alexa Fluor 488 (Hsp70-488) and Bag3 the capillary was pre-conditioned with 1 M NaOH, H2O and UltraTrol LN (Target Discovery; Palo Alto, CA) for 3 min each followed by introduction of dextran sieving matrix (180 mM boric acid, 200 mM Tris, 1 mM EDTA, 13.8 mM SDS, 7% w/v 1.5–2.8 MDa dextran, 10% w/v glycerol) at 40 psi for 10 min. Samples were injected electrokinetically at 15 kV for 1 min and electrophoresed with applied fields of 567 V/cm. Capillaries were regenerated by flushing with H2O followed by preconditioning when a shift in migration time was observed, usually after 1 hour of use.
Isothermal Titration Calorimetry
Titrations were performed on a NanoITC 2G (TA Instruments; New Castle, DE). Data were collected using Nano ITCRun software and a dissociation constant value was calculated using NanoAnalyze software (TA instruments). The syringe contained 6 μM FITC-lysozyme for titration into 0.2 μM anti-lysozyme in the cell.
Results and Discussion
CE-LIF of Interacting Proteins
Hsp70, Hsp90 and lysozyme were fluorescently labeled to allow for sensitive LIF detection. We initially attempted separation of the free proteins and complexes with the binding partners by CE-LIF without cross-linking (Figure 1 and S-1). For all three examples of interacting pairs, detection of complexes was difficult without cross-linking.
Figure 1.

Electropherograms with (red trace) or without (black trace) cross-linking of protein complexes of (A) Hsp70-488-Bag3, (B) Hsp90-488 dimer, and (C) FITC-lysozyme-anti-lysozyme. Separation was performed with (A,B) dextran gel and (C) free solution with pH 10, 10 mM borate electrophoresis buffer. Cross-linking was with 1% formaldehyde for 10 min in HEPES buffer. Table S-1 provides resolution between free and complex peaks for the cross-linked electropherograms.
The Hsp70-Bag3 interaction has previously been identified as a difficult PPI for CE method development due to adsorption of Bag3 to fused silica capillary around pH 7.4 We found adsorption of Bag3 to be a persistent problem, even at pH 10, so that no discernible complex peaks were observed in samples containing Hsp70-488 and Bag3 when using free solution CE (Figure S-1A). Bag3 adsorption is likely a result of it being intrinsically disordered which is common with PPI targets.36 (Previous study used capillaries covalently modified with a perfluorinated alkylating agent to prevent adsorption of Bag3.) With free solution electrophoresis of Hsp90-488 at pH 10, protein adsorption was not observed; however, monomeric Hsp90-488 was not readily resolved from the dimeric form suggesting very similar electrophoretic mobilities of dimer and monomer (Figure S-1B). A free solution CE separation of FITC-lysozyme-anti-lysozyme at pH 10, in the absence of cross-linking, results in multiple unresolved peaks possibly due to dissociation and association occurring on the time scale of the separation (Figure 1C).
A potentially better approach to separation of protein complexes is capillary gel electrophoresis (CGE) in presence of SDS, e.g. using an entangled polymer solution as a sieving media (SDS-CGE).37 SDS-CGE facilitates predictable protein separation based on size; however, the denaturing conditions disrupt PPI so that Hsp70-Bag3 and Hsp90 dimer protein complexes could not be detected. Indeed, only free protein was detected for mixtures of these interacting proteins separated by SDS-CGE (Figure 1). A similar effect was observed for FITC-lysozyme-anti-lysozyme (Figure S-2). The results with the non-cross-linked complexes illustrate the different challenges of developing APCE assays for PPI.
Cross-linking Conditions
To overcome the challenges associated with detection of non-covalent interactions by CE, interacting proteins were cross-linked prior to electrophoresis. Covalent cross-linking of interacting proteins with formaldehyde allowed for the direct detection of free protein and protein complex using the denaturing gel separation for the chaperone complexes (Figure 1A,B) and a high pH electrophoresis buffer for FITC-lysozyme-anti-lysozyme (Figure 1C). This result shows that cross-linking facilitates detection of interacting proteins by free solution CE or SDS-CGE separations.
The effect of cross-linking conditions, such as reaction time and formaldehyde concentration, on amount of complex detected was determined for the Hsp70-Bag3, lysozyme-anti-lysozyme and Hsp90 homodimer (Figure 2, Table S-2,3,4). In this study, the amount of complex formed was quantified as the complex peak area as a percentage of total peak area to account for any artifacts from instability of the laser source or injection variability. It has previously been reported that different PPIs require different cross-linking conditions;38 however, most formaldehyde cross-linking assays, such as chromatin immunoprecipitation and mass spectrometry, utilize between 10 and 20 min of cross-linking with 0.05–1% formaldehyde.39,40 A range of reaction conditions can be easily tested with CE separation to determine conditions that favor high yields for a particular PPI. Cross-linker concentration and reaction time are considered largely complimentary with a general increase in yield expected for an increase in either.28,29,38
Figure 2.
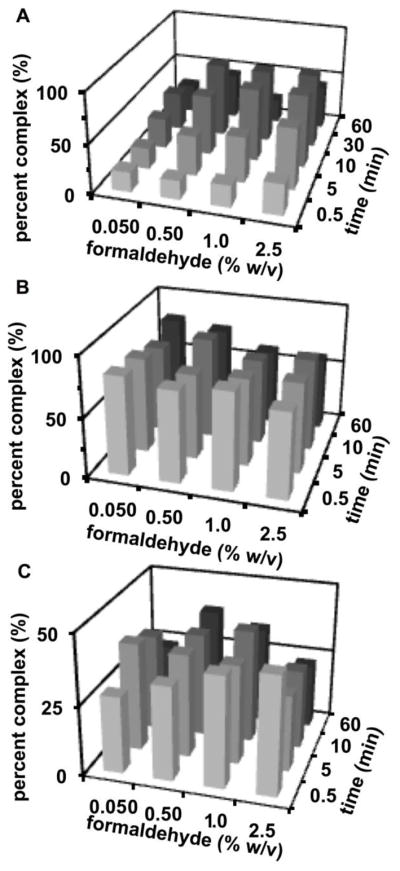
Dependence of (A) 25 nM Hsp70-488 and 100 nM Bag3 (B) 50 nM Hsp90-488 and (C) 10 nM FITC-lysozyme and 20 nM antibody on dimer complex detected on concentration of formaldehyde and cross-linking reaction time.
An increase in Hsp70-Bag3 complex peak area was detected with increasing formaldehyde concentration or time up to 2.5% formaldehyde or 30 min of cross-linking, respectively, while FITC-lysozyme-anti-lysozyme appeared relatively insensitive to the times assayed with 0.5 to 1% formaldehyde. The amount of Hsp90 dimer complex observed was fairly stable for all reaction times and concentrations assayed. Interestingly, the amount of complex detected decreased if the cross-linking reaction was allowed to proceed for 60 min. A trend toward decreasing apparent yields of complex at high cross-linking times has been previously reported by mass spectrometry.38 This effect may be due to the formation of higher molecular weight aggregates, which are not injected onto the gel columns. For all complexes, the amount of complex observed was within a range of 13% for 10 min of cross-linking reaction with 0.5–2.5% formaldehyde. These results show that, at least for these proteins, finding conditions for maximal complex formation is straightforward and the results will be stable over a wide range of conditions.
To favor high yields of cross-linking and short reaction times, an intermediate cross-linking reaction condition of 10 min and 1% formaldehyde was chosen for further assay testing. To minimize artifacts when cross-linking high concentrations of protein28,29, interacting proteins were investigated in the nanomolar to low micromolar concentration range. This concentration range is also more useful for determining quantitative binding data for many proteins.
PXCE fundamentally requires cross-linkable residues in the protein-protein interaction site. Although formaldehyde is efficient and reacts with many residues, other PPIs may benefit from different cross-linkers with longer spacer arms or more controlled reactivity.41 Information about the PPI site may facilitate the choice of cross-linker.
Determination of Binding Affinities
To determine if PXCE allows for quantitative affinity information to be obtained, saturation binding assays were performed and the data fit by non-linear regression to determine Kd (Figure 3). Hsp70-488 and Bag3 were found to interact with a Kd = 25 ± 5 nM by PXCE. Previously the Kd for this pair has been reported as 23 ± 8 nM with Hsp70–488 by APCE and 15 ± 2 nM with unlabeled Hsp70 by ITC, both without cross-linking.4 Similar Kd values were found using 5, 10 and 20 min cross-linking reactions (Figure S-3, Table S-5). Hsp90-488 was found to form a homodimer with Kd = 2.6 ± 0.3 nM by PXCE. The Kd of Hsp90 homodimerization was previously reported to be 60 ± 12 nM by size exclusion chromatography. In this technique, association and dissociation occur over the timescale of the separation, and the elution time is used as an indicator of the degree of dimerization.42 A dissociation constant for the Hsp90 homodimer could not be obtained by ITC possibly due to the limitations with quantifying homodimers with nanomolar Kd values by ITC. The Kd value determined for FITC-lysozyme-anti-lysozyme by PXCE was determined to be 24 ± 3 nM by PXCE and 17 ± 2 nM by ITC with 1:2 antibody to FITC-lysozyme stoichiometry (Figure S-4). Thus, for Hsp70-Bag3 and FITC-lysozyme-anti-lysozyme PXCE gave Kd values similar to ITC, which used non-cross-linked proteins. The largest discrepancy was for Hsp90 dimerization and may be due to differences in the conditions used for the interacting proteins.
Figure 3.

Determination of dissociation constant (Kd) for (A,B) Hsp70-488-Bag3 (C,D) Hsp90-488 dimer and (E,F) FITC-lysozyme-antibody. Electropherograms for (A) 25 nM Hsp70-488 at increasing concentrations of Bag3, (C) 1, 20 and 100 nM Hsp90-488 and (E) 10 nM FITC-lysozyme with increasing concentrations of monoclonal antibody (mAb). Non-linear regression of (B) 25 ± 5 nM for Hsp70-488-Bag3, (D) 2.6 ± 0.3 nM for Hsp90-488 and (F) determined a Kd 24 ± 3 nM FITC-lysozyme-antibody. Error bars are standard deviation (n = 3).
To determine if PXCE is useful for ranking PPI affinities, a competitive binding experiment was carried out with Hsp70 proteins containing mutations in key residues within the Bag3 interaction site that have previously been reported to inhibit Hsp70-Bag1 interactions (Figure 4A).43 Unlabeled wild type and mutant Hsp70 were titrated into a fixed concentration of Bag3 and Hsp70–488 to determine the affinity of unlabeled Hsp70 for Bag3. In these experiments, adding unlabeled Hsp70 variants decreased the complex peak area for Hsp70–488 allowing quantification. A higher concentration of mutant Hsp70 than wild type Hsp70 was required to compete with the Hsp70–488 for Bag3 binding (Figure 4). The data were normalized to positive (no Bag3 added) and negative controls (no unlabeled Hsp70) to quantify the percent inhibition. The inhibitory constant (Ki) was determined to be 9 ± 2 nM for unlabeled wild type Hsp70, 160 ± 60 nM for Hsp70 E,D 283, 292 A,A and 400 ± 200 nM for Hsp70 R,R 258,262 A,A by PXCE. The inhibitory constant for the unlabeled wild type Hsp70 is lower than the Kd found for the Hsp70–488, in agreement with the Kd = 15 ± 2 nM previously reported by ITC for the unlabeled Hsp70-Bag3.4 For comparison, inhibitory constants were determined by FCPIA with similar Ki = 15 ± 8 nM determined for the wild type and 190 ± 30 for the E,D 283, 292 A,A mutant and 900 ± 300 for the R,R 258, 262 A,A mutant. In FCPIA one binding partner is immobilized on a bead while the other is fluorescently labeled, the binding partners are incubated and a flow cytometer is used to determine bead associated fluorescence. There are multiple differences in the PXCE and FCPIA assay formats including different fluorescent labels and the requirement of FCPIA to immobilize one of the binding partners on a bead, which could interfere with the PPI affinity. Despite these assay differences, the rank order of FCPIA and PXCE for the wild type and mutant interactions is the same (Figure 4B). Kis were also in good agreement with the largest discrepancy being a factor of two higher Ki for the Hsp70 R,R 258 262 A,A mutant by FCPIA.
Figure 4.

Determination of inhibition constant (Ki) of Hsp70 proteins by (A) PXCE. Increasing concentration of unlabeled Hsp70 (Wild-Type), Hsp70 E,D 283, 292 A,A or Hsp70 R,R 258, 262, A,A with 25 nM Hsp70-488 and 50 nM Bag3. Error bars are standard deviation (n = 3). (B) Comparison of Ki values obtained by PXCE and FCPIA.
Quantification of PPI Small Molecule Inhibitors
We next examined the possibility of using PXCE to determine the inhibition of PPI by small molecules. We tested 3 compounds that are known to inhibit the Hsp70-Bag3 interaction: JG-311, JG-98, and JG-231.44–46 As shown in Figure 5, PXCE allowed detection of protein complex inhibition for these compounds. IC50 values were determined to be 1.3 ± 0.1 μM, 800 ± 200 nM and 400 ± 200 nM for JG-231, JG-98 and JG-311, respectively. The small molecules were all found to have similar IC50 values with similar associated errors by dose response using both PXCE and FCPIA assays (Figure S-5).
Figure 5.

Quantification of small molecule inhibitors by PXCE. Electropherograms of (A) Hsp70-Bag3 negative control and Hsp70-488-Bag3 in the presence of fluorescent inhibitor. (B) Dose response curves for JG-231, JG-98 and JG-311. Log(IC50) values were determined to be −5.9 ± 0.1 for JG-231, −6.1 ± 0.3 for JG-98 and −6.3 ± 0.2 for JG-311. JG-258 was used as negative control and 1 μM unlabeled Hsp70 was used as a positive control. Error bars are range of two trials. (C) Comparison of IC50 values obtained by PXCE and FCPIA.
These experiments also illustrate a potential advantage of PXCE for screening of new modulators of PPI. All of the tested molecules are fluorescent. Fluorescent drug molecules can interfere with detection of PPI modulators in many fluorescence assays. PXCE has the advantage of identifying potentially interfering fluorescent small molecules on the basis of extra peaks and allows for separation of the fluorescent small molecules from free protein and protein complex. Using a gel electrophoresis separation, small molecule compounds migrated much more rapidly than the high molecular weight proteins (Figure 5A) allowing for quantification of inhibitor potency despite fluorescence of small molecule. The denaturing gel conditions likely also promoted dissociation of the small molecule from the protein allowing for separation without quantitative interference. The limiting factor is that strongly fluorescent molecules can dominate the electropherograms at high concentrations. In this case, the assay allowed for quantification of free and bound protein peaks for up to 50 μM JG-311, JG-98 and JG-231.
Implications and Limitations
This work demonstrates the utility of formaldehyde cross-linking for nanomolar interaction affinities. It has been reported that equilibrium shifts are minimized and quantitative information is attainable with cross-linking for high affinity complexes when free proteins that react with the cross-linker do not then bind and cross-link to other proteins and when high efficiency cross-linkers are used in large excess.47 Cross-linking of rapidly dissociating PPIs may be challenging as it has been reported that such PPIs are not readily captured with formaldehyde cross-linking.48 Faster reacting reagents may be preferable for such cases to eliminate the chance of complex dissociation or additional complex formation. Despite these caveats, the data presented suggest that formaldehyde with PXCE will be broadly useful for different types of proteins and for different applications such as affinity determinations and detection of small molecule modulators.
This work demonstrated the application of PXCE to dimers and immunocomplexes; however, it may be possible to investigate more complex interactions using PXCE. In many cases multimeric complexes are of interest and alter protein function. As long as the proteins remain within the size range of gel used, it should be possible to detect and quantify the various complexes. PXCE may also be useful for studies of amyloid aggregates which have previously been investigated by CE.7,49
Conclusion
PXCE allows for quantifying protein-protein interaction affinities of target complexes including the lysozyme-anti-lysozyme immunocomplex, Hsp70-Bag3 heterodimer and Hsp90 homodimers. CE of protein complexes that are not cross-linked, as in traditional APCE, can be limited by the difficulty of developing methods that maintain complexes but also allow separations. Use of cross-linking allows for simplified method development compared to APCE by making PPIs amenable to harsh separation conditions and separation times longer than the typical time-scale of the PPI. This development should make PXCE, with the associated advantages of speed, low sample consumption, resolution, and quantification, applicable to a wider range of proteins and accessible to more labs. CE-LIF allows for sensitive detection of PPIs with the requirement of having binding partners be fluorescently labeled for LIF detection. Quantitative information can be obtained for small molecule modulators including fluorescent molecules suggesting that PXCE may be a valuable strategy for characterizing such molecules. CE, especially in microchip format, has the advantage of providing rapid separations, suggesting the possibility of use of PXCE for high-throughput screening.28,50
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01GM102236 to R.T.K. and NIH grant NS059690 to J.E.G.
Footnotes
Supporting information for this article includes electropherograms demonstrating the challenges with separating complexes of interest by free solution or gel electrophoresis, additional saturation binding curves for the Hsp70-Bag3 interaction, isothermal calorimetry and flow cytometry protein interaction assays for comparison with PXCE results.
References
- 1.Heegaard NHH. Electrophoresis. 2009;30:229. [Google Scholar]
- 2.Shimura K, Karger BL. 1994;66(1):9. doi: 10.1021/ac00073a004. [DOI] [PubMed] [Google Scholar]
- 3.Hafner FT, Kautz RA, Iverson BL, Tim RC, Karger BL. Anal Chem. 2000;72(23):5779. doi: 10.1021/ac000853+. [DOI] [PubMed] [Google Scholar]
- 4.Rauch JN, Nie J, Buchholz TJ, Gestwicki JE, Kennedy RT. Anal Chem. 2013;85:9824. doi: 10.1021/ac4023082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimura K, Waki T, Okada M, Toda T, Kimoto I, Kasai KI. Electrophoresis. 2006;27(10):1886. doi: 10.1002/elps.200500239. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen JT, Østergaard J, Houen G, Heegaard NHH. Electrophoresis. 2008;29(8):1723. doi: 10.1002/elps.200700618. [DOI] [PubMed] [Google Scholar]
- 7.Picou RA, Schrum DP, Ku G, Cerqua RA, Kheterpal I, Gilman SD. Anal Biochem. 2012;425(2):104. doi: 10.1016/j.ab.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Schultz NM, Kennedy RT. Anal Chem. 1993;65(9):3161. [Google Scholar]
- 9.Lassen KS, Bradbury ARM, Rehfeld JF, Heegaard NHH. Electrophoresis. 2008;29(12):2557. doi: 10.1002/elps.200700908. [DOI] [PubMed] [Google Scholar]
- 10.Wan QH, Le XC. Anal Chem. 1999;71(19):4183. doi: 10.1021/ac9902796. [DOI] [PubMed] [Google Scholar]
- 11.Mohamadi MR, Kaji N, Tokeshi M, Baba Y. Anal Chem. 2007;79(10):3667. doi: 10.1021/ac0623890. [DOI] [PubMed] [Google Scholar]
- 12.Shi M, Zhao S, Huang Y, Liu YM, Ye F. J Chromatogr B Anal Technol Biomed Life Sci. 2011;879(26):2840. doi: 10.1016/j.jchromb.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 13.Babu CVS, Chung BC, Lho DS, Yoo YS. J Chromatogr A. 2006;1111(2):133. doi: 10.1016/j.chroma.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 14.Berezovski M, Krylov SN. J Am Chem Soc. 2002;124(46):13674. doi: 10.1021/ja028212e. [DOI] [PubMed] [Google Scholar]
- 15.Thuy Tran N, Taverna M, Miccoli L, Angulo JF. Electrophoresis. 2005;26(16):3105. doi: 10.1002/elps.200400091. [DOI] [PubMed] [Google Scholar]
- 16.Liyanage R, Krylova SM, Krylov SN. J Chromatogr A. 2013;1322:90. doi: 10.1016/j.chroma.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Xing J, Tan W, Lam M, Carnelley T, Weinfeld M, Le XC. Anal Chem. 2002;74(15):3714. doi: 10.1021/ac0201979. [DOI] [PubMed] [Google Scholar]
- 18.Yang PL, Whelan RJ, Mao YW, Lee AWM, Carter-Su C, Kennedy RT. Anal Chem. 2007;79(4):1690. doi: 10.1021/ac061936e. [DOI] [PubMed] [Google Scholar]
- 19.de Jong S, Epelbaum N, Liyanage R, Krylov SN. Electrophoresis. 2012;33(16):2584. doi: 10.1002/elps.201200153. [DOI] [PubMed] [Google Scholar]
- 20.German I, Buchanan DD, Kennedy RT. Anal Chem. 1998;70(21):4540. doi: 10.1021/ac980638h. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Wang Z, Li XF, Le XC. Angew Chemie - Int Ed. 2006;45(10):1576. doi: 10.1002/anie.200503345. [DOI] [PubMed] [Google Scholar]
- 22.Lauer HH, McManigill D. Anal Chem. 1986;58:166. [Google Scholar]
- 23.Mccormick RM. Anal Chem. 1988;60(21):2322. doi: 10.1021/ac00172a003. [DOI] [PubMed] [Google Scholar]
- 24.Verzola B, Gelfi C, Righetti PG. J Chromatogr A. 2000;874(2):293. doi: 10.1016/s0021-9673(00)00090-x. [DOI] [PubMed] [Google Scholar]
- 25.Bushey MM, Jorgenson JW. J Chromatogr. 1989;480:301. doi: 10.1016/s0021-9673(01)84299-0. [DOI] [PubMed] [Google Scholar]
- 26.Green JS, Jorgenson JW. J Chromatogr. 1989;478:63. [Google Scholar]
- 27.Muskotál A, Kokol V. Electrophoresis. 2010;31(6):1097. doi: 10.1002/elps.200900525. [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Flynn GC. J Chromatogr B. 2009;877(27):3012. doi: 10.1016/j.jchromb.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 29.Sutherland BW, Toews J, Kast J. J Mass Spectrom. 2008;43(7):699. doi: 10.1002/jms.1415. [DOI] [PubMed] [Google Scholar]
- 30.Blair LJ, Sabbagh JJ, Dickey CA. Expert Opin Ther Targets. 2015;18(10):1219. doi: 10.1517/14728222.2014.943185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Assimon VA, Gillies AT, Rauch JN, Gestwicki JE. Curr Pharm Des. 2013;19(3):404. doi: 10.2174/138161213804143699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang L, Thompson AD, Ung P, Carlson HA, Gestwicki JE. J Biol Chem. 2010;285(28):21282. doi: 10.1074/jbc.M110.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyata Y, Chang L, Bainor A, Mcquade TJ, Walczak CP, Zhang Y, Larsen MJ, Kirchhoff P, Gestwicki JE. J Biomol Screen. 2010;15(10):1211. doi: 10.1177/1087057110380571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shemetov AA, Gusev NB. Arch Biochem Biophys. 2011;513(1):1. doi: 10.1016/j.abb.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Shackman JG, Watson CJ, Kennedy RT. J Chromatogr A. 2004;1040(2):273. doi: 10.1016/j.chroma.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 36.Uversky VN. Expert Opin Drug Discov. 2012;7(6):475. doi: 10.1517/17460441.2012.686489. [DOI] [PubMed] [Google Scholar]
- 37.Ganzler K, Greve KS, Cohen AS, Karger BL, Guttman A, Cooke NC. Anal Chem. 1992;64(22):2665. doi: 10.1021/ac00046a003. [DOI] [PubMed] [Google Scholar]
- 38.Vasilescu J, Guo X, Kast J. Proteomics. 2004;4(12):3845. doi: 10.1002/pmic.200400856. [DOI] [PubMed] [Google Scholar]
- 39.Klockenbusch C, O’Hara JE, Kast J. Anal Bioanal Chem. 2012;404(4):1057. doi: 10.1007/s00216-012-6065-9. [DOI] [PubMed] [Google Scholar]
- 40.Solomon MJ, Larsen PL, Varshavsky A. Cell. 1988;53(6):937. doi: 10.1016/s0092-8674(88)90469-2. [DOI] [PubMed] [Google Scholar]
- 41.Mattson G, Conklin E, Desai S, Nielander G, Savage MD, Morgensen S. Mol Biol Rep. 1993;17(3):167. doi: 10.1007/BF00986726. [DOI] [PubMed] [Google Scholar]
- 42.Richter K, Muschler P, Hainzl O, Buchner J. J Biol Chem. 2001;276(36):33689. doi: 10.1074/jbc.M103832200. [DOI] [PubMed] [Google Scholar]
- 43.Briknarová K, Takayama S, Brive L, Havert ML, Knee DA, Velasco J, Homma S, Cabezas E, Stuart J, Hoyt DW, Satterthwait AC, Llinás M, Reed JC, Ely KR. Nat Struct Biol. 2001;8(4):349. doi: 10.1038/86236. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Srinivasan SR, Connarn J, Ahmad A, Young ZT, Kabza AM, Zuiderweg ERP, Sun D, Gestwicki JE. ACS Med Chem Lett. 2013;4(11):1042. doi: 10.1021/ml400204n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Colvin T, Rauch JN, Acosta-Alvear D, Kampmann M, Dunyak B, Hann B, Aftab BT, Murnane M, Cho M, Walter P, Weissman JS, Sherman MY, Gestwicki JE. Mol Cancer Ther. 2015;14(3):642. doi: 10.1158/1535-7163.MCT-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, Shishido T, Chen LB. Cancer Res. 1996;56(3):538. [PubMed] [Google Scholar]
- 47.Mädler S, Seitz M, Robinson J, Zenobi R. J Am Soc Mass Spectrom. 2010;21(10):1775. doi: 10.1016/j.jasms.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 48.Schmiedeberg L, Skene P, Deaton A, Bird A. PLoS One. 2009;4(2):e4636. doi: 10.1371/journal.pone.0004636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pryor NE, Moss MA, Hestekin CN. Electrophoresis. 2014:1814. doi: 10.1002/elps.201400012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bousse L, Mouradian S, Minalla a, Yee H, Williams K, Dubrow R. Anal Chem. 2001;73(6):1207. doi: 10.1021/ac0012492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.