

Abstract
The common chemical and biological properties of antisense oligonucleotides provide the opportunity to identify and characterize chemical class effects across species. The chemical class that has proven to be the most versatile and best characterized is the 2′-O-methoxyethyl chimeric antisense oligonucleotides. In this report we present an integrated safety assessment of data obtained from controlled dose-ranging studies in nonhuman primates (macaques) and healthy human volunteers for 12 unique 2′-O-methoxyethyl chimeric antisense oligonucleotides. Safety was assessed by the incidence of safety signals in standardized laboratory tests for kidney and liver function, hematology, and complement activation; as well as by the mean test results as a function of dose level over time. At high doses a number of toxicities were observed in nonhuman primates. However, no class safety effects were identified in healthy human volunteers from this integrated data analysis. Effects on complement in nonhuman primates were not observed in humans. Nonhuman primates predicted safe doses in humans, but over predicted risk of complement activation and effects on platelets. Although limited to a single chemical class, comparisons from this analysis are considered valid and accurate based on the carefully controlled setting for the specified study populations and within the total exposures studied.
Introduction
Over the past 25 years, antisense technology has been developed and validated as a new platform for target validation, drug discovery, and development.1,2,3,4,5,6,7,8 The technology has proven to be versatile. Essentially all cellular RNAs including nuclear retained noncoding RNAs, pre mRNAs, cytoplasmic noncoding RNAs and mRNAs have been shown to be approachable with antisense oligonucleotides (ASOs).1,9 ASOs can be designed to exploit a variety of terminating mechanisms after hybridizing to the target RNA.5,6,7 Target RNAs may be degraded by recruitment of RNase H1 (DNA-like ASOs) or Ago2 (siRNA or ssRNA). ASOs can be designed to inhibit translation or alter splicing and recently, ASOs have been designed to alter splicing to increase the production of proteins, in effect, serving as agonists.4,10
Although, off-target effects could potentially occur both because of hybridization to mismatched sites in nucleic acids and interactions with proteins, it has proven quite straightforward to avoid most off-target effects and thus ASOs can be highly selective.11 Consequently, ASOs are ideal agents to alter a single member of closely related gene families and even mutant versus normal gene products.12 Thus, they have proven to be ideal agents for target validation and may be optimal agents in the treatment of many diseases.
The antisense drug discovery process is substantially more efficient than other technologies. The efficiencies derive from several attributes. The factors that determine the affinity of the interaction with targeted RNAs (Watson-Crick hybridization) are well understood and can be used to create very robust, rapid and efficient screening systems. RNA structure and intermediary metabolism are well understood and modern sequencing methods are enabling even more detailed understanding of the diversity of RNAs and their structures in cells. Factors that may alter the ability of ASOs to interact with their cognate sites (RNA structure, protein binding, and RNA metabolism) are sufficiently understood to be incorporated into screening processes. Very rapid and inexpensive processes are also available to precisely characterize target specificity at the RNA level.
As therapeutic agents, ASOs do have limitations, they have a relatively slow onset of action, and so are not candidates for ultra-acute diseases such as myocardial infarction or stroke. Though ASOs distribute broadly, some tissues e.g., skeletal muscle, are low accumulators and require higher doses. Additionally, ASOs do not cross an intact blood brain barrier, so ASOs to treat CNS diseases must be given intrathecally.13,14 Adverse events such as local cutaneous reactions at the injection site and flu-like symptoms have also been observed and have resulted in some patients discontinuing therapy.15 However advances in screening, have supported the development of ASOs with minimal injection site reactions and flu-like symptoms observed in clinical trials.16,17,18,19 These advances include more extensive screening of sites in the target RNA in conjunction with (i) usage of algorithms to filter out sequences and structural motifs, such as CpG, that have been identified as problematic, and sequences that are complementary to nontarget RNAs, and (ii) incorporation of in vitro and in vivo evaluations designed to identify sequences that may produce an exaggerated proinflammatory effect.20,21,22,23
Numerous chemical classes of ASOs have been created with several having advantageous properties, such as greater potency, fewer toxicity and tolerability issues.5 Within a chemical class of ASOs, all drugs have similar structures, physical and chemical properties, including similar lengths, molecular weight, charge, solubility, presence of nucleobases, phosphorothioate moieties, same 2′-modifications, and stability. These shared physical-chemical attributes result in drugs with similar pharmacokinetics, potencies, adverse events, and tolerability.22,24,25,26,27 Consequently, investments in process chemistry, formulations, analytical methods, and manufacturing and development of advances such as an integrated safety database can be amortized across an entire pipeline.28
These common properties also afford the opportunity to identify and characterize chemical class effects across species. The chemical class that has proven to be the most useful and well characterized is the 2′-O- methoxyethyl (2′MOE) chemical class.8,22,24,25,26,27,28 We have taken advantage of this attribute to create an integrated safety database that includes data from 2′MOE ASOs studied in monkeys (the primary nonrodent toxicology species) and humans.29 2′MOE ASOs designed to recruit RNase H1 have a 2′-deoxynucleotide gap plus 2′MOE wings (see Supplementary Figure S1). These compounds are referred to as 2′MOE chimeric ASOs.
In this report, we summarize the safety of the 2′MOE chimeric ASOs that we have studied in normal human volunteers and compare that experience to observations in the nonhuman primate (NHP), cynomolgus and rhesus macaques. As a first step, in the current publication, clinical data are limited to those from normal human volunteer studies to exclude disease related effects. In subsequent reports we will examine longer-term treatments in various patient groups. Safety was assessed using standardized laboratory tests for markers of injury or function of organs, tissues, and cells considered as potential targets of drug toxicity in humans based on (i) the generic disposition of the 2′MOE chimeric ASOs, (ii) prior observations from individual clinical trials on one or more of the ASO drugs, (iii) other toxicities identified and characterized in nonclinical NHP studies, and (iv) to clarify the central results. Assessments of both nonclinical and clinical data sets were based on the incidence of clinically-meaningful signals and the mean results by dose and exposure for each selected laboratory test.
Although the NHP has proven to be an effective toxicological model for ASOs, monkeys appear to be more sensitive to ASO-induced complement activation than humans and other species.22,30,31 Chronic low level activation can lead to depletion of complement, sensitivity to infectious disease and damage to the vascular system and kidney.22 Fortunately, none of these effects have been observed in humans, and particular attention has focused on complement activation in clinical trials of ASOs. Because other publications have described these effects in detail, the current publication will focus on a comparison of ASO effects on complement split products in the monkey and healthy human volunteers. Additionally, ASOs are well known to induce transient increases in activated partial thromboplastin time by inhibiting the intrinsic tenase complex.22,32 These effects are not cumulative and have not been associated with any clinical sequelae, so will not be considered here.
To focus on the chemical class-related adverse events, data associated with a target-specific effect were excluded, such as ALT increases caused by mipomersen (ASO 4, see Supplementary Table S1), as these results are clearly related to rapid reduction of the target, apoB-100.33,34,35 In fact, the only observations excluded in this translational analysis were the hepatic transaminase test results for two ASOs, one with ALT increases associated with apoB-100 reduction (ASO 4) and the other with ALT increases associated with glucagon receptor reduction (ASO 11). Thus the data reported represent a broad unbiased report of our experience with adverse effects that can be considered chemical class-related adverse effects. To demonstrate an ASO that does produce a change in laboratory parameters, albeit target related, the effects of mipomersen on liver function in normal volunteers are summarized.
Results
Data were derived from the nonclinical studies in NHPs and randomized placebo-controlled dose-ranging phase 1 trials in healthy human volunteers for twelve 2′MOE chimeric ASOs (Figure 1 and Supplementary Table S1). The nonclinical study population of NHPs was comprised of 710 monkeys (658 cynomolgus and 52 rhesus macaque) of near equal distribution between sexes (333 female and 377 male), ranging in age from 2 to 8 years, and 2–8 kg in weight. The clinical study population of healthy volunteers was comprised of 750 human subjects (179 women and 571 men), predominantly white (84.4%) who ranged in age from 18 to 70 years (mean age, 41.3 ± 13.0) and were marginally overweight (mean body mass index, 26.0 ± 3.3). Demographics and baseline characteristics of the clinical study population are summarized by dose level in Supplementary Table S2.
Figure 1.

Subject data distribution and sequence diversity of second generation 2′-O-methoxyethyl (2′MOE) chimeric antisense oligonucleotides (ASOs) included in the translational safety database analysis. (a) subject data distribution by molecular target for each of the 12 ASOs under study (see also, Supplementary Table S1), and (b) positional frequency of nucleotide residues in the gap-aligned 2′MOE chimeric ASO sequences.52 Gap design indicates wing (5′ and 3′) and gap nucleotide span. Number in parenthesis indicates the number of ASOs per design.
Sample size and sequence diversity
To determine if the number of 2′MOE chimeric ASOs evaluated is sufficient to support conclusions about potential class effects, we took two approaches. First, the ASOs were independently selected after extensive screening of sites in each target RNA.20,21 So in effect, they represent a “random” sample of sequence space within the RNAs tested. We found no significant over representation of sub-sequences common to the ASO sequences studied, and only a handful of pentamer and hexamer sequence motifs appeared to be over-represented in our sample set relative to the sequence space of screened ASOs (see Supplementary Table S3). Second, we examined the sequences studied to see if there was significant bias. There were very few biases at any site for any nucleotide in the gap-aligned sequences (Figure 1), again emphasizing the “randomness” of the 2′MOE chimeric ASO collection.
Safety laboratory tests
The current integrated analysis included laboratory tests for kidney (serum creatinine, blood urea nitrogen (BUN), glomerular filtration rate, and urine protein), liver (alanine transaminase, aspartate transaminase (AST), total bilirubin, alkaline phosphatase, albumin, and prothrombin), hematology (platelets, absolute neutrophil count ), lymphocytes, hemoglobin, and hematocrit) and complement (complement factor 3 (C3), complement split products Bb and C5a). To evaluate the generic effects of 2′MOE chimeric ASOs on liver, two ASOs (ASO 4 and ASO 11, directed against apoB100 and GCGR, respectively) were excluded from the transaminase analyses (ALT and AST) because of target-specific liver effects.33,36
Nonclinical evaluation of the specified 2′MOE chimeric ASOs included data from both nonGLP and Good Laboratory Practice (GLP) studies (n = 20) in NHPs with treatment periods up to 13 weeks. Treatment periods for the respective randomized placebo-controlled studies (n = 15) in healthy human volunteers ranged up to 6 weeks. For the clinical evaluation, mean results by dose number were based on data from studies with a multidose regimen, where study drug was administered every other day in the first week of dosing, with or without weekly once dosing thereafter (see Supplementary Figure S2).
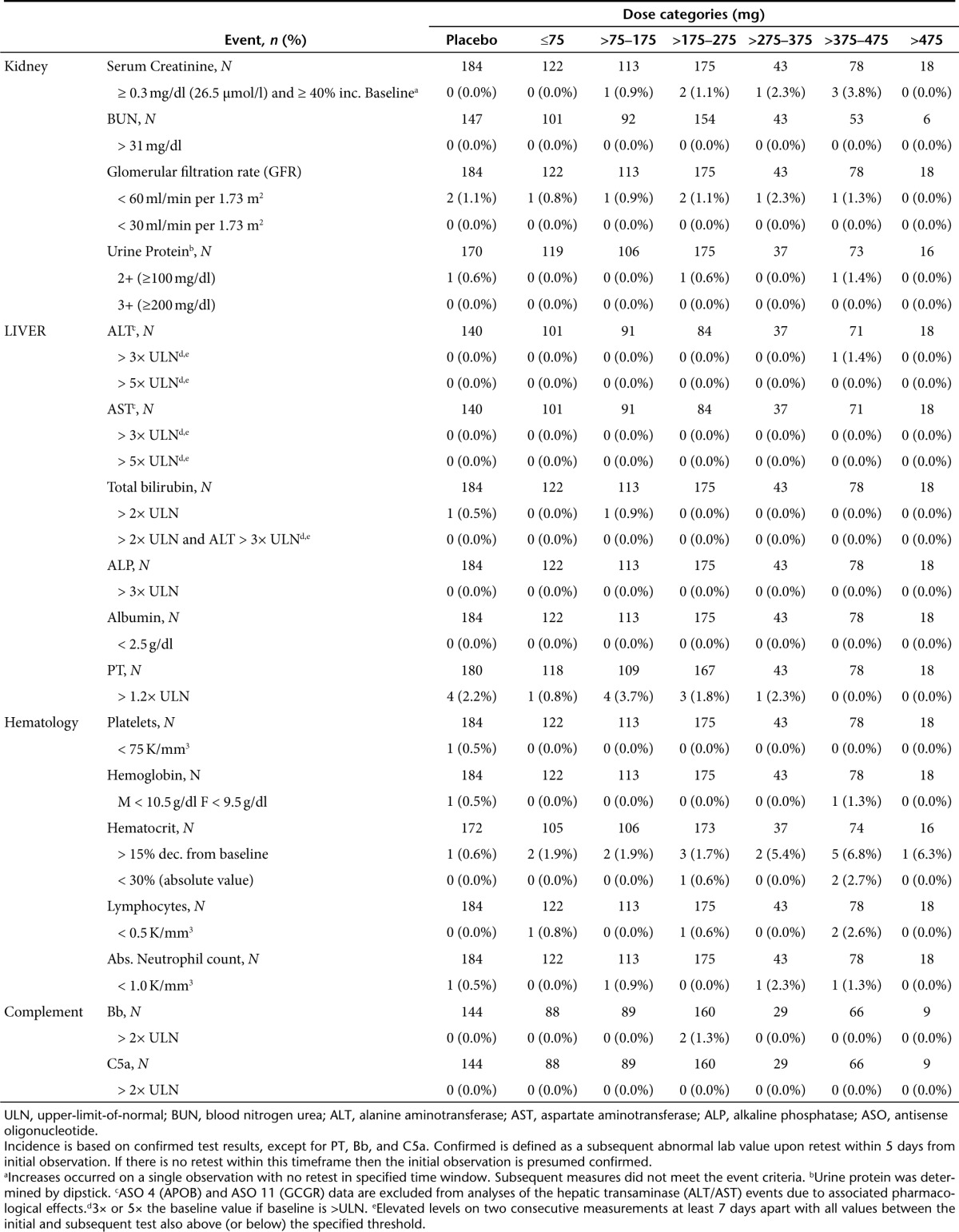
Kidney. In NHPs, no dose-dependent changes in renal parameters were identified (Figure 2a,c). Even at the highest doses studied, i.e., >24 mg/kg/wk, no remarkable changes in mean serum creatinine or BUN were observed. Similarly in healthy human volunteers, no meaningful changes in serum creatinine or BUN were observed. In both species, mean levels were statistically different at certain doses and time points compared with the control group (see Supplementary Table S4), but these changes in mean levels were small and transient. Furthermore, the incidence of an increase in creatinine (>0.3 mg/dl and 1.4-fold increase from baseline) or BUN (>1× upper-limit-of-normal (ULN)) in ASO-treated NHPs, was comparable with the controls (Table 1). Although a dose-dependent trend was observed in the incidence of an increase in creatinine in humans (Table 2), further investigation of the individual subject data found that these increases occurred in a single observation that was not confirmed by a repeat test. There were no dose-dependent changes in urinary protein. The incidence of 2 + (100 mg/dl) was 0.3% in ASO-treated subjects compared with 0.6% in placebo-treated subjects. No dose-dependent changes were evident in the mean levels of creatinine or BUN, or mean glomerular filtration rate, over time (Figure 2b,d,e). Thus, we conclude that there is no evidence of drug-associated effects on renal function in NHPs or normal humans.
Figure 2.

Kidney laboratory test results from nonhuman primates and healthy human volunteers treated with multiple doses up to 13 and 6 weeks, respectively, by dose group. (a,b) Serum creatinine levels in nonhuman primates (ULN = 1.1 mg/dl) and healthy human volunteers (LLN = 0.5 and ULN = 1.4 mg/dl); (c,d) Blood nitrogen urea (BUN) levels in nonhuman primates (ULN = 32 mg/dl) and healthy human volunteers (LLN = 5 and ULN = 22 mg/dl); (e) Glomerular filtration rate (GFR) in healthy human volunteers. GFR was derived using the MDRD equation: 170 × (serum creatinine (mg/dl))−0.999 × (age)−0.176 × (0.762, if subject is female) × (1.180, if subject is black) × (serum urea nitrogen (mg/dl))−0.170 × (serum albumin (g/dl))0.318. Data presented is the mean ± SE. Each data point represents data from at least 10 subjects, four ASOs and four independent studies. Dashed lines indicate the laboratory test reference range values and yellow y-axis markers indicate the event criteria. A tabulated summary of results is provided in Supplementary Table S4. ULN, upper-limit-of-normal; LLN, lower-limit-of-normal; ASO, antisense oligonucleotide.
Table 1. Incidence of abnormal laboratory test results in nonhuman primates.
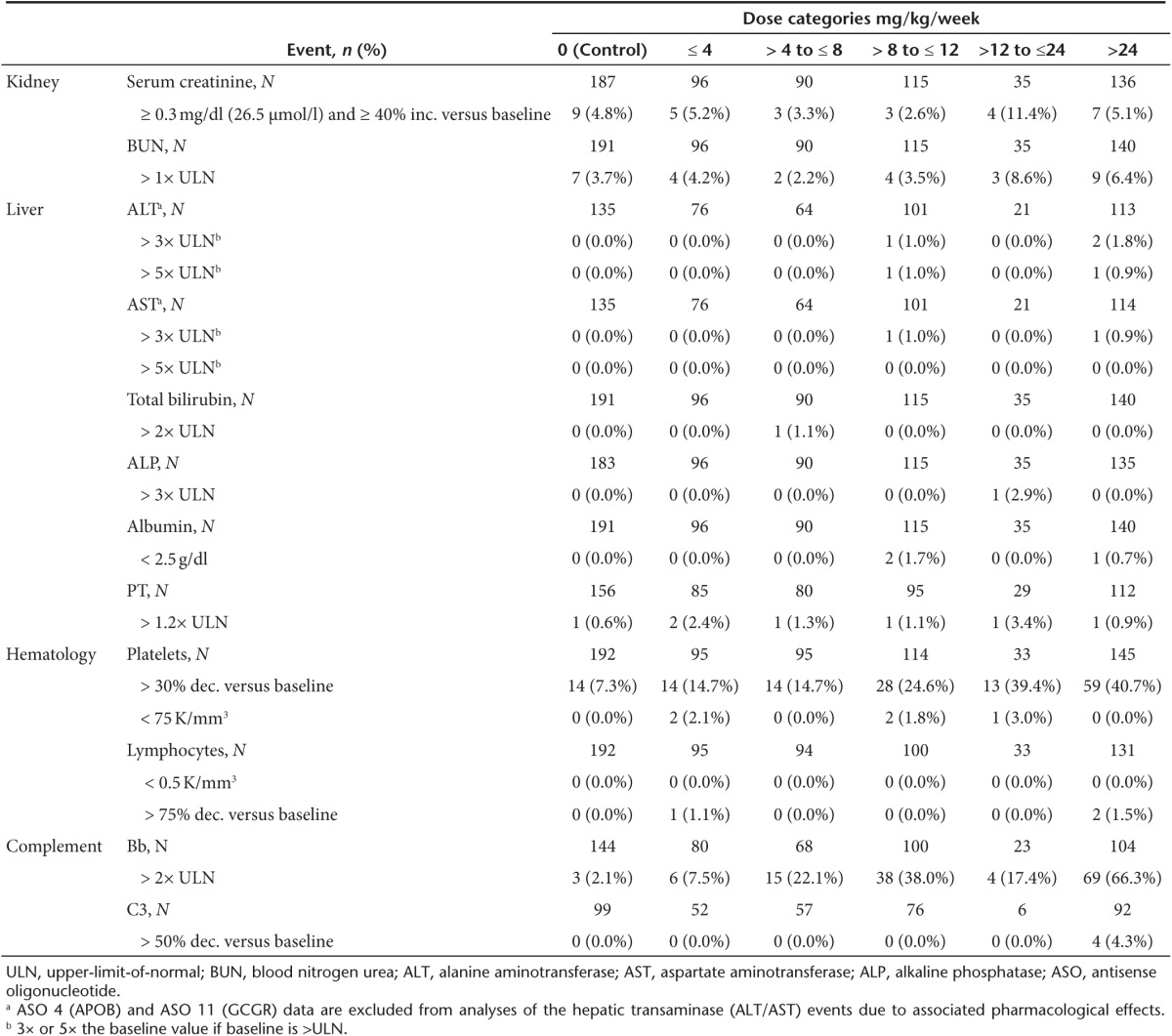
Table 2. Incidence of abnormal laboratory test results in healthy human volunteers.
Liver. There was no marked effect of ASO treatment on liver function over time or by dose in NHPs (Figure 3 and Supplementary Table S5). The minimal incidence of clinically meaningful increases in ALT (3 of 375, 0.8%), AST (2 of 376, 0.5%), or total bilirubin (1 of 476, 0.2%) had no apparent dose relationship (Table 1). At >24 mg/kg/wk there were two monkeys from one study with ALT levels >3× ULN, one of which reached >5× ULN. The monkey observed to have ALT elevations >5× ULN in the >8–12 mg/kg/wk dose group was treated with a different ASO, from the single monkey with an elevation in AST >3× ULN in the >8–12 mg/kg/wk group and the single monkey with an elevation in total bilirubin >2× ULN in the >4–8 mg/kg/wk group. NHPs were treated with different 2′MOE chimeric ASOs, so there was no pattern of observations with a specific sequence, and thus, there is no dose relationship or sequence pattern that is evident. Thus, these data argue against the possibility of a 2′MOE chimeric ASO class effect on liver function in the NHP.
Figure 3.

Liver laboratory test results from nonhuman primates and healthy human volunteers treated with multiple doses up to 13 and 6 weeks, respectively, by dose group. (a,b) Alanine aminotransferase (ALT) levels in nonhuman primates (ULN = 105 U/L) and healthy human volunteers during (LLN = 6 and ULN = 41 U/L); (c,d) Aspartate aminotransferase (AST) levels in nonhuman primates (ULN = 120 U/L) and healthy human volunteers (LLN = 9 and ULN = 34 U/L); (e,f) Total bilirubin levels in nonhuman primates (ULN = 0.6 mg/dl) and healthy human volunteers (LLN = 0.1 and ULN = 1.1 mg/dl); (g,h) Alkaline phosphatase (ALP) levels in nonhuman primates (ULN = 133 U/dl) and healthy human volunteers (LLN = 37and ULN = 116 U/L); (i,j) Serum albumin levels in nonhuman primates (ULN = 5.2 g/dl) and healthy human volunteers (LLN = 3.5, ULN = 5.3 g/dl). Data presented is the mean ± SE. Each data point represents data from at least 10 subjects, four ASOs and four independent studies. Dashed lines indicate the laboratory test reference range values and yellow y-axis markers indicate the event criteria. A tabulated summary of results is provided in Supplementary Table S5. ULN, upper-limit-of-normal; LLN, lower-limit-of-normal; ASO, antisense oligonucleotide.
In humans, there was a single incidence of ALT above 3× ULN in the ASO-treated groups, and none above 5× ULN (Table 2). There were no cases of ALT >3× ULN associated with elevated total bilirubin (>2× ULN) in the ASO-treated groups even including the data from ASO 4 (mipomersen) and ASO 11 studies. The incidence of abnormal results for synthetic function, such as albumin and prothrombin, was similar between placebo and ASO-treated groups. Looking more closely at mean ALT values a small increase in the mean ALT levels was observed over the course of treatment in the higher dose groups (>175 mg) of the multidose regimen (Figure 3). These increases were occasionally statistically different from placebo (see Supplementary Table S5), but mean levels remained within the range of normal with continued dosing. Importantly, as shown in Table 2, these slight increases in mean ALT values were not caused by large excursions by a few subjects who experienced more severe elevations. Furthermore, there was no discernable association between maximum ALT observations measured over the course of studies and dose level, or total exposure (see Supplementary Figure S3).
To confirm that our assessment would have identified a liver signal, we show results from a phase 1 healthy volunteer study with mipomersen,33 where a direct correlation was observed between the maximum reduction of apoB-100 and maximum ALT levels (see Supplementary Figure S4). Other studies on mipomersen also demonstrated that the ALT increase was correlated with the rate and extent of apoB-100 reduction.34,35 Thus, evaluation of liver transaminases identified an ASO that reduces a target which has been genetically and biologically confirmed to be required for clearance of triglycerides from liver, and in deficiency states is associated with changes in liver homeostasis, lipid metabolism, and increases in ALTs.37,38
Hematology. Based on evaluation of the mean platelet counts (Figure 4a) and respective summary statistics (see Supplementary Table S6), there are no obvious dose-dependent reductions in platelets observed in NHPs. However, results in Table 1 show that there is an increased incidence of >30% reduction of platelets compared with baseline (<0.7× BSLN) at high doses (>12 mg/kg/wk, i.e., 20, 40 or 50 mg/kg/wk). In nonGLP screening studies we have evaluated ~ 40 2′MOE chimeric ASOs at a dose of 40 mg/kg/wk and identified only a few ASOs that produced sustained effects on platelet counts (see Supplementary Figure S5a). Thus, there is no apparent class effect on platelets, but individual sequences may be problematic in the NHP22and perhaps humans.
Figure 4.

Hematology laboratory test results from nonhuman primates and healthy human volunteers treated with multiple doses up to 13 and 6 weeks, respectively, by dose group. (a,b) Platelet counts in nonhuman primates (ULN = 677 K/mm3) and healthy human volunteers (LLN = 140 and ULN = 400 K/mm3); (c,d) Lymphocyte counts in nonhuman primates (ULN = 10 K/mm3) and healthy human volunteers (LLN = 1.0 and ULN = 5.0 K/mm3); (e-g) Hemoglobin (Hb), hematocrit (HCT), and absolute neutrophil count (ANC) in healthy human volunteers only (Hb, LLN = 13, ULN = 17 g/dl; HCT, LLN = 39, ULN = 51%, ANC, LLN = 1.4, ULN = 8.0 K/mm3). Data presented is the mean ± SE. Each data point represents data from at least 10 subjects, four ASOs and four independent studies. Dashed lines indicate the laboratory test reference range values and yellow y-axis markers indicate the event criteria. A tabulated summary of results is provided in Supplementary Table S6. ULN, upper-limit-of-normal; LLN, lower-limit-of-normal; ASO, antisense oligonucleotide.
Additionally, as shown in Table 1, on occasion individual NHPs have displayed reductions in platelets (<75 K/mm3). These events are sporadic and not obviously dose or sequence related. However, as the incidence of this finding is low, the numbers of NHP evaluated does not support a firm conclusion about dose relatedness. An example of this phenomenon is shown in the results from one study where a profound reduction in platelet counts (<75 K/mm3) was observed in a single monkey during the course treatment (see Supplementary Figure S5b). Studies are underway to understand the mechanisms underlying the acute platelet decreases in monkeys; however, they appear to be highly idiosyncratic and are likely not associated with myelosuppression, or thromboembolic phenomena.
In contrast, in human studies, we observed no remarkable reduction in platelet counts over the course of treatment (Figure 4b) and no apparent dose or total exposure relationship (see Supplementary Table S6). The only incidence of platelet counts below 75,000 per µl occurred in a placebo treated subject (Table 2). Obviously analyses of data from studies in patients treated for longer periods of time on multiple 2′MOE ASOs are required to complete the assessment, but to date in long term studies on mipomersen, no clinically meaningful effect on platelets has been observed.39
No dose-dependent effects on other hematologic parameters were observed in the NHPs (Figure 4c) or in humans (Figure 4d–g). Although statistically significant differences were observed between ASO-treated groups and control, or placebo, in lymphocyte counts (see Supplementary Table S6), the changes were small; and as shown in the incidence table, were not considered clinically meaningful since the results were not confirmed upon retest (Table 2). A slight decline in hemoglobin and hematocrit was observed in the clinical studies, and associated with frequent phlebotomy (Figure 4e,f).
Complement. The interaction of 2′MOE chimeric ASOs with Factor H (alternative complement pathway) in NHPs has been well described.30,31 The transient inhibition of Factor H upon 2′MOE ASO binding results in activation of the alternate complement pathway and increased production of complement split products Bb and C3a. In NHPs, there was a dose-dependent increase in the incidence of elevated complement Bb (>2× ULN) following 2′MOE ASO treatment (Table 1). This increase in complement split product is an acute response, as reflected by observations in the 24 hour period after subcutaneous injection (Figure 5a). The effect on Bb translated to overall reductions in total complement C3 at >24 mg/kg/wk, but at lower incidence.
Figure 5.

Complement activation during the 24-hour period after subcutaneous injection of study drug in nonhuman primates and healthy human volunteers treated with multiple doses up to 13 and 6 weeks, respectively, by dose group. (a,b) Complement split product, Bb, in nonhuman primates (ULN = 2.8 μg/ml) and healthy human volunteers (ULN = 1.49 μg/ml). Data presented is the mean ± SE. Each data point represents data from at least 10 subjects and four ASOs. Dashed gray lines indicate the upper limit of the reference range and the yellow y-axis markers indicate the event criteria. ULN, upper-limit-of-normal; ASO, antisense oligonucleotide.
There was no evidence of complement activation in ASO treated human subjects as measured by the incidence of increases in the complement split products, Bb and C5a, above 2× ULN (Table 2). Finally, no acute class effect was identified in the 24-hour period after s.c. injection in normal volunteers, as shown in Figure 5b for complement split product Bb.
More recently discovered 2′MOE chimeric ASOs are more potent and better tolerated than mipomersen
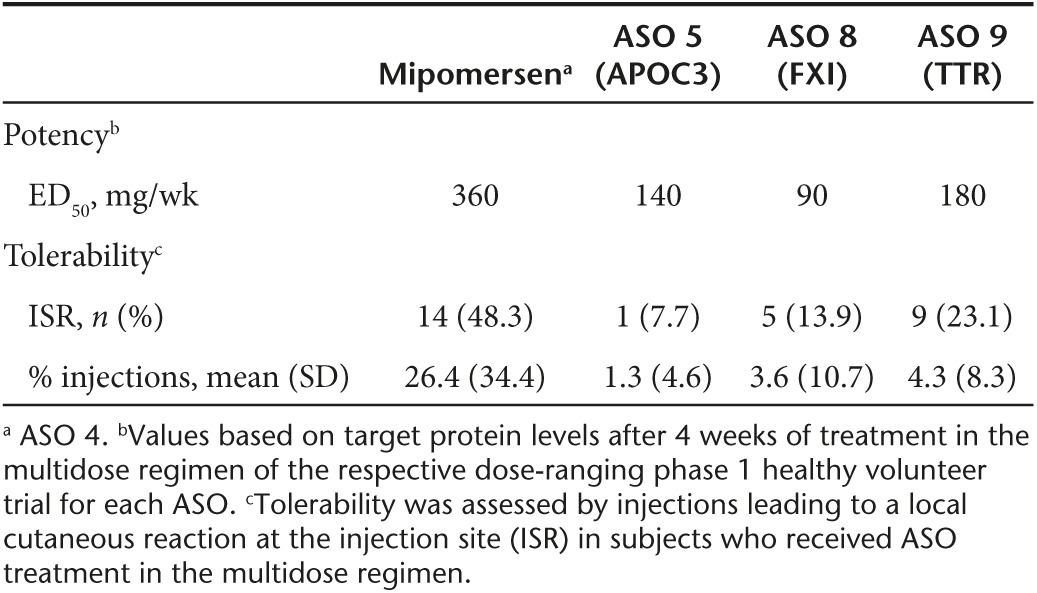
During the last decade, advances in ASO screening methods have led to identification of more potent and better tolerated drugs for development. To test the translation of these improvements, we compared the phase 1 data of mipomersen (the first second generation 2′MOE chimeric ASO to progress through late stages of development to approval) with data of later, more contemporary second generation 2′MOE chimeric ASO drugs (Table 3). ASOs were administered subcutaneous (s.c.) at a concentration of 150–250 mg/ml using a half inch, 29 or 30 gauge needle. From this assessment we found that potency increased from twofold to fourfold, based on the estimated median effective dose (ED50) in humans. Tolerability also improved relative to mipomersen, as indicated by a 52–84% reduction in the incidence of local reactions at the injection site (ISR) and 84–95% reduction in the mean percentage of injections which led to an ISR. There were no dose discontinuations based on these events. Tentatively, we attribute these observations to identifying more optimal sites in target RNAs and the use of approaches to avoid proinflammatory sequences. These advances in screening are beyond the scope of this manuscript, but will be reported elsewhere.
Table 3. Advances in ASO screening methods increase potency and improve tolerability.
Discussion
In this initial report, we demonstrate usage of an integrated safety database that we have created to characterize potential class-related adverse effects associated with 2′MOE chimeric ASOs in NHPs and normal human volunteers. NHPs were chosen specifically for the purpose of this investigation because of the pharmacokinetic similarities in dose-exposure relationships and this is considered the most relevant species for safety assessment. Much has been published regarding the observed class effects for 2′MOE ASOs for specific representative compounds.22 This integrated evaluation of our safety database allows us to assess the behavior of multiple compounds now within this class of ASO across a full dose range. Integrating NHP and human data then supports an evaluation of the predictive value of NHPs with regard to safety in humans. For examples, liver and kidney are known to be the primary organs of distribution in NHPs.24 Based on this distribution, kidney is one of the primary target organs for histologic changes in monkeys, but the functional effects are generally limited.40 Consistent with these reports, no class effects were identified in NHPs or humans for measures of liver or kidney function. This result is confirmed by comparing the influence of dose or total exposure on mean values of these analytes as well as comparing the incidence of abnormal values by dose or total exposure. Nor were there effects on hematologic parameters that cannot be explained by frequent phlebotomy. Further, this analysis confirms that complement activation does not occur at the doses tested in humans, but does occur in NHPs.
Identification of optimal ASOs for both potency and tolerability to advance into the clinic is a detailed and highly structured process.20,21 This empirical approach takes advantage of RNA structure and other properties to avoid problematic sequences, such as CpG motifs and G quartets. We determined that within the constraints imposed by rational screening processes, the sequences included in the integrated analysis are not biased. We also computed the probability of finding each subsequence of the selected ASOs in the pool of screened ASO sequences for each respective target RNA, and found no remarkable bias or over representation of subsequences. To confirm that ASOs that result in adverse events would be identified in this data set, we compared the results obtained with this sample of ASOs to mipomersen, an ASO with clearly defined target-specific adverse effects on the liver.33,34,35,39 Finally, our phase 1 data showed that advances in the ASO screening process have resulted in an increase in potency and better tolerability with the more recent 2′MOE ASOs in development,16,17,18,19,41,42 compared with mipomersen.
Another critical goal of the current analysis was to determine if the safety profile in NHP toxicology studies of an ASO was predictive of safety in normal human volunteers. This is particularly important since it is known that NHPs are more sensitive to complement activation than humans.30,31 Thus it is gratifying that, overall, NHPs were reasonably predictive of human responses to ASOs. However, there were several important differences. In NHPs, the effects on platelets of 2′MOE ASOs were more substantial than in humans. Additionally, remarkable reductions in platelets (<75 K/mm3) have been observed sporadically in NHPs that are not clearly dose or sequence related. Although data in normal volunteers do not allow us to exclude the possibility that reduction in platelets observed at higher doses in NHPs might be encountered at high doses in humans, studies in patients with cancer have been conducted at doses as high as 1,200 mg/wk and platelet reductions were minimal.43,44,45 Thus, it appears that NHPs may over predict the propensity of 2′MOE chimeric ASOs to reduce platelets in humans. Some of this difference may be accounted for by the enhanced sensitivity of NHPs to complement activation, but more work is needed to better understand this phenomenon.46
To date, over 6,000 humans have been treated with 2′MOE ASOs. Additionally, mipomersen is now in its fourth year of commercial use. So the experience with this class of agents is substantial and encouraging with regard to safety. The creation of our safety database supports comparisons between ASOs of the 2′MOE class and published reports of other chemical classes.47,48
Clearly, the analysis presented has limitations. Though substantial, the number of ASOs studied is still relatively limited and focused on a single chemical class of ASOs. Second, the total exposure in subjects is limited. However, the fact that the NHP studies and the human studies were conducted with normal individuals in carefully controlled trials assures that the comparisons are likely valid and that conclusions about chemical class related adverse events are likely accurate within the total exposures studied. Future analyses in patient populations treated for longer periods of time should support evaluating the interplay between this chemical class and disease states and other medications and the effects of prolonged exposure to 2′MOE ASOs on organ function. These will be the subjects of future reports.
During the review of this manuscript, we reported that severe thrombocytopenic events occurred in two placebo-controlled phase 3 studies with two different 2′MOE chimeric ASOs, IONIS-TTRRx and volanesorsen. Although, moderate thrombocytopenia has been reported for a 2′MOE chimeric ASO in patients with multiple sclerosis,49 we have never previously experienced severe thrombocytopenic events in any clinical trial including long term studies. While these trials remain blinded and investigations to understand the causes of these events are still in progress, we do know that the events are not due to prolonged exposure as all events occurred 4–9 months after initiation of therapy. In fact, many patients have been treated longer than 6 months with these two 2′MOE chimeric ASOs. Even more recently, a long term study of the natural history of patients with familial chylomicronemia syndrome,50 showed that familial chylomicronemia syndrome patients experience very substantial oscillations in platelet counts with platelet counts below 50,000/μl observed often. So familial chylomicronemia syndrome itself may be a significant contributor to the thrombocytopenia observed in the volanesorsen trial. Transthyretin (TTR) amyloidosis patients also experience decline in platelets, but the declines are less significant than observed in familial chylomicronemia syndrome patients. In fact, 8% of patients entering the phase 3 trial with IONIS-TTRRx have grade 1 thrombocytopenia, so the disease may be a minor contributor to the observations. Obviously there are many factors such as infections, concomitant drugs administered, and the diseases themselves that may have contributed to these events. When the investigations into mechanisms have matured, we will report the results. In the meantime, we are monitoring patients more frequently for thrombocytopenia.
Obviously, making definitive predictions about the behavior of a specific 2′MOE ASO based on the behavior of the chemical class is inappropriate. Different sequences may be identified that have specific adverse events and there can always be target related toxicities. However, such a database does provide useful information that supports more prudent decisions about development that can lead to lower risk for patients in clinical trials. We believe that this is the first example of such an approach being taken and look forward to the development of similar databases for other classes of ASO drugs that share common chemical and mechanistic features.
Materials and Methods
Database. Nonclinical study data were imported from an Oracle database using a Java web application designed to ensure the recorded data were validated and standardized across lab test parameters, study protocols, and investigators. The database was populated with individual data where a numerical value was generated. Imported text files were converted into SAS datasets for further analysis by a SAS program (SAS Institute, Cary, NC).
Clinical study data were imported from individual study data sets into one SAS dataset for each laboratory test.29
Assessment of sample size. We assembled the aggregate 1,051 screened ASO sequences for each ASO target RNA studied in this work. These sequences are biased, but not exclusively, selected to have binding sites within the exons of each of the desired targets and subject to a variety of algorithms and heuristics that we have found historically to render more active and tolerable ASOs. Using these sequences we computed the background frequency of every subsequence of length 2–15. Then for every subsequence found within the ASOs included in the current integrated safety database analysis, we computed the probability of finding N copies of each subsequence within the aggregate set of screened ASO sequences using a bionomial test. This test was parameterized with the probability of finding a copy being equal to the observed frequency in the set of screened ASOs.
Nonclinical data. The current analysis utilized data from 12 GLP studies in cynomolgus monkeys and 8 nonGLP studies in either cynomolgus or rhesus macaque monkeys. All studies were performed in Association for Assessment of Animal Laboratory Care (AALAC) accredited facilities with Institutional Animal Care and Use Committee (IACUC) approval, and were conducted in accordance with international regulations on use of animals in research.
Route of administration and dose regimens were comparable across the 2′MOE chimeric ASOs tested. In general, each study consisted of 4–5 dose groups (3–5 animals/sex/group) including a saline treated control group. The route of administration was predominantly by s.c. injection, with 1 hour i.v. infusion used in a few studies. Typically, the dose regimen consisted of 3–4 doses during the first week of treatment followed by once or twice weekly dose administration (see Supplementary Table S7).
To profile the safety of each 2′MOE chimeric ASO and identify potential safety effects in humans, multiples of the anticipated human therapeutic doses were evaluated. Maintenance doses ranged from 1 to 50 mg/kg/wk, which translated from 1 to 25× the human efficacious dose, respectively, based on plasma drug exposure (area under the curve AUC). The study duration ranged up to 13 weeks of repeat-dose treatment. In some studies, a subset of animals (n = 2/sex/group) were maintained on study for up to 13 weeks in a treatment-free period after the primary termination time point to determine the potential reversibility of effects observed during the treatment phase of the study.
Data were collected at least once before the first dose (up to 3 weeks prior), and at several time points during the dosing and recovery phases of the study.
Clinical data. Clinical studies were performed in compliance with the guidelines of Good Clinical Practice and Declaration of Helsinki. All human subjects gave written informed consent. Evaluable subjects received at least one dose of study drug.
All 2′MOE chimeric ASOs were initially evaluated in humans as a single dose, and all studies were dose-ranging and included a placebo-control group. Study dose regimens are shown in Supplementary Table S8. The route of study drug administration was predominantly by subcutaneous injection (615 subjects received study drug by s.c. injection, 100 subjects by i.v. infusion; and 35 subjects by s.c. and i.v.). Samples were collected prior to dosing for standard laboratory tests. Samples for complement split products and prothrombin were also collected at several time points within a 24 hour period after certain dose times.
The median effective dose (ED50) was estimated from the 5-point dose response curves for ASOs that had pharmacological data on the respective target protein and were evaluated in a multidose regimen with a treatment period of at least 4 weeks. Estimates were derived from data collected after 4 weeks of dosing, and therefore, reflect pre-steady state conditions.25,26
Percentage of injections leading to an injection site reaction (ISR) was calculated as follows for each subject: (A/B)*100, where A = number of injections with an ISR after injection, and B = total number of injections. ISR was defined as injection site erythema, injection site swelling, injection site pruritus, injection site pain or tenderness which started the day of s.c. injection, and persists (start to stop) for 2 days or more.
Statistics. Data are presented by the incidence of events and descriptive summary statistics of laboratory test results. All study data was included for analysis of the incidence of events. The Baseline was defined as the last nonmissing value prior to the first dose. An event was defined as data falling outside the normal range or reaching the specified threshold, as defined by protocol stopping rules, standard reporting, or Grade 3 criteria provided by the FDA in Guidance to Industry for healthy adults and adolescents.51 Differences between groups were assessed on the multidose regimen test results. ASO-treated dose groups were compared with control or placebo group at each week (nonclinical) or dose (clinical) using analysis of covariance with the baseline value as a covariate.
The ULN for nonclinical data was calculated as the (mean + 2*SD) of the baseline values of NHPs in the data set (see Supplementary Table S9). For the clinical data, the ULN for the liver transaminase tests was the larger value of the baseline result and the ULN provided by the associated laboratory of origin. The over-time analysis of laboratory test results included all study data up to 10 days after the last dose. The lower-limit-of-normal and ULN displayed for healthy human volunteers in the mean results over time figures represent the respective median values (see Supplementary Table S10).
SUPPLEMENTARY MATERIAL Figure S1. Second generation 2'MOE chimeric ASO (a) design and (b) mechanism of action. Figure S2. 2'MOE chimeric ASOs included in the clinical multi-dose regimen analysis. Figure S3. Effect of (a) dose level and (b) total dose exposure on maximum ALT levels in humans. Figure S4. Direct correlation between maximum apoB reduction and log-transformed maximum ALT levels in subjects administered mipomersen (r = 0.58, P = 0.002). Figure S5. Low incidence of (a) moderate sustained and (b) severe sporadic decreases of platelet counts in NHPs treated with a 2'MOE chimeric ASO. Table S1. 2'MOE chimeric ASOs evaluated in the integrated safety database analysis. Table S2. Demographics of subjects in randomized placebo-controlled healthy human volunteer studies. Table S3. Limited over representation of subsequences in selected 2'MOE Chimeric ASOs. Table S4. Kidney test results over time by dose category. Table S5. Liver test results over time by dose category. Table S6. Hematology test results over time by dose category. Table S7. Non-clinical study dose regimens. Table S8. Clinical study dose regimens. Table S9. Upper limit of reference range for nonclinical lab tests. Table S10. Range of normal for clinical lab tests.
Acknowledgments
We thank C. F. B., J. S., S. G. H., and S. T. for their critical review of the manuscript; N. P. for technical support; T. R. for graphics support; and D. P. for administrative assistance, all from Ionis Pharmaceuticals. All authors are employees of Ionis Pharmaceuticals (Carlsbad, CA).
Supplementary Material
References
- Beaudet, AL and Meng, L (2016). Gene-targeting pharmaceuticals for single-gene disorders. Hum Mol Genet 25(R1): R18–R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClorey, G and Wood, MJ (2015). An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr Opin Pharmacol 24: 52–58. [DOI] [PubMed] [Google Scholar]
- Lee, RG, Crosby, J, Baker, BF, Graham, MJ and Crooke, RM (2013). Antisense technology: an emerging platform for cardiovascular disease therapeutics. J Cardiovasc Transl Res 6: 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo, F, Hua, Y, Krainer, AR and Bennett, CF (2012). Antisense-based therapy for the treatment of spinal muscular atrophy. J Cell Biol 199: 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, CF and Swayze, EE (2010). RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol 50: 259–293. [DOI] [PubMed] [Google Scholar]
- Crooke, ST, Vickers, T, Lima, W and Wu, H (2007). Mechanisms of antisense drug action, an introduction. In: Crooke, ST (ed.). Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp. 3–46. [Google Scholar]
- Lima, W, Wu, H and Crooke, ST (2007). The RNase H mechanism. In: Crooke, ST (ed.) Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp. 47–74. [Google Scholar]
- Bennett, CF (2007). Pharmacological properties of 2′-O-methoxyethyl-modified oligonucleotides. In: Crooke, ST (ed.). Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp. 273–303. [Google Scholar]
- Meng, L, Ward, AJ, Chun, S, Bennett, CF, Beaudet, AL and Rigo, F (2015). Towards a therapy for Angelman syndrome by targeting a long noncoding RNA. Nature 518: 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriboga, CA, Swoboda, KJ, Darras, BT, Iannaccone, ST, Montes, J, De Vivo, DC et al. (2016). Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 86: 890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima, WF, Vickers, TA, Nichols, J, Li, C and Crooke, ST (2014). Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS One 9: e101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell, AL, Skotte, NH, Kordasiewicz, HB, Østergaard, ME, Watt, AT, Carroll, JB et al. (2014). In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther 22: 2093–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardee, GE, Tillman, LG and Geary RS (2007). Routes and formulations for delivery of antisense oligonucleotides. In: Crooke, ST (ed). Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp. 217–236. [Google Scholar]
- Miller, TM, Pestronk, A, David, W, Rothstein, J, Simpson, E, Appel, SH et al. (2013). An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12: 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovingh, K, Besseling, J and Kastelein, J (2013). Efficacy and safety of mipomersen sodium (Kynamro). Expert Opin Drug Saf 12: 569–579. [DOI] [PubMed] [Google Scholar]
- Graham, MJ, Lee, RG, Bell, TA 3rd, Fu, W, Mullick, AE, Alexander, VJ et al. (2013). Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res 112: 1479–1490. [DOI] [PubMed] [Google Scholar]
- Gaudet, D, Alexander, VJ, Baker, BF, Brisson, D, Tremblay, K, Singleton, W et al. (2015). Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med 373: 438–447. [DOI] [PubMed] [Google Scholar]
- Jones, NR, Pegues, MA, McCrory, MA, Singleton, W, Bethune, C, Baker, BF et al. (2012). A selective inhibitor of human C-reactive protein translation is efficacious in vitro and in C-reactive protein transgenic mice and humans. Mol Ther Nucleic Acids 1: e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, MS, Hughes, SG, Singleton, W, Yamashita, M and Genovese, MC (2015). Results of a proof of concept, double-blind, randomized trial of a second generation antisense oligonucleotide targeting high-sensitivity C-reactive protein (hs-CRP) in rheumatoid arthritis. Arthritis Res Ther 17: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freier, SM and Watt, AT (2007). Basic principles of antisense drug discovery. In: Crooke, ST (ed). Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp. 117–142. [Google Scholar]
- Crooke, RM and Graham, MJ (2013). Modulation of lipoprotein metabolism by antisense technology: preclinical drug discovery methodology. Methods Mol Biol 1027: 309–324. [DOI] [PubMed] [Google Scholar]
- Henry, SP, Kim, T, Kramer-Strickland, K, Zanardi, TA, Fey, RA and Levin, AA (2007). Toxicologic properties of 2′-O-methoxyethyl chimeric antisense inhibitors in animals and man. In: Crooke, ST (ed). Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp 327–363. [Google Scholar]
- Burel, SA, Han, SR, Lee, HS, Norris, DA, Lee, BS, Machemer, T et al. (2013). Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic Acid Ther 23: 213–227. [DOI] [PubMed] [Google Scholar]
- Geary, RS, Yu, RZ, Siwkowski, A and Levin, AA (2007). Pharmacokinetic/pharmacodynamic properties of phosphorothioate 2′-O-(2-methoxyethyl)-modified antisense oligonucleotides in animals and man. In: Crooke, ST (ed). Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp. 305–326. [Google Scholar]
- Geary, RS (2009). Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol 5: 381–391. [DOI] [PubMed] [Google Scholar]
- Yu, RZ, Grundy, JS and Geary, RS (2013). Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol 9: 169–182. [DOI] [PubMed] [Google Scholar]
- Kwoh, TJ (2007). An overview of the clinical safety experience of first- and second-generation antisense oligonucleotides. In: Crooke, ST (ed). Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp. 365–399. [Google Scholar]
- Capaldi, DC and Scozzari, AN (2007). Manufacturing and analytical processes for 2′-O-(2-methoxyethyl)-modified oligonucleotides. In: Crooke, ST (ed). Antisense Drug Technology Principles, Strategies, and Applications, 2nd edn. CRC Press: New York, pp. 401–434. [Google Scholar]
- Smith, D, Schulz, D, Kloss, G and Cheng, W (2010). Considerations for building an integrated safety database using SAS. http://www.lexjansen.com/pharmasug/2010/AD/AD15.pdf.
- Shen, L, Frazer-Abel, A, Reynolds, PR, Giclas, PC, Chappell, A, Pangburn, MK et al. (2014). Mechanistic understanding for the greater sensitivity of monkeys to antisense oligonucleotide-mediated complement activation compared with humans. J Pharmacol Exp Ther 351: 709–717. [DOI] [PubMed] [Google Scholar]
- Henry, SP, Jagels, MA, Hugli, TE, Manalili, S, Geary, RS, Giclas, PC et al. (2014). Mechanism of alternative complement pathway dysregulation by a phosphorothioate oligonucleotide in monkey and human serum. Nucleic Acid Ther 24: 326–335. [DOI] [PubMed] [Google Scholar]
- Sheehan, JP and Lan, HC (1998). Phosphorothioate oligonucleotides inhibit the intrinsic tenase complex. Blood 92: 1617–1625. [PubMed] [Google Scholar]
- Kastelein, JJ, Wedel, MK, Baker, BF, Su, J, Bradley, JD, Yu, RZ et al. (2006). Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation 114: 1729–1735. [DOI] [PubMed] [Google Scholar]
- Akdim, F, Tribble, DL, Flaim, JD, Yu, R, Su, J, Geary, RS et al. (2011). Efficacy of apolipoprotein B synthesis inhibition in subjects with mild-to-moderate hyperlipidaemia. Eur Heart J 32: 2650–2659. [DOI] [PubMed] [Google Scholar]
- Stein, EA, Dufour, R, Gagne, C, Gaudet, D, East, C, Donovan, JM et al. (2012). Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation 126: 2283–2292. [DOI] [PubMed] [Google Scholar]
- Kelly, RP, Garhyan, P, Raddad, E, Fu, H, Lim, CN, Prince, MJ et al. (2015). Short-term administration of the glucagon receptor antagonist LY2409021 lowers blood glucose in healthy people and in those with type 2 diabetes. Diabetes Obes Metab 17: 414–422. [DOI] [PubMed] [Google Scholar]
- Sankatsing, RR, Fouchier, SW, de Haan, S, Hutten, BA, de Groot, E, Kastelein, JJ et al. (2005). Hepatic and cardiovascular consequences of familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol 25: 1979–1984. [DOI] [PubMed] [Google Scholar]
- Ahmed, A and Keeffe, EB (1998). Asymptomatic elevation of aminotransferase levels and fatty liver secondary to heterozygous hypobetalipoproteinemia. Am J Gastroenterol 93: 2598–2599. [DOI] [PubMed] [Google Scholar]
- Santos, RD, Duell, PB, East, C, Guyton, JR, Moriarty, PM, Chin, W et al. (2015). Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur Heart J 36: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, SP, Johnson, M, Zanardi, TA, Fey, R, Auyeung, D, Lappin, PB et al. (2012). Renal uptake and tolerability of a 2′-O-methoxyethyl modified antisense oligonucleotide (ISIS 113715) in monkey. Toxicology 301: 13–20. [DOI] [PubMed] [Google Scholar]
- Gaudet, D, Brisson, D, Tremblay, K, Alexander, VJ, Singleton, W, Hughes, SG et al. (2014). Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 371: 2200–2206. [DOI] [PubMed] [Google Scholar]
- Büller, HR, Bethune, C, Bhanot, S, Gailani, D, Monia, BP, Raskob, GE et al.; FXI-ASO TKA Investigators. (2015). Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med 372: 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, DS, Kurzrock, R, Oh, Y, Wheler, J, Naing, A, Brail, L et al. (2011). A phase 1 dose escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clin Cancer Res 17: 6582–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, KN, Siu, LL, Hirte, H, Hotte, SJ, Knox, J, Kollmansberger, C et al. (2008). A phase I study of OGX-011, a 2′-methoxyethyl phosphorothioate antisense to clusterin, in combination with docetaxel in patients with advanced cancer. Clin Cancer Res 14: 833–839. [DOI] [PubMed] [Google Scholar]
- Chi, KN, Eisenhauer, E, Fazli, L, Jones, EC, Goldenberg, SL, Powers, J et al. (2005). A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J Natl Cancer Inst 97: 1287–1296. [DOI] [PubMed] [Google Scholar]
- Frazier, KS (2015). Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist's perspective. Toxicol Pathol 43: 78–89. [DOI] [PubMed] [Google Scholar]
- Bianchini, D, Omlin, A, Pezaro, C, Lorente, D, Ferraldeschi, R, Mukherji, D et al. (2013). First-in-human phase I study of EZN-4176, a locked nucleic acid antisense oligonucleotide to exon 4 of the androgen receptor mRNA in patients with castration-resistant prostate cancer. Br J Cancer 109: 2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho, T, Adams, D, Silva, A, Lozeron, P, Hawkins, PN, Mant, T et al. (2013). Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 369: 819–829. [DOI] [PubMed] [Google Scholar]
- Limmroth, V, Barkhof, F, Desem, N, Diamond, MP and Tachas, G; ATL1102 Study Group (2014). CD49d antisense drug ATL1102 reduces disease activity in patients with relapsing-remitting MS. Neurology 83: 1780–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet, D (2016). Lessons learned from emerging therapies for severe hypertriglyceridaemia. 84th EAS Congress (29 May – 1 June), Innsbruck, Austria.
- US Food and Drug Administration (2007). Guidance for industry: Toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials (September 2007). http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/vaccines/ucm074775.htm.
- Crooks, GE, Hon, G, Chandonia, JM and Brenner, SE (2004). WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.