

Abstract
Background
Runs of homozygosity (ROH) are consecutive homozygous genotypes, which may result from population inbreeding or consanguineous marriages. ROH enhance the expression of recessive traits.
Methods
We mapped ROH in a case control study of women delivering at term compared to women delivering at or before 34 weeks gestation. Gene sets known to be important in risk of preterm birth were examined for their overlap with identified ROH segments.
Results
While we found no evidence of increased burden of ROH or copy number variations in mothers delivering at or before 34 weeks compared to term, we identified 424 genome-wide 50 kb segments with significant difference in abundance of overlapping ROH segments in cases versus controls, p<0.05. These regions overlap 199 known genes. We found preterm birth associated genes (CXCR4, MYLK, PAK1) and genes shown to have an evolutionary link to preterm (CXCR4, PPP3CB, C6orf57, DUSP13, and SLC25A45) with significant differences in abundance of overlapping ROH blocks in cases versus controls, p<0.001.
Conclusion
We conclude, while we found no significant burden of runs of homozygosity, we did identify genomic regions with significantly greater abundance of ROH blocks in women delivering preterm that overlapped genes known to be involved in preterm birth.
INTRODUCTION
Human genetic variation contributes to our individual characteristics, our susceptibility to disease and our individual responses to medications. This genetic variation can take the form of single nucleotide polymorphisms, copy number variations, chromosomal duplications and chromosomal loss. Runs of homozygosity (ROH) are an important part of genetic variation(1). ROH are stretches of consecutive homozygous genotypes, which may result from identity by descent, population inbreeding and/or cultural support for consanguineous marriages. Abundant ROH enhance the expression of recessive traits. Evolutionary pressures and purifying selection purge deleterious variants, but elevate the frequency of haplo-types surrounding a favored allele. Recent reports have shown that long runs of homozygosity are enriched for deleterious variants(2). This is especially true for ROH that span several megabases. The ability to do homozygosity mapping through examination of ROH has been enhanced by the availability of high-density genotyping arrays and has contributed to our understanding of homozygosity in both inbred and outbred populations. Reports using this approach in large numbers of individuals with very low evidence for inbreeding (up to 10 generations) have shown that ROH up to 4 Mb is common(1, 2).
Despite significant advances in the care of low birth weight infants, preterm birth remains a leading cause of newborn morbidity, mortality and hospitalization in the first year of life(3). Moreover, despite numerous attempts at intervention, the incidence of prematurity has shown minimal improvement over the last two decades. The risk factors associated with prematurity are many, including: adverse socio-demographic factors, race/ethnicity, infection, stress, trauma and prior history of a premature birth. The leading etiology is idiopathic. A large number of clinical/epidemiologic studies have examined the individual and collective contribution of each of these factors(4–6). There is substantial evidence for a genetic contribution to the risk of preterm birth(4–11). Twin studies suggest heritability is 36–40%, however differences in gestational age used and other details cloud the precision of those estimates(9, 10). Large epidemiological analyses drawn from population based studies support a maternal origin for the genetic contribution(s) to risk of preterm birth, with little contribution by paternal or fetal genetic factors(7, 12–14). A family history of preterm birth and inter-pregnancy interval of <18 months also increase the risk of prematurity(7, 10, 15). Population-based studies of consanguinity have suggested that there may be an increased risk of autosomal recessive inheritance associated with preterm birth(16, 17). One study found cousin marriage to result in a 1.6-fold increase risk of spontaneous preterm birth before 33 weeks gestation(16). A similar study found an odds ratio of 1.5 (CI, 1.2–1.9) of increased risk of preterm birth when mother and father are related(17). Whether one or several of these genetic etiologies converge via a final common pathway is unclear.
In an attempt to better understand the contribution of structural and genomic variation to the genetic etiology of preterm birth, we analyzed a large genome wide association study of preterm birth for structural variations (copy number variations, insertions and deletions) and for large genomic stretches of runs of homozygosity. We analyzed high-density genotyping data from the gene environment association studies initiative (GENEVA) funded by the trans-NIH Genes Environment and Health Initiative. The GENEVA study included 4,000 Danish women and children from the Danish National Birth Cohort, for which phenotype and genotype information from a genome-wide association study are available. In order to study a more parsimonious group of preterm births which were more likely to be due to genetic disorders than environmental causes, we restricted our analysis to mothers that delivered at less than 34 weeks gestation. Thus, we focused on the genotype data from mothers who delivered at term (n=1018) and those who delivered prematurely (<34 weeks gestation n=454). We found no association with copy number variations and preterm birth. While we saw no evidence of significant burden of runs of homozygosity in preterm birth, there are significant genomic regions with ROH associated with genes known to be in involved in preterm birth.
RESULTS
Geneva Dataset
This study is a secondary analysis of genotype data from a genome-wide study of preterm birth known as Genome-Wide Association Studies of Prematurity and Its Complications, dbGaP Study Accession: phs000103.v1.p1. All phenotypic and genotype information deposited into dbGAP and were made available to us. The data represent a genome-wide case/control study using approximately 1,000 preterm mother-child pairs from the Danish National Birth Cohort (DNBC) along with 1,000 control pairs where the child was born at ~40 weeks’ gestation. This study employed whole genome genotyping, on the ILLUMINA Human 660W-Quad_v1_A array. No significant univariate genome-wide associations were found related to preterm birth and this data is available as supplementary material (see Supplementary File S1 online). We used the genotype data from the mothers who delivered at term (n=1018) and those who delivered prematurely (<34 weeks gestation n=454) to carry out homozygosity mapping.
Population Structure and Relatedness
Principal component and relatedness analysis of the original Geneva dataset were employed to confirm that any identified associations were not due to unrecognized family structure with the dataset and this data is available as supplementary material (see Supplementary File S1 online). The relatedness estimates identified one unexpected duplicate that involved a misidentified gender subject, which was been removed from the data set. Also unexpected were two half-sib-like pairs. Otherwise 3 full siblings, 2 half siblings, and 10 first cousins were detected. For each pair of the families associated with one of the two unexpected half-sib-like pairs, only one mother was picked to be included in further analyses. The population structure using principal components analysis (PCA) was conducted on the original 3,867 mother-child subjects from the GENEVA project, along with HapMap anchor controls (CEU, YRI, CHB and JPT). All of the study samples cluster near the CEU HapMap anchor point as expected and this data is available as supplementary material (see Supplementary Figure S2 online). Because of the inclusion criteria requirement that the infant’s parents and all four grandparents must be of Danish origin, no additional analysis of within population stratification was conducted.
Structural Variation and CNV Analysis
We examined the data for univariate association of CNVs with preterm birth and to adjust for the spurious appearance of ROH due to regions of hemizygosity. CNV analysis was performed with the Illumina Bead Studio Program and PennCNV(18). Using PLINK to perform association testing on segmental CNV data from the PennCNV variants, we did not identify any significant segregation of CNVs (indels, duplications) between the preterm birth cases and the term controls. We did identify a total of 90 hemizygous CNVs from 78 mothers that were used to correct for spurious ROH. We also found that 16 of the ROH blocks (minimum segment of 1000kb or larger) overlapped with CNV blocks.
Homozygosity Burden
The burden of homozygosity was analyzed for three categories of minimum final segment length (1000 kb, 2000 kb, 3000 kb). For each minimum segment length, the proportion of individuals with at least one ROH, the number of ROHs per individual and the total length ROHs per individual were compared between the case and control cohorts, with case-control status determined by gestational age (GA). Control (individuals with GA greater or equal to 38 weeks) vs case (individuals with GA less than or equal to 34 weeks) status was analyzed with logistic regression using differences in the proportion, total number and total length of ROH as predictors and maternal age as a covariate. The total lengths of ROHs per individual were also calculated and the means are reported for four mutually exclusive gestational age categories.
We did not find a statistically significant association between the proportion of individuals with at least one ROH and case/control status for any of the ROH minimum length categories (Table 1). Additionally, when considering only individuals with at least one minimum ROH length segment, while the average number of ROH per individual in the cases was greater than that of the controls (ratio > 1), this finding did not reach a statistically significant association with case/control status for any of the minimum ROH length categories (Table 2).
Table 1.
| Minimum segment length | Proportion of cases with ROH | Proportion of controls with ROH | Ratio | Logistic regression coefficient | P-value |
|---|---|---|---|---|---|
| 1,000 kb | 1 | 1 | 1 | – | – |
| 2,000 kb | 0.733 | 0.689 | 1.06 | −0.2181 | 0.084 |
| 3,000 kb | 0.182 | 0.16 | 1.14 | −0.1609 | 0.279 |
Table 2.
| Minimum segment length | Average number ROH in cases | Average number ROH in controls | Ratio | Logistic regression coefficient | P-value |
|---|---|---|---|---|---|
| 1,000 kb | 18.008 | 17.655 | 1.02 | −0.015 | 0.192 |
| 2,000 kb | 2.081 | 1.923 | 1.08 | −0.036 | 0.254 |
| 3,000 kb | 1.602 | 1.515 | 1.06 | −0.017 | 0.747 |
Despite the average total length of ROH showing a general trend for increasing length with decreasing gestational weeks, neither regression nor ANOVA demonstrated a statistically significant change that was gestational week dependent (data not shown). In addition, there was no statistically significant association between total length of ROH and the case /control status as defined above via logistic regression, Table 3. We used PLINK to compute genome wide homozygosity in order to determine if there was more than expected overall homozygosity and whether there was a difference between preterm and term deliveries. For both cases and controls there was very little excess in homozygosity and we found no statistical association between the statistic and case/control status (data not shown).
Table 3.
| Minimum segment length | Average total ROH in cases | Average total ROH in controls | Ratio | Logistic regression coefficient | P-value |
|---|---|---|---|---|---|
| 1,000 kb | 25,530,560 | 24,627,939 | 1.04 | −7.489×10−9 | 0.151 |
| 2,000 kb | 5,800,931 | 5,205,360 | 1.11 | −6.456×10−9 | 0.346 |
| 3,000 kb | 7,452,289 | 6,634,542 | 1.12 | −3.952×10−9 | 0.632 |
ROH Mapping and Analysis
We next carried out mapping and analysis for ROH blocks with a minimum segment length of 2000 kb to access whether any local genomic regions were predictive of case-control status. A schematic of our methods appears in Figure 1. We found 424 50 kb segments with significant differences in the abundance of overlapping ROH blocks between the cases and the controls. The results are shown as a −log P Manhattan plot in Figure 2 with a threshold for p<0.05 shown. These were distributed randomly across the genome, Figure 3. We found that 88% of these regions had an overabundance of overlapping ROH in preterm mothers vs term mothers, a result not consistent with chance alone (binomial test p-value < .0001). While the p values for the individual 50 kb regions were not corrected for multiple comparisons, the results point to regions of interest or overlapping known genes associated with preterm birth. We mapped these significant 50 kb windows to the list of all known genes and found 199 known genes and 43 other transcripts including open reading frames, microRNA and lncRNA overlapping these regions and this data is available as supplementary material (see Supplementary Table S3 online). A list of the coordinates of these significant 424 50kb segments and the coordinates of their overlapping ROH blocks is available as supplementary material (see Supplementary Table S4 online).
Figure 1. Schematic of ROH Analysis.
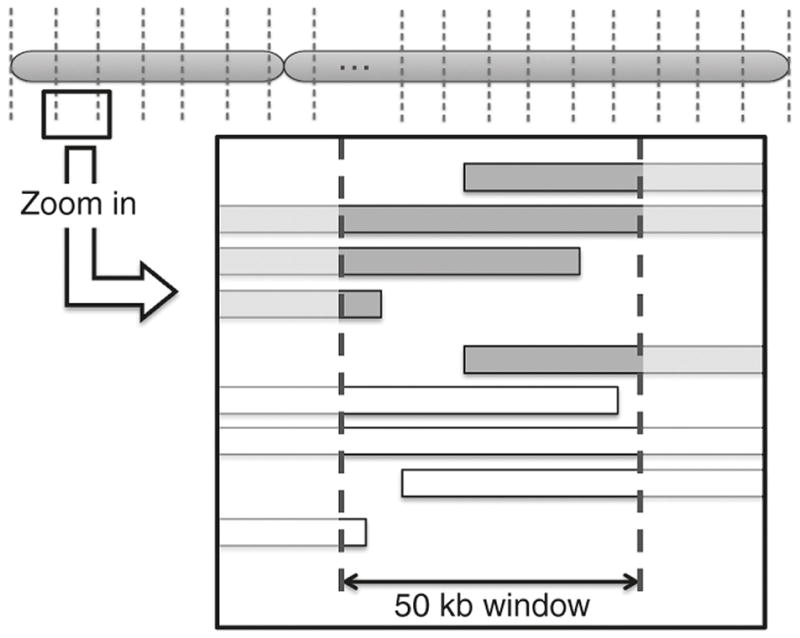
Each chromosome (chr 1-22) was split into 50 kb wide windows. The number of minimum final segments of 2000 kb ROH blocks from cases and controls that fell into these sectors was analyzed for differential abundance by Fisher’s exact test. Cases were represented in gray colored bars and controls were represented in white colored bars.
Figure 2. Manhattan Plot of p-values for genome wide 50 kb windows.
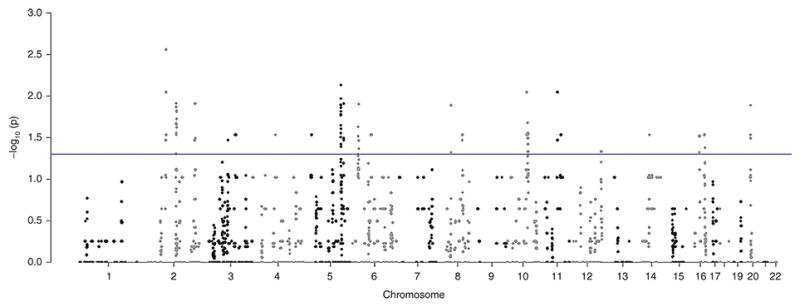
The vertical axis shows the −log p value for comparison by Fisher’s exact test of the number of ROH blocks nested within between preterm birth and control patients. Each dot in the graph represents the midpoint of 50 kb window for cases (34 weeks and less) and controls (38 weeks and above). The horizontal line shows the threshold for comparison of the abundance of blocks where the −log p is for p <0.05, above which difference in number of ROH blocks is significant.
Figure 3. Chromosomal distribution of ROH and 50 kb significant windows.
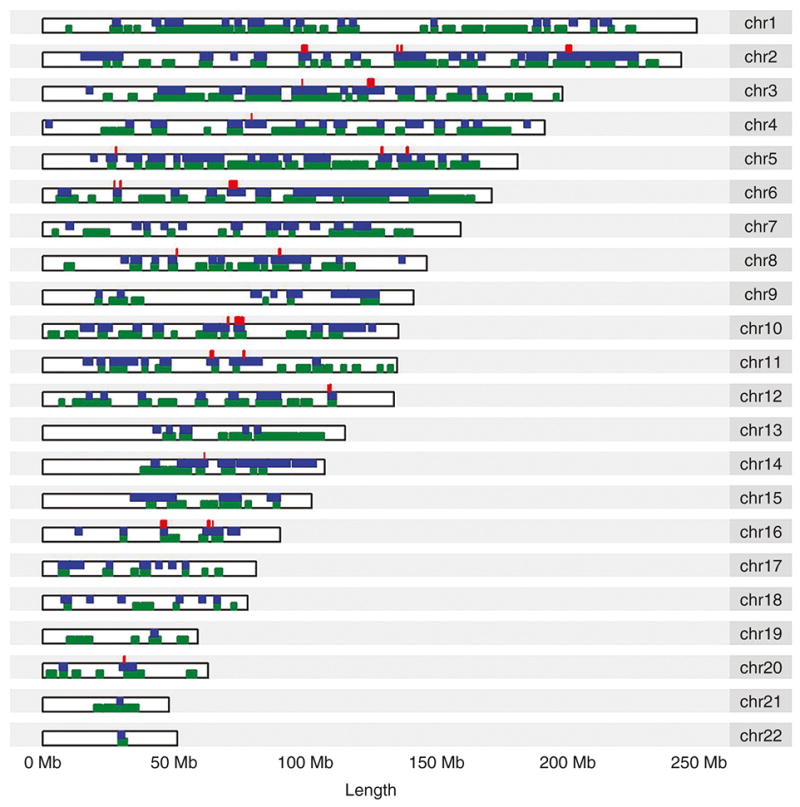
Green colored regions represent presence of a 2000 kb or greater ROH in at least one TERM patient. Blue colored regions represent presence of a 2000 kb or greater ROH in at least one PRETERM patient. 424 50 kb significant windows are marked in red.
Preterm Birth Associated Gene Mapping
From our previous work we found 30 pathways associated with preterm birth with high confidence values (FDR, <0.05) that contained 329 genes(19). We analyzed this gene set for overlap with all ROH blocks greater than 2000kb. Since 7 of the genes were from chromosome X, we excluded these and analyzed the remaining 322 genes. We found 3 preterm birth associated genes for which the overlapping ROH blocks were significantly more abundant in mothers delivering preterm. These genes (CXCR4, MYLK, PAK1) are shown in Table 4. We also examined the overlap with a mammalian gene set previously shown to have an evolutionary link to preterm birth with the ROH segments(20). We found ROH blocks with significantly greater abundance in preterm mothers overlapping CXCR4, PPP3CB, C6orf57, DUSP13, and SLC25A45 (Table 5) (20).
Table 4.
| Gene symbol | Name | HGNC ID | Chr | Abundance | P-value |
|---|---|---|---|---|---|
| CXCR4 | Chemokine (C-X-C motif) receptor 4 | 2561 | 2 | > Case | 0.007 |
| PAK1 | P21 protein (Cdc42/Rac)-activated kinase 1 | 8590 | 11 | > Case | 0.02 |
| MYLK | Myosin light chain kinase | 7590 | 3 | > Case | 0.02 |
Table 5.
| Gene symbol | Name | HGNC ID | Chr | Abundance | P-value | |
|---|---|---|---|---|---|---|
| Gain of function | CXCR4 | Chemokine (C-X-C motif) receptor 4 | 2561 | 2 | > Case | 0.007 |
| PPP3CB | Protein phosphatase 3, catalytic subunit, beta isozyme | 9315 | 10 | > Case | 0.027 | |
| C6orf57 | Chromosome 6 open reading frame 57 | 20957 | 6 | > Case | 0.029 | |
| Loss of function | DUSP13 | Dual specificity phosphatase 13 | 19681 | 10 | > Case | 0.032 |
| SLC25A45 | Solute carrier family 25, member 45 | 27442 | 11 | > Case | 0.033 |
Discussion
Autosomal recessive inheritance can be investigated through homozygosity mapping or examination of large genomic segments for ROH. Previous population-based studies of consanguinity have demonstrated that there may be an increased risk of autosomal recessive inheritance associated with preterm birth(16, 17, 21, 22). Fine mapping of ROH has been made possible by high density SNP genotyping arrays. Homozygosity mapping has allowed identification of important genomic regions and recent studies have shown increased runs of homozygosity in intellectual disabilities associated with simplex autism(23). Using this approach we carried out a secondary analysis of a large genome wide association study for preterm birth using genotype data from the GENEVA Project. We sought evidence for structural variation (CNVs) and long runs of homozygosity in association with preterm birth. We used a restricted definition for preterm birth (<34 weeks) in order to avoid spurious associations. While we found no significant burden of runs of homozygosity, we did identify genomic regions with significantly greater abundance of ROH blocks in women delivering preterm, albeit we did not correct for multiple comparisons, which overlapped genes known to be involved in preterm birth (Figure 2)
Out of the 322 genes identified in dbPTB(19), we found 3 genes (CXCR4, PAK1, MYLK) that overlapped with ROH blocks which showed significant abundance in cases versus controls. CXCR4 encodes a CXC chemokine receptor specific for stromal cell-derived factor-1. CXCR4 has been shown to be up regulated in labor and is related to inflammatory response(24). SDF1 needs CXCR4 as a receptor and it is “probable that the activation of CXCR4 by SDF1 is one of the sources of maternal-fetal immune tolerance”(25). SDF1 is upregulated during cytotrophoblast fusion to the syncytiotrophoblast.(26) It also facilitates trophoblast invasion into endometrium and enhances VEGF expression, which is crucial for placental angiogenesis(25). NFKB, an important inflammatory signaling molecule in the placenta, also leads to activation of CXCR4. Chemokines such as CXCR4 regulate decidual leukocyte recruitment during labor(27, 28).
PAK1 encodes a family member of serine/threonine p21-activating kinases, known as PAK proteins. PAK1 is only present in pregnant myometrial tissue(29). PAKS have been shown to regulate uterine contractility and/or load bearing during pregnancy(29). PKN1, is an alias of PAK1; an increase of PKN1 is associated with an increase in GTP RHOA (regulator of actin-myosin in uterine smooth muscle cells) in spontaneous preterm labor(30). Increases in contractile activity in term and preterm labor may be due to increase in RHO activity or RHO related proteins(30). Up-Regulation of myometrial RHO effector proteins (PKN1 and DIAPH1) is associated with increased GTP-RHOA in spontaneous preterm labor (29, 30).
MYLK, a member of the immunoglobulin gene superfamily, is expressed in muscle that encodes myosin light chain kinase. MYLK is involved in generation of smooth muscle tissue. Interestingly, studies show down regulation of MYLK along with the disparate regulation of MYL9, suggesting a mechanistic role of S-nitrosylation in preterm labor (31). This has been substantiated in a review of the human uterine smooth muscle S-nitrosoproteome fingerprint in pregnancy, labor and preterm labor(31).
These results are interesting from an evolutionary point of view. Risk alleles for preterm birth that deleteriously affect reproductive fitness should be eliminated by purifying selection(32) . This phenomenon occurs more rapidly with greater efficiency of elimination for dominant alleles, allowing recessive alleles to persist in the population. This effect is called directional dominance(33). Directional dominance has been investigated particularly for behavioral traits where such observations have been made for schizophrenia and other forms of intellectual disability(33). We saw significant overlap of ROH blocks with genes associated with a mammalian gene set previously shown to have an evolutionary link to pre-term birth(20). Interestingly, CXCR4 was identified both by mapping the genes linked evolutionarily to preterm birth and the genes identified by pathway-based analysis of a preterm birth GWAS(19, 20). PPP3CB is another of the evolutionarily linked genes. PPP3CB (also known as calcineurin A beta) is involved in the calcium influx dependent dephosphorylation of MEF-2A (which is involved in muscle development, neuronal differentiation, cell growth control)(31). Increased calcineurin A mRNA levels were found during pregnancy and studies have found the presence and activation of calcineurin/NFAT signaling pathway just before labor in mice(34). In addition to the overlap with dbPTB genes and mammalian genes shown to have an evolutionary link to preterm birth, we found 199 genes overlapping the 50 kb significant windows.
Our study has both strengths and limitations. We examined a large collection of patients. This was a homogeneous population with very little substructure or stratification. The patients were individually genotyped on a high density array. We used a strict definition for prematurity (<34 weeks gestation) to avoid association with the more recent increases in late preterm infants. Our data were corrected for hemizygosity. We saw no association of copy number variations with preterm birth in this population. The limitations of the study include that this was a solely Danish population and we did not have a replication set.
We conclude that while we found no significant burden of runs of homozygosity, we did identify genomic regions with significantly greater abundance of ROH blocks in women delivering preterm that overlapped genes known to be involved in preterm birth. These data will be useful for future analysis of variants identified by deep sequencing or other strategies. They will help to prioritize candidate genes for further testing and/or biological validation.
METHODS
Case - Control Descriptions and Definitions
The Danish National Birth Cohort (DNBC) followed over 100,000 pregnancies beginning in the first trimester and has extensive biological material and epidemiologic data on health outcomes in both mother and child. Informed consent was obtained from all mothers as part of the DNBC data collection (35). The Gene Environment Association Studies initiative (GENEVA), funded by the trans-NIH Genes, Environment, and Health Initiative (GEI), dbGaP Study Accession: phs000103.v1.p1. consists of data for approximately 4000 Danish women and children from the DNBC, including phenotype and genotype information from approximately 1,000 mother-child pairs with mostly spontaneous onset of preterm labor or preterm premature rupture of membranes (PPROM) and 1,000 term birth mother-child pairs. Global inclusion criteria were applied to cases and controls, consisting of singleton gestation, live birth, child free of congenital abnormalities, absence of maternal conditions known to be associated with preterm delivery or often requiring early delivery of the baby (placenta previa, placental abruption, polyhydramnios, isoimmunization, placental insufficiency, pre-eclampsia/eclampsia). In addition, for inclusion in the GENEVA study, the child’s parents and all four grandparents had to have been born in Denmark (except in 24 cases with one or two grandparents from other Nordic countries). For our interests, we limited our study to the genotype data of mothers that fell into two phenotypic groups: Controls includes mothers that went in labor at 38 weeks of gestation and later and Cases includes mothers that went into labor at 34 weeks of gestation and earlier. This analysis was approved by Institutional Review Board at Women & Infants Hospital of Rhode Island.
Genotyping
Genome-wide SNP genotyping was performed using the Illumina Human 660 W-Quad_v1_A, (Illumina, San Diego, CA) (n=560,768 SNPs). According to the data set release as well as the quality control and quality assurance policies of the GENEVA consortium(36), genotypes were not reported for any SNP which had a call rate less than 85% or which had more than 1 replicate error as defined with the HapMap control samples. Identical-by-descent (IBD) coefficients were estimated using 107,014 au-tosomal SNPs with missing call rate < 5% and no closer than 15 kb apart(36).
Population Structure and Relatedness
Population structure was examined by principal components analysis (PCA), as described by Patterson et al.(37), using independent, autosomal SNPs with missing call rates < 5% and MAF > 5%. To select independent SNPs, we utilized PLINK’s LD pruning function(38). For the first round of short-range LD pruning we used a fifty SNP window with a shift of five SNPs, and pairwise genetic correlation with a threshold of 0.2. In a second round to remove long-range LD, we took the average number of SNPs over 5Mb from the output of the first round as our window (s = 131), again with an iteration of five SNPs and a threshold of 0.2. The resulting 69,877 SNPs were used to generate the principal components.
The relatedness between each pair of participants was evaluated by estimation of three coefficients corresponding to the probability that two (k2), one (k1) or zero (k0) pairs of alleles are identical-by-descent (IBD). IBD coefficients were estimated using 107,014 autosomal SNPs with missing call rate < 5% and no closer than 15 kb apart. All participants were analyzed together and all pairs of participants with a kinship coefficients > 1/64 were recorded.
Runs of Homozygosity
We used the Runs of Homozygosity (ROH) program in PLINK version 1.07 to identify autosomal ROH(38). The PLINK ROH algorithm is especially well-suited for SNP genotyping data analysis(39). Based on comparative studies among the ROH methods, PLINK was judged effective not only for SNP genotyping arrays but also on whole exome sequencing based data for long (≤1500 kb) ROH. We analyzed a range of minimum final segment lengths of ROH blocks including 1000 kb, 2000 kb and 3000 kb. Data from Chromosomes X and Y were excluded. We allowed for one heterozygous and five missing calls per window in order to prevent underestimating ROH as a result of an occasional genotyping error or missing genotype. In addition, the minimum density accepted was 50 (1 SNP per 50 kb) and the largest gap accepted was 1000 kb. In order to identify ROH that may be common in the general population, we used publicly available HapMap individual female genotypes from 30 CEU family trios (CEPH Utah residents with ancestry from Northern and Western Europe)(40). We used the genotype data from females only and ran ROH analysis to identify the possible common regions of ROH. We did not exclude any of these HapMap overlapping regions from our data, but instead we report those regions that overlap the previously identified genes associated with preterm birth from this Danish data set.
CNV Analysis
Since hemizygous deletions can mistakenly be designated as ROH, we analyzed copy number variations (CNV) to identify CNV regions that overlap with ROH blocks. We used Illumina’s Bead Studio Program and PennCNV for CNV analysis. The results from the two different programs were largely similar. We used custom Perl scripts to identify the overlapping regions along the genome. For association testing we used PLINK to perform permutation-based test of association on segmental CNV data for cases and controls using the PennCNV variants.
Homozygosity Burden Analysis
The burden of autosomal homozygosity was analyzed by calculating the proportion of individuals with at least one ROH, the total number of ROH per individual, and the total length of ROH per individual for a given minimum final segment length. To determine the presence of statistically significant burden we used logistic regression with preterm/term status as the dependent variable for each of these predictors including maternal age at delivery as a covariate. Univariate analysis of the genotype data has demonstrated that the mother’s age at delivery did not have any apparent effect and thus was left out of subsequent analysis. There was no significant difference in infants’ gender and minimal difference in BMI as reported in GENEVA quality control document.
ROH Mapping and Analysis
We mapped and analyzed the abundance of ROH both genome-wide and by comparison to specific gene sets. First, we divided each chromosome (chr 1-22) into 50 kb windows and mapped the ROH blocks that overlapped with these windows. Any overlap was included and an ROH could be counted as overlapping multiple 50 kb windows. We applied Fisher exact test for each 50 kb window to compare the abundance of overlapping ROH in cases vs. controls. If a section carried at least 2 ROH blocks from cases or controls we included the section in the calculations. We used UCSC Table Browser to map significant 50 kb windows to all genes which have any overlap.
We next analyzed two gene sets associated with prematurity for their overlap with our identified ROH(19, 20). For a given set of genes, the length of each gene from build Hg18 plus 5 kb upstream and 5 kb downstream extensions was used to map overlap between the ROH blocks and the genes of interest. To be consistent in our approach, we again applied the rule: if at least two ROH blocks from cases or controls overlapped the gene we applied Fisher exact test to determine the significance of differential abundance in overlap between cases and controls for the gene.
Supplementary Material
Acknowledgments
Funding Statement: This work was supported by the National Foundation March of Dimes Prematurity, White Plains, NY, Initiative grant number 21-FY14-154, and National Institutes of Health Grants, Bethesda, Maryland, NIH-5T35HL094308-02, NIH-NCRR P20 RR018728 and NIH P20GM103537. XZ was supported by a developmental project award from the Atlantic Coast Sexually Transmitted Infection Cooperative Research Center (AC STI CRC), from the National Institutes of Health (U19 NIH/NIAID U19AI113170). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of this manuscript.
We thank Vincent Lynch (University of Chicago) for the mammalian gene set linked to evolutionary risk of preterm birth (20). XZ was supported by a developmental project award from the Atlantic Coast Sexually Transmitted Infection Cooperative Research Center (AC STI CRC), from the National Institutes of Health (U19 NIH/NIAID U19AI113170)
Footnotes
Disclosure: We have no disclosures to declare.
Data Access
The results are based on SNP genotype data from the GENEVA study of the Danish National Birth Cohort, available in dbGaP PHS 000103.v1.p1. The genes used for the analysis are based on the Database for Preterm Birth(41).
References
- 1.McQuillan R, Leutenegger AL, Abdel-Rahman R, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szpiech ZA, Xu J, Pemberton TJ, et al. Long runs of homozygosity are enriched for deleterious variation. Am J Hum Genet. 2013;93:90–102. doi: 10.1016/j.ajhg.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muglia LJ, Katz M. The enigma of spontaneous preterm birth. N Engl J Med. 2010;362:529–535. doi: 10.1056/NEJMra0904308. [DOI] [PubMed] [Google Scholar]
- 4.Crider KS, Whitehead N, Buus RM. Genetic variation associated with preterm birth: a HuGE review. Genet Med. 2005;7:593–604. doi: 10.1097/01.gim.0000187223.69947.db. [DOI] [PubMed] [Google Scholar]
- 5.Pennell CE, Jacobsson B, Williams SM, et al. Genetic epidemiologic studies of preterm birth: guidelines for research. Am J Obstet Gynecol. 2007;196:107–118. doi: 10.1016/j.ajog.2006.03.109. [DOI] [PubMed] [Google Scholar]
- 6.Plunkett J, Muglia LJ. Genetic contributions to preterm birth: implications from epidemiological and genetic association studies. Ann Med. 2008;40:167–195. doi: 10.1080/07853890701806181. [DOI] [PubMed] [Google Scholar]
- 7.Boyd HA, Poulsen G, Wohlfahrt J, Murray JC, Feenstra B, Melbye M. Maternal contributions to preterm delivery. Am J Epidemiol. 2009;170:1358–1364. doi: 10.1093/aje/kwp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clausson B, Lichtenstein P, Cnattingius S. Genetic influence on birthweight and gestational length determined by studies in offspring of twins. BJOG. 2000;107:375–381. doi: 10.1111/j.1471-0528.2000.tb13234.x. [DOI] [PubMed] [Google Scholar]
- 9.Treloar SA, Macones GA, Mitchell LE, Martin NG. Genetic influences on premature parturition in an Australian twin sample. Twin Res. 2000;3:80–82. doi: 10.1375/136905200320565526. [DOI] [PubMed] [Google Scholar]
- 10.Ward K, Argyle V, Meade M, Nelson L. The heritability of preterm delivery. Obstet Gynecol. 2005;106:1235–1239. doi: 10.1097/01.AOG.0000189091.35982.85. [DOI] [PubMed] [Google Scholar]
- 11.Witkin SS, Vardhana S, Yih M, Doh K, Bongiovanni AM, Gerber S. Polymorphism in intron 2 of the fetal interleukin-1 receptor antagonist genotype influences midtrimester amniotic fluid concentrations of interleukin-1beta and interleukin-1 receptor antagonist and pregnancy outcome. Am J Obstet Gynecol. 2003;189:1413–1417. doi: 10.1067/s0002-9378(03)00630-6. [DOI] [PubMed] [Google Scholar]
- 12.Little J. Invited commentary: maternal effects in preterm birth--effects of maternal genotype, mitochondrial DNA, imprinting, or environment? Am J Epidemiol. 2009;170:1382–1385. doi: 10.1093/aje/kwp326. [DOI] [PubMed] [Google Scholar]
- 13.Svensson AC, Sandin S, Cnattingius S, et al. Maternal effects for preterm birth: a genetic epidemiologic study of 630,000 families. Am J Epidemiol. 2009;170:1365–1372. doi: 10.1093/aje/kwp328. [DOI] [PubMed] [Google Scholar]
- 14.Weinberg CR, Shi M. The genetics of preterm birth: using what we know to design better association studies. Am J Epidemiol. 2009;170:1373–1381. doi: 10.1093/aje/kwp325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnstone F, Inglis L. Familial trends in low birth weight. Br Med J. 1974;3:659–661. doi: 10.1136/bmj.3.5932.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mumtaz G, Nassar AH, Mahfoud Z, et al. Consanguinity: a risk factor for preterm birth at less than 33 weeks’ gestation. Am J Epidemiol. 2010;172:1424–1430. doi: 10.1093/aje/kwq316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obeidat BR, Khader YS, Amarin ZO, Kassawneh M, Al Omari M. Consanguinity and adverse pregnancy outcomes: the north of Jordan experience. Matern Child Health J. 2010;14:283–289. doi: 10.1007/s10995-008-0426-1. [DOI] [PubMed] [Google Scholar]
- 18.Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uzun A, Dewan AT, Istrail S, Padbury JF. Pathway-based genetic analysis of preterm birth. Genomics. 2013;101:163–170. doi: 10.1016/j.ygeno.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynch VJ, Leclerc RD, May G, Wagner GP. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat Genet. 2011;43:1154–1159. doi: 10.1038/ng.917. [DOI] [PubMed] [Google Scholar]
- 21.Abbas HA, Yunis K. The effect of consanguinity on neonatal outcomes and health. Hum Hered. 2014;77:87–92. doi: 10.1159/000362125. [DOI] [PubMed] [Google Scholar]
- 22.Bellad MB, Goudar SS, Edlavitch SA, et al. Consanguinity, prematurity, birth weight and pregnancy loss: a prospective cohort study at four primary health center areas of Karnataka, India. J Perinatol. 2012;32:431–437. doi: 10.1038/jp.2011.115. [DOI] [PubMed] [Google Scholar]
- 23.Gamsiz ED, Viscidi EW, Frederick AM, et al. Intellectual disability is associated with increased runs of homozygosity in simplex autism. Am J Hum Genet. 2013;93:103–109. doi: 10.1016/j.ajhg.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephen GL, Lui S, Hamilton SA, et al. Transcriptomic profiling of human choriodecidua during term labor: inflammation as a key driver of labor. Am J Reprod Immunol. 2015;73:36–55. doi: 10.1111/aji.12328. [DOI] [PubMed] [Google Scholar]
- 25.Rzepka R, Dolegowska B, Rajewska A, Kwiatkowski S. On the significance of new biochemical markers for the diagnosis of premature labour. Mediators Inflamm. 2014;2014:251451. doi: 10.1155/2014/251451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaleel MA, Tsai AC, Sarkar S, Freedman PV, Rubin LP. Stromal cell-derived factor–1 (SDF-1) signalling regulates human placental trophoblast cell survival. Mol Hum Reprod. 2004;10:901–909. doi: 10.1093/molehr/gah118. [DOI] [PubMed] [Google Scholar]
- 27.Hamilton SA, Tower CL, Jones RL. Identification of chemokines associated with the recruitment of decidual leukocytes in human labour: potential novel targets for preterm labour. PLoS One. 2013;8:e56946. doi: 10.1371/journal.pone.0056946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marvin KW, Keelan JA, Eykholt RL, Sato TA, Mitchell MD. Use of cDNA arrays to generate differential expression profiles for inflammatory genes in human gestational membranes delivered at term and preterm. Mol Hum Reprod. 2002;8:399–408. doi: 10.1093/molehr/8.4.399. [DOI] [PubMed] [Google Scholar]
- 29.Moore F, Da Silva C, Wilde JI, Smarason A, Watson SP, Lopez Bernal A. Up-regulation of p21- and RhoA-activated protein kinases in human pregnant myometrium. Biochem Biophys Res Commun. 2000;269:322–326. doi: 10.1006/bbrc.2000.2290. [DOI] [PubMed] [Google Scholar]
- 30.Lartey J, Smith M, Pawade J, Strachan B, Mellor H, Lopez Bernal A. Up-regulation of myometrial RHO effector proteins (PKN1 and DIAPH1) and CPI-17 (PPP1R14A) phosphorylation in human pregnancy is associated with increased GTP-RHOA in spontaneous preterm labor. Biol Reprod. 2007;76:971–982. doi: 10.1095/biolreprod.106.058982. [DOI] [PubMed] [Google Scholar]
- 31.Ulrich C, Quilici DR, Schlauch KA, Buxton IL. The human uterine smooth muscle S-nitrosoproteome fingerprint in pregnancy, labor, and preterm labor. Am J Physiol Cell Physiol. 2013;305:C803–816. doi: 10.1152/ajpcell.00198.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charlesworth D, Willis JH. The genetics of inbreeding depression. Nat Rev Genet. 2009;10:783–796. doi: 10.1038/nrg2664. [DOI] [PubMed] [Google Scholar]
- 33.Keller MC, Simonson MA, Ripke S, et al. Runs of homozygosity implicate autozygosity as a schizophrenia risk factor. PLoS Genet. 2012;8:e1002656. doi: 10.1371/journal.pgen.1002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tabata C, Ogita K, Sato K, et al. Calcineurin/NFAT pathway: a novel regulator of parturition. Am J Reprod Immunol. 2009;62:44–50. doi: 10.1111/j.1600-0897.2009.00710.x. [DOI] [PubMed] [Google Scholar]
- 35.Olsen J, Melbye M, Olsen SF, et al. The Danish National Birth Cohort--its background, structure and aim. Scand J Public Health. 2001;29:300–307. doi: 10.1177/14034948010290040201. [DOI] [PubMed] [Google Scholar]
- 36.Laurie CC, Doheny KF, Mirel DB, et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet Epidemiol. 2010;34:591–602. doi: 10.1002/gepi.20516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and populationfbased linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pippucci T, Magi A, Gialluisi A, Romeo G. Detection of runs of homozygosity from whole exome sequencing data: state of the art and perspectives for clinical, population and epidemiological studies. Hum Hered. 2014;77:63–72. doi: 10.1159/000362412. [DOI] [PubMed] [Google Scholar]
- 40.International HapMap C. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 41.Uzun A, Laliberte A, Parker J, et al. dbPTB: a database for preterm birth. Database (Oxford) 2012;2012:bar069. doi: 10.1093/database/bar069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.