

Abstract
SCFSkp2/Cks1 ubiquitinates Thr187-phosphorylated p27 for degradation. Over-expression of Skp2 coupled with under-expression of p27 are frequent characteristics of cancer cells. When the role of SCFSkp2/Cks1 mediated p27 ubiquitination in cancer was specifically tested by p27 Thr187-to-Ala knockin (p27T187A KI), it was found dispensable for KrasG12D induced lung tumorigenesis but essential for Rb1 deficient pituitary tumorigenesis. Here we identify pRb and p53 doubly deficient (DKO) prostate tumorigenesis as a context in which p27 ubiquitination by SCFSkp2/Cks1 is required for p27 down-regulation. p27 protein accumulated in prostate when p27T187A KI mice underwent DKO prostate tumorigenesis. p27T187A KI or Skp2 knockdown (KD) induced similar degrees of p27 protein accumulation in DKO prostate cells, and Skp2 KD did not further increase p27 protein in DKO prostate cells that contained p27T187A KI (AADKO prostate cells). p27T187A KI activated an E2F1-p73-apoptosis axis in DKO prostate tumorigenesis, slowed disease progression, and significantly extended survival. Querying co-occurrence relationships among RB1, TP53, PTEN, NKX3-1, and MYC in TCGA of prostate cancer identified co-inactivation of RB1 and TP53 as the only statistically significant co-occurrences in metastatic castration resistant prostate cancer (mCRPC). Together, our study identifies Skp2/Cks1 pocket inhibitors as potential therapeutics for mCRPC. Procedures for establishing mCRPC organoid cultures from contemporary patients were recently established. An Skp2/Cks1 pocket inhibitor preferentially collapsed DKO prostate tumor organoids over AADKO organoids, which spontaneously disintegrated over time when DKO prostate tumor organoids grew larger, setting the stage to translate mouse model findings to precision medicine in the clinic on the organoid platform.
Keywords: p27 phosphorylation and accumulation, Skp2/Cks1 pocket inhibitors, advanced prostate cancer, prostate cancer organoids
Introduction
Levels of p27, a cyclin-dependent kinase inhibitor, can be regulated by mRNA expression, protein synthesis, and protein degradation. One down-regulation mechanism is its ubiquitination by SCFSkp2/Cks1 ubiquitin ligase, which prepares p27 for degradation in the proteasomes (1). SCFSkp2/Cks1-mediated p27 ubiquitination requires its phosphorylation on Thr187. Replacing Thr187 with alanine (p27T187A) to render this position unphosphorylable prevents p27 binding to and ubiquitination by SCFSkp2/Cks1 (2, 3). This is because T187p and a conserved E185 together mediate p27 binding to a pocket formed jointly by Skp2 and the accessory protein Cks1 (4–6). To test the physiological significance of this biochemical mechanism, p27T187A/T187A (p27T187A KI) mice were generated (7). p27T187A KI mice do not show p27 protein accumulation, demonstrating that p27 ubiquitination by SCFSkp2/Cks1 is dispensable for p27 degradation in normal physiology.
p27 is a haplo-insufficient tumor suppressor (8), whose reduced expression is frequently associated with aggressive cancers (9). Knockout of p27 induces spontaneous pituitary tumorigenesis and can accelerate tumorigenesis induced by a large number of oncogenic events in numerous tissues, suggesting that stabilizing p27 can be therapeutic for many types of cancer. However, when p27 KO accelerated lung tumorigenesis in KrasG12D/+ mice, p27T187A KI did not have a protective effect (10), suggesting that p27 ubiquitination by SCFSkp2/Cks1 is dispensable for p27 degradation in lung tumorigenesis by KrasG12D. On the other hand, p27 KO accelerated pituitary melanotroph tumorigenesis in Rb1+/− mice (11) and p27T187A KI blocked it (12). Thus, the role of SCFSkp2/Cks1 in mediating p27 degradation can be highly critical in promoting tumorigenesis but only in specific contexts.
Finding additional tumorigenic contexts in which SCFSkp2/Cks1 plays a critical role in promoting tumorigenesis could be challenging, since most tissues resist tumorigenesis by deletion of Rb1 alone. It is however evident that, once the susceptible contexts are identified, inhibition of SCFSkp2/Cks1 is technically feasible (13, 14), affects a highly select sub-group of SCFSkp2 substrates, and is forecasted by the p27T187A KI mice to be non-toxic. Here we identify pRb and p53 doubly deficient (DKO) prostate tumorigenesis as an additional susceptible context. We further make the case for translating mouse model findings to clinical metastatic castration resistant prostate cancer (mCRPC) on the organoid platform.
Results
p27T187A KI accumulates p27 in pRb and p53 doubly deficient prostate
Based on our previous findings that Skp2 KO and p27T187A KI accumulated p27 in Rb1 KO melanotroph tumorigenesis (15) and Skp2 KO accumulated p27 in Rb1 and Trp53 DKO prostate tumorigenesis (16), we hypothesized that p27T187A KI could also accumulate p27 in DKO prostate tumorigenesis. To test this hypothesis, we generated mice of p27+/+, p27T187A/T187A, and PB-Cre4;Rb1lox/lox;Trp53lox/lox on p27+/+ or p27T187A/T187A background to determine p27 expression in their prostate epithelium by IHC. At 5–6 months of age, co-deletion of Rb1 and Trp53 induced neoplastic lesions only in proximal prostate acini. We show p27 staining in distal prostate acini to compare the four genotypes in normal appearing tissues (Figure 1Aa,c,e,g). Similar p27 staining were observed in p27+/+ and p27T187A/T187A prostates (Figure 1Ab,d). When Rb1 and Trp53 were co-deleted, p27 staining reduced in p27+/+ prostate (Figure 1Af), but increased in p27T187A/T187A prostates (Figure 1Ah). p27 Western blot of the prostate ventral and anterior lobes provided similar findings (Figure 1B). This Western blot also revealed increased Skp2 protein levels in DKO prostate in p27+/+ and p27T187A/T187A mice. This finding resembles similar findings in human prostate cancer and explains in part the reduction of p27 in p27+/+ prostate but increase of p27 in p27T187A/T187A prostate (please see Discussion for more details).
Figure 1. p27T187A/T187A mice accumulated p27 protein in DKO prostate.
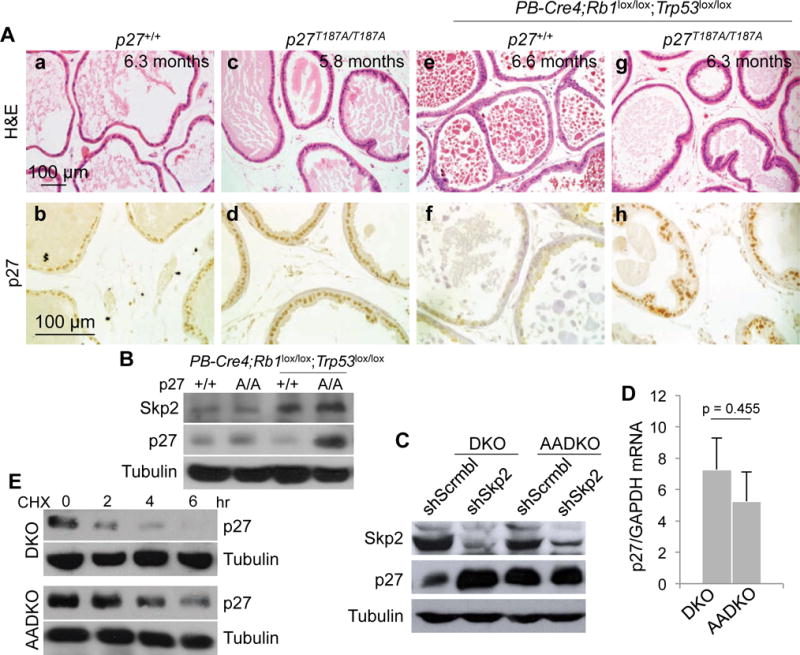
(A) Representative (from ten mice of each genotype) H&E and p27 IHC staining of consecutive prostate sections of the four genotypes as marked. (B) Protein levels were determined by Western blot of prostate ventral and anterior lobes of the indicated genotypes. A/A is abbreviated from T187A/T187A. (C) DKO and AADKO prostate cells were transduced with lentiviruses expressing the indicated shRNA (Scrmbl, scrambled sequences). Following antibiotic selection, the transduced cells were subject to Western blot for the indicated proteins. The same cells were subject to RT-qPCR to determine levels of p27 mRNA relative to GAPDH mRNA (D) and CHX chase to determine p27 protein stability compared to α-tubulin (E). Error bars indicate s.e.m. of the means of three samples. p value is by two-sided t test.
To learn the molecular basis for this requirement for p27 Thr187 phosphorylation, we established early passage prostate cells from DKO prostate in p27+/+ mice (called DKO cells) and in p27T187A/T187A mice (AADKO cells). We found that Skp2 knockdown in DKO cells increased p27 protein levels while AADKO cells intrinsically contained similarly increased levels of p27 (Figure 1C). RT-qPCR and CHX experiments suggest that p27 accumulation in AADKO prostate cells was not due to increased expression of p27 mRNA (Figure 1D) but was due to increased protein stability (Figure 1E). If p27T187A KI prevented p27 ubiquitination by SCFSkp2/Cks1 to accumulate p27, knockdown of Skp2 in p27T187A KI cells would not further increase p27, and this proved true (Figure 1C). These genetic and molecular findings suggest that ubiquitination of p27T187p by SCFSkp2/Cks1 became rate-limiting in preventing p27 accumulation in prostate when Rb1 and Trp53 were co-deleted in it.
p27T187A KI activates a p27-E2F1-p73-apoptosis axis in DKO prostate tumorigenesis
Skp2 KO or p27T187A KI inhibited pRb deficient pituitary melanotroph tumorigenesis by E2F1 induced apoptosis (Figure S1A) (15, 17). To determine whether this E2F1 induced apoptosis could remain effective when Trp53 is additionally deleted, we determined the contribution of p73, an E2F1 target gene and a p53 family member, to this apoptosis. As shown in Figure S1B. Rb1 deletion did not increase apoptosis, measured as sub-G1 cells, in Skp2 WT MEFs, but increased them to 19% of the population in Skp2 KO MEFs. Combining knockdown of p73 reduced sub-G1 cells to 7–8% of the population (Figure S1C), demonstrating that p73 contributed about half of the E2F1 induced apoptosis in this context.
We next investigated whether p27T187A KI activated this p27-E2F1-p73 axis in AADKO prostate tumor cells. We found that Skp2 knockdown in DKO cells increased p73 protein. AADKO cells intrinsically contained a similarly increased level of p73 protein, which Skp2 knockdown did not further increase (Figure 2A). Thus, effects of Skp2 knockdown and p27T187A KI on p73 protein mirrored those on p27 protein. At mRNA levels however, p27T187A KI significantly increased expression of p73 mRNA in AADKO cells compared to DKO cells (Figure 2B), different from p27 (Figure 1D). The increased p73 mRNA expression was accompanied by significantly increased occupancy of E2F1 and p73 promoters (both are typical E2F target genes) but not GAPDH promoter (a housekeeping gene) by E2F1 in AADKO cells compared to DKO cells (Figure 2C, left). E2F4 occupancy on E2F1 and p73 promoters did not increase in AADKO cells (Figure 2C, middle), which is consistent with the fact that E2F4 does not bind cyclin A. ChIP with normal IgG did not enrich E2F1 or p73 promoter sequences in AADKO cells (Figure 2C, right).
Figure 2. p27T187A KI activated an E2F1-p73 axis.
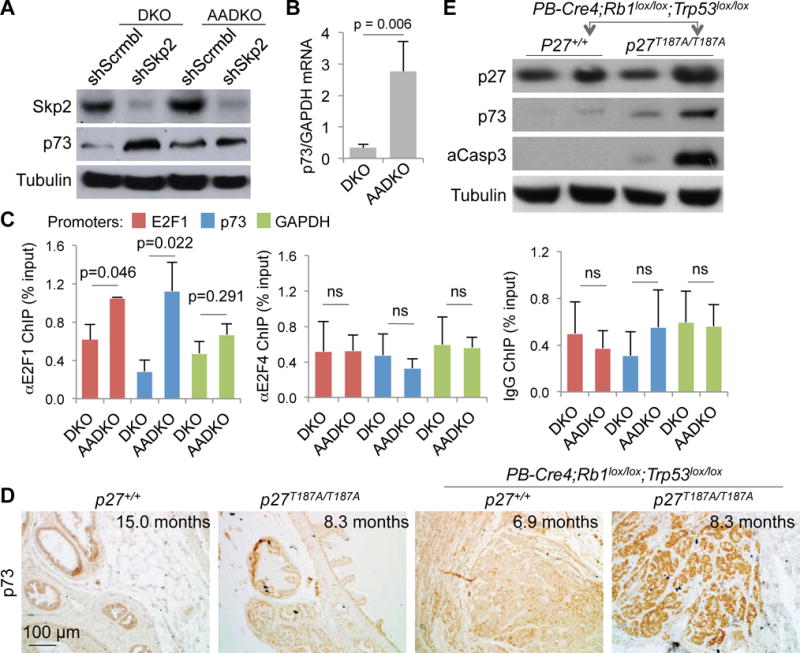
(A) DKO and AADKO prostate cells treated with the indicated shRNA (as in Figure 1C) were subject to Western blot. (B) The same cells were used for RT-qPCR to determine levels of p73 mRNA relative to GAPDH mRNA. (C) ChIP assay using antibody for E2F1 (left), E2F4 (middle), or normal IgG as control, to determine recruitment of E2F1 and E2F4 onto E2F1, p73, and GAPDH promoters in DKO and AADKO prostate cells. Error bars indicate s.e.m. of the means of three samples. p values are by two-sided t test. ns, not significant (p > 0.05). (D) Representative (of three, see also Figure S2) p73 IHC staining of prostate sections of the four genotypes as indicated. (E) Protein levels were determined by Western blot with prostate ventral and anterior lobes of the indicated genotypes.
We next determined p73 protein expression in prostate tissues. Of the four genotypes examined, we found robust anti-p73 staining only in p27T187A/T187A;PB-Cre4;Rb1lox/lox;Trp53lox/lox prostate (Figure 2D). Western blot of prostate tissues confirmed the staining findings (Figure 2E). p73 protein similarly increased in Skp2−/−;PB-Cre4;Rb1lox/lox;Trp53lox/lox prostate (Figure S2).
The Western blot also revealed accumulation of cleaved caspase 3 in p27T187A/T187A;PB-Cre4;Rb1lox/lox;Trp53lox/lox prostate tissues (Figure 2E), suggesting that the increased p73 in this genotype induced apoptosis in the absence of p53. We next used TUNEL staining to demonstrate that DKO neoplastic lesions contained more apoptosis in p27T187A/T187A mice than in p27+/+ mice (Figure 3A,B,C). p73 can induce apoptosis independent of p53 likely because the TAp73 isoform activates similar target genes for cell cycle arrest (p21) or apoptosis (PUMA, BAX) as p53 (see (18) for a recent review). To determine whether p73 contributed to this apoptosis, we determined the effects of p73 knockdown (Figure 3D) in AADKO prostate tumor cells. In untreated AADKO cell culture, 8.8% of the population showed sub-G1 DNA content. Knockdown of p73 reduced the sub-G1 population to 1.4%, which was confirmed by a 2nd shp73 (Figure 3E). We further show that DKO prostate tumor cells contained little sub-G1 cells (0.6%), which was increased to 17% by Skp2 knockdown, additional knockdown of p73 reduced it to background levels (Figure 3F). These findings delineate a p27-E2F1-p73-apoptosis pathway activated by p27T187A KI in DKO prostate tumorigenesis (Figure S3).
Figure 3. p27T187A KI increased apoptosis in DKO prostate tumorigenesis and p73 played a major role in the apoptosis.
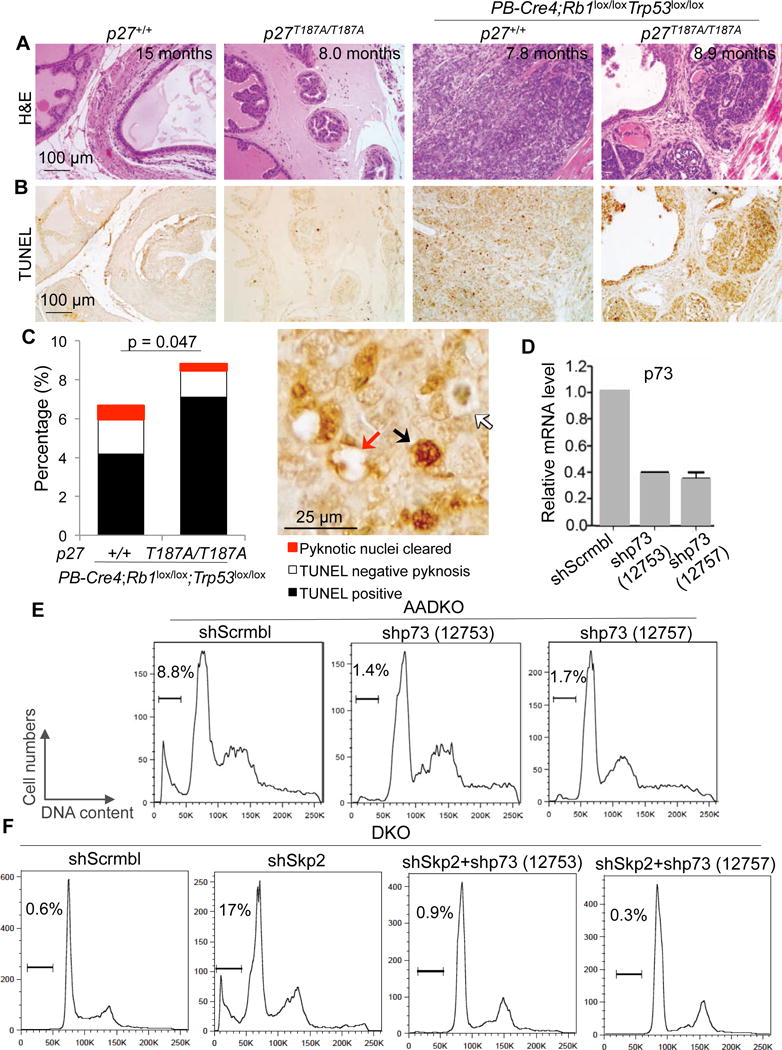
(A and B) Consecutive prostate sections of four genotypes, as indicated, stained with H&E or TUNEL. (C) Quantification of apoptosis. Arrows in the photograph demonstrate how total apoptosis was quantified. About 200 cells on each section/mouse were counted. The bar graph was based on the average of quantifications from three mice. p value is by two-sided t test comparing the combined apoptosis in p27+/+ and p27T187A/T187A mice. (D) RT-qPCR to determine knockdown efficiencies by two shp73 constructs. Error bars are s.e.m. (E and F) Propidium iodide based DNA content FACS (representative of two experiments) to detect and quantify apoptosis as sub-G1 cell% in the population, as marked above the brackets.
p27T187A KI significantly delays progression of DKO prostate tumorigenesis to lethality
DKO prostate tumorigenesis is invasive, metastatic, castration resistant, and becomes lethal within 7–10 months (19), but cannot progress beyond PIN stages in Skp2 KO mice (16). Skp2 KO mice are significantly smaller and less fertile (20), which are not seen in p27T187A KI mice (7). We next determined how DKO prostate tumorigenesis progressed in p27T187A KI mice.
In addition to inducing p53-independent apoptosis via a p27-E2F1-p73 axis, p27T187A KI inhibited cell proliferation in DKO prostate tumorigenesis, as measured by Ki67 (Figure S4A,C) and pHH3 (Figure S4B,D) staining. At 5–7 months of age, 50% of PB-Cre4;Rb1lox/lox;Trp53lox/lox mice contained gross prostate tumors but none of the p27T187A/T187A;PB-Cre4;Rb1lox/lox;Trp53lox/lox mice did so; by 8–9 months, 80% of PB-Cre4;Rb1lox/lox;Trp53lox/lox mice contained gross prostate tumors compared to 50% of p27T187A/T187A;PB-Cre4;Rb1lox/lox;Trp53lox/lox mice (Figure 4A,B). When characterized by survival, 2 of 30 PB-Cre4;Rb1lox/lox;Trp53lox/lox mice were alive pass 9 months, while 13 of 38 p27T187A/T187A;PB-Cre4;Rb1lox/lox;Trp53lox/lox mice survived pass 10 months (Figure 4C). Thus, remarkably, the innocuous p27T187A mutation can delay progression of the aggressive DKO prostate tumorigenesis to significantly extend survival.
Figure 4. Progression of DKO prostate tumorigenesis was inhibited in p27T187A/T187A mice.
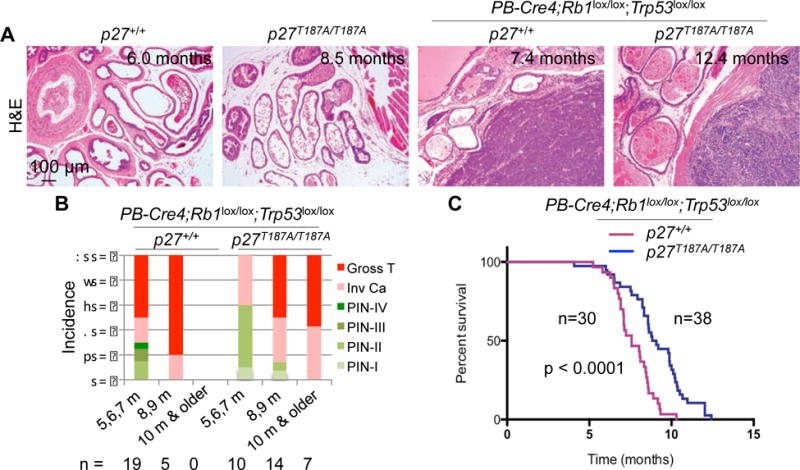
(A) Representative H&E stained prostate sections of the indicated genotypes at the indicated ages. (B) Pathological diagnoses of prostate lesions as PIN of four stages, invasive carcinomas, and gross tumors. Numbers of mice examined in each age group are indicated below the chart. (C) Kaplan-Meier survival analysis comparing the p27+/+ and p27T187A/T187A cohorts of mice undergoing DKO prostate tumorigenesis. Hazard ratio = 2.514, 95% confidence interval = 2.077 to 6.222, p is by log-rank test.
Co-occurrences of RB1 and TP53 inactivation is statistically significant in mCRPC
To determine how often both RB1 and TP53 are genetically inactivated in human prostate cancer, we queried the genetic status of RB1, TP53, PTEN, NKX3-1, and MYC (five known prostate cancer drivers) in TCGA of prostate cancer on cBioPortal (21) (Table 1). Inactivation of RB1 and TP53 did not co-occur in three cohorts of primary prostate cancer but co-occurred with statistical significance in two of the three metastatic castration resistant prostate cancer (mCRPC) cohorts. Many of the co-occurred inactivation are likely biallelic since they were detected together with shallow deletions of RB1 and TP53 (Figure S5). None of the other possible pairs among these five genes showed statistically significant co-occurrences for their alterations. Inactivation of TP53 and activation of MYC co-occurred with statistical significance in one primary prostate cancer cohorts but the co-occurrences lost statistical significance with disease progression to mCRPC.
Table 1.
Pairwise co-occurrence relationships among RB1, TP53, PTEN, NKX3-1 and MYC in primary prostate cancer and metastatic castration-resistant prostate cancer (mCRPC)
| Primary prostate cancer | mCRPC | |||||
|---|---|---|---|---|---|---|
| Studies | MSKCC, 20101 | Broad/Cornell 2012 | TCGA, 2015 | MSKCC, 20101 | Michigan, 20122 | Robinson et al, 2015 |
| Specimen #s | 157 | 109 | 333 | 28 | 502 | 150 |
| RB1 inactivation3 | 3.2% | 0.0% | 0.9% | 10% | 29% | 8.6% |
| TP53 inactivation3 | 1.9% | 6.4% | 7.5% | 10% | 54% | 50% |
| PTEN inactivation3 | 5.7% | 7.3% | 17% | 39% | 50% | 40% |
| NKX3-1 inactivation3 | 3.8% | 0.0% | 16% | 3.6% | 18% | 3.3% |
| MYC activation4 | 40% | 1.8% | 13% | 50% | 25% | 19% |
| Co-occurrence5 | PPt, RM, PM, PtM | PPt | PM, RM, RPt, PPt | RP, RN, PN, RPt, | RM, PPt, NM, RN, PN, RPt, RP | RP, PPt, NM, PM, RPt, PN, |
| Statistic significance6 | p = 0.033 | p = 0.023 | p = 0.039 | |||
This study included both primary prostate cancer and mCRPC.
mCRPC specimens were obtained at autopsy.
Inactivation of RB1, TP53, PTEN, and NKX3-1 is queried for HOMDEL MUT.
Activation of MYC is queried for AMP MUT EXP > 2 (larger than 2 SD from the mean).
Tendency for co-occurrence is by Log Odds Ratio; R, P, Pt, N, and M are short for RB1, TP53, PTEN, NKX3-1, and MYC, respectively, to indicate the pairs.
p value is by Fisher Exact Test. p < 0.05 is considered statistically significant, which is highlighted by bold font. Other pairs show tendencies with p values between 0.083 and 0.575. Tendency pairs with p values between 0.631 (the next higher value) to 0.907 (the highest) are not shown.
RB1 and TP53 often incur Shallow Deletions, suggesting biallelic inactivation for some Mutation samples, as shown by the Oncoprints for two studies in Figure. S5.
About one quarter of the patients develop metastatic prostate cancer after prostatectomy or at presentation. These cases are treated with androgen deprivation therapies, but invariably the cancer would relapse in the form of mCRPC. Inhibitors of residual androgen synthesis, such as abiraterone acetate, 2nd generation AR antagonist, such as enzalutamide, and chemotherapies with taxane can be effective but the efficacies are not durable. Currently, mCRPCs kill 27,000 men annually in US alone. Our TCGA analyses suggest that DKO prostate tumorigenesis could serve as a mouse model for mCRPC to identify pressingly needed new therapy strategies.
A small molecule Skp2/Cks1 pocket inhibitor inhibits DKO and DU 145 cells
Using a structure-based in silico screen, small molecules that occupy Skp2/Cks1 pocket to inhibit its interaction with p27T187p were identified (13) (Figure 5A). We employed one of such inhibitors, Compound 1 (C1), to determine the pharmacologic effects of inhibiting the Skp2/Cks1-p27T187p interaction in DKO prostate tumor cells. We also tested C1 on AADKO prostate tumor cells, in which the Skp2/Cks1-p27T187p interaction is not present, to genetically interrogate its mechanism of action. There are six human metastatic prostate cancer cell lines in CCLE (Cancer Cell Line Encyclopedia (22)); one of them, DU145, is hormone-insensitive and contains truncating mutation K715* in RB1 and inactivating mutation V274F in TP53. We included DU145 as a model for human mCRPC to test the effects of C1 (Figure 5B). At 1.25 μM, C1 reduced DKO cell proliferation by 80% relative to DMSO vehicle control but reduced AADKO cell proliferation by only 20%. This selectivity for DKO cells continued when C1 concentration was increased to 2.5 μM. AADKO cells proliferated significantly more slowly than DKO cells in the absence of C1 (Figure 5C), but C1 at 5.0 μM still reduced AADKO proliferation by 75% relative to DMSO control (Figure 5B). We found that C1 treatment increased p27 only in DKO cells but increased p21 in both DKO and AADKO cells (Figure 5D). These findings support the designed mechanism of action of C1 and reveal a therapeutic advantage in pharmacological targeting of the Skp2/Cks1 pocket over p27T187A KI (Figure 5A). In DU145 cells, C1 inhibited proliferation and increased p27 and p21 to a degree comparable to DKO prostate tumor cells (Fig. 5B,C,D), suggesting that inhibition of the Skp2/Cks1 pocket is a potential therapeutic strategy for mCRPC (also see Discussion).
Figure 5. A specific inhibitor of SCFSkp2/Cks1 selectively inhibited DKO prostate tumor cells and DU145 cells in monolayer cultures.
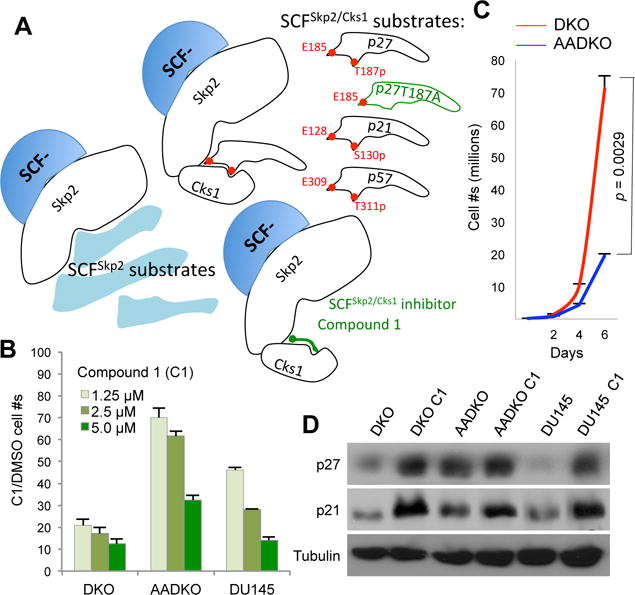
(A) The designed inhibition mechanism for SCFSkp2/Cks1 inhibitor Compound 1 (C1) compared to p27T187A KI (p57 is a predicted substrate). (B) Proliferation chart showing cell numbers relative to the vehicle (DMSO) following treatment with Skp2/Cks1 pocket inhibitor Compound 1 (C1) at three concentrations after 2 days in monolayer cultures of the indicated cells. (C) p27T187A KI inhibited proliferation of DKO prostate tumor cells in monolayer culture (representative of three independent experiments). Error bars are s.e.m. of the means of triplet plates. p value is by two-sided t test. Cell numbers were counted in triplicate plates every two days for six days. (D) Western blot following treatment with C1 (5 μM) for 2 days.
Skp2/Cks1 pocket inhibitor inhibits DKO prostate tumor organoids
Recently, six organoid lines from mCRPC biopsies from contemporary patients were reported, 5 of them contain mutations and/or gene deletions in both RB1 and TP53 (23). Unlike monolayer cultures, organoids respond to therapeutics in acini-like structures. To translate mouse model findings to clinical mCRPC on the same organoid platform, we next generated DKO and AADKO prostate tumor organoids (Figure 6A,B). At day 15 after plating, DKO and AADKO cells generated organoids with similar efficiencies and size ranges (Figure 6E,G,H). Longitudinal monitoring revealed most DKO organoids grew larger while most AADKO organoids spontaneously disintegrated into debris piles over a six-day period (Figure 6C). As such, a prominent difference between DKO and AADKO organoid cultures is the 1.9 fold increase in debris piles in the latter in untreated cultures (Figure 6F,H). These characteristics differed from monolayer cultures, where AADKO cells proliferated much more slowly than DKO cells (Figure 5C), and demonstrate the ability of DKO and AADKO organoid cultures to more closely model autochthonous DKO and AADKO prostate tumors.
Figure 6.
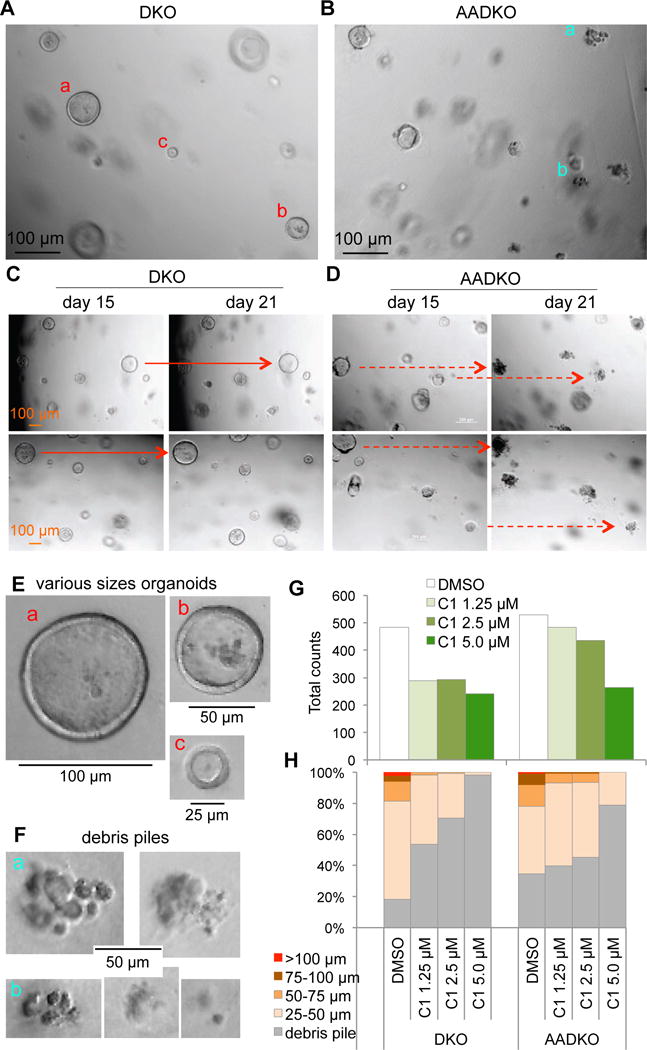
A specific inhibitor of SCFSkp2/Cks1 selectively inhibited DKO prostate tumor cell in organoid cultures. (A and B) Organoid cultures of DKO and AADKO prostate tumor cells after 15 days in culture. (C and D) The same areas of the organoid cultures were photographed on day 15 and day 21. Solid red arrows point to some examples of organoids growing larger, dashed red arrows point to some examples of organoids disintegrated into debris piles. (E) Organoids of various sizes in (A, a, b, and c in red) were cropped out and shown at the original photograph size. (F) Debris piles in (B, a and b in blue) were cropped out and shown. (G) Counting of organoids and debris piles of various sizes following treatment with C1 at various concentrations for 15 days with vehicle (DMSO) control. (H) Fractions of organoids of the indicated sizes and debris piles.
We then tested the effects of C1 on DKO and AADKO prostate tumor organoids. At 1.25 μM, C1 reduced organoid forming efficiency by 1.7 fold for DKO organoids compared to 1.1 fold for AADKO organoids (Figure 6G); and increased debris piles 2.9 fold for DKO organoids compared to 1.1 fold for AADKO organoids (Figure 6H). This selectivity for DKO organoids largely disappeared when C1 concentrations were increased to 5.0 μM, although about 20% of small AADKO organoids remained in AADKO culture. These organoid findings provide further genetic and functional validation for the therapeutic potential of Skp2/Cks1 pocket inhibitors for pRb and p53 deficient prostate cancer.
Discussion
Prostate cancer is one of the major cancer types that showed Skp2 overexpression coupled with p27 under-expression. A tissue microarray study of 4 normal prostate samples, 74 high grade prostatic intraepithelial neoplasm (HGPIN), and 622 primary prostate cancer specimens (24) documented significant Skp2 overexpression in HGPIN over normal tissue, in primary prostate cancer over HGPIN, and Skp2 overexpression significantly correlated with p27 under-expression (Table S1).
pRb inhibits Skp2 binding to p27 (25), represses Skp2 mRNA expression via E2F (26, 27), and promotes Skp2 protein degradation via APC/CCdh1 (28). Pten also inhibits Skp2 expression but the mechanisms are not known. Pten overexpression in human glioblastoma cells decreased Skp2 expression while Pten KO MEFs show increased Skp2 expression (29). To determine whether these findings could explain Skp2 overexpression in primary prostate cancer, we queried RB1, TP53, PTEN; and Skp2 in a cohort of 333 primary prostate cancer specimens in cBioPortal (Table S2). Inactivation of TP53 or PTEN tend to co-occur with activation of Skp2, but neither reached statistical significance. Inactivation of RB1 does not co-occur with Skp2 activation, likely because RB1 is inactivated in only 0.9% of the samples in this cohort,. When we made the same queries to a cohort of 150 mCRPC specimens, in which inactivation of RB1 and activation of Skp2 both become more frequent, they co-occurred with statistical significance. These TCGA findings provide one explanation for overexpression of Skp2 in prostate cancer and might explain the inhibition of Rb1 and Trp53 DKO prostate tumorigenesis or inhibition of Pten KO prostate tumorigenesis by Skp2 KO (16, 30). Skp2 has a growing list of substrates in addition to p27; p27T187A KI can test the contribution of lacking p27 ubiquitination by SCFSkp2/Cks1 to the effects of Skp2 KO. In Rb1 deficient pituitary tumorigenesis, p27T187A KI largely phenocopied Skp2 KO to block this tumorigenesis (12, 15). On the other hand, p27T187A KI did not inhibit KrasG12D/+ induced lung tumorigenesis. In this study, we determined the effects of p27T187A KI on DKO prostate tumorigenesis, which represents a more challenging tumorigenic context, since co-deletion of Rb1 and Trp53 wound disable most of the antitumor mechanisms in the cells.
Co-deletion of Rb1 and Trp53 confers dependency on SCFSkp2/Cks1 to prevent p27 protein accumulation in prostate
p27 can be ubiquitinated by several ubiquitin ligases, including SCFSkp2/Cks1, Pirh2 (31), KPC1 (32), Cul4 (33), and WWP1 (34). Combined activities of these ligases, rather than any single ligase, likely determine the stability of p27 protein. pRb inhibits Skp2 via at least three mechanisms (described in the previous section); pRb loss may thereby greatly enhance Skp2 activity, leading to greatly enhanced p27 ubiquitination by SCFSkp2/Cks1 in Rb1 deficient pituitary melanotroph tumorigenesis. In this context, p27 protein degradation became dependent on Thr187 phosphorylation; p27T187A KI therefore can inhibit Rb1 deficient pituitary tumorigenesis (12, 15).
In another context, DNA damage-activated p53 stimulates expression of Pirh2 and KPC1 as typical p53 target genes to degrade p27 in mouse embryonic fibroblasts (16). Deletion of Trp53 in prostate however did not induce p27 protein accumulation (our unpublished results), likely because p53 is not activated in prostate development and small reductions in Pirh2 and KPC1 can be compensated by other p27 ubiquitin ligases. Co-deletion of Trp53 and Rb1 reduced p27 protein (Fig. 1Ae,f), likely because the other p27 ubiquitin ligases now included increased (Fig. 1B) and activated Skp2 due to pRb loss. In this context of increased and activated Skp2 and reduced Pirh2 and KPC1, p27T187A KI accumulated p27 protein (Figure 1A and B). Our unpublished results further show that deletion of Rb1 in Skp2 KO or p27T187A KI prostate also did not accumulate p27, likely because p53 is activated by Rb1 deletion to stimulate expression of Pirh2 and KPC1. These findings, together with the results from cultured DKO and AADKO prostate cells (Figure 1C to E) provide the first combined genetic and biochemical evidence for the relevance of SCFSkp2/Cks1-mediated p27 ubiquitination in preventing p27 accumulation in vivo (please also see Introduction).
p27T187A KI identifies substrate-specific inhibitors of protein degradation for mCRPC
With FDA approval for the use of a proteasome inhibitor, bortezomib, to treat multiple myeloma, targeting ubiquitin-mediated protein degradation has become a validated cancer therapy strategy (35, 36). The current challenge is to add specificity to this strategy, since unspecific inhibition of protein degradation generates serious side-effects, which could explain at least in part the negative outcome of a phase II trial of bortezomib for castration resistant metastatic prostate cancer (37). In the ubiquitination mediated protein degradation hierarchy, inhibitors of the Skp2/Cks1 pocket are perhaps the most substrate selective, affecting only p27 and p21 (and possibly p57 by sequence prediction). Our findings that p27T187A significantly inhibited progression of DKO prostate tumorigenesis to extend survival provide a basis for identifying prostate cancer patients for treatment with such inhibitors. Indeed, in the above mentioned phase II trial of bortezomib, one of 24 evaluable patients with castration resistant metastatic prostate cancer achieved the primary end point (defined as no increase in PSA from baseline and no radiographic progression at 12 weeks) (37). It is regretful that the genetic status of RB1 and TP53 in this patient’s prostate cancer was not determined and documented in the pre-TCGA era. It is highly likely that many mCRPC patients could be indicated for therapy with Skp2/Cks1 pocket inhibitors since RB1 and TP53 inactivation co-occurred statistically significantly in mCRPC based on the most recently available TCGA of primary prostate cancer and mCRPC (Table 1).
Our findings with p27T187A KI also provided genetic support for the mechanisms of action of the currently available inhibitor compound 1 (C1), since it inhibited DKO prostate tumor cells far more effectively than AADKO prostate tumor cells (in which the interaction between Skp2/Cks1 pocket and p27 is already prevented) and, furthermore, demonstrated the superiority of such inhibitors over p27T187A KI, since C1 at higher concentration can still inhibit AADKO prostate tumor cells. Since higher concentrations of C1 induced similar p21 accumulation in DKO and AADKO cells, C1 may increase p27 and p21, likely p57 and other yet to be identified substrates of the Skp2/Cks1 pocket to exert stronger inhibition of the Skp2/Cks1 pocket than p27T187A KI. However, it is important to note that regulation of p21 and p27 protein degradation are by different as well as shared mechanisms. While SCFSkp2/Cks1 can promote degradation of p27 and p21, p21 degradation is also promoted by MDM2/MDMX (38), APC/CCdc20 (39), CRL4Cdt2 (40), and CRL2LRR1 (41), which are not known to promote p27 degradation. Furthermore, although the comparison between DKO and AADKO prostate tumor cells support the designed action mechanism of C1, the likelihood of its having other substrates, off-target effects, or both, still exists. This is especially true in human prostate cancer cell lines, such as DU145, which likely contain far more genetic and epigenetic alterations and overall far more heterogeneous than targeted mouse prostate cancer cells, such as DKO and AADKO cells.
Inhibition of DKO prostate tumorigenesis by p27T187A KI is notably weaker than that by Skp2 KO, the latter blocked DKO prostate tumorigenesis inside the PIN stage (16). It is therefore of great interest to test inhibitors that target Skp2 as a whole, such as the more recently reported inhibitor Compound 25, which blocks Skp2-Skp1 interaction in the SCFSkp2 ligase (42). The broadening of substrate spectrum would however likely result in undesirable side effects. In this respect, Skp2 KO mice are significantly smaller and less fertile while p27T187A KI mice are virtually normal.
Translating mouse model findings to clinical precision medicine on organoid platform
DU145 cell line was established about 40 years ago (43), and is the only prostate cancer cell line containing inactivating mutations in RB1 and TP53. Extended monolayer culture of established cancer cell lines can encourage secondary genomic alterations that are not present in the original cancer specimens (44, 45), and monolayer culture bears no resemblance to the prostate when testing therapeutic effects of potential therapeutics. Organoid cultures are emerging as an alternative since cancer cell organoids demonstrate stable genomic landscapes faithful to the original cancer specimens, and respond to therapeutics in acinus-like structures (46, 47). Prostate cancer organoid cultures can be generated from mouse models (48, 49), and we have now established organoid cultures from DKO and AADKO prostate tumor models. We provide evidence that the proliferation and survival characteristics of DKO and AADKO organoids more closely resemble those in DKO and AADKO autochthonous prostate tumors than DKO and AADKO monolayer cells. While monolayer AADKO cells simply proliferated much more slowly than DKO monolayer cells, their respective organoids formed with similar efficiency. The difference is in the expansion of the organoids: when DKO organoids were growing larger, AADKO organoids spontaneously disintegrated into debris piles. In this scenario, the inhibitor C1 collapsed DKO organoids into debris piles, mimicking AADKO organoids.
Six recently established mCRPC organoid lines from contemporary patients contained frequent inactivation of RB1 and TP53 while maintaining stable genomic landscapes faithful to the biopsy specimens that they were derived from (23). Based on this success, more mCRPC organoid lines from contemporary patients are being generated potentially reaching the scale of a biobank, as recently reported for colorectal cancer (47). Testing potential therapeutics on prostate cancer organoids established from mouse prostate cancer models and human clinical cancer specimens side-by-side could increase the predictive value of the observed effects in selecting candidates for further development and in guiding clinical trial designs.
Materials and Methods
Mice
PB-Cre4 mice (50), Rb1lox/lox mice (51), Trp53lox/lox mice (52), and p27T187A/T187A mice (7) were described previously. All male mice used for experiments are on FVB, C57BL6J, and 129Sv hybrid background. Animals were allocated to experimental groups based on their genotypes and ages. Investigators were not blinded to the genotypes and ages. We did not estimate sample sizes before the experiments. All samples were included in analysis when control samples validated the experiments. Sample sizes were chosen when we obtained a p value, which was based on at least three biological replicates. Mice were maintained under pathogen-free conditions in the Albert Einstein College of Medicine animal facility. Mouse experiments protocols were reviewed and approved by Einstein Animal Care and Use Committee, conforming to accepted standards of humane animal care.
IHC, IF, TUNEL Assay and microscopy
Tissue sections and staining have been described previously (16). Briefly, paraffin embedded prostate tissues were sectioned at 5 μm thickness. Slides were deparaffinized, hydrated, and incubated in a steamer for 20 minutes in sodium citrate buffer (Vector Labs, H3301) for antigen retrieval. Sections were first treated with 3% H2O2 to quench endogenous peroxidase, washed several times, blocked with 10% normal goat serum, and then incubated in primary antibodies at 4°C overnight. For immunohistochemistry (IHC) and immunofluorescence (IF) staining, antibodies included: phospho-histone H3 (Cell Signaling Technology, #9701), Ki67 (Vector Labs, SP6), rabbit anti-p27 (Abcam, ab92741) and p73 (Abcam, ab40658). TUNEL staining was performed with an Apoptosis Detection Kit (Millipore, S7100). IHC staining was counterstained with Harris Hematoxylin (Poly Scientific R&D Corp, S212), and IF staining were counterstained with DAPI (Sigma-Aldrich, D-9564). Images were visualized with a Nikon Eclipse Ti-U microscope and captured with Olympus DP71 camera and DP Controller software (3.2.1.276), and saved with DP manager software (3.1.1.208). The images were further processed by Adobe Photoshop. For staining quantification, pictures ere taken under 400× magnificence, about 300–400 cells were counted in each image and 3 images from each mouse were counted. The data were presented as means of 3 mice.
Early Passage Prostate Cancer Cell Cultures, DU145 Cell Line, Cell Proliferation Measurement, Organoid Cultures, and Treatments
Primary prostate tumor cells were prepared from 0.3 cm × 0.3 cm tumor tissues, which were minced and dissociated in collagenase A in DMEM for 2 hours at 37°C. Primary prostate tumor cells were cultured in DMEM containing 10% FBS and their genotypes were confirmed by PCR genotyping of Rb1, Trp53, p27, and PB-Cre4. DU145 cell line was obtained from ATCC and we did not perform STR profiling on this line. DU145 was cultured in DMEM containing 10% FBS. Measurement of cell proliferation was performed as previously described (16).
Prostate tumor cell organoid cultures were generated based on published protocols (48, 49). Quantification in Figure 5 was based on photographs of ten 10x lens fields, each field yielding 3–5 photographs focusing on consecutive planes inside Matrigel layers. Two duplicate wells were counted in this manner.
Knockdown of Skp2 or p73 was by lenti-shRNA vectors obtained from the Einstein shRNA core facility (shSkp2: 5′-GCAAGACTTCTGAACTGCTAT-3′; shp73 (12753): 5′-GCGCCTGTCATCCCTTCCAAT-3′ and shp73 (12757): 5′-CAGCCTTTGGTTGACTCCTAT-3′); controlled by scrambled shRNA (shScrmbl). Generation of lentiviral stocks and transduction of cells were as described (53). Successful lentiviral transduction was ensured by puromycin (#BP2956-100, Fisher Scientific) resistance, followed by mRNA (RT-qPCR) and protein (Western blots) measurements.
Compound 1 (C1, which inhibits interaction between Skp2/Cks1 and p27T187p (13)) was purchased from Xcess Biosciences Inc. (#432001-69-9). Prostate cancer cells were plated overnight and then treated with C1 for 2 days at the indicated concentrations.
Western Blot, CHX, RT-qPCR, ChIP Assays
Western blot, CHX, RT-qPCR, ChIP experiments were described previously (16, 17). Antibodies for Western blots are Skp2 (Santa Cruz Biotechnology, H435), p21 (Santa Cruz Biotechnology, SC-397), p27 (BD Bioscience, #610242), p73 (Abcam, ab40658), activated Caspase 3 (Cell Signaling Technology, #9661), and α-tubulin (Sigma-Aldrich, T6074). RT-qPCR primers for p27 are sense: 5′-GCGGTGCCTTTAATTGGGTCT and antisense: 5′-GGCTTCTTGGGCGTCTGC T; p73 sense: 5′-AACGCCGAGCATCAATCC) and antisense: 5′-AGCCCAGACTCTGAGCACTT; GAPDH sense (5′-GGTTGTCTCCTGCGACTTCA and antisense 5′-GGTGGTCCAGGGTTTCTTAC. ChIP antibodies are E2F1 (Santa Cruz Biotechnology, SC-193X), E2F4 (Santa Cruz Biotechnology, SC-866X), IgG (Santa Cruz Biotechnology, SC-2027). ChIP primer sequences are: E2F1: sense 5-CTTTGGAGGTGAGCCTGAAGAG-3′, antisense 5′-GGGTCTGGCGAAGCGAACA-3′; p73: sense 5′-TGAGAGTGCGGTTCTATTGGC-3′, antisense 5′-GCCCTGAACATCTGCGTCTC-3′; GAPDH: sense 5′-GAGTTCTGGGAGTCTCGTGG-3′, antisense 5′-CTCTTCGGGTGGTGGTTCA-3′.
Flow Cytometry Analysis for sub-G1 Cell Populations
Cells were plated for 16 hours. Cells were washed with PBS, trypsinized with 0.25% trypsin-EDTA at 37°C for 3–5 minutes, and resuspended with 0.5 ml PBS. Then cells were fixed with ice-cold 80% EtOH at 4°C overnight. The fixed cells were spun down and resuspended in 0.5 ml 0.25 mg/ml RNAse A and 30 μg/ml propidium iodide in PBS. Cells were filtered using 40 μm cell strainer before DNA content analysis using DXP10 Calibur at Einstein FACS core facility. Data were analyzed using Flowjo software.
Statistical Analyses
In Kaplan-Meier survival analysis, p value, hazard ratio and 95% confidence interval of hazard ratio were analyzed by log-rank test (GraphPad Prism 6 Software). p values for differences in Ki67, phospho-histone H3, and TUNEL positive cells, cell proliferation, RT-qPCR, and ChIP between indicated samples were analyzed by Student’s t-test. All statistical analyses are two-sided. p<0.05 is considered as statistically significant. We have not determined whether test data conform to normal distribution and we did not determine variance for any group of data.
Supplementary Material
Acknowledgments
This work was supported by NIH grants RO1CA127901and RO1CA131421 (LZ), Albert Einstein Comprehensive Cancer Research Center (5P30CA13330) and Liver Research Center (5P30DK061153) provided core facility support. HZ was a recipient of DOD PCRP Postdoctoral Fellowship (PC121837), and LZ was a Irma T. Hirschl Career Scientist Award recipient. We thank Dr. James Roberts for providing the p27T187A KI mice.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends in cell biology. 2012 Jan;22(1):33–41. doi: 10.1016/j.tcb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 1997 Sep 1;16(17):5334–44. doi: 10.1093/emboj/16.17.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999 May 1;13(9):1181–9. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, et al. The cell-cycle regulatory protein Cks1 is required for SCFSkp2-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;3:321–4. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 5.Spruck C, Strohmaier H, Watson M, Smith APL, Ryan A, Krek W, et al. A CDK-independent function of mammalian Cks1: targeting of SCFSkp2 to the CDK inhibitor p27Kip1. Mol Cell. 2001;7:639–50. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- 6.Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, et al. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005 Oct 7;20(1):9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakidis TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature. 2001 Sep 20;413(6853):323–7. doi: 10.1038/35095083. [DOI] [PubMed] [Google Scholar]
- 8.Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–80. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nature reviews Cancer. 2008 Apr;8(4):253–67. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 10.Timmerbeul I, Garrett-Engele CM, Kossatz U, Chen X, Firpo E, Grunwald V, et al. Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc Natl Acad Sci U S A. 2006 Sep 19;103(38):14009–14. doi: 10.1073/pnas.0606316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park MS, Rosai J, Nguyen HT, Capodieci P, Cordon-Cardo C, Koff A. p27 and Rb are on overlapping pathways suppressing tumorigenesis in mice. Proc Natl Acad Sci (USA) 1999;96:6382–7. doi: 10.1073/pnas.96.11.6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao H, Bauzon F, Bi E, Yu JJ, Fu H, Lu Z, et al. Substituting Threonine187 With Alanine in p27Kip1 Prevents Pituitary Tumorigenesis By Two-Hit Loss Of Rb1 And Enhances Humoral Immunity In Old Age. J Biol Chem. 2015 Jan 12; doi: 10.1074/jbc.M114.625350. published online Jan 12, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chemistry & biology. 2012 Dec 21;19(12):1515–24. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nature reviews Drug discovery. 2014 Dec;13(12):889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Bauzon F, Ji P, Xu X, Sun D, Locker J, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nat Genet. 2010 Jan;42(1):83–8. doi: 10.1038/ng.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao H, Bauzon F, Fu H, Lu Z, Cui J, Nakayama K, et al. Skp2 Deletion Unmasks a p27 Safeguard that Blocks Tumorigenesis in the Absence of pRb and p53 Tumor Suppressors. Cancer Cell. 2013 Nov 11;24(5):645–59. doi: 10.1016/j.ccr.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu Z, Bauzon F, Fu H, Cui J, Zhao H, Nakayama K, et al. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat Commun. 2014;5:3463. doi: 10.1038/ncomms4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Candi E, Agostini M, Melino G, Bernassola F. How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: regulators and effectors. Human mutation. 2014 Jun;35(6):702–14. doi: 10.1002/humu.22523. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006 Aug 15;66(16):7889–98. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 20.Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000;19:2069–81. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013 Apr 2;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012 Mar 29;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014 Sep 25;159(1):176–87. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang G, Ayala G, De Marzo A, Tian W, Frolov A, Wheeler TM, et al. Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clin Cancer Res. 2002 Nov;8(11):3419–26. [PubMed] [Google Scholar]
- 25.Ji P, Jiang H, Rekhtman K, Bloom J, Ichetovkin M, Pagano M, et al. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol Cell. 2004 Oct 8;16(1):47–58. doi: 10.1016/j.molcel.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Wang C. F-box protein Skp2: a novel transcriptional target of E2F. Oncogene. 2005 Apr 27;25(18):2615–27. doi: 10.1038/sj.onc.1209286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yung Y, Walker JL, Roberts JM, Assoian RK. A Skp2 autoinduction loop and restriction point control. J Cell Biol. 2007 Aug 27;178(5):741–7. doi: 10.1083/jcb.200703034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Binne UK, Classon MK, Dick FA, Wei W, Rape M, Kaelin WG, Jr, et al. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat Cell Biol. 2007 Feb;9(2):225–32. doi: 10.1038/ncb1532. [DOI] [PubMed] [Google Scholar]
- 29.Mamillapalli R, Gavrilova N, Mihaylova VT, Tsvetkov LM, Wu H, Zhang H, et al. PTEN regulates the ubiquitin-dependent degradation of the CDK inhibitor p27(KIP1) through the ubiquitin E3 ligase SCF(SKP2) Curr Biol. 2001 Feb 20;11(4):263–7. doi: 10.1016/s0960-9822(01)00065-3. [DOI] [PubMed] [Google Scholar]
- 30.Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010 Mar 18;464(7287):374–9. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hattori T, Isobe T, Abe K, Kikuchi H, Kitagawa K, Oda T, et al. Pirh2 promotes ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. Cancer Res. 2007 Nov 15;67(22):10789–95. doi: 10.1158/0008-5472.CAN-07-2033. [DOI] [PubMed] [Google Scholar]
- 32.Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004 Dec;6(12):1229–35. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 33.Miranda-Carboni GA, Krum SA, Yee K, Nava M, Deng QE, Pervin S, et al. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev. 2008 Nov 15;22(22):3121–34. doi: 10.1101/gad.1692808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao X, Xue L, Han L, Ma L, Chen T, Tong T. WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) delays cellular senescence by promoting p27(Kip1) degradation in human diploid fibroblasts. J Biol Chem. 2011 Sep 23;286(38):33447–56. doi: 10.1074/jbc.M111.225565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bassermann F, Eichner R, Pagano M. The ubiquitin proteasome system – implications for cell cycle control and the targeted treatment of cancer. Biochimica et biophysica acta. 2014 Jan;1843(1):150–62. doi: 10.1016/j.bbamcr.2013.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008 Mar 15;14(6):1649–57. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 37.Morris MJ, Kelly WK, Slovin S, Ryan C, Eicher C, Heller G, et al. A phase II trial of bortezomib and prednisone for castration resistant metastatic prostate cancer. The Journal of urology. 2007 Dec;178(6):2378–83. doi: 10.1016/j.juro.2007.08.015. discussion 83–4. [DOI] [PubMed] [Google Scholar]
- 38.Jin Y, Lee H, Zeng SX, Dai MS, Lu H. MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. EMBO J. 2003 Dec 1;22(23):6365–77. doi: 10.1093/emboj/cdg600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amador V, Ge S, Santamaria PG, Guardavaccaro D, Pagano M. APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Molecular cell. 2007 Aug 3;27(3):462–73. doi: 10.1016/j.molcel.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Havens CG, Walter JC. Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Molecular cell. 2009 Jul 10;35(1):93–104. doi: 10.1016/j.molcel.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Starostina NG, Simpliciano JM, McGuirk MA, Kipreos ET. CRL2(LRR-1) targets a CDK inhibitor for cell cycle control in C. elegans and actin-based motility regulation in human cells. Dev Cell. 2010 Nov 16;19(5):753–64. doi: 10.1016/j.devcel.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell. 2013 Aug 1;154(3):556–68. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stone KR, Mickey DD, Wunderli H, Mickey GH, Paulson DF. Isolation of a human prostate carcinoma cell line (DU 145) International journal of cancer Journal international du cancer. 1978 Mar 15;21(3):274–81. doi: 10.1002/ijc.2910210305. [DOI] [PubMed] [Google Scholar]
- 44.Gillet JP, Varma S, Gottesman MM. The clinical relevance of cancer cell lines. Journal of the National Cancer Institute. 2013 Apr 3;105(7):452–8. doi: 10.1093/jnci/djt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato T, Clevers H. SnapShot: Growing Organoids from Stem Cells. Cell. 2015 Jun 18;161(7):1700–e1. doi: 10.1016/j.cell.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 47.van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015 May 7;161(4):933–45. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2014 Sep 25;159(1):163–75. doi: 10.1016/j.cell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, Bergren SK, et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol. 2014 Oct;16(10):951–61. 1–4. doi: 10.1038/ncb3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ, et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mechanisms of development. 2001 Mar;101(1–2):61–9. doi: 10.1016/s0925-4773(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 51.Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003 Jul 10;424(6945):223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 52.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001 Dec;29(4):418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 53.Sun D, Melegari M, Sridhar S, Rogler CE, Zhu L. A multi-miRNA hairpin method that improves gene knockdown efficiency and provides linked multi-gene knockdown. BioTechniques. 2006;41:59–63. doi: 10.2144/000112203. [DOI] [PubMed] [Google Scholar]
- 54.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010 Jul 13;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012 Jun;44(6):685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.TCGA. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015 Nov 5;163(4):1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012 Jul 12;487(7406):239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015 May 21;161(5):1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.