

Abstract
Objective Signal transducer and activator of transcription (STAT) proteins regulate key cellular fate decisions including proliferation and apoptosis. STAT3 overexpression induces tumor growth in multiple neoplasms. STAT3 is constitutively activated in chordoma, a tumor with a high recurrence rate despite maximal surgical and radiation treatment. We hypothesized that a novel small molecule inhibitor of STAT3 (FLLL32) would induce significant cytotoxicity in sacral and clival chordoma cells.
Methods Sacral (UCh1) and clival (UM-CHOR-1) chordoma cell lines were grown in culture (the latter derived from primary tumor explants). FLLL32 dosing parameters were optimized using cell viability assays. Antitumor potential of FLLL32 was assessed using clonal proliferation assays. Potential mechanisms underlying observed cytotoxicity were examined using immunofluorescence assays.
Results FLLL32 induced significant cytotoxicity in UCh1 and UM-CHOR-1 chordoma cells, essentially eliminating all viable cells, correlating with observed downregulation in activated, phosphorylated STAT3 upon administration of FLLL32. Mechanisms underlying the observed cytotoxicity included increased apoptosis and reduced cellular proliferation through inhibition of mitosis.
Conclusion As a monotherapy, FLLL32 induces potent tumor kill in vitro in chordoma cell lines derived from skull base and sacrum. This effect is mediated through inhibition of STAT3 phosphorylation, increased susceptibility to apoptosis, and suppression of cell proliferation.
Keywords: chordoma, FLLL32, sacrum, skull base, STAT3
Introduction
Chordomas are rare tumors that account for 1 to 4% of all bone malignancies. Histologically, these tumors are generally low grade but demonstrate clinically malignant behavior evidenced by tissue invasion. Clinically, chordomas are locally aggressive and have a high propensity for recurrence, progressing in similar fashion to other malignant tumors.1 Population-based epidemiologic studies using the Surveillance, Epidemiology, and End Results database indicate an incidence of 0.08 per 100,000 people, predominantly in adult men, with a peak incidence at 50 to 60 years of age.1 2 3 A survival analysis of more than 400 cases suggests a median survival of 6.29 years in patients with chordoma. Survival is approximately 67.6% at 5 years but declines rapidly to 39.9 and 13.1% at 10 and 20 years, respectively.2 In the subset of patients with a skull base chordoma, median survival is significantly worse, ranging from 12 to 36 months.4
Chordomas are derived from undifferentiated notochordal remnants that exist throughout the axial skeleton. Consequently, these tumors can occur at the skull base, in the mobile spine, and in the sacrum. Incidence at each of these sites is equally distributed.1 Chordomas occurring at the skull base are particularly problematic due to the close proximity to critical bony, vascular, and neural structures. This feature markedly compromises the ability to achieve complete en bloc surgical resection, which is the mainstay of primary tumor treatment. The aim of surgical therapy is maximal resection in the context of neurological preservation. Failure to achieve complete resection results in recurrence rates that are approximately fourfold higher than for cases in which the ideal en bloc total resection is achieved.5 Difficulty with accurate assessment of surgical margins further complicates surgical resection. Indeed, complete en bloc resection is attainable in less than 50% of skull base chordomas.1 Regardless of whether complete resection is accomplished, recurrence rates remain significant.
Radiotherapy has long been used as part of the management strategy for chordomas. The use of conventional radiotherapy as the primary modality for treatment has proven to be ineffective, yielding dismal control rates. Conventional radiation therapy at doses of 40 to 60 Gy yielded 5-year local control of only 10 to 40%.6 7 8 The utility of conventional ionizing radiation remains limited, primarily because chordomas are relatively radioresistant, requiring high doses of radiation approaching 70 Gy, while residing close to highly radiation-sensitive structures such as the spinal cord, brain stem, and cranial nerves. This limits the ability to deliver effective doses without inducing significant toxicity.3 Advances in radiation technology, particularly the use of targeted photons and the introduction of hadron-based therapy (carbon ions, protons, helium), have allowed local delivery of very high doses of radiation and have optimized local control.9 10 11 12 Adjuvant care currently entails proton- or hadron-based radiotherapy, intensity-modulated radiotherapy, or stereotactic radiosurgery.
Tumor recurrence rates remain high at 16 to 40% at 10 years, even in the context of total or near-total excision followed by adjuvant radiation.13 Skull base chordomas are more likely to recur than those centered elsewhere in the axial skeleton. In a meta-analysis of skull base chordomas, the recurrence rate was 68% with an average disease-free interval of 45 months (median, 23 months).14 Reoperation for resection is often attempted in cases of recurrence. However, as expected, this is associated with poorer outcomes,15 emphasizing the importance of aggressive upfront surgical resection.
Chemotherapeutics have been used in an attempt to reduce the high recurrence rates associated with chordomas despite maximal surgery and adjuvant radiotherapy. Unfortunately, chordomas are not susceptible to conventional chemotherapy.16 17 18 19 Molecular and genetic profiling have been used to identify potential targets for novel therapeutics, though no consistent oncogenic driver has yet been identified in chordomagenesis.20 21 These therapeutic agents may reduce recurrence rates by eliminating the radioresistant cells that survive surgery and radiotherapy.
Topical or systemic administration of a possible antiproliferative agent as an adjunct to surgical resection is an attractive candidate for study provided the toxicity profile is acceptable. Effective molecular therapies could potentially reduce the incidence of disease recurrence, thereby avoiding morbid and less successful revision surgery. Two signal transducer and activator of transcription (STAT)3 inhibitors have demonstrated efficacy in the treatment of chordoma previously. One such study demonstrated that CDDO-Me inhibits the expression and activation of both STAT3 and the oncoprotein Src in vitro.22 Another study from the same laboratory showed chordoma cytotoxicity in vitro with the combination of SD-1029, a STAT3 inhibitor, with doxorubicin or cisplatin.23 The current study investigates the effect of FLLL32, a curcumin-based Janus kinase (JAK2)/STAT3 inhibitor,24 on chordoma cells in vitro in terms of efficacy as a monotherapy.
Methods
Cell Lines
The chordoma cell line UCh1 is of sacral origin and is made available to researchers through the Chordoma Foundation. UCh1 was grown on gel-coated flasks in 4:1 IMDM/RPMI + 10% FBS media. The chordoma cell line UM-CHOR-1 is a primary culture derived from a clival lesion and was developed in our laboratories at the University of Michigan. UM-CHOR-1 expresses brachyury and meets all validation criteria as outlined by the Chordoma Foundation for verification of chordoma cell line status. UM-CHOR-1 is adherent to cell culture flasks and was grown in 4:1 IMDM/RPMI + 10% FBS media. Cultured cells were maintained in 90% N2/5% CO2 normoxic conditions at 37°C.
Cell Viability Assay
Resazurin cell viability assays were performed to establish the optimal FLLL32 STAT3 inhibitor (courtesy of James R. Fuchs, PhD) concentration for subsequent assays. UCh1 cells were plated at a concentration of 2,500 cells per well on gel-coated 48-well plates and allowed to grow for 24 hours. Serial dilutions of FLLL32 at 10, 5, 2.5, 1.25, 0.6, 0.3, and 0 µM were used to treat these cells. All assays were performed in duplicate. Alamar Blue Cell Proliferation Reagent (Life Technologies, Grand Island, New York, United States) (30 µL of 10% reagent) was added to each well, and the cells then incubated for 1 hour. A plate spectrophotometer was used to detect fluorescence at 585 nm. The IC50 for FLLL32 was subsequently determined using standard technique. Based on this result, FLLL32 (5 µM) was used for all experiments.
Resazurin cell viability assays were also performed to evaluate the cytotoxicity of FLLL32 on chordoma cell lines in vitro. Cells were plated at a concentration of 2,500 cells per well on 48-well plates. Cells were then treated with 5 µM FLLL32 or equal volume of media containing 0.1% dimethyl sulfoxide (DMSO) vehicle as a control. All assays were performed in triplicate. Then, 30 µL of 10% Alamar Blue was added to each well, incubated for 1 hour, and fluorescence was detected at 585 nm.
Clonal Proliferation Assay
Clonal proliferation assays were performed to evaluate the cytotoxicity of FLLL32 on chordoma cell lines in vitro. Cells were plated at a concentration of 10,000 cells per well on 48-well plates and allowed to grow for 3 days. Cells were then treated with 5 µM FLLL32 or equal volume of media containing 0.1% DMSO vehicle as a control. All assays were performed in triplicate. As described earlier, 30µL of 10% Alamar Blue was added to each well, incubated for 1 hour, and fluorescence was detected at 585 nm.
Western Blot Assay
Cells were lysed in RIPA buffer (Sigma-Aldrich, St. Louis, Missouri, United States). Protein concentration was estimated using a BCA Protein Assay kit (Pierce Biotechnology, Rockford, Illinois, United States). Cell lysates (25–75 µg) were electrophoresed on 4 to 12% Bis-Tris polyacrylamide gels (Invitrogen, Carlsbad, California, United States) and transferred to Immobilon-PSQ polyvinylidene difluoride membranes (Millipore, Bedford, Massachusetts, United States). Membranes were blocked with phosphate-buffered saline + 0.1% Tween-20 (PBS-T) containing 5% skim milk, and then incubated with primary antibodies overnight at 4°C. The primary antibodies used for the analysis included 1:500 anti-STAT3 mouse mAb (Cell Signaling Technology, Danvers, Massachusetts, United States) and 1:1,000 antiphosphorylated STAT (pSTAT)3 (Y705) rabbit mAb (Abcam, Cambridge, Massachusetts, United States). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primary antibodies (6C5; Fitzgerald Industries, Acton, Massachusetts, United States) were used as loading control.
Fluorescence Immunocytochemistry
After treatment with FLLL32 or vehicle control, fluorescence immunocytochemistry was performed for Ki-67, cleaved caspase 3 (CC3), and pSTAT3 (Y705) protein expression. Cells were plated on 24-well plates at a density of 10,000 cells per well and allowed to grow for 24 hours. Cells were then treated with either 5 µM FLLL32 or equal volume of media containing 0.1% DMSO vehicle for 4 days. Cells were then washed with PBS and fixed using 4% paraformaldehyde in PBS at room temperature for 15 minutes. Cells were permeabilized with 0.1% Triton X-100 detergent (Sigma-Aldrich) in PBS for 30 minutes at room temperature, washed with PBS, and incubated in SuperBlock blocking buffer (Thermo Scientific, Rockford, Illinois, United States) for 1 hour. Primary antibody incubation was performed overnight for 18 hours at 4°C. Dilutions in PBS + 0.1% Triton X-100 were as follows: 1:500 anti-Ki-67 mouse monoclonal antibody (mAb) (DAKO, Glostrup, Denmark), 1:500 anti-CC3 rabbit mAb (Cell Signaling Technology), 1:500 anti-STAT3 mouse mAb (Cell Signaling Technology), 1:500 anti-pSTAT3 (S727) rabbit mAb (Abcam), and 1:500 anti-pSTAT3 (Y705) rabbit mAb (Abcam). After washing with PBS, a final 60-minute incubation with a 1:500 dilution of cyanin (Cy) three-conjugated goat antimouse or Cy2-conjugated goat antirabbit secondary antibodies (Jackson Immunoresearch Laboratories, West Grove, Pennsylvania, United States) diluted in PBS was performed at room temperature. After washing with PBS, cells were then counterstained and mounted using 4,6-diamidino-2-phenylindole (DAPI) (ProLong Gold with DAPI; Invitrogen) and visualized under fluorescence microscopy. Five high-power fields were photographed and counted. Quantification of staining was performed in terms of fraction of immunoreactive cells relative to DAPI-staining cells per high-powered field.
Statistical Methods
Cell viability and clonal proliferation experiments were performed in triplicate and repeated once for each condition. Western blot and fluorescence immunocytochemistry experiments were repeated twice for each condition for confirmation of our findings. Descriptive parametric statistics were calculated using GraphPad Prism software (GraphPad Software, Inc., La Jolla, California, United States). Two-tailed Student t-tests were performed to compare means. Statistical significance for these tests was set at p = 0.05. Graphic representation of the data was created using Microsoft Excel software (Microsoft Corporation, Redmond, Washington, United States).
Results
FLLL32 Monotherapy Induces Cytotoxicity and Inhibits Proliferation in Chordoma Cells
Cell viability assays using resazurin were performed every 48 hours over the course of 8 days to assess the cytotoxic effect of FLLL32-induced STAT3 inhibition on UCh1 chordoma cells. Proliferation assays (Fig. 1A, B) showed UCh1 and UM-CHOR-1 growth arrest after approximately 2 days, with a significantly diminished population of viable cells following treatment with FLLL32 (p < 0.05). The cell viability of UCh1 treated with FLLL32 relative to untreated controls at day 8 was 16.6%. Cells treated with vehicle control continued to grow without evidence of inhibition. These findings suggest that FLLL32-mediated STAT3 inhibition is potently cytotoxic to chordoma cells.
Fig. 1.
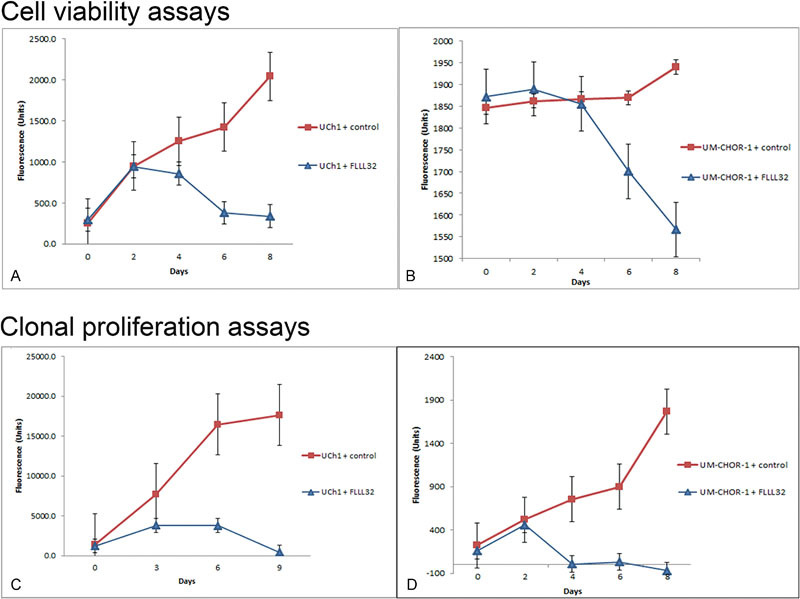
FLLL32 inhibits cellular proliferation and exhibits cytotoxicity. (A) Resazurin cell viability assays were performed on the UCh1 cell line in vitro. Cells were plated at a concentration of 2,500 cells per well, then treated with 5 µM FLLL32 or equal volume of 0.1% DMSO vehicle as a control. Cell viability in FLLL32-treated cells was dramatically reduced, relative to the normal growth pattern exhibited by control cells. Growth arrest was witnessed after approximately 2 days, with a significantly diminished population of viable cells following treatment with FLLL32 (p < 0.01). (B) Resazurin cell viability assays were performed on the UM-CHOR-1 cell line in vitro. Cell viability in FLLL32-treated cells was significantly reduced, relative to the normal growth pattern exhibited by control cells. Growth arrest was witnessed after approximately 2 days, with a significantly diminished population of viable cells following treatment with FLLL32 (p < 0.05). (C) Clonal proliferation assays were performed to evaluate the cytotoxicity of FLLL32 on the UCh1 cell line in vitro. Cells were plated at a concentration of 10,000 cells per well and allowed to grow for 72 days. Cells were then treated with 5 µM FLLL32 or equal volume 0.1% DMSO vehicle as a control. Cytotoxicity was demonstrated after 3 days in FLLL32-treated cells. Complete arrest of cell proliferation was witnessed in UCh1 cells with a decline of cell numbers to 2.6% in the treated cells by day 9 (p < 0.01). (D) Clonal proliferation assays were performed to evaluate the cytotoxicity of FLLL32 on the UM-CHOR-1 cell line in vitro. Though not immediate, cytotoxicity was demonstrated after 2 days in FLLL32-treated cells. Within 24 hours of initiating treatment with FLLL32 (day 4), no viable cells could be detected. This effect was sustained for the remainder of the assay period and was statistically significant when compared with untreated controls (p < 0.01). DMSO, dimethyl sulfoxide.
Clonogenic assays are considered a more accurate method for evaluating cell viability and proliferation and are superior to colorimetric assays in this regard.25 As a result, after confirming the potential efficacy of FLLL32 for inducing chordoma cell line cytotoxicity using colorimetric cell viability studies, we performed clonal proliferation assays (Fig. 1C). UCh1 cell colonies were allowed to proliferate for 72 hours before the addition of FLLL32. The STAT3 inhibitor induced complete arrest of cell proliferation in UCh1 with a decline of cell numbers to 2.6% in the treated cells by day 9 (p < 0.01). This extremely low number of viable cells is, essentially, below the detection threshold of the plate spectrophotometer, suggesting complete or near complete eradication of UCh1 cells by FLLL32.
Clonogenic assays were also performed in the clival chordoma UM-CHOR-1 cell line after allowing 72 hours for establishment of cell growth before treatment with FLLL32 (Fig. 1D). As with the UCh1 cell line, UM-CHOR-1 cells were highly susceptible to STAT3 inhibition. Indeed, within 24 hours of initiating treatment with FLLL32 (day 4), no viable cells could be detected. This effect was sustained for the remainder of the assay period and was statistically significant when compared with untreated controls (p < 0.01).
FLLL32 Inhibits STAT3 Phosphorylation at the Y705 Residue
STAT3 expression in UCh1 and UM-CHOR-1 cells was evaluated by Western blot analysis, with and without FLLL32 treatment (Fig. 2). There was no appreciable difference in total STAT3 expression. pSTAT3 is the active form of STAT3 protein. FLLL32 is thought to inhibit STAT3 phosphorylation at the critical Y705 residue [pSTAT3(Y)]. In contrast to total STAT3 expression, there was a marked decrease in pSTAT3(Y) expression with FLLL32 treatment compared with control, as detected by Western blot analysis.
Fig. 2.
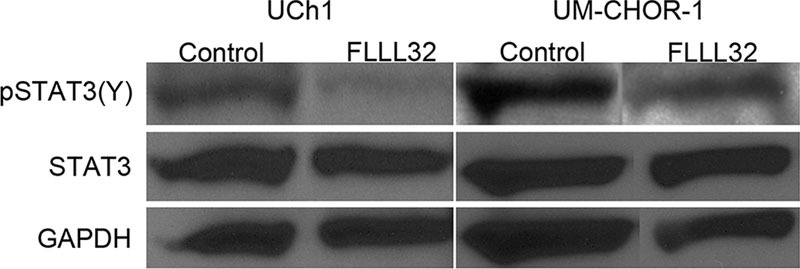
FLLL32 inhibits pSTAT3(Y) but not STAT3 expression. Western blot analysis demonstrates a relative paucity of pSTAT3(Y) expression in FLLL32-treated cells, using both the UCh1 and UM-CHOR-1 cell lines, when compared with control cells. Total STAT3 expression, however, did not change with FLLL32 treatment. GAPDH was used as loading control. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; pSTAT, phosphorylated signal transducer and activator of transcription; STAT, signal transducer and activator of transcription.
Fluorescence immunocytochemistry was performed using a pSTAT3 antibody specific for the S727 residue [pSTAT3(S)]. No difference in STAT3 (Fig. 3A) or pSTAT3(S) (Fig. 3B) staining was noted between FLLL32-treated and control UM-CHOR-1 cells, while pSTAT3(Y) staining was notably less with FLLL32 treatment (Fig. 3C). Accordingly, expression of pSTAT3(Y) was significantly reduced with FLLL32 treatment in UCh1 (Fig. 4A) and UM-CHOR-1 (Fig. 5A) chordoma cell lines (p < 0.05).
Fig. 3.
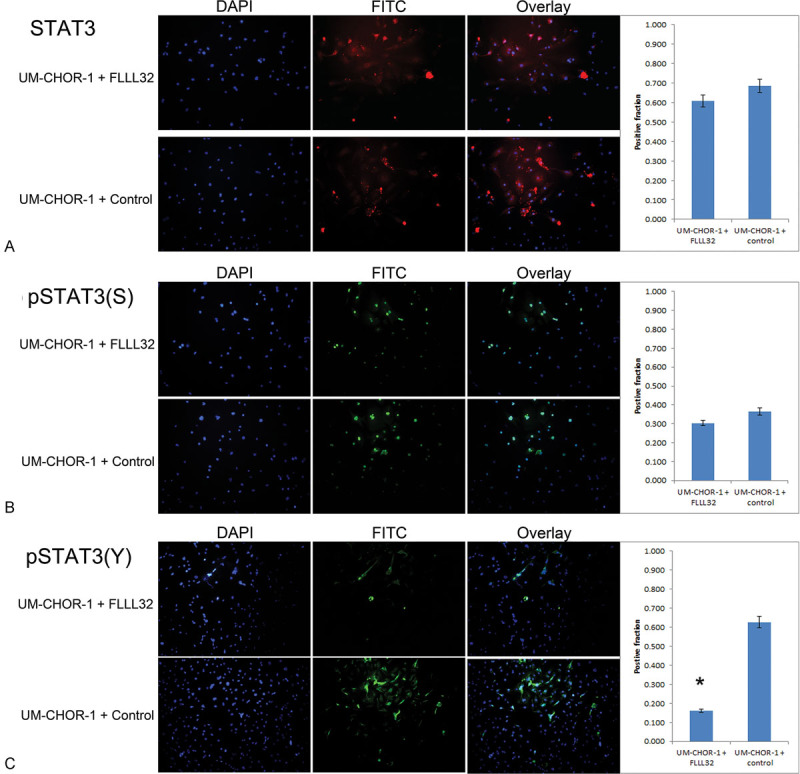
FLLL32 inhibits STAT3 phosphorylation at the Y705 residue. (A) STAT3 immunofluorescence staining was performed on UM-CHOR-1 cells treated with FLLL32 or with equal volume 0.1% DMSO vehicle as a control. Of cells treated with FLLL32, 60.8% exhibited positive staining, compared with 68.7% of control-treated cells, showing no significant difference in total STAT3 expression. (B) Staining for pSTAT3(S) residue was not significantly different between FLLL32-treated (30.4%) and control-treated (36.6%) UM-CHOR-1 cells. (C) Staining for pSTAT3(Y), however, showed a significant reduction with FLLL32 treatment (16.1%), relative to control-treated (62.7%) UM-CHOR-1 cells (p < 0.05). (*Statistically significant.) DMSO, dimethyl sulfoxide; pSTAT, phosphorylated signal transducer and activator of transcription; STAT, signal transducer and activator of transcription.
Fig. 4.
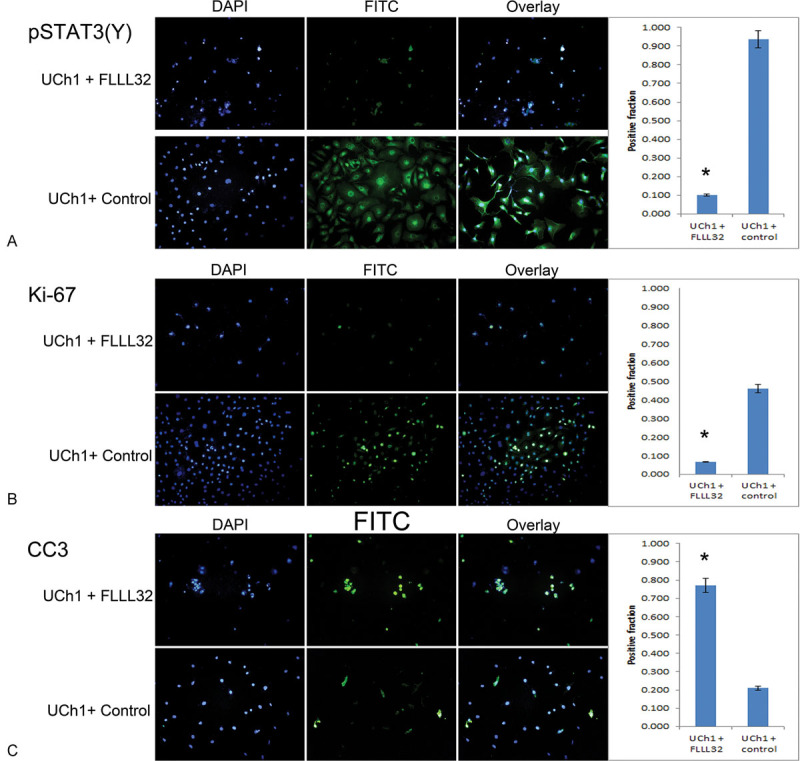
FLLL32 inhibition of pSTAT3 (Y705) results in reduced cellular proliferation and increased apoptosis in UCh1 cells. (A) Immunofluorescence staining was performed on UCh1 cells treated with FLLL32 or with equal volume 0.1% DMSO vehicle as a control. Of cells treated with FLLL32, 10.0% exhibited positive staining, compared with 93.8% of control-treated cells; as such, a significant reduction in pSTAT3(Y) expression was seen in the FLLL32-treated cells (p < 0.05). (*Statistically significant.) (B) Ki-67 staining was significantly higher in control-treated UCh1 cells (46.0%) relative to FLLL32-treated cells (6.7%), indicating decreased proliferative activity with FLLL32 treatment (p < 0.05). (C) CC3 staining was significantly higher in FLLL32-treated UCh1 cells (77.1%) relative to control-treated cells (21.1%), indicating increased apoptosis induced by FLLL32 treatment (p < 0.05). DMSO, dimethyl sulfoxide; pSTAT, phosphorylated signal transducer and activator of transcription.
Fig. 5.
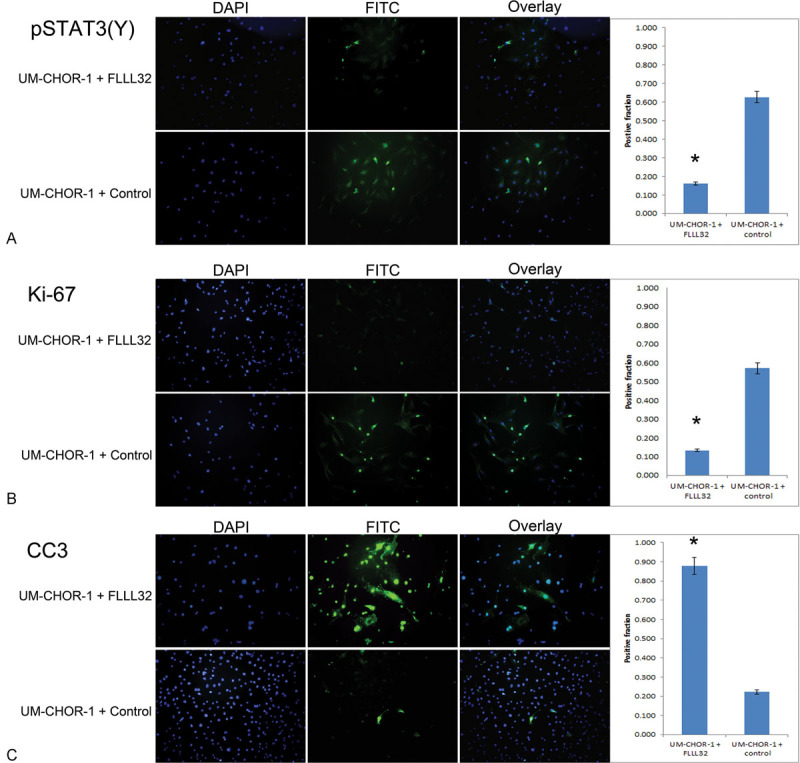
FLLL32 inhibition of pSTAT3 (Y705) results in reduced cellular proliferation and increased apoptosis in UM-CHOR-1 cells. (A) Immunofluorescence staining was performed on UM-CHOR-1 cells treated with FLLL32 or with equal volume 0.1% DMSO vehicle as a control. Of cells treated with FLLL32, 16.1% exhibited positive staining compared with 62.7% in control-treated cells, showing a significant reduction in pSTAT3(Y) expression in the FLLL32-treated cells (p < 0.05). (*Statistically significant.) (B) Ki-67 staining was significantly higher in control-treated UM-CHOR-1 cells (57.1%) relative to FLLL32-treated cells (13.3%), indicating decreased proliferative activity with FLLL32 treatment (p < 0.05). (C) CC3 was significantly higher in FLLL32-treated UM-CHOR-1 cells (87.9%) relative to control-treated cells (22.2%), indicating increased apoptosis induced by FLLL32 treatment (p < 0.05). CC3, cleaved caspase 3; DMSO, dimethyl sulfoxide; pSTAT, phosphorylated signal transducer and activator of transcription.
FLLL32 Treatment Results in Reduced Cellular Proliferation and Increased Apoptosis
Fluorescence immunocytochemistry was performed to quantify the effects of FLLL32 treatment on UCh1 (Fig. 4) and UM-CHOR-1 (Fig. 5) cells in vitro. Antigen Ki-67 is a nuclear protein and a marker associated exclusively with cellular proliferation. Ki-67 is closely associated with the G1, S, G2, and mitosis phases of the cell cycle. FLLL32 treatment induced a significant reduction in Ki-67 immunofluorescence relative to untreated controls in both UCh1 (Fig. 4B) and UM-CHOR-1 (Fig. 5B) cell lines (p < 0.05). This result correlated with the observed reduction in cell proliferation demonstrated in clonogenic assays.
The cysteine-aspartic acid protease family proteins are activated sequentially in the initiation and execution of apoptosis. CC3 is a key component of the apoptotic cascade, and levels of this protein correlate with the degree of apoptosis. CC3 immunofluorescence was significantly increased with FLLL32 treatment in both UCh1 (Fig. 4C) and UM-CHOR-1 (Fig. 5C) cell lines relative to untreated controls (p < 0.05). This suggests that apoptosis is a contributing mechanism to the reduction in cellular proliferation seen on clonogenic assays following treatment of chordoma cell lines with FLLL32.
Discussion
The current clinical paradigm for the management of chordomas consists of aggressive surgical resection followed by adjuvant radiotherapy. The totality of surgical resection is often limited by the need to limit neurovascular morbidity. Consequently, macroscopic or microscopic disease often persists beyond the initial surgical resection. Although newer adjuvant radiotherapy modalities, particularly hadron-based therapy, have shown efficacy in reducing recurrence rates, the persistence of radio-resistant chordoma cells in the radiated wound bed have resulted in high recurrence rates associated with this tumor, particularly at the skull base.13 14
Research efforts in recent years have focused on characterizing the molecular profile of chordoma with a view to identifying drug targets. Molecular signaling pathways involved in the pathogenesis of chordoma include the platelet-derived growth factor receptor (PDGFR) tyrosine kinase pathway. This pathway is mediated by PI3K/AKT and mTORC1, transforming growth factor α, and basic fibroblast growth factor.3 Molecular profiling has demonstrated a high proportion of tumors with overexpression of PDGFR and kinase tyrosine (KIT).26 Imatinib, a tyrosine-kinase inhibitor specific for PDGFR and KIT, has been tested against chordoma in a sizeable number of patients, including in a phase II clinical trial.27 28 Enrolled patients had advanced disease and demonstrated nondimensional tissue responses with hypodensity and decreased contrast uptake on computed tomography. Liquefaction of tumor was reported in some patients with symptom improvement. However, the objective complete response rate was only 2%, a single patient out of the cohort of 50. A further nine patients achieved a minor response of less than 30% diameter decrease. Disease stabilization was achieved in 72% of cases. Median progression-free survival was 9 months with a median overall survival of 34 months in this advanced-disease population.27
Other molecular targets include components of the epidermal growth factor receptor (EGFR) pathway including PI3K, AKT, TSC, and mTOR. EGFR upregulation has been demonstrated in approximately 70% of chordomas, likely contributing to nuclear instability and cell proliferation.4 29 30 EGFR inhibitors have not been used in formal clinical trials, and suggestions of efficacy are based on responses from only a handful of patients. Rapamycin, an mTOR inhibitor, has been postulated as a potential therapy as up to 65% of chordomas might be susceptible to inhibition of mTOR.31 The identification of the transcription factor, brachyury, as a constant molecular marker for chordoma has introduced an exciting new avenue by which chordoma might be approached.32 33
The modest efficacy of the aforementioned small molecule inhibitors indicates a need to identify molecular targets that are perhaps more critical to tumor progression. STAT proteins are key cytoplasmic transcription factors. The role of STAT proteins in critical cell fate decisions such as cell growth, differentiation, and apoptosis, as well as metastasis and immune evasion, makes them attractive targets for antitumor therapy (Fig. 6).34 Constitutive activation of STAT3 has been reported in a large variety of human neoplasms including lung, breast, colon, pancreas, brain, head and neck, renal, prostate, skin, and hematogenous cancers.35 36 37 38 39 STAT proteins contain multiple domains including a DNA-binding site, Src homology-2 (SH2) domains, and a critical tyrosine residue (Y705) situated in the C-terminal domain.34 Cytokine and growth factors induce receptor dimerization and transphosphorylation by binding to cell surface receptors. STAT proteins are then recruited to the activated cell surface receptors via their SH2 domain and are, in turn, activated through phosphorylation of the critical Y705 residue by upstream kinases.35 The key STAT-activating phosphorylation step can also be initiated by the JAK family of cytoplasmic tyrosine kinases. Transmembrane growth factor receptors, such as the EGFR, harbor intrinsic tyrosine kinase activity are able to phosphorylate STAT independently. Once activated, STAT monomers are able to dimerize through their SH2 domains in a process initiated and stabilized by the key Y705 residue. The activated STAT dimers translocate to the nucleus and bind to specific DNA-response elements in target genes to modulate cell growth, survival, differentiation, metastasis, and immune evasion.34 40
Fig. 6.
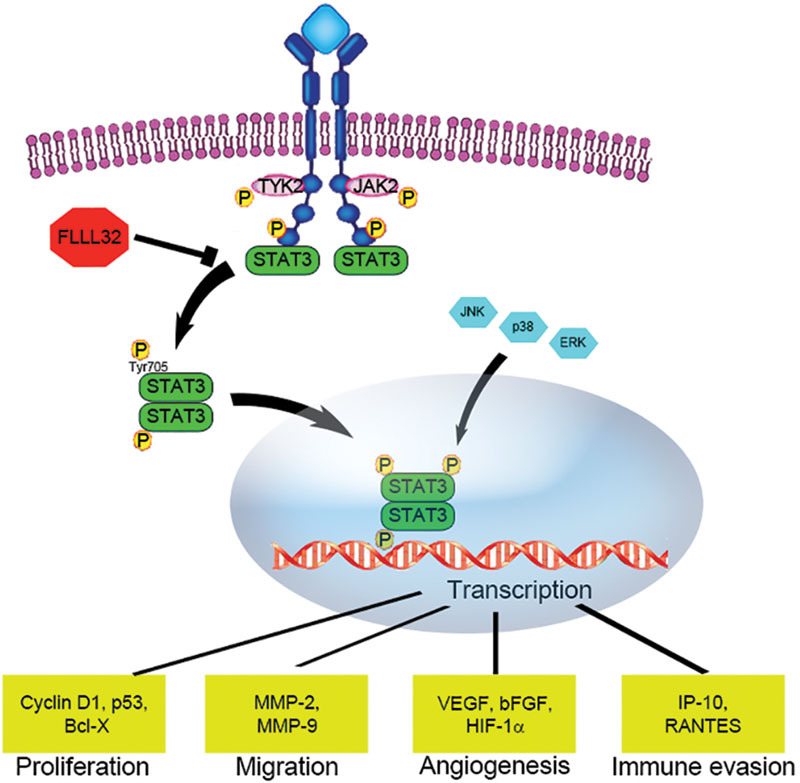
STAT3 activation is associated with cellular proliferation, inhibition of apoptosis, cell migration, and immune modulation. Cytoplasmic STAT3 monomers dimerize when phosphorylated at the tyrosine 705 (Y705) residue by Janus kinases associated with cytokine receptors, and then translocate to the nucleus, where the homodimers activate target gene transcription. STAT, signal transducer and activator of transcription.
STAT3 can also be phosphorylated at serine 727 (S727) by members of the mitogen-activated protein kinases and c-Jun N-terminal kinase families.41 While phosphorylated Y705 is generally believed to be essential for STAT3 transcriptional activity, the function of phosphorylated S727 remains controversial, having been reported to cause both down- and upregulatory effects on STAT3 transcriptional activity,42 and in regulating the homeostasis of embryonic stem cells.43 Our pSTAT3(S) staining experiment demonstrates that FLLL32 affects STAT3 phosphorylation only at the Y705 residue.
Recently, an analysis of 70 chordoma samples derived from different patients indicated that STAT3 was constitutively activated in all samples.23 The level of pSTAT3 directly correlated with disease recurrence, metastasis, and survival. Inhibition of STAT3 decreased chordoma cell proliferation in vitro in three derivative cell lines, and this effect was amplified when the inhibitor was used with conventional chemotherapeutics.23 These results suggest that STAT3 may drive both tumor growth and metastatic potential in chordoma. In both of the UM-CHOR-1 and UCh1 cell lines examined in the current study, there was evidence of STAT3 activation with phosphorylation on the Y705 residue. These cell lines should be useful in examining the effect of STAT3 activation in chordomas.
FLLL32 is a small molecule STAT3 pathway inhibitor derived from curcumin, a naturally occurring phenol found in the spice, turmeric.36 FLLL32 selectively targets JAK2-mediated activation of STAT3 by blocking the Y705 residue. This novel therapeutic also has a downstream effect on STAT3 itself, blockading the SH2 dimerization site, thereby preventing assembly of STAT3 monomers into a functional dimer.44 This prevents STAT3-mediated upregulation of downstream apoptosis inhibitors,45 cell cycle activators,46 and proangiogenic factors.47 This molecular pathway is particularly attractive as a therapeutic target because it lies at the convergence of multiple extracellular signaling pathways involved in cell survival.48
The previously studied STAT3 inhibitor CDDO-Me is theorized to bind directly to STAT3 by a mechanism dependent on the alkylation of Cys259, thus inhibiting the formation of STAT3 dimers.22 In contrast, SD-1029 inhibits tyrosyl phosphorylation of STAT3 and the JAK2 isoenzyme.49 It is unclear whether these STAT3 inhibitors function similarly, though expression of STAT3 and pSTAT3, as well as the antiapoptotic proteins Bcl-xL and MCL-1, were inhibited after CDDO-Me and SD-1029 treatment. In addition, poly (adenosine diphosphate ribose) polymerase cleavage, an apoptosis-associated biochemical event, was detected after treatment with both CDDO-Me and SD-1029.
In this study, a validated sacral chordoma cell line and a novel clival chordoma cell line were used to demonstrate the efficacy of FLLL32 as a cytotoxic agent. FLLL32 was capable of inhibiting STAT3 phosphorylation on the Y705 residue in both cell lines. Functional assays, including gold standard clonogenic studies, demonstrated a clear antiproliferative effect. This effect was so pronounced that both sacral (UCh1) and clival (UM-CHOR-1) cells were eliminated within days of initiating treatment. Additional immunofluorescence studies provided a preliminary evaluation of the mechanisms underlying the cytotoxic effect of FLLL32, which appear to include activation of caspase-3-mediated apoptosis, and suppression of cell mitosis and proliferation.
The need for effective adjuvant chemotherapy in the management of chordoma is undisputed as the rate of recurrence remains high despite advances in surgery through the advent of endoscopic endonasal approaches and in radiation with the introduction of hadron-based therapies. The results of our study indicate that, given an acceptable toxicity profile, further study of FLLL32 in the treatment of chordoma is warranted. An in vivo flank model of chordoma has been introduced32 and will be used to test FLLL32 both topically and systemically. Of particular interest is the development of topical delivery strategies for FLLL32. As chordoma recurs focally at sites of persistent nests of neoplastic cells, the topical administration of a STAT3 inhibitor into the surgical resection bed could potentially reduce the risk of local recurrence, meriting further investigation.
Conclusion
FLLL32 is an effective antitumor agent against UCh1 sacral and UM-CHOR-1 clival chordoma in vitro. In vivo studies using a murine flank model are indicated for further study. This STAT3 inhibitor may prove to be a viable adjuvant treatment option against chordoma.
Acknowledgments
This project was supported by Award Number F32NS074744 from the National Institute of Neurological Disorders and Stroke. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health.
Footnotes
Conflict of Interest The authors have no conflict of interest to report pertaining to the materials or methods used in this study or the findings specified in this article.
References
- 1.Walcott B P, Nahed B V, Mohyeldin A, Coumans J V, Kahle K T, Ferreira M J. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13(2):e69–e76. doi: 10.1016/S1470-2045(11)70337-0. [DOI] [PubMed] [Google Scholar]
- 2.McMaster M L, Goldstein A M, Bromley C M, Ishibe N, Parry D M. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12(1):1–11. doi: 10.1023/a:1008947301735. [DOI] [PubMed] [Google Scholar]
- 3.Williams B J, Raper D M, Godbout E. et al. Diagnosis and treatment of chordoma. J Natl Compr Canc Netw. 2013;11(6):726–731. doi: 10.6004/jnccn.2013.0089. [DOI] [PubMed] [Google Scholar]
- 4.Stacchiotti S, Casali P G. Systemic therapy options for unresectable and metastatic chordomas. Curr Oncol Rep. 2011;13(4):323–330. doi: 10.1007/s11912-011-0176-x. [DOI] [PubMed] [Google Scholar]
- 5.Di Maio S, Temkin N, Ramanathan D, Sekhar L N. Current comprehensive management of cranial base chordomas: 10-year meta-analysis of observational studies. J Neurosurg. 2011;115(6):1094–1105. doi: 10.3171/2011.7.JNS11355. [DOI] [PubMed] [Google Scholar]
- 6.Forsyth P A, Cascino T L, Shaw E G. et al. Intracranial chordomas: a clinicopathological and prognostic study of 51 cases. J Neurosurg. 1993;78(5):741–747. doi: 10.3171/jns.1993.78.5.0741. [DOI] [PubMed] [Google Scholar]
- 7.Catton C, O'Sullivan B, Bell R. et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41(1):67–72. doi: 10.1016/s0167-8140(96)91805-8. [DOI] [PubMed] [Google Scholar]
- 8.Cummings B J, Hodson D I, Bush R S. Chordoma: the results of megavoltage radiation therapy. Int J Radiat Oncol Biol Phys. 1983;9(5):633–642. doi: 10.1016/0360-3016(83)90228-6. [DOI] [PubMed] [Google Scholar]
- 9.Martin J J, Niranjan A, Kondziolka D, Flickinger J C, Lozanne K A, Lunsford L D. Radiosurgery for chordomas and chondrosarcomas of the skull base. J Neurosurg. 2007;107(4):758–764. doi: 10.3171/JNS-07/10/0758. [DOI] [PubMed] [Google Scholar]
- 10.Goitein M, Cox J D. Should randomized clinical trials be required for proton radiotherapy? J Clin Oncol. 2008;26(2):175–176. doi: 10.1200/JCO.2007.14.4329. [DOI] [PubMed] [Google Scholar]
- 11.Austin-Seymour M, Munzenrider J, Goitein M. et al. Fractionated proton radiation therapy of chordoma and low-grade chondrosarcoma of the base of the skull. J Neurosurg. 1989;70(1):13–17. doi: 10.3171/jns.1989.70.1.0013. [DOI] [PubMed] [Google Scholar]
- 12.Amichetti M, Cianchetti M, Amelio D, Enrici R M, Minniti G. Proton therapy in chordoma of the base of the skull: a systematic review. Neurosurg Rev. 2009;32(4):403–416. doi: 10.1007/s10143-009-0194-4. [DOI] [PubMed] [Google Scholar]
- 13.Koutourousiou M, Snyderman C H, Fernandez-Miranda J, Gardner P A. Skull base chordomas. Otolaryngol Clin North Am. 2011;44(5):1155–1171. doi: 10.1016/j.otc.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Jian B J, Bloch O G, Yang I. et al. Adjuvant radiation therapy and chondroid chordoma subtype are associated with a lower tumor recurrence rate of cranial chordoma. J Neurooncol. 2010;98(1):101–108. doi: 10.1007/s11060-009-0068-1. [DOI] [PubMed] [Google Scholar]
- 15.Smoll N R, Gautschi O P, Radovanovic I, Schaller K, Weber D C. Incidence and relative survival of chordomas: the standardized mortality ratio and the impact of chordomas on a population. Cancer. 2013;119(11):2029–2037. doi: 10.1002/cncr.28032. [DOI] [PubMed] [Google Scholar]
- 16.Yang C, Hornicek F J, Wood K B. et al. Characterization and analysis of human chordoma cell lines. Spine. 2010;35(13):1257–1264. doi: 10.1097/BRS.0b013e3181c2a8b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chugh R, Dunn R, Zalupski M M. et al. Phase II study of 9-nitro-camptothecin in patients with advanced chordoma or soft tissue sarcoma. J Clin Oncol. 2005;23(15):3597–3604. doi: 10.1200/JCO.2005.02.170. [DOI] [PubMed] [Google Scholar]
- 18.Hof H, Welzel T, Debus J. Effectiveness of cetuximab/gefitinib in the therapy of a sacral chordoma. Onkologie. 2006;29(12):572–574. doi: 10.1159/000096283. [DOI] [PubMed] [Google Scholar]
- 19.Singhal N, Kotasek D, Parnis F X. Response to erlotinib in a patient with treatment refractory chordoma. Anticancer Drugs. 2009;20(10):953–955. doi: 10.1097/CAD.0b013e328330c7f0. [DOI] [PubMed] [Google Scholar]
- 20.Le L P, Nielsen G P, Rosenberg A E. et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PLoS ONE. 2011;6(5):e18846. doi: 10.1371/journal.pone.0018846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choy E, MacConaill L E, Cote G M. et al. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PLoS ONE. 2014;9(7):e101283. doi: 10.1371/journal.pone.0101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C, Hornicek F J, Wood K B. et al. Blockage of Stat3 with CDDO-Me inhibits tumor cell growth in chordoma. Spine. 2010;35(18):1668–1675. doi: 10.1097/BRS.0b013e3181c2d2b4. [DOI] [PubMed] [Google Scholar]
- 23.Yang C, Schwab J H, Schoenfeld A J. et al. A novel target for treatment of chordoma: signal transducers and activators of transcription 3. Mol Cancer Ther. 2009;8(9):2597–2605. doi: 10.1158/1535-7163.MCT-09-0504. [DOI] [PubMed] [Google Scholar]
- 24.Lin L, Deangelis S, Foust E. et al. A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol Cancer. 2010;9:217. doi: 10.1186/1476-4598-9-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vega-Avila E, Pugsley M K. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc West Pharmacol Soc. 2011;54:10–14. [PubMed] [Google Scholar]
- 26.Tamborini E, Miselli F, Negri T. et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res. 2006;12(23):6920–6928. doi: 10.1158/1078-0432.CCR-06-1584. [DOI] [PubMed] [Google Scholar]
- 27.Stacchiotti S, Longhi A, Ferraresi V. et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol. 2012;30(9):914–920. doi: 10.1200/JCO.2011.35.3656. [DOI] [PubMed] [Google Scholar]
- 28.Casali P G, Messina A, Stacchiotti S. et al. Imatinib mesylate in chordoma. Cancer. 2004;101(9):2086–2097. doi: 10.1002/cncr.20618. [DOI] [PubMed] [Google Scholar]
- 29.Börgel J, Olschewski H, Reuter T, Miterski B, Epplen J T. Does the tuberous sclerosis complex include clivus chordoma? A case report. Eur J Pediatr. 2001;160(2):138. doi: 10.1007/s004310000645. [DOI] [PubMed] [Google Scholar]
- 30.Kimmell K T, Dayoub H, Stolzenberg E D, Sincoff E H. Chordoma in the lateral medullary cistern in a patient with tuberous sclerosis: a case report and review of the literature. Surg Neurol Int. 2010;1:13. doi: 10.4103/2152-7806.63908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Presneau N, Shalaby A, Idowu B. et al. Potential therapeutic targets for chordoma: PI3K/AKT/TSC1/TSC2/mTOR pathway. Br J Cancer. 2009;100(9):1406–1414. doi: 10.1038/sj.bjc.6605019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu W, Mohyeldin A, Shah S R. et al. Generation of chordoma cell line JHC7 and the identification of brachyury as a novel molecular target. J Neurosurg. 2011;115(4):760–769. doi: 10.3171/2011.5.JNS11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vujovic S, Henderson S, Presneau N. et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209(2):157–165. doi: 10.1002/path.1969. [DOI] [PubMed] [Google Scholar]
- 34.Jing N, Tweardy D J. Targeting Stat3 in cancer therapy. Anticancer Drugs. 2005;16(6):601–607. doi: 10.1097/00001813-200507000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Darnell J E Jr. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2(10):740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 36.Lin L, Hutzen B, Zuo M. et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010;70(6):2445–2454. doi: 10.1158/0008-5472.CAN-09-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grandis J R, Drenning S D, Zeng Q. et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A. 2000;97(8):4227–4232. doi: 10.1073/pnas.97.8.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 39.Song J I, Grandis J R. STAT signaling in head and neck cancer. Oncogene. 2000;19(21):2489–2495. doi: 10.1038/sj.onc.1203483. [DOI] [PubMed] [Google Scholar]
- 40.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19(21):2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 41.Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19(21):2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- 42.Qin H R, Kim H J, Kim J Y. et al. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008;68(19):7736–7741. doi: 10.1158/0008-5472.CAN-08-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang G, Yan H, Ye S, Tong C, Ying Q L. STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem Cells. 2014;32(5):1149–1160. doi: 10.1002/stem.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bill M A, Fuchs J R, Li C. et al. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol Cancer. 2010;9:165. doi: 10.1186/1476-4598-9-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naka T, Narazaki M, Hirata M. et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387(6636):924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- 46.Takemoto S, Mulloy J C, Cereseto A. et al. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc Natl Acad Sci U S A. 1997;94(25):13897–13902. doi: 10.1073/pnas.94.25.13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niu G, Wright K L, Huang M. et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21(13):2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 48.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 49.Duan Z, Bradner J E, Greenberg E. et al. SD-1029 inhibits signal transducer and activator of transcription 3 nuclear translocation. Clin Cancer Res. 2006;12(22):6844–6852. doi: 10.1158/1078-0432.CCR-06-1330. [DOI] [PubMed] [Google Scholar]