

Abstract
Patients with chronic kidney disease (CKD) are at increased risk for bone fractures compared with the general population. Repression of the Wnt/β‐catenin signaling pathway is associated with bone abnormalities. Inhibition of glycogen synthase kinase (GSK)‐3β, a critical component of the Wnt/β‐catenin signaling pathway, increases bone volume through accumulation of β‐catenin. It remains unknown whether inhibition of GSK‐3β increases bone volume in CKD. The present in vivo study examined the effects of GSK‐3β inhibition on bone volume in CKD mice. Wild‐type mice were divided into three groups. One group was fed a control diet (CNT) and the other two groups were fed a diet containing 0.2% adenine and given water with or without lithium chloride (LiCl), a GSK‐3 inhibitor (CKD, CKD+LiCl, respectively). GSK‐3β heterozygous knockout mice were fed a diet containing 0.2% adenine (CKD‐GSK‐3β +/−). After 6 weeks, trabecular and cortical bone volumes of the femur were analyzed using microcomputed tomography. CKD mice developed azotemia, hyperphosphatemia, and hyperparathyroidism, followed by a decrease in cortical bone volume without any change in trabecular bone volume. Serum levels of urea nitrogen, phosphate, and parathyroid hormone were comparable among the three groups of CKD mice. Trabecular bone volume increased in CKD‐GSK‐3β +/− and CKD+LiCl mice compared with CNT and CKD mice. However, there were no significant differences in cortical bone volume among the three groups of CKD mice. The results suggest that inhibition of GSK‐3β increases trabecular bone volume but not cortical bone volume in adenine‐induced uremic mice with uncontrolled hyperparathyroidism.
Keywords: Bone fracture, chronic kidney disease, glycogen synthase kinase‐3β, Wnt/β‐catenin signaling pathway
Introduction
The incidence of bone fractures is high in patients with chronic kidney disease (CKD) (Alem et al. 2000; Nickolas et al. 2006; Ensrud et al. 2007; Fried et al. 2007), and this increased incidence is associated with high rates of hospitalization, disability, and mortality in patients with CKD (Mittalhenkle et al. 2004; Nitsch et al. 2009; Tentori et al. 2014). Studies have shown that impaired bone quality and quantity in CKD are ascribed to decreased bone strength, which ultimately enhances the risk for bone fractures (Bacchetta et al. 2010; Malluche et al. 2011; Nickolas et al. 2011, 2013; West et al. 2015; Ott 2016). Secondary hyperparathyroidism, retention of uremic toxins, acidemia, hypogonadism, increased oxidative stress and inflammation, and malnutrition may partly contribute to impairment in bone quality and quantity (Nickolas et al. 2008; Kazama et al. 2013; Drüeke and Massy 2016). However, the pathogenesis behind the increased incidence of bone fractures among the CKD population still remains unclear. Identification of pathways that critically regulate bone pathology in CKD patients is required to prevent uremia‐related bone fractures.
The Wnt/β‐catenin signaling pathway plays a pivotal role in osteogenesis through the differentiation of mesenchymal stem cells into osteoblasts (Baron and Kneissel 2013). Binding of Wnt agonists to the receptor comprising frizzled and low‐density lipoprotein receptor‐related protein 5/6 phosphorylates glycogen synthase kinase (GSK)‐3β and results in prevention of ubiquitin‐mediated proteolysis of β‐catenin in the cytosol, leading to the translocation of β‐catenin into the nucleus and promotion of transcription of osteogenesis‐related genes. Because the activity of the Wnt/β‐catenin signaling pathway largely depends on the amount of β‐catenin, GSK‐3β plays a key role in regulating this pathway. We previously reported that GSK‐3β heterozygous knockout (GSK‐3β +/−) mice displayed accelerated bone development and regeneration through the Wnt/β‐catenin signaling pathway (Arioka et al. 2013). GSK‐3 inhibitors have been reported to increase bone mass and strength and promote bone healing in diseased rodents (Clément‐Lacroix et al. 2005; Kulkarni et al. 2006; Marsell et al. 2012; Sisask et al. 2013; Arioka et al. 2014). Collectively, these results suggest that GSK‐3β is a promising target for the development of novel bone anabolic agents.
Recent study has shown that repression of the Wnt/β‐catenin signaling pathway is associated with bone abnormalities in CKD (Sabbagh et al. 2012). The study revealed that phosphorylated β‐catenin‐positive osteocytes increased in patients with CKD compared with participants without CKD. More recently, animal experiments have shown that inhibition of Wnt antagonists, which act upstream of GSK‐3β, increases bone volume in CKD models (Fang et al. 2014; Moe et al. 2015). These results suggest that activation of the Wnt/β‐catenin signaling pathway has the potential to increase bone volume in CKD. However, it remains unclear whether direct inhibition of GSK‐3β increases bone volume in CKD.
This study sought to determine whether inhibition of GSK‐3β increases bone volume in CKD. To this end, we used an adenine‐induced CKD model, GSK‐3β +/− mice, and lithium chloride (LiCl), a known inhibitor of GSK‐3 (Ryves and Harwood 2001) in our in vivo model and evaluated the effects of GSK‐3β repression on bone volume and parameters assessed using microcomputed tomography (micro‐CT). We used GSK‐3β +/− mice in the study as GSK‐3β homozygous knockout mice show the embryonic lethality phenotype because of hepatocyte apoptosis and ventricular septal defects (Hoeflich et al. 2000; Kerkela et al. 2008).
Materials and Methods
Ethical considerations and animal care
The study protocol was approved by the Committee of Ethics on Animal Experiments of Kyushu University (A26‐213‐0). Animal handling and procedures were carried out in compliance with the Guidelines for Animal Experiments, Kyushu University, and Law (No. 105) and Notification (No. 6) of the Japanese Government. Mice were housed in a climate‐controlled space on a 12‐h day/night cycle and allowed free access to food and water. All synthetic rodent diets were purchased from Oriental Yeast Co., Ltd (Tokyo, Japan).
Generation of GSK‐3β +/− mice
GSK‐3β +/− mice were generated as described previously by Kimura et al. (2008). Briefly, using homologous recombination, we flanked the exon‐encoding catalytic domain of GSK‐3β with LoxP elements. Floxed GSK‐3β mice were crossed with mice expressing Cre recombinase under the control of the EIIa promoter, and their progeny were crossed with C57BL/6 mice. Heterozygous knockout of a GSK‐3β allele was confirmed by PCR using mouse genomic DNA, as described previously by Kimura et al. (2008).
Experimental protocol
Eight‐week‐old male wild‐type C57BL/6 mice (n = 24) were randomly divided into three groups. Control mice (CNT, n = 8) were fed a control diet (calcium 1.0%, phosphate 1.2%; Oriental Yeast Co., Ltd, Tokyo, Japan) for 6 weeks. The remaining 16 mice were fed a diet containing 0.2% adenine (adenine‐diet) for 6 weeks. Eight mice were not treated (CKD, n = 8) and the other eight mice were treated with LiCl (Wako, Osaka, Japan) in drinking water (150 mg/L) for 6 weeks (CKD+LiCl, n = 8). For the fourth group, GSK‐3β +/− mice (n = 8) were fed an adenine‐diet for 6 weeks (CKD‐GSK‐3β +/−) to determine the effect of GSK‐3β haploinsufficiency on bone volume and properties. The adenine‐induced CKD mouse model was used to recapitulate uremia‐related bone abnormalities because adenine‐induced uremic rat and mouse models show chronic progressive tubulointerstitial nephritis caused by accumulation of 2,8‐dihydroxyadenine crystals in renal tubules and interstitia (Yokozawa et al. 1986; Jia et al. 2013).
One day before euthanasia, mice were housed in metabolic cages for 24 h, and food and water intake and urine volume were recorded. Mice were euthanized on day 42, and their blood and femurs collected. Blood was clotted at room temperature for 1 h and the obtained serum was separated by centrifugation at 3000 × g and stored at −30°C until analysis. The left femur was immersed in 70% ethanol and stored at 4°C until analysis.
Biochemical parameters
Serum concentrations of albumin, urea nitrogen, sodium, calcium, and phosphate were measured with an automated analyzer (Hitachi, Tokyo, Japan). Serum levels of intact parathyroid hormone (PTH) (Immutopics International, San Clemente, CA), osteocalcin (Biomedical Technologies, Stoughton, MA), and tartrate‐resistant acid phosphatase‐5b (TRACP‐5b) (Immunodiagnostic Systems, Gaithersburg, MD) were determined using commercially available mouse ELISA kits. The kits were used according to the manufacturer's instructions, and their qualities were within analytical levels.
Determination of bone volume and parameters by micro‐CT
Morphological analysis of mouse femurs was performed using a micro‐CT system (Skyscan 1076 scanner; Skyscan, Konitich, Belgium), as described previously (Bouxsein et al. 2010). Briefly, scanning conditions were set to 48 kV, 201 μA, and 9 μm for one scan image. Three‐dimensional reconstruction of images was performed with InstaRecon/NRecon software (Skyscan). Two regions were quantitatively analyzed in mice: the cortical bone region from 2.0 to 2.5 mm above the growth plate at the distal metaphysis; and the trabecular bone region from 0.1 to 1.1 mm above the growth plate at the distal metaphysis. We calculated the following parameters: bone volume/total volume; trabecular number; trabecular thickness; trabecular separation; cortical thickness; cortical bone area; and total bone area. For each parameter, micro‐CT‐derived standard bone morphometry nomenclature, symbols, and units were used (Bouxsein et al. 2010).
Statistical analysis
All statistical analyses were performed using JMP version 10.0 software (SAS Institute, Tokyo, Japan). Data are presented as mean ± SEM. Differences among groups were compared using one‐way analysis of variance, followed by Tukey–Kramer tests. For all tests, a two‐tailed P‐value <0.05 was considered statistically significant.
Results
Adenine‐induced CKD mice develop azotemia, hyperphosphatemia, and hyperparathyroidism
Body weight at week 6 was significantly lower in CKD, CKD‐GSK‐3β +/−, and CKD+LiCl mice than in CNT mice. There were no significant differences in body weight among the three CKD groups. Food intake was significantly lower in CKD mice than in CNT mice, while there were no significant differences in food intake among the three CKD groups (Table 1). Serum levels of albumin were comparable among the four groups. Serum levels of urea nitrogen, phosphate, and intact PTH were significantly higher in CKD, CKD‐GSK‐3β +/−, and CKD+LiCl mice than in CNT mice, and were comparable among the three CKD groups. The serum calcium concentration was significantly higher in CKD+LiCl mice than in CNT mice, and there were no significant differences among the three CKD groups (Table 2).
Table 1.
Physical parameters of each group after 6 weeks of treatment
| CNT | CKD | CKD‐GSK‐3β +/− | CKD+LiCl | |
|---|---|---|---|---|
| Body weight, g | 27.6 ± 1.0 | 18.5 ± 0.3* | 18.0 ± 0.7* | 20.3 ± 0.2* |
| Food intake, g/day | 2.8 ± 0.1 | 2.2 ± 0.1* | 2.2 ± 0.2 | 2.5 ± 0.2 |
| Water intake, mL/day | 5.1 ± 0.4 | 7.8 ± 0.3 | 7.6 ± 1.4 | 24.9 ± 2.1*, †, ‡ |
| Urine volume, mL/day | 1.3 ± 0.2 | 3.9 ± 0.5 | 3.2 ± 0.5 | 16.3 ± 1.8*, †, ‡ |
CNT, mice fed a control diet; CKD, mice fed a diet containing 0.2% adenine; CKD‐GSK‐3β +/−, GSK‐3β +/− mice fed a diet containing 0.2% adenine; CKD+LiCl, mice fed a diet containing 0.2% adenine and given water containing LiCl (0.15 mg/mL). Data are mean ± SEM, and were compared using one‐way ANOVA followed by the Tukey–Kramer test. A two‐tailed P‐value <0.05 was considered statistically significant. *P < 0.05 versus CNT, † P < 0.05 versus CKD, ‡ P < 0.05 versus CKD‐GSK‐3β +/−. CKD, chronic kidney disease; CNT, control; GSK‐3β +/−, glycogen synthase kinase‐3 beta heterozygous knockout; LiCl, lithium chloride.
Table 2.
Serum biochemistries of each group after 6 weeks of treatment
| CNT | CKD | CKD‐GSK‐3β +/− | CKD+LiCl | |
|---|---|---|---|---|
| Albumin, g/dL | 3.2 ± 0.1 | 3.2 ± 0.1 | 3.0 ± 0.1 | 3.3 ± 0.1 |
| Urea nitrogen, mg/dL | 25.3 ± 1.5 | 59.1 ± 1.7* | 63.9 ± 3.5* | 54.3 ± 4.8* |
| Sodium, mEq/L | 147.3 ± 1.3 | 148.5 ± 2.5 | 149.2 ± 2.5 | 148.6 ± 1.2 |
| Calcium, mg/dL | 8.5 ± 0.2 | 9.1 ± 0.2 | 8.9 ± 0.3 | 9.7 ± 0.2* |
| Phosphate, mg/dL | 9.3 ± 0.5 | 15.6 ± 1.1* | 16.2 ± 1.6* | 15.3 ± 0.9* |
| Intact PTH, pg/mL | 430 ± 134 | 2948 ± 403* | 2942 ± 467* | 2919 ± 381* |
| Osteocalcin, ng/mL | 9.4 ± 2.9 | 101.2 ± 17.8* | 140.8 ± 23.1* | 76.9 ± 8.7*, ‡ |
| TRACP‐5b, U/L | 2.1 ± 0.5 | 9.0 ± 1.3* | 9.8 ± 1.6* | 8.2 ± 0.7* |
CNT, mice fed a control diet; CKD, mice fed a diet containing 0.2% adenine; CKD‐GSK‐3β +/−, GSK‐3β +/− mice fed a diet containing 0.2% adenine; CKD+LiCl, mice fed a diet containing 0.2% adenine and given water containing LiCl (0.15 mg/mL). Data are mean ± SEM, and were compared using one‐way ANOVA followed by the Tukey–Kramer test. A two‐tailed P‐value <0.05 was considered statistically significant. *P < 0.05 versus CNT, ‡ P < 0.05 versus CKD‐GSK‐3β +/−. CKD, chronic kidney disease; CNT, control; GSK‐3β +/−, glycogen synthase kinase‐3 beta heterozygous knockout; LiCl, lithium chloride; PTH, parathyroid hormone; TRACP‐5b, tartrate‐resistant acid phosphatase‐5b.
Bone turnover markers increase in adenine‐induced CKD mice
Serum levels of osteocalcin and TRACP‐5b were significantly higher in CKD, CKD‐GSK‐3β +/−, and CKD+LiCl mice than in CNT mice. The serum concentration of osteocalcin was significantly lower in CKD+LiCl mice than in CKD‐GSK‐3β +/− mice. The serum levels of TRACP‐5b was comparable among the three CKD groups (Table 2).
LiCl treatment induces polyuria and polydipsia
As shown in Table 1, water intake and urine volume at week 6 were significantly higher in CKD+LiCl mice than in CNT, CKD, and CKD‐GSK‐3β +/− mice. Serum levels of sodium were comparable among the four groups.
Uremia affects cortical bone volume
Representative coronal and cross‐sectional images of femurs from each group are shown in Figure 1. Quantitative morphological parameters for trabecular bone volume are shown in Figure 2. Bone volume/tissue volume, trabecular number, and trabecular thickness were not decreased in CKD mice compared with CNT mice (Fig. 2A–C). There were no significant differences in trabecular separation between CNT and CKD mice (Fig. 2D). Figure 3 shows quantitative morphological parameters for cortical bone volume. Cortical thickness, cortical bone area, and cortical bone area/total bone area were significantly reduced in CKD mice compared with CNT mice (Fig. 3A, B, and D). There were no significant differences in total bone area between CNT and CKD mice (Fig. 3C).
Figure 1.
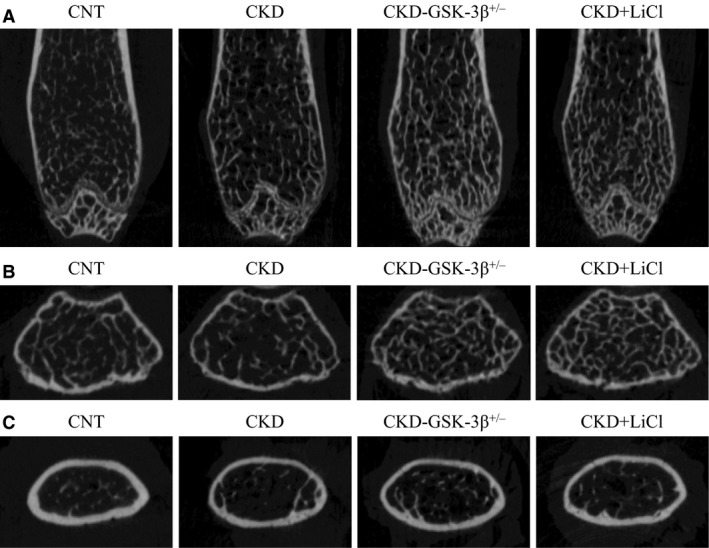
Representative microcomputed tomography images of femurs. (A) Longitudinal images of femurs. (B) Cross‐sectional images of femurs in trabecular bone area. (C) Cross‐sectional images of femurs in cortical bone area. CKD, chronic kidney disease; CNT, control; GSK‐3β +/−, glycogen synthase kinase‐3 beta heterozygous knockout; LiCl, lithium chloride.
Figure 2.
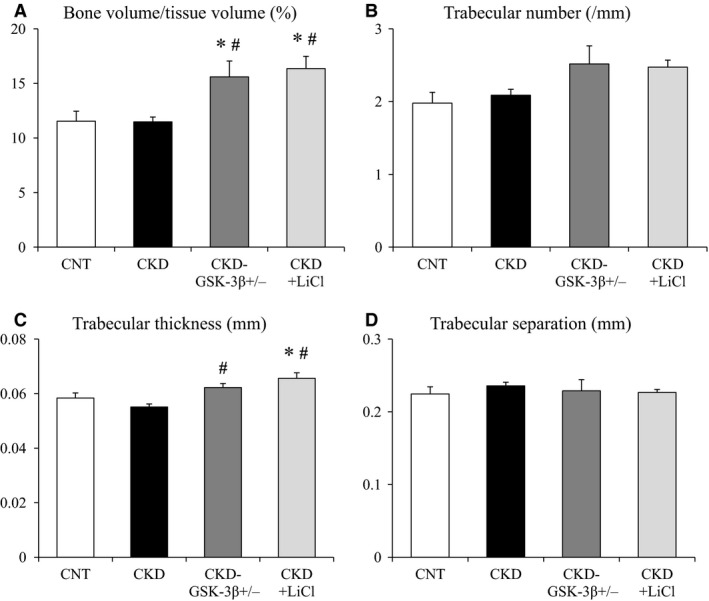
Quantitative morphological parameters for trabecular bone. (A) Bone volume/tissue volume (%), (B) Trabecular number (/mm), (C) Trabecular thickness (mm), and (D) Trabecular separation (mm). Data are mean ± SEM, and were compared using one‐way ANOVA followed by the Tukey–Kramer test. A two‐tailed P‐value <0.05 was considered statistically significant. *P < 0.05 versus CNT, # P < 0.05 versus CKD. CKD, chronic kidney disease; CNT, control; GSK‐3β +/−, glycogen synthase kinase‐3 beta heterozygous knockout; LiCl, lithium chloride.
Figure 3.
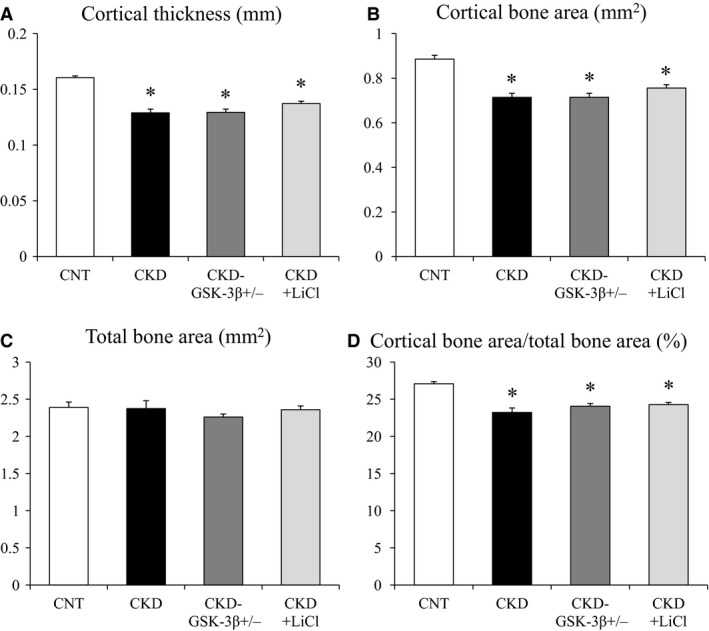
Quantitative morphological parameters for cortical bone. Cortical thickness (mm), (B) Cortical bone area (mm2), (C) Total bone area (mm2), and (D) Cortical bone area/total bone area (%). Data are mean ± SEM, and were compared using one‐way ANOVA followed by the Tukey–Kramer test. A two‐tailed P‐value <0.05 was considered statistically significant. *P < 0.05 versus CNT. CKD, chronic kidney disease; CNT, control; GSK‐3β +/−, glycogen synthase kinase‐3 beta heterozygous knockout; LiCl, lithium chloride.
GSK‐3β inhibition increases trabecular bone volume but does not affect cortical bone volume in CKD mice
Bone volume/tissue volume significantly increased in CKD‐GSK‐3β +/− and CKD+LiCl mice compared with CNT and CKD mice (Fig. 2A). Trabecular thickness was higher in CKD‐GSK‐3β +/− and CKD+LiCl mice than in CKD mice. In CKD+LiCl mice, in particular, trabecular thickness was elevated, even compared with CNT mice (Fig. 2C). Trabecular number tended to be high in CKD‐GSK‐3β +/− and CKD+LiCl mice, but there were no significant differences (Fig. 2B). Trabecular separation was comparable among the four groups (Fig. 2D). Cortical thickness, cortical bone area, and cortical bone area/total bone area were not significantly rescued in CKD‐GSK‐3β +/− and CKD+LiCl mice compared with CKD mice (Fig. 3A, B, and D). Total bone area was comparable among the four groups (Fig. 3C).
Discussion
This study is the first to show the impact of GSK‐3β inhibition on bone volume in CKD mice. By using GSK‐3β +/− mice and LiCl, we revealed that inhibition of GSK‐3β increased trabecular bone volume in adenine‐induced uremic mice without affecting kidney function and mineral parameters, but did not improve the decrease in cortical bone volume. These findings suggest that GSK‐3β inhibitor serves as a bone anabolic agent that increases trabecular bone volume and potentially reduces the risk for bone fractures even in CKD.
In this study, GSK‐3β inhibition increased trabecular bone volume. Although cortical bone mass is a major determinant of bone strength and has been shown to be associated with bone fracture risk in patients on hemodialysis (Jamal et al. 2006), recent clinical studies have revealed that not only cortical bone volume but also trabecular bone volume is associated with the risk for fractures (Nickolas et al. 2010; Cejka et al. 2011; Bucur et al. 2015). Furthermore, bone volume/tissue volume has been shown to reflect trabecular bone connectivity, a component of bone quality (Kazama et al. 2010). Therefore, our results suggest that an increase in trabecular bone volume by GSK‐3β inhibition potentially reduces the risk for bone fractures in CKD. Notably, in our adenine‐induced CKD model, trabecular bone volume was not decreased in CKD group compared with CNT group, indicating that the increase in trabecular bone volume by GSK‐3β inhibition might be achieved independently of CKD‐related bone abnormalities. In this regard, we can only conclude that inhibition of GSK‐3β increases trabecular bone volume even under the CKD condition such as hyperphosphatemia and hyperparathyroidism. However, given that few bone anabolic agents are available especially in CKD, our data are promising and should be confirmed by other CKD models with decreased trabecular bone volume.
GSK‐3β +/− mice with normal kidney function showed increases in trabecular and cortical bone volume in a previous study (Arioka et al. 2013). In this study, however, inhibition of GSK‐3β increased trabecular bone volume, but did not rescue the decrease in cortical bone volume in adenine‐induced CKD mice. Similar findings were observed in CKD rats treated with anti‐sclerostin antibody, a known activator of the Wnt/β‐catenin signaling pathway (Moe et al. 2015). In that study, the researchers found that treatment with anti‐sclerostin antibody increased trabecular bone volume in CKD rats with low PTH but had no effect on cortical bone volume in CKD rats with low and high serum PTH levels. These results indicate that the magnitude of the serum PTH level influences the effects of the Wnt/β‐catenin signaling pathway on bone volume in CKD. It has also been reported that continuous elevation of PTH results in a reduction of bone volume (Frolik et al. 2003). In addition, phosphate and uremic toxin have been shown to suppress osteoblastic differentiation and induce osteoblast dysfunction (Tanaka et al. 2013; Okamoto et al. 2014). Taken together, the catabolic effect induced by sustaining high levels of PTH and osteoblast dysfunction by uremia might have exceeded the anabolic effect of GSK‐3β inhibition on cortical bone volume in our CKD mice. Further studies are necessary to determine whether GSK‐3β inhibition increases cortical bone volume in CKD model with controlled levels of PTH.
Differences in the effects of CKD on cortical and trabecular bone are an intriguing subject. One clinical study reported that CKD patients showed decreased cortical bone volume, which negatively correlated with the serum PTH level (Nickolas et al. 2013). Another study reported that the trabecular microarchitecture was impaired and trabecular bone volume decreased in CKD patients without severe hyperparathyroidism (Bacchetta et al. 2010). Notably, differential effects of hyperparathyroidism on trabecular and cortical bone were observed in a rat model of CKD (Miller et al. 1998). In that study, trabecular bone volume decreased in CKD rats with mild‐to‐moderate hyperparathyroidism, while it increased in CKD rats with severe hyperparathyroidism. Cortical porosity was positively associated with the serum PTH level. These results indicate that differences between changes in trabecular bone volume and cortical bone volume may be partly explained by the degree of hyperparathyroidism and bone turnover in CKD. As adenine‐induced CKD mice developed moderate‐to‐severe hyperparathyroidism, it is plausible that cortical bone volume decreased and trabecular bone volume did not change in our CKD mice. However, the precise mechanisms of changes in bone volume in CKD still remain unknown.
Serum bone metabolism markers are often used to estimate bone metabolism because bone volume is mainly determined by the balance between bone formation and resorption (Chu et al. 2003; Shidara et al. 2008). In this study, we measured serum levels of osteocalcin, a marker of bone formation, and TRACP‐5b, a marker of bone resorption. Both of them were increased in the three CKD groups compared with control group, showing high bone turnover induced by severe hyperparathyroidism as evidenced by high serum PTH levels. These changes in bone turnover markers also indicated that combined measurement of these two different bone metabolism markers could not explain the differential effect of GSK‐3β inhibition on cortical and trabecular bone volume in our study. This is partly because serum levels of bone turnover makers reflect the total summation of the systemic bone metabolism. Further experiments such as bone histomorphometric analyses are required to determine the differential impact of GSK‐3β inhibition on cortical and trabecular bone metabolism in CKD.
In this study, there were no significant differences between GSK‐3β haploinsufficiency and LiCl administration in terms of effect on bone volume. GSK‐3 has two isoforms, GSK‐3α and GSK‐3β (Woodgett 1990). Although homology of the two isoforms is 98% in the catalytic domain, their function is not identical. GSK‐3β homozygous knockout mice exhibit the embryonic lethality phenotype (Hoeflich et al. 2000; Kerkela et al. 2008), whereas GSK‐3α homozygous knockout mice are viable. In the Wnt/β‐catenin signaling pathway, functional redundancy of the two isoforms has been reported (Doble et al. 2007), and an increase in bone volume was observed in both GSK‐3α homozygous knockout mice (Zhou et al. 2013) and GSK‐3β +/− mice (Arioka et al. 2013). Because lithium inhibits both GSK‐3α and GSK‐3β, GSK‐3β +/− mice are not always similar to mice treated with LiCl. Although isoform‐specific inhibition is a challenging task, further studies are necessary to determine the differential effect of each GSK‐3 isoform‐specific inhibition on bone volume in CKD.
GSK‐3 is associated with the pathophysiology of several diseases, such as nervous system disorders, deranged glucose metabolism, nephrogenic diabetes insipidus, acute kidney injury, inflammation, cardiac hypertrophy, abnormal bone formation, vascular calcification, and cancer (Rao et al. 2010; Wang et al. 2010; Bao et al. 2012; Howard et al. 2012; Takahashi‐Yanaga 2013; Nørregaard et al. 2015). In this study, mice treated with LiCl developed polyuria and polydipsia, while GSK‐3β +/− mice did not. The difference may be explained by the fact that the effect of lithium on the ability to concentrate urine is mediated by dual inhibition of GSK‐3α and GSK‐3β (Rao et al. 2010; Nørregaard et al. 2015). In GSK‐3β +/− mice, residual GSK‐3β and GSK‐3α might maintain the ability to concentrate urine. With regard to the effects on kidney function, inhibition of GSK‐3 had no positive or negative effects in the adenine‐induced model. Kidney damage induced by adenine might exceed the renal effects of GSK‐3 inhibition (Wang et al. 2010; Bao et al. 2012; Howard et al. 2012). These results suggest that inhibition of this kinase is potentially double‐edged and involves a risk for unfavorable effects, especially if used long‐term.
Previous studies have shown that lithium affects parathyroid gland, kidney, and other organs and has the potential to cause several adverse effects such as hypercalcemia, hyperparathyroidism, chronic renal interstitial fibrosis, renal impairment, and nephrogenic diabetes insipidus (Rao et al. 2005; McKnight et al. 2012; Walker et al. 2013; Shine et al. 2015). Although there were no significant differences in kidney function and serum levels of PTH between CKD and CKD+LiCl mice, we could not exclude the possibility that lithium affected kidney and parathyroid function, thereby indirectly affecting bone metabolism in our study. In this regard, in patients with CKD, treatment with LiCl may not be practical because of its renal toxicity (Walker et al. 2013; Shine et al. 2015), potential induction of secondary hyperparathyroidism, and the difficulties in its dose adjustment. Thus, therapeutic approaches that exert bone‐specific inhibitory effects on GSK‐3β are more ideal, especially in the CKD population.
This study has several limitations. First, the effects of GSK‐3β inhibition on bone strength were not evaluated. Second, although we previously found a decrease in the expression of GSK‐3β and an increase in the expression of β‐catenin in bone marrow cells from GSK‐3β +/− mice (Arioka et al. 2013), we did not assess the expression of GSK‐3β or β‐catenin in GSK‐3β +/− mice and CKD mice with LiCl treatment. Third, the effects of GSK‐3 isoform‐specific inhibition on uremic bone volume should be clarified in future studies. Because the GSK‐3β homozygous knockout shows the embryonic lethality phenotype, osteocyte or osteoblast‐specific GSK‐3β deletion will be necessary. Forth, only male mice were studied in this study. Given that there is a crosstalk between GSK‐3β signaling and estrogen signaling pathway and the prevalence of osteoporosis in women with CKD is high (Klawansky et al. 2003; Yin et al. 2015), future studies should be directed toward the impact of GSK‐3β inhibition on bone in female mice and more specifically, in ovariectomized mouse model. Lastly, we determined the effects of GSK‐3β inhibition on the trabecular bone volume in only one CKD model. Because the effects of CKD on trabecular bone volume were not consistent among different CKD models (Lau et al. 2013; Ferrari et al. 2014; Liu et al. 2014), further studies are needed to examine the impact of GSK‐3β inhibition on the trabecular bone volume in other CKD models such as 5/6th nephrectomy.
In conclusion, this study found that inhibition of GSK‐3β increased trabecular bone volume, but did not rescue the decrease in cortical bone volume in adenine‐induced uremic mice. These results suggest that an inhibitor of GSK‐3β may be a potential candidate as a bone anabolic agent in the CKD population. Further studies are needed to determine the beneficial effects of GSK‐3β blocking on bone quality, quantity, strength, and fracture risk in CKD patients.
Conflict of Interest
None declared.
Acknowledgments
We thank Dr. Akihiko Takashima (Department of Aging Neurology, National Center for Geriatrics and Gerontology, Oobu, Japan) for providing GSK‐3β +/− mice for this study. We also thank Edanz Editing (http://www.edanzediting.co.jp/) for their English editing of our manuscript.
Tatsumoto N., Arioka M., Yamada S., Takahashi‐Yanaga F., Tokumoto M., Tsuruya K., Kitazono T., Sasaguri T.. Inhibition of GSK‐3β increases trabecular bone volume but not cortical bone volume in adenine‐induced uremic mice with severe hyperparathyroidism. Physiol Rep, 4 (21), 2016, e13010, doi: 10.14814/phy2.13010
Funding Information
This study was supported by KAKENHI from the Ministry of Education, Culture, Sports, Science and Technology of Japan to MA (15K18990); and by a grant from The Kidney Foundation, Japan (JKFB15‐37).
References
- Alem, A. M. , Sherrard D. J., Gillen D. L., Weiss N. S., Beresford S. A., Heckbert S. R., et al. 2000. Increased risk of hip fracture among patients with end‐stage renal disease. Kidney Int. 58:396–399. [DOI] [PubMed] [Google Scholar]
- Arioka, M. , Takahashi‐Yanaga F., Sasaki M., Yoshihara T., Morimoto S., Takashima A., et al. 2013. Acceleration of bone development and regeneration through the Wnt/β‐catenin signaling pathway in mice heterozygously deficient for GSK‐3β . Biochem. Biophys. Res. Commun. 440:677–682. [DOI] [PubMed] [Google Scholar]
- Arioka, M. , Takahashi‐Yanaga F., Sasaki M., Yoshihara T., Morimoto S., Hirata M., et al. 2014. Acceleration of bone regeneration by local application of lithium: Wnt signal‐mediated osteoblastogenesis and Wnt signal‐independent suppression of osteoclastogenesis. Biochem. Pharmacol. 90:397–405. [DOI] [PubMed] [Google Scholar]
- Bacchetta, J. , Boutroy S., Vilayphiou N., Juillard L., Guebre‐Egziabher F., Rognant N., et al. 2010. Early impairment of trabecular microarchitecture assessed with HR‐pQCT in patients with stage II‐IV chronic kidney disease. J. Bone Miner. Res. 25:849–857. [DOI] [PubMed] [Google Scholar]
- Bao, H. , Ge Y., Zhuang S., Dworkin L. D., Liu Z., and Gong R.. 2012. Inhibition of glycogen synthase kinase‐3β prevents NSAID‐induced acute kidney injury. Kidney Int. 81:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron, R. , and Kneissel M.. 2013. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 19:179–192. [DOI] [PubMed] [Google Scholar]
- Bouxsein, M. L. , Boyd S. K., Christiansen B. A., Guldberg R. E., Jepsen K. J., and Müller R.. 2010. Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. J. Bone Miner. Res. 25:1468–1486. [DOI] [PubMed] [Google Scholar]
- Bucur, R. C. , Panjwani D. D., Turner L., Rader T., West S. L., and Jamal S. A.. 2015. Low bone mineral density and fractures in stages 3‐5 CKD: an updated systematic review and meta‐analysis. Osteoporos. Int. 26:449–458. [DOI] [PubMed] [Google Scholar]
- Cejka, D. , Patsch J. M., Weber M., Diarra D., Riegersperger M., Kikic Z., et al. 2011. Bone microarchitecture in hemodialysis patients assessed by HR‐pQCT. Clin. J. Am. Soc. Nephrol. 6:2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, P. , Chao T. Y., Lin Y. F., Janckila A. J., and Yam L. T.. 2003. Correlation between histomorphometric parameters of bone resorption and serum type 5b tartrate‐resistant acid phosphatase in uremic patients on maintenance hemodialysis. Am. J. Kidney Dis. 41:1052–1059. [DOI] [PubMed] [Google Scholar]
- Clément‐Lacroix, P. , Ai M., Morvan F., Roman‐Roman S., Vayssière B., Belleville C., et al. 2005. Lrp5‐independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc. Natl. Acad. Sci. U. S. A. 102:17406–17411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble, B. W. , Patel S., Wood G. A., Kockeritz L. K., and Woodgett J. R.. 2007. Functional redundancy of GSK‐3alpha and GSK‐3beta in Wnt/beta‐catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 12:957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drüeke, T. B. , and Massy Z. A.. 2016. Changing bone patterns with progression of chronic kidney disease. Kidney Int. 89:289–302. [DOI] [PubMed] [Google Scholar]
- Ensrud, K. E. , Lui L. Y., Taylor B. C., Ishani A., Shlipak M. G., Stone K. L., et al. ; Osteoporotic Fractures Research Group . 2007. Renal function and risk of hip and vertebral fractures in older women. Arch. Intern. Med. 167:133–139. [DOI] [PubMed] [Google Scholar]
- Fang, Y. , Ginsberg C., Seifert M., Agapova O., Sugatani T., Register T. C., et al. 2014. CKD‐induced wingless/integration1 inhibitors and phosphorus cause the CKD‐mineral and bone disorder. J. Am. Soc. Nephrol. 25:1760–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari, G. O. , Ferreira J. C., Cavallari R. T., Neves K. R., dos Reis L. M., Dominguez W. V., et al. 2014. Mineral bone disorder in chronic kidney disease: head‐to‐head comparison of the 5/6 nephrectomy and adenine models. BMC Nephrol. 15:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried, L. F. , Biggs M. L., Shlipak M. G., Seliger S., Kestenbaum B., Stehman‐Breen C., et al. 2007. Association of kidney function with incident hip fracture in older adults. J. Am. Soc. Nephrol. 18:282–286. [DOI] [PubMed] [Google Scholar]
- Frolik, C. A. , Black E. C., Cain R. L., Satterwhite J. H., Brown‐Augsburger P. L., Sato M., et al. 2003. Anabolic and catabolic bone effects of human parathyroid hormone (1‐34) are predicted by duration of hormone exposure. Bone 33:372–379. [DOI] [PubMed] [Google Scholar]
- Hoeflich, K. P. , Luo J., Rubie E. A., Tsao M. S., Jin O., and Woodgett J. R.. 2000. Requirement for glycogen synthase kinase‐3beta in cell survival and NF‐kappaB activation. Nature 406:86–90. [DOI] [PubMed] [Google Scholar]
- Howard, C. , Tao S., Yang H. C., Fogo A. B., Woodgett J. R., Harris R. C., et al. 2012. Specific deletion of glycogen synthase kinase‐3β in the renal proximal tubule protects against acute nephrotoxic injury in mice. Kidney Int. 82:1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal, S. A. , Gilbert J., Gordon C., and Bauer D. C.. 2006. Cortical pQCT measures are associated with fractures in dialysis patients. J. Bone Miner. Res. 21:543–548. [DOI] [PubMed] [Google Scholar]
- Jia, T. , Olauson H., Lindberg K., Amin R., Edvardsson K., Lindholm B., et al. 2013. A novel model of adenine‐induced tubulointerstitial nephropathy in mice. BMC Nephrol. 14:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazama, J. J. , Koda R., Yamamoto S., Narita I., Gejyo F., and Tokumoto A.. 2010. Cancellous bone volume is an indicator for trabecular bone connectivity in dialysis patients. Clin. J. Am. Soc. Nephrol. 5:292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazama, J. J. , Iwasaki Y., and Fukagawa M.. 2013. Uremic osteoporosis. Kidney Int. Suppl. 3:446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkela, R. , Kockeritz L., Macaulay K., Zhou J., Doble B. W., Beahm C., et al. 2008. Deletion of GSK‐3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Invest. 118:3609–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, T. , Yamashita S., Nakao S., Park J. M., Murayama M., Mizoroki T., et al. 2008. GSK‐3beta is required for memory reconsolidation in adult brain. PLoS ONE 3:e3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klawansky, S. , Komaroff E., Cavanaugh P. F. Jr, Mitchell D. Y., Gordon M. J., Connelly J. E., et al. 2003. Relationship between age, renal function and bone mineral density in the US population. Osteoporos. Int. 14:570–576. [DOI] [PubMed] [Google Scholar]
- Kulkarni, N. H. , Onyia J. E., Zeng Q., Tian X., Liu M., Halladay D. L., et al. 2006. Orally bioavailable GSK‐3alpha/beta dual inhibitor increases markers of cellular differentiation in vitro and bone mass in vivo. J. Bone Miner. Res. 21:910–920. [DOI] [PubMed] [Google Scholar]
- Lau, W. L. , Linnes M., Chu E. Y., Foster B. L., Bartley B. A., Somerman M. J., et al. 2013. High phosphate feeding promotes mineral and bone abnormalities in mice with chronic kidney disease. Nephrol. Dial. Transplant. 28:62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Song W., Boulanger J. H., Tang W., Sabbagh Y., Kelley B., et al. 2014. Role of TGF‐β in a mouse model of high turnover renal osteodystrophy. J. Bone Miner. Res. 29:1141–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malluche, H. H. , Mawad H. W., and Monier‐Faugere M. C.. 2011. Renal osteodystrophy in the first decade of the new millennium: analysis of 630 bone biopsies in black and white patients. J. Bone Miner. Res. 26:1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsell, R. , Sisask G., Nilsson Y., Sundgren‐Andersson A. K., Andersson U., Larsson S., et al. 2012. GSK‐3 inhibition by an orally active small molecule increases bone mass in rats. Bone 50:619–627. [DOI] [PubMed] [Google Scholar]
- McKnight, R. F. , Adida M., Budge K., Stockton S., Goodwin G. M., and Geddes J. R.. 2012. Lithium toxicity profile: a systematic review and meta‐analysis. Lancet 379:721–728. [DOI] [PubMed] [Google Scholar]
- Miller, M. A. , Chin J., Miller S. C., and Fox J.. 1998. Disparate effects of mild, moderate, and severe secondary hyperparathyroidism on cancellous and cortical bone in rats with chronic renal insufficiency. Bone 23:257–266. [DOI] [PubMed] [Google Scholar]
- Mittalhenkle, A. , Gillen D. L., and Stehman‐Breen C. O.. 2004. Increased risk of mortality associated with hip fracture in the dialysis population. Am. J. Kidney Dis. 44:672–679. [PubMed] [Google Scholar]
- Moe, S. M. , Chen N. X., Newman C. L., Organ J. M., Kneissel M., Kramer I., et al. 2015. Anti‐sclerostin antibody treatment in a rat model of progressive renal osteodystrophy. J. Bone Miner. Res. 30:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolas, T. L. , McMahon D. J., and Shane E.. 2006. Relationship between moderate to severe kidney disease and hip fracture in the United States. J. Am. Soc. Nephrol. 17:3223–3232. [DOI] [PubMed] [Google Scholar]
- Nickolas, T. L. , Leonard M. B., and Shane E.. 2008. Chronic kidney disease and bone fracture: a growing concern. Kidney Int. 74:721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolas, T. L. , Stein E., Cohen A., Thomas V., Staron R. B., McMahon D. J., et al. 2010. Bone mass and microarchitecture in CKD patients with fracture. J. Am. Soc. Nephrol. 21:1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolas, T. L. , Cremers S., Zhang A., Thomas V., Stein E., Cohen A., et al. 2011. Discriminants of prevalent fractures in chronic kidney disease. J. Am. Soc. Nephrol. 22:1560–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolas, T. L. , Stein E. M., Dworakowski E., Nishiyama K. K., Komandah‐Kosseh M., Zhang C. A., et al. 2013. Rapid cortical bone loss in patients with chronic kidney disease. J. Bone Miner. Res. 28:1811–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch, D. , Mylne A., Roderick P. J., Smeeth L., Hubbard R., and Fletcher A.. 2009. Chronic kidney disease and hip fracture‐related mortality in older people in the UK. Nephrol. Dial. Transplant. 24:1539–1544. [DOI] [PubMed] [Google Scholar]
- Nørregaard, R. , Tao S., Nilsson L., Woodgett J. R., Kakade V., Yu A. S., et al. 2015. Glycogen synthase kinase 3α regulates urine concentrating mechanism in mice. Am. J. Physiol. Renal. Physiol. 308:F650–F660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, T. , Taguchi M., Osaki T., Fukumoto S., and Fujita T.. 2014. Phosphate enhances reactive oxygen species production and suppresses osteoblastic differentiation. J. Bone Miner. Metab. 32:393–399. [DOI] [PubMed] [Google Scholar]
- Ott, S. M. 2016. Bone strength: more than just bone density. Kidney Int. 89:16–19. [DOI] [PubMed] [Google Scholar]
- Rao, R. , Zhang M. Z., Zhao M., Cai H., Harris R. C., Breyer M. D., et al. 2005. Lithium treatment inhibits renal GSK‐3 activity and promotes cyclooxygenase 2‐dependent polyuria. Am. J. Physiol. Renal. Physiol. 288:F642–F649. [DOI] [PubMed] [Google Scholar]
- Rao, R. , Patel S., Hao C., Woodgett J., and Harris R.. 2010. GSK3beta mediates renal response to vasopressin by modulating adenylate cyclase activity. J. Am. Soc. Nephrol. 21:428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryves, W. J. , and Harwood A. J.. 2001. Lithium inhibits glycogen synthase kinase‐3 by competition for magnesium. Biochem. Biophys. Res. Commun. 280:720–725. [DOI] [PubMed] [Google Scholar]
- Sabbagh, Y. , Graciolli F. G., O'Brien S., Tang W., dos Reis L. M., Ryan S., et al. 2012. Repression of osteocyte Wnt/β‐catenin signaling is an early event in the progression of renal osteodystrophy. J. Bone Miner. Res. 27:1757–1772. [DOI] [PubMed] [Google Scholar]
- Shidara, K. , Inaba M., Okuno S., Yamada S., Kumeda Y., Imanishi Y., et al. 2008. Serum levels of TRAP5b, a new bone resorption marker unaffected by renal dysfunction, as a useful marker of cortical bone loss in hemodialysis patients. Calcif. Tissue Int. 82:278–287. [DOI] [PubMed] [Google Scholar]
- Shine, B. , McKnight R. F., Leaver L., and Geddes J. R.. 2015. Long‐term effects of lithium on renal, thyroid, and parathyroid function: a retrospective analysis of laboratory data. Lancet 386:461–468. [DOI] [PubMed] [Google Scholar]
- Sisask, G. , Marsell R., Sundgren‐Andersson A., Larsson S., Nilsson O., Ljunggren O., et al. 2013. Rats treated with AZD2858, a GSK3 inhibitor, heal fractures rapidly without endochondral bone formation. Bone 54:126–132. [DOI] [PubMed] [Google Scholar]
- Takahashi‐Yanaga, F. 2013. Activator or inhibitor? GSK‐3 as a new drug target. Biochem. Pharmacol. 86:191–199. [DOI] [PubMed] [Google Scholar]
- Tanaka, H. , Iwasaki Y., Yamato H., Mori Y., Komaba H., Watanabe H., et al. 2013. p‐Cresyl sulfate induces osteoblast dysfunction through activating JNK and p38 MAPK pathways. Bone 56:347–354. [DOI] [PubMed] [Google Scholar]
- Tentori, F. , McCullough K., Kilpatrick R. D., Bradbury B. D., Robinson B. M., Kerr P. G., et al. 2014. High rates of death and hospitalization follow bone fracture among hemodialysis patients. Kidney Int. 85:166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, R. J. , Leader J. P., Bedford J. J., Gobe G., Davis G., Vos F. E., et al. 2013. Chronic interstitial fibrosis in the rat kidney induced by long‐term (6‐mo) exposure to lithium. Am. J. Physiol. Renal. Physiol. 304:F300–F307. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Havasi A., Gall J., Bonegio R., Li Z., Mao H., et al. 2010. GSK3beta promotes apoptosis after renal ischemic injury. J. Am. Soc. Nephrol. 21:284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, S. L. , Lok C. E., Langsetmo L., Cheung A. M., Szabo E., Pearce D., et al. 2015. Bone mineral density predicts fractures in chronic kidney disease. J. Bone Miner. Res. 30:913–919. [DOI] [PubMed] [Google Scholar]
- Woodgett, J. R. 1990. Molecular cloning and expression of glycogen synthase kinase‐3/factor A. EMBO J. 9:2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, X. , Wang X., Hu X., Chen Y., Zeng K., and Zhang H.. 2015. ERβ induces the differentiation of cultured osteoblasts by both Wnt/β‐catenin signaling pathway and estrogen signaling pathways. Exp. Cell Res. 335:107–114. [DOI] [PubMed] [Google Scholar]
- Yokozawa, T. , Zheng P. D., Oura H., and Koizumi F.. 1986. Animal model of adenine‐induced chronic renal failure in rats. Nephron 44:230–234. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Freeman T. A., Ahmad F., Shang X., Mangano E., Gao E., et al. 2013. GSK‐3α is a central regulator of age‐related pathologies in mice. J. Clin. Invest. 123:1821–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]