

Abstract
QseC is a membrane-bound histidine sensor kinase found in Gram-negative pathogens and is involved in the regulation of bacterial virulence. LED209, a QseC-specific inhibitor, significantly inhibits the virulence of several pathogens and partially protects infected mice from death by blocking QseC. However, the mechanism of its antibacterial effects remains unclear. In this experiment, a Salmonella Typhimurium (S. Typhimurium) and macrophage co-culture system was utilized to investigate possible mechanisms underlying the antimicrobial effects of the QseC inhibitor. QseC blockade inhibited the expression of QseC-dependent virulence genes, including flhDC, sifA, and sopB, in S. Typhimurium, leading to inhibition of swimming motility, invasion capacity, and replication capacity of the pathogens. Release of lactate dehydrogenase (LDH) from S. Typhimurium-infected macrophages was significantly inhibited by blocking QseC. Activated caspase-1 and IL-1β levels were suppressed, and intracellular bacterial count was reduced in infected macrophages. QseC blockade effectively reduced the virulence of S. Typhimurium, inhibited S. Typhimurium-induced pyroptosis of macrophages, and promoted elimination of intracellular bacteria from infected macrophages. Thus, the antibacterial effects of QseC inhibitor are mediated via enhancement of intracellular killing of S. Typhimurium in macrophages.
The rapid and global distribution of antimicrobial-resistant organisms is one of the leading causes of death in patients with severe infections and has become a significant challenge to humans1,2. Certain types of Gram-negative bacteria have become resistant to all available antibiotic drugs, contributing to increased morbidity and mortality, difficulty in controlling infectious illness, and escalating healthcare costs3,4. On the other hand, discovery and development of new antibiotics has rapidly declined, and the number of new antibiotics approved by the US Food and Drug Administration has decreased in recent years. Consequently, the arsenal of antibiotics is shrinking, weakening our ability to treat infectious diseases5,6. Therefore, developing new antibiotic agents with low probabilities of inducing resistance is of paramount importance.
Anti-quorum sensing is a promising theoretical strategy to prevent bacterial infections because this type of targeting may reduce resistance selection. Many Gram-negative bacteria carry QseC, a highly conserved membrane histidine sensor kinase, to identify environmental cues and regulate the expression of virulence factors. Upon sensing both host-derived adrenergic signals and the bacterial aromatic signal autoinducer-3, QseC autophosphorylates and subsequently phosphorylates a transcription factor, namely, QseB, which activates the transcription of key virulence genes7. Blockade of QseC by LED209, a selective inhibitor of QseC, significantly inhibits QseC-mediated activation of virulence-related gene expression and partially protects mice from death following infection with Salmonella typhimurium or Francisella tularensis8. However, LED209 only inhibits bacterial virulence and does not suppress S. Typhimurium growth. The fundamental mechanism underlying the in vivo protective effects of LED209 remains unclear.
The macrophage inflammasome, a critical component of the innate immune response, is a large multiprotein complex that recognizes invading pathogens in the cytosol and enables autocatalytic activation of caspase-1. Activation of this protein triggers maturation and release of the pro-inflammatory cytokines IL-1β and IL-18 as well as pyroptosis, a pro-inflammatory form of macrophage cell death9,10. Previous studies reported that the macrophage NLRC4 inflammasome can detect S. Typhimurium flagellin. Moreover, caspase-1-induced pyroptosis of macrophages is an innate immune response to fight intracellular bacteria11. However, excessive cell pyroptosis can cause immunological diseases and septic shock in the host12,13.
We hypothesized that QseC inhibitors may suppress inflammasome over-activation and macrophage pyroptosis by inhibiting bacterial virulence release and enhancing the clearance of S. Typhimurium. In this experiment, we investigated the possible antimicrobial mechanisms of Br-LED209, a LED209-derived QseC inhibitor, with a co-culture system of S. Typhimurium and macrophages. Br-LED209 effectively blocked the QseC of S. Typhimurium and suppressed the expression of its virulence genes. After that, QseC blockade inhibited S. Typhimurium-induced pyroptosis of macrophages and promoted the elimination of intracellular bacteria, which were possibly mediated by inhibiting excessive activation of inflammasomes in the infected macrophages.
Materials and Methods
Bacterial strains
S. Typhimurium (XJ76218), enterohemorrhagic Escherichia coli O157:H7 (EHEC) (XJ76330), and Shigella flexneri (XJ76116) were obtained from Xijing Affiliated Hospital of the Fourth Military Medical University (Xi’an, China). All of these strains were isolated from feces of patients.
Synthesis and characterization of Br-LED209
Ara-acetylaminobenzene sulfonyl chloride and para-bromoaniline were used as raw materials to obtain 4-amino-N-(4-bromophenyl)benzenesulfonamide through acylation and deacylation. The resulting product and phenyl isothiocyanate were then used to yield N-(4-bromophenyl)-4-(3-phenyl thiourea)benzenesulfonamide, named Br-LED209 in short. The synthesis route and chemical structure of Br-LED209 is shown in Figure S1. The chemical structure of Br-LED209 was confirmed by mass spectrometry (MS), 1H NMR, and 13C NMR (data not shown). Br-LED209 is a light yellow powder with melting point between 116 °C and 120 °C. The purity of Br-LED209 is higher than 95%. Br-LED209 was dissolved in DMSO and the final concentration of DMSO in all of the in vitro experiments was 1% (v/v). DMSO at the same concentration was used as control in all the experiments.
Construction of the qseC mutant strain
An isogenic S. Typhimurium qseC mutant was constructed using λ red mutagenesis as described previously8. Briefly, a qseC PCR product was generated using primers showed in Table 1 and pKD3 as template, and gel-purified. The PCR product was electroporated into S. Typhimurium containing pKD46, which were then incubated at 37 °C for 1 hour, and plated on media containing 30 μg/ml chloramphenicol overnight at 37 °C. The resultant colonies were patched for chloramphenicol resistance and ampicillin sensitivity, and PCR verified for the absence of the gene. Plasmid pCP20, which encodes a resolvase, was electroporated into the mutant strain, and the resulting colonies were patched for chloramphenicol sensitivity. The chloramphenicol cassette was resolved from the mutant and a non-polar isogenic qseC mutant was created.
Table 1. primers and plasmids used for qseC mutation in this study.
| Primer | Sequence | |||
|---|---|---|---|---|
| qseC up | Forward | 5′-CACAGTGCCATAACGGCAACG-3′ | ||
| Reverse | 5′-GAAGCAGCTCCAGCCTACACTCATGCGTCACCCAGGGTGT-3′ | |||
| qseC down | Forward | 5′-CTAAGGAGGATATTCATATGCAGAGACTTTTGCCAAAAACGC-3′ | ||
| Reverse | 5′-GAGGACGGCCTGACGGTGATGT-3′ | |||
| cm | Forward | 5′-ACACCCTGGGTGACGCATGAGTGTAGGCTGGAGCTGCTTC-3′ | ||
| Reverse | 5′-GCGTTTTTGGCAAAAGTCTCTGCATATGAATATCCTCCTTAG-3′ | |||
| Plasmids | Description | |||
| pKD3 | pANTSγ derivative containing FRT-flanked chloramphenicol resistance | |||
| pKD46 | A red recombinase expression plasmid | |||
| pCP20 | An ampicillin and CmR plasmid that shows temperature-sensitive replication and thermal induction of FLP synthesis | |||
Bacterial susceptibility and growth assay
To analyze bacterial growth, one isolated single colony was inoculated into 2 ml of Luria–Bertani (LB) broth and cultured at 37 °C with shaking overnight. The overnight culture was diluted to 1:100 in 4 ml of fresh LB broth and then cultured at 37 °C. Br-LED209 was added to bacterial cultures to obtain final concentrations of 10, 50, and 200 μM. Bacterial cultures were collected for cell growth analysis by measuring OD600 absorbance every 1 h, and growth curves were generated according to the values of OD600 absorbance. The minimum inhibitory concentrations (MICs) were determined by a microdilution assay with broth microdilution guidelines published by the Clinical and Laboratory Standards Institute. Levofloxacin (LEV) was used as the positive antibiotic control.
Ethics statement
The experimental and animal care procedures were approved by the Animal Care and Use Committee of the Fourth Military Medical University. All procedures were carried out in strict accordance with the approved guidelines.
Protective effects of Br-LED209 on S. Typhimurium-infected mice
Eight- to ten-week-old male BALB/c mice weighing 18–22 g were used in this study. Infection was induced by intraperitoneal administration of 1.0 × 108 colony-forming units (CFUs) of wild-type (WT) S. Typhimurium or the qseC mutant in 0.4 ml of LB broth. The mice were treated orally with 20 mg/kg Br-LED209 three hours before and after infection. Livers and spleens were harvested at 8, 16 and 24 hours after infection. The samples were homogenized, and the supernatants were plated on agar plates for evaluating bacterial burden in the infected organs. The morphologies of livers and spleens of infected mice were examined by observing hematoxylin and eosin-stained sections of the respective tissues.
Quantitative real-time RT-PCR
To evaluate the QseC blocking efficiency of Br-LED209, the expression of virulence genes in WT S. Typhimurium and the qseC mutant was detected by quantitative real-time PCR. In brief, RNA was extracted from an overnight culture grown aerobically in LB medium in the absence or presence of 200 μM Br-LED209 using a bacterial RNA isolation kit (Tiangen) according to the manufacturer’s protocol. The primers used in the real-time assays are listed in Table 2. Real-time RT-PCR was performed with SYBR Premix Ex TaqII (Takara) following the manufacturer’s instruction. The rpoA (RNA polymerase subunit A) gene was used as the endogenous control. Data was collected using the Bio-Rad CFX Manager 2.1 software. Data were normalized to levels of rpoA and analyzed by the comparative critical threshold (CT) method. Expression levels in different groups were compared by the relative quantification method. Real-time data are expressed below as fold changes compared with the WT group in the absence of Br-LED209.
Table 2. Primers for real-time PCR used in this study.
| Gene | Primer sequence | |
|---|---|---|
| flhDC | Forward | 5′-GTCAAACCGGAAATGACAAACTAA-3′ |
| Reverse | 5′-ACCCTGCCGCAGATGGT-3′ | |
| sifA | Forward | 5′-GTTGTCTAATGGAACCGATAATATCG-3′ |
| Reverse | 5′-CTACCCCCTCCCTTCGACAT-3′ | |
| sopB | Forward | 5′-CGGGTACCGCGTCAATTTC-3′ |
| Reverse | 5′-TGGCGGCGAACCCTATAAA-3′ | |
| rpoA | Forward | 5′-GCGCTCATCTTCTTCCGAAT-3′ |
| Reverse | 5′-CGCGGTCGTGGTTATGTG-3′ | |
Motility assay
To evaluate the effect of Br-LED209 on the flagella motility of S. Typhimurium, S. Typhimurium (WT and qseC mutant) was cultured in LB medium for 12 h. Subsequently, 1 μl aliquot of the culture was spotted onto 0.3% agar plates with or without 200 μM Br-LED209, and halo sizes were measured at 6, 12 and 18 hours after incubation at 37 °C14.
HeLa cell invasion and intracellular bacteria replication assay
HeLa cells were plated in 96-well culture dishes at a concentration of 2 × 104 cells/well and infected with WT S. Typhimurium or the qseC mutant at a multiplicity of infection (MOI) of 50:1 for 1 h. The cells were then treated with 100 μg/ml gentamicin for 1 h to kill extracellular bacteria. The cell culture medium was replaced with 10 μg/ml gentamicin for the remainder of the experiment. The cells were then lysed with 1% Triton X-100 at the indicated time points, and the lysates were diluted and plated on agar plates to determine the number of CFUs14. Fold increase was calculated as the ratio of intracellular bacteria at 6 and 12 hours to that at 1 hour.
Macrophage release lactate dehydrogenase (LDH) assay
Male BALB/c mice weighing 18–22 g were sacrificed by cervical dislocation. Resident peritoneal macrophages were harvested as described previously with slight modification15. In brief, the peritoneal cavity was lavaged with 5 ml of ice-cold sterile PBS. The buffer containing resident peritoneal cells was slowly withdrawn. Cells were collected by centrifugation (1000 × g for 5 min) and plated in a culture flask containing Dulbecco’s minimal essential medium (Hyclone) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The peritoneal macrophages were allowed to adhere for 3 h (37 °C, 5% CO2) and then washed with PBS to remove unattached cells.
The macrophages were plated in 96-well culture dishes (Falcon, Franklin Lakes, NJ, USA) at a concentration of 5 × 104 cells/well. Experiments were performed as described previously with slight modifications16,17,18. Briefly, S. Typhimurium were grown overnight in LB at 37 °C. The overnight culture was subsequently diluted at 1:50 into fresh LB and grown for 3–4 h at 37 °C before infection. Macrophages were infected with WT S. Typhimurium or the qseC mutant diluted in fresh DEME at MOI of 25:1. Images of macrophages were taken under a light microscope at 1 h after infection. LDH released in the supernatant was detected using a cytotoxicity detection kit (Roche). Data on detected LDH were used to calculate the pyroptotic rate of infected macrophages based on the following equation: [(experimental release − spontaneous release)/(maximum release − spontaneous release)] × 100, where spontaneous release is from the cytoplasm of uninfected macrophages, and maximum release is that obtained by lysis of macrophages with a solution of 0.1% Triton X-100.
Fluorescence microscopy
For the fluorescence microscope assay, macrophages seeded on a glass slide were infected with WT S. Typhimurium or the qseC mutant at MOI of 25:1 for 1 h and washed twice with PBS. The macrophages were stained with DAPI and the membrane impermeant dye propidium iodide (PI). The percentage of PI-positive cells was determined by counting cell numbers in four random visual fields.
Western blot
The caspase-1 p10 subunit and processed interleukin (IL)-1β released into the culture supernatant from macrophages were collected and precipitated with 10% TCA (vol/vol) for 1 h on ice. Precipitated proteins were pelleted at 20,000× g for 15 min at 4 °C, washed with ice-cold acetone, air-dried, resuspended in SDS-PAGE sample buffer, and heated to 95 °C for 10 min. Proteins were loaded and separated on 15% SDS-polyacrylamide gel and then transferred to polyvinylidene difluoride membranes (Millipore Corporation, Billerica, MA, USA). Western blots were performed with rabbit anti-mouse caspase-1 antibody (Epitomics, EPR4321) diluted to 1:5000 and goat anti-mouse IL-1β antibody (R&D Systems, AF-401-NA) diluted to 1:2000. Cell lysates were probed with anti-β-actin antibodies (Sigma, Cat. No. A2066) diluted to 1:5000.
Macrophage infection assay
Macrophage infection experiments were performed as described previously with some modifications19,20,21. In brief, macrophages were plated in 96-well culture dishes at a concentration of 5 × 104 cells/well. S. Typhimurium were cultured at 37 °C with shaking overnight and were opsonized in DMEM and 10% normal mouse serum for 20 min. The macrophages were infected with opsonized WT S. Typhimurium or the qseC mutant at MOI of 25:1 for 30 min. Then the cells were washed with PBS and treated with 100 μg/ml gentamicin for 1 h to kill extracellular bacteria. The cell culture medium was replaced with 10 μg/ml gentamicin for the remainder of the experiment. To count the number of intracellular bacteria, macrophages were washed three times with PBS and lysed with 1% Triton X-100 for 10 min. Bacteria were diluted and plated on LB medium plates to determine the number of CFU at indicated time points.
Statistical analysis
Data are shown as mean ± SD. One-way ANOVA and two-way ANOVA were used to evaluate statistical significance. A probability value of P < 0.05 was considered statistically significant.
Results
Bacterial growth was not inhibited by Br-LED209 in vitro
To assess whether Br-LED209 influences the growth of bacteria in vitro, S. Typhimurium, S. flexneri, and EHEC were cultured in LB medium in the absence or presence of Br-LED209 at different concentrations. The growth curve demonstrated that Br-LED209 did not inhibit growth of the three strains in vitro at concentrations up to 200 μM. Meanwhile, levofloxacin, which was used as a positive control, completely inhibited the growth of all three tested strains at two fold dilutions of MIC values used on each strain (Fig. 1A–C). The results of MIC assay showed that Br-LED209 did not have significant bactericidal effects at a concentration of 256 μg/ml in vitro (Fig. 1D).
Figure 1. Br-LED209 has no influence on bacterial growth in vitro.

Growth curves of Salmonella Typhimurium (A) Enterohemorrhagic Escherichia coli (EHEC) (B) Shigella flexneri (C) were drawn every hour at OD600 absorbance in the absence or presence of Br-LED209 at different concentrations. The antibiotic levofloxacin (LEV) was used as positive control. (D) Minimal inhibitory concentration (MIC) of Br-LED209.
S. Typhimurium-infected mice were protected in vivo by blocking QseC
To evaluate the protective effects of Br-LED209 in vivo, BALB/c mice were infected by intraperitoneal administration of WT S. Typhimurium or the qseC mutant at 1.0 × 108 CFUs. The mice were treated twice orally with Br-LED209 (20 mg/kg) at 3 h before and after infection. Livers and spleens were harvested at 8, 16 and 24 hours after infection, homogenized and then plated on agar plates for bacterial counts. The results showed that bacterial numbers in the liver and spleen were dramatically lower in Br-LED209-treated mice and qseC mutant-infected mice than the corresponding values in WT-infected mice (Fig. 2A,B).
Figure 2. Bacterial burden and pathological damages in organs of infected mice were reduced by blocking QseC.

(A,B) BALB/c mice were infected by intraperitoneal administration of 1.0 × 108 CFUs of WT S. Typhimurium or the qseC mutant. The mice were treated twice orally with Br-LED209 (20 mg/kg) at 3 h before and after infection. Livers and spleens were harvested at 8, 16 and 24 hours after infection, homogenized and then plated on agar plates for bacterial counts. (*P < 0.05, **P < 0.01, ***P < 0.001 vs. WT in two-way ANOVA, n = 5) (C) Morphological characteristics of liver and spleen in BALB/c mice were compared among groups infected with WT S. Typhimurium, the qseC mutant or Br-LED209. Scale bars: 100 μm.
Livers and spleens were harvested at 24 hours after infection and stained with hematoxylin and eosin. The morphological structure of liver tissue samples exhibited obvious hepatocyte edema and vacuolar degeneration in the WT-infected group. Besides, the WT-infected group showed significant congestion in hepatic sinusoid and central vein. No obvious hepatocyte edema, vacuolar degeneration and congestion were observed in the Br-LED209-treated and the qseC mutant-infected groups (Fig. 2C). In spleen tissue, splenic corpuscles demolished and disappeared, red pulp widened and white pulp atrophied in the WT-infected group. Meanwhile, obvious congestion was observed in the red pulp and white pulp area of the WT-infected group. However, hyperemia of red pulp and white pulp was drastically reduced in the Br-LED209-treated group and the qseC mutant-infected group (Fig. 2C).
Swimming motility of S. Typhimurium was inhibited by blocking QseC
Flagella-mediated motility is fundamental to S. Typhimurium pathogenesis. For this reason, expression of the flhDC gene, which encodes the master regulator of flagellum biosynthesis, was evaluated by quantitative real-time PCR. Data were normalized to levels of rpoA and were calculated as fold changes compared to the WT group. The results showed that flhDC expression in S. Typhimurium was dramatically decreased in the presence of Br-LED209 at 200 μM and also the qseC mutant compared with the WT strain (Fig. 3A). Next, a motility assay was performed to examine whether swimming motility of S. Typhimurium was affected. The diameter of motility halos was significantly reduced in the presence of Br-LED209 at 200 μM and also in the qseC mutant compared with the WT strain (Fig. 3B,C). These motility experiments were performed in triplicate, and the halo diameters reflect the average of these experiments. These results suggested that motility inhibition of S. Typhimurium was related to QseC blockade.
Figure 3. The swimming motility of S. Typhimurium was inhibited by blocking QseC.

(A) Expression of the mobility-related gene flhDC was evaluated. RNA was extracted from an overnight culture grown aerobically in LB medium in the absence or presence of Br-LED209. Real-time reverse transcription (RT)-PCR was performed. The rpoA gene was used as the endogenous control. Data were normalized to levels of rpoA and were calculated as fold changes compared to the WT group. (***P < 0.001 vs. WT in one-way ANOVA, n = 3) (B,C) Swimming motility assay. About 1 μl of WT bacteria or the qseC mutant was spotted onto agar plates with or without 200 μM Br-LED209, and halo sizes were measured at 6, 12 and 18 hours after incubation at 37 °C. (*P < 0.05, ***P < 0.001 vs. WT; #P < 0.05 vs. WT+Br-LED209 in two-way ANOVA, n = 3).
The invasion and replication capacities of S. Typhimurium were inhibited by blocking QseC
To evaluate the invasion and replication capacity of S. Typhimurium, the expression of invasion- and replication-associated genes sopB and sifA in S. Typhimurium were measured by quantitative real-time PCR. Data were normalized to levels of rpoA and calculated as fold changes compared with the WT group. The results showed that QseC blockade inhibited the expression of sopB and sifA in S. Typhimurium (Fig. 4A,B). To further investigate the down-regulation of invasion-associated genes in response to QseC blockade, the invasion capacity and replication capacity of S. Typhimurium on epithelial cells were investigated. HeLa cells were infected with WT S. Typhimurium or the qseC mutant at the MOI of 50:1 for 1, 6, or 12 h. The cells were lysed with 1% Triton X-100, and the lysates were diluted and plated on agar plates to determine the CFU. Fold-increase was calculated as a ratio of the intracellular bacteria count at 6 and 12 hours to that at 1 hour. The results demonstrated that QseC blockade significantly decreased the numbers of intracellular bacteria at each time point (Fig. 4C,D), suggesting that the invasion and replication capacities of S. Typhimurium were inhibited by blocking QseC.
Figure 4. The invasion and replication capacities of S. Typhimurium were inhibited by blocking QseC.

(A,B) Expression of the invasion- and replication-related genes sopB and sifA was evaluated in S. Typhimurium. RNA was extracted from an overnight culture grown aerobically in LB medium in the absence or presence of Br-LED209, and RT-PCR was performed. The rpoA gene was used as the endogenous control. Data were normalized to levels of rpoA and were calculated as fold changes compared to the WT group. (***P < 0.001 vs. WT in one-way ANOVA, n = 3) (C) HeLa cells were infected with S. Typhimurium for 1 h. The cells were treated with 100 μg/ml gentamicin for 1 h to kill extracellular bacteria. Then the cells were lysed and bacteria were diluted and plated on LB medium plates to determine the number of CFU. (***P < 0.001 vs. WT in one-way ANOVA, n = 3) (D) After treatment with 100 μg/ml gentamicin to kill extracellular bacteria, the cell culture medium was replaced with 10 μg/ml gentamicin for the remainder of the experiment. Cells were lysed at the indicated time points and intracellular bacteria were counted. Fold-increase was calculated as the ratio of intracellular bacterial count at 6 and 12 hours to that 1 hour. (***P < 0.001 vs. WT in two-way ANOVA, n = 3).
Inflammasome activation and pyroptosis of infected macrophages was inhibited by blocking QseC
Br-LED209 did not kill S. Typhimurium in vitro (Fig. 1), but eliminated them from the internal organs of infected mice (Fig. 2). We hypothesized that the clearance of bacteria from infected mice by Br-LED209 might be mediated by antimicrobial innate immune response. Macrophages play crucial role in controlling S. Typhimurium infection. Therefore, we investigated whether the function of macrophages was affected by S. Typhimurium infection. Overnight culture of S. Typhimurium were diluted at 1:50 into fresh LB and grown for 3–4 h at 37 °C before the infection. Macrophages were infected with WT S. Typhimurium or the qseC mutant at MOI of 25:1. Images of macrophages were taken under a light microscope at 1 h after infection. The images showed that a high percentage of macrophages infected with S. Typhimurium were swollen, and many of the infected cells had died at 1 h after infection. In the Br-LED209-treated group and the qseC mutant-infected group, macrophages were protected and no significant cell death was observed (Fig. 5A). LDH released in the supernatant was detected 1 h after infection using a cytotoxicity detection kit. Results showed that the amount of released LDH decreased significantly after QseC blockade (Fig. 5B). To further confirm these results, we stained the infected macrophages with DAPI and PI and calculated the percentage of PI-positive cells by counting cell numbers in four random visual fields. We detected strong PI signals in S. Typhimurium-infected macrophages. QseC blockade significantly reduced the number of PI-positive cells (Fig. 5C,D). The inflammasome can recognize invading pathogens, activate caspase-1 and subsequently trigger inflammatory cell death known as pyroptosis. To further confirm the manner of death of infected macrophages, the expression of activated caspase-1 and IL-1β were detected by Western blot. Data showed that QseC blockade significantly suppressed the production of the caspase-1 p10 and IL-1β p17 forms of macrophages (Fig. 5E), which indicated that inflammasome activation had been significantly inhibited and macrophage pyroptosis had been prevented.
Figure 5. Inflammasome activation and pyroptosis of infected macrophages was inhibited by blocking QseC.

(A) S. Typhimurium were grown overnight in LB and subsequently diluted at 1:50 into fresh LB and grown for 3-4 h at 37 °C. Macrophages were infected with WT S. Typhimurium or the qseC mutant diluted in fresh DEME at MOI of 25:1. Morphological characteristics of macrophages after S. Typhimurium infection. Light microscope images were taken at 1 hour post-infection. Scale bars: 20 μm. (B) LDH released from S. Typhimurium-infected macrophages was measured. The pyroptotic rate of macrophages was calculated based on the equation in methods. (***P < 0.001 vs. WT in one-way ANOVA, n = 3) (C,D) Membrane permeability of macrophages after S. Typhimurium infection. Macrophages were stained with DAPI and the membrane impermeant dye propidium iodide (PI). Scale bars: 20 μm. The percentage of PI-positive cells was determined by counting cell numbers in four random visual fields. (***P < 0.001 vs. WT in one-way ANOVA) (E) Macrophages were infected with S. Typhimurium. Activated caspase-1 and IL-1β were precipitated from macrophage supernatants and detected by Western blot.
Elimination of intracellular bacteria from macrophages was enhanced by blocking QseC
Next, the capacity of the macrophages to kill bacteria was evaluated. Since inflammasome-activating S. Typhimurium induced rapid macrophage pyroptosis within 1 hour of infection (Fig. 5), intracellular bacterial burden could not be measured under this condition. Thus, another condition was used to study macrophage-killing capacity according to many previously published studies19,20,21. S. Typhimurium were cultured overnight and opsonized in DMEM containing 10% normal mouse serum for 20 min before infection. The numbers of intracellular vital bacteria were counted at 1, 8, and 16 hours after infection. The data showed no difference in the number of recovered bacteria among the different treatment groups at one hour after infection. However, the number of bacteria recovered from macrophages significantly decreased after Br-LED209 treatment or infection with qseC mutant bacteria at 8 and 16 hours after infection (Fig. 6). The results indicated that QseC blockade could promote the clearance of intracellular Salmonella by inhibiting intracellular replication of bacteria and increasing the killing capacity of macrophages.
Figure 6. Elimination of intracellular bacteria from macrophages was enhanced by blocking QseC.
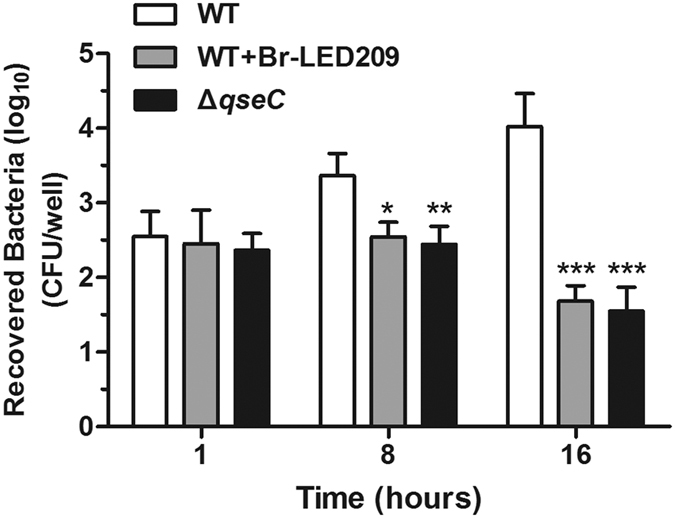
S. Typhimurium were cultured at 37 °C with shaking overnight and opsonized in DMEM and 10% normal mouse serum for 20 min. Macrophages were infected with opsonized WT S. Typhimurium or the qseC mutant at MOI of 25:1 for 30 min. The cells were washed with PBS and treated with 100 μg/ml gentamicin for 1 h to kill extracellular bacteria. Then, cell culture medium was replaced with 10 μg/ml gentamicin for the remainder of the experiment. To count the number of intracellular bacteria, macrophages were lysed with 1% Triton X-100. Bacteria were diluted and plated on LB medium plates to calculate CFU at the indicated time points. (*P < 0.05, **P < 0.01, ***P < 0.001 vs. WT in two-way ANOVA, n = 3).
Discussion
QseC is a promising target for developing broad-spectrum antimicrobials because of its broad distribution and high degree of conservation among different bacterial species22. LED209 is a potent QseC inhibitor that blocks the expression of virulence genes and suppresses the pathogenicity of Gram-negative bacteria8,22. Thus, LED209 is a promising antimicrobial agent. However, LED209 only exerts protective effects against S. Typhimurium infection in a mouse model and does not affect bacterial growth in vitro. The mechanism underlying this phenomenon remains unclear.
In this study, Br-LED209, which is a LED209 derivative, was synthesized to explore the mechanism of its protective effects in vivo. The efficacy of Br-LED209 at inhibiting QseC was then evaluated in vitro and in vivo. Results showed that Br-LED209 did not kill S. Typhimurium in vitro (Fig. 1). However, bacterial burden in the liver and spleen of infected mice was reduced significantly and pathological damage of these infected organs was alleviated after Br-ED209 treatment in vivo (Fig. 2). These results indicated that Br-LED209 retained its ability to block QseC.
QseC regulates multiple virulence factors involved in the pathogenesis of S. Typhimurium. As such, downstream genes regulated by QseC would be affected if the protective effects of Br-LED209 were mediated by blocking QseC. The flhDC gene encodes the master regulator of flagellum biosynthesis in S. Typhimurium, and regulates bacterial motility and facilitates bacterial invasion23. The sopB gene encoding SopB participates in the invasion of nonphagocytic cells, early maturation of Salmonella-containing vacuole (SCV), regulation of SCV trafficking, and inhibition of SCV–lysosome fusion24,25. The sifA gene is required for SCV membrane integrity and SCV maintenance. This gene permits the survival and replication of S. Typhimurium in macrophages and inhibits lysosome function20,26. All of these three genes are tightly regulated by QseC and are particularly important in S. Typhimurium pathogenesis. Therefore, the expression and virulence of these genes were investigated. Results indicated that QseC blockade significantly inhibited the expression of QseC-regulated virulence genes including flhDC, sifA, and sopB in S. Typhimurium. Moreover, QseC blockade suppressed flagellar motility as well as the invasion and replication capacities of S. Typhimurium in epithelial cells (Figs 3 and 4).
Macrophages are key innate immune cells that play a critical role in controlling bacterial infection. Inflammasome signaling of macrophages and caspase-1-induced pyroptotic cell death are innate immune effector mechanisms against many bacterial species11,27,28,29,30. Pyroptosis removes the replication niche of intracellular bacteria and promotes bacterial clearance through the NADPH oxidase system of recruited neutrophils11. Previous studies have reported that caspase-1 activation clears pathogens such as Salmonella28, Shigella29, and Legionella30, and controls infection.
However, systemic or excessive activation of inflammasome signaling and pyroptosis in vivo may be deleterious to bacterial clearance12,13. For example, Shigella infection activated NLR inflammasomes, promoted macrophage cell death, and secured its own dissemination31. In a murine model of acute pneumonia, pyroptosis of alveolar macrophage by inflammasome signaling induction impaired Pseudomonas aeruginosa clearance and increased mortality. Conversely, restricting inflammasome activation enhanced bacterial clearance and decreased pathology32. NLRC4 inflammasome-induced pyroptotic signaling have been reported to cause enhanced vascular permeability, septic shock, and rapid death in mice after the cytosolic delivery of bacterial flagellin33. Ayres et al. found that inflammasome activation in a mouse model of Escherichia coli resulted in IL-1β–driven lethal immunopathology, which could not be tolerated by the host34. Inhibiting inflammasome signaling and pyroptosis by caspase-1 specific inhibitors or caspase-1 and NLRC4 knockout also significantly alleviated tissue damage, decreased bacterial burden, and increased the survival rate of infected mice32,33,34,35.
Many studies have proposed that pyroptosis may benefit the host during infection but may be detrimental during overwhelming infection or sepsis12,13. At the early onset of infection, bacteria hide in macrophages and replicate themselves until bacterial load is sufficient to subvert immune defense. Under such condition, pyroptosis is beneficial to the host because bacteria will be released from macrophages and subsequently killed by neutrophils. However, during overwhelming infection, inflammasome-induced pyroptosis releases a large number of pathogens from macrophages for dissemination throughout the host. The recruited neutrophils will fail to kill all bacteria and effectively control infection. Therefore, repeated rounds of pyroptosis can lead to severe inflammatory changes and significant damage to host tissues36.
Low bacterial quantities or localized infections induce inflammasome signaling and pyroptosis, and positively promote bacterial clearance11,28,29,30. High bacterial quantities or severe sepsis cause lethal inflammasome activation and pyroptosis31,32,34,37,38. In the present study, we performed intraperitoneal injection of 108 CFUs of S. Typhimurium into mice. Such a high bacterial load could lead to severe and overwhelming sepsis-like infection. QseC blockade significantly suppressed flagellar gene expression in S. Typhimurium and subsequently inhibited caspase-1 activation, IL-1β release, and macrophage pyroptosis (Fig. 5), promoted the antimicrobial activity of macrophages to eliminate intracellular bacteria (Fig. 6), and alleviated inflammatory damage of infected tissues (Fig. 2).
The defensive function of inflammasome activation and pyroptosis as well as the role of IL-1β in the clearance of microbial infections are controversial. Pyroptosis is a caspase-1-dependent form of programmed cell death. Caspase-1 has a critical function in the cleavage of pro-IL-1β and in triggering inflammatory response39. IL-1β may induce the upregulation of adhesion molecules in the endothelium to mediate neutrophil recruitment and clear bacteria40. However, IL-1β is one of the major proinflammatory cytokines in sepsis41,42. While a low amount of these cytokines is critical for cell-mediated bacterial killing, excessive production of cytokines can lead to severe immunopathology43,44.
In summary, over-activation of inflammasomes can induce excessive macrophage pyroptosis and severe tissue damage. Our results demonstrated that QseC blockade significantly inhibited the pyroptosis of infected macrophages and improved the clearance of S. Typhimurium from infected macrophages.
Additional Information
How to cite this article: Li, Z. et al. Pyroptosis of Salmonella Typhimurium-infected macrophages was suppressed and elimination of intracellular bacteria from macrophages was promoted by blocking QseC. Sci. Rep. 6, 37447; doi: 10.1038/srep37447 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos 81273555, 81471997, 81473252 and 81673477).
Footnotes
Author Contributions Z.L., Z.H. and X.X.L. designed the experiments; X.S. synthesized and characterized the compounds; Z.L., Q.Z., X.Y.X., Y.Z. F.D. and D.Q. performed the experiments and analysed the data; and X.X.L., Z.H. and Z.L. wrote the paper. All authors reviewed the manuscript.
References
- Organization W. H. Antimicrobial Resistance: Global Report on Surveillance 2014. http://www.who.int/drugresistance/documents/surveillancereport/en/ (2014). doi:10.1038/509S2a (2014).
- Hede K. Antibiotic resistance: An infectious arms race. Nature 509, S2–S3, doi: 10.1038/509S2a (2014). [DOI] [PubMed] [Google Scholar]
- Walsh T. R. & Toleman M. A. The emergence of pan-resistant Gram-negative pathogens merits a rapid global political response. J Antimicrob Chemother 67, 1–3, doi: 10.1093/jac/dkr378 (2012). [DOI] [PubMed] [Google Scholar]
- Hawkey P. M. Multidrug-resistant Gram-negative bacteria: a product of globalization. J Hosp Infect 89, 241–247, doi: 10.1016/j.jhin.2015.01.008 (2015). [DOI] [PubMed] [Google Scholar]
- Kinch M. S., Patridge E., Plummer M. & Hoyer D. An analysis of FDA-approved drugs for infectious disease: antibacterial agents. Drug Discov Today 19, 1283–1287, doi: 10.1016/j.drudis.2014.07.005 (2014). [DOI] [PubMed] [Google Scholar]
- Piddock L. J. The crisis of no new antibiotics--what is the way forward? Lancet Infect Dis 12, 249–253, doi: 10.1016/S1473-3099(11)70316-4 (2012). [DOI] [PubMed] [Google Scholar]
- Clarke M. B., Hughes D. T., Zhu C., Boedeker E. C. & Sperandio V. The QseC sensor kinase: a bacterial adrenergic receptor. Proc Natl Acad Sci USA 103, 10420–10425, doi: 10.1073/pnas.0604343103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko D. A. et al. Targeting QseC signaling and virulence for antibiotic development. Science 321, 1078–1080, doi: 10.1126/science.1160354 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K. & Tschopp J. The inflammasomes. Cell 140, 821–832, doi: 10.1016/j.cell.2010.01.040 (2010). [DOI] [PubMed] [Google Scholar]
- Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol 11, 213–220, doi: 10.1038/nri2936 (2011). [DOI] [PubMed] [Google Scholar]
- Miao E. A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11, 1136–1142, doi: 10.1038/ni.1960 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aachoui Y., Sagulenko V., Miao E. A. & Stacey K. J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol 16, 319–326, doi: 10.1016/j.mib.2013.04.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I. & Miao E. A. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev 265, 130–142, doi: 10.1111/imr.12287 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira C. G., Weinshenker D. & Sperandio V. QseC mediates Salmonella enterica serovar typhimurium virulence in vitro and in vivo. Infect Immun 78, 914–926, doi: 10.1128/IAI.01038-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M. Collecting resident or thioglycollate-elicited peritoneal macrophages. Methods Mol Biol 1031, 37–40, doi: 10.1007/978-1-62703-481-4_4 (2013). [DOI] [PubMed] [Google Scholar]
- Broz P., von Moltke J., Jones J. W., Vance R. E. & Monack D. M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483, doi: 10.1016/j.chom.2010.11.007 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao E. A. et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7, 569–575, doi: 10.1038/ni1344 (2006). [DOI] [PubMed] [Google Scholar]
- Miao E. A. et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107, 3076–3080, doi: 10.1073/pnas.0913087107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuzon C. R. et al. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 19, 3235–3249, doi: 10.1093/emboj/19.13.3235 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumell J. H., Rosenberger C. M., Gotto G. T., Marcus S. L. & Finlay B. B. SifA permits survival and replication of Salmonella typhimurium in murine macrophages. Cell Microbiol 3, 75–84 (2001). [DOI] [PubMed] [Google Scholar]
- Odendall C. et al. The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe 12, 657–668, doi: 10.1016/j.chom.2012.09.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis M. M. et al. QseC inhibitors as an antivirulence approach for Gram-negative pathogens. MBio 5, e02165, doi: 10.1128/mBio.02165-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecher B. et al. Motility allows S. Typhimurium to benefit from the mucosal defence. Cell Microbiol 10, 1166–1180, doi: 10.1111/j.1462-5822.2008.01118.x (2008). [DOI] [PubMed] [Google Scholar]
- Hernandez L. D., Hueffer K., Wenk M. R. & Galan J. E. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304, 1805–1807, doi: 10.1126/science.1098188 (2004). [DOI] [PubMed] [Google Scholar]
- Bakowski M. A. et al. The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe 7, 453–462, doi: 10.1016/j.chom.2010.05.011 (2010). [DOI] [PubMed] [Google Scholar]
- McGourty K. et al. Salmonella inhibits retrograde trafficking of mannose-6-phosphate receptors and lysosome function. Science 338, 963–967, doi: 10.1126/science.1227037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T., Fink S. L. & Cookson B. T. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7, 99–109, doi: 10.1038/nrmicro2070 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Tejero M. et al. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med 203, 1407–1412, doi: 10.1084/jem.20060206 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansonetti P. J. et al. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 12, 581–590 (2000). [DOI] [PubMed] [Google Scholar]
- Molofsky A. B. et al. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med 203, 1093–1104, doi: 10.1084/jem.20051659 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S. et al. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc Natl Acad Sci USA 111, E4254–E4263, doi: 10.1073/pnas.1324021111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen T. S. & Prince A. S. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest 123, 1630–1637, doi: 10.1172/JCI66142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J. et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111, doi: 10.1038/nature11351 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres J. S., Trinidad N. J. & Vance R. E. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med 18, 799–806, doi: 10.1038/nm.2729 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z. et al. Antimicrobial cathelicidin peptide LL-37 inhibits the pyroptosis of macrophages and improves the survival of polybacterial septic mice. Int Immunol 28, 245–253, doi: 10.1093/intimm/dxv113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. J. & Jo E. K. NLRP3 inflammasome and host protection against bacterial infection. J Korean Med Sci 28, 1415–1423, doi: 10.3346/jkms.2013.28.10.1415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raupach B., Peuschel S. K., Monack D. M. & Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun 74, 4922–4926, doi: 10.1128/IAI.00417-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A. et al. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am J Respir Crit Care Med 174, 1003–1010, doi: 10.1164/rccm.200604-546OC (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry N. A. et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774, doi: 10.1038/356768a0 (1992). [DOI] [PubMed] [Google Scholar]
- Franchi L. et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol 13, 449–456, doi: 10.1038/ni.2263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. G. et al. Circulating interleukin-1 and tumor necrosis factor in septic shock and experimental endotoxin fever. J Infect Dis 161, 79–84 (1990). [DOI] [PubMed] [Google Scholar]
- Girardin E., Grau G. E., Dayer J. M., Roux-Lombard P. & Lambert P. H. Tumor necrosis factor and interleukin-1 in the serum of children with severe infectious purpura. N Engl J Med 319, 397–400, doi: 10.1056/NEJM198808183190703 (1988). [DOI] [PubMed] [Google Scholar]
- Cook D. N., Pisetsky D. S. & Schwartz D. A. Toll-like receptors in the pathogenesis of human disease. Nat Immunol 5, 975–979, doi: 10.1038/ni1116 (2004). [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. Blocking IL-1 in systemic inflammation. J Exp Med 201, 1355–1359, doi: 10.1084/jem.20050640 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.