

Abstract
It is still controversial whether comprehensive genome screening of target molecules by next generation sequencing (NGS) is needed to increase clinical efficacy of investigational drugs or accelerate drug development, although several studies are being carried out. Therefore, we performed a prospective study to evaluate the feasibility of comprehensive gene screening in this setting. Our findings indicate that actionable alterations were identified in 45% of the analyzed patients, most frequently in those with breast cancer. Common actionable alterations were found in PIK3CA mutation, BRCA2 mutation, ERBB2 amplification, and CCND1 amplification. In total, 22% of the analyzed patients could be entered into phase I clinical trials, and 8% of them were treated with “matched” drugs. Among patients who received matched therapies, response and disease control rates were 33 and 78%, respectively. On the other hand, in the patients who received “non-matched” therapy, the objective response rate was 6%. We believe this data indicates that NGS-based molecular pre-screening is a potent platform for use before patient entry into phase I trials.
Electronic supplementary material
The online version of this article (doi:10.1186/s12943-016-0553-z) contains supplementary material, which is available to authorized users.
Keywords: Next-generation sequencing, Phase I trial, Molecular pre-screening, Molecular targeted therapy, Driver mutation
Findings
Background
Recent molecular characterization studies showed that many alterations occurred in oncogenes or tumor suppressor genes that might become therapeutic targets, for instance, PIK3CA [1], AKT1 [2], FGFR1 [3–5], and BRCA1/2 [6]. Screening for such targetable genetic alterations will aid in identifying subpopulations of patients who will benefit from specific treatments.
In phase I trials, information on genomic alterations in tumors is quite helpful to allow each patient entry to a suitable clinical trial, in which the molecular targeted drug is theoretically matched to the alterations. If we were to find super-responders in these trials, the genomic alterations would be recognized as specific biomarkers to predict the response to the investigational drug.
Therefore, we conducted a prospective cohort study to investigate the feasibility of NGS-based pre-screening to identify genomic alterations in patients considering entry into phase I clinical trials (Additional file 1: Supplementary Methods) [7–15]. We named this study the “Trial of Oncopanel for Introduction into Clinical Study-Phase 1 (TOPICS-1).”
Results
Registration and sequencing
From July 2013 to October 2014, 183 patients were recruited for the study (Fig. 1 and Additional file 2: Table S1). Fifty-two patients were omitted from sequencing analysis. The major reasons were low-quality DNA (22 patients) and insufficient tumor tissue (21 patients). Less common reasons included insufficient DNA quantities (3 patients) and other factors (6 patients) (Additional file 2: Table S1). The success rates were higher in surgical samples (93/125, 74.4%) than in biopsy samples (38/58, 65.5%). As a result sequencing was performed in 131 patients. The types of cancer were as follows: 35 breast (27%), 35 gastric (27%), 21 ovarian (16%), 12 lung (9%), 8 bile duct (6%), 5 cervical (4%), 4 thymic (3%), 3 endometrial (2%), 3 colon (2%), 2 sarcomas (2%), and 3 others (2%) (Fig. 1). Thirty (79%) of 38 biopsied samples and 83 (89%) of 93 surgical samples were primary tumor lesions. The other biopsy samples were derived from metastases in the liver (4 patients), lymph nodes (2 patients), and skin (2 patients) (Additional file 2: Table S1).
Fig. 1.
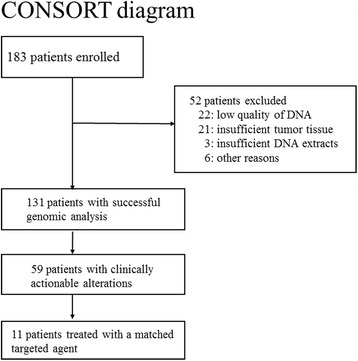
Registered and analyzed tumors in the TOPICS-1 study. The diagnoses of patients who underwent genomic testing. Each diagnosis included a variety of different histologic subtypes
Detected genomic alterations
We identified 1.42 mutations (Additional file 3: Table S5) and 0.26 amplifications (Additional file 4: Table S6) per patient. In addition, a fusion gene (CD74-ROS1) was found in 1 patient. Of the 131 patients, 104 (79%) had at least 1 genomic alteration, and 59 (45%) had at least 1 actionable genomic alteration (Additional file 5: Table S7). The most frequent genomic alterations were TP53 mutations (59 [46%] of 131 patients), PIK3CA mutations (15 patients [11%]), ERBB2 amplifications and mutations (12 patients [9%]), BRCA2 mutations (8 patients [6%]), CCND1 amplifications (8 patients [6%]), KRAS mutations (8 patients [6%]), MYC amplifications (7 patients [5%]), and ARID1A mutations (5 patients [4%]) (Fig. 2). Among them, we considered PIK3CA mutations, ERBB2 amplifications and mutations, BRCA2 mutations, and CCND1 amplifications as actionable. Other actionable alterations were less common, including BRCA1 mutations (4 patients [2%]), EGFR amplifications (2 patients [2%]), AKT1 mutations (2 patients [2%]), MDM2 amplifications (2 patients [2%]), FGFR1 amplifications (2 patients [2%]), and a ROS1 fusion (1 patient [1%]).
Fig. 2.
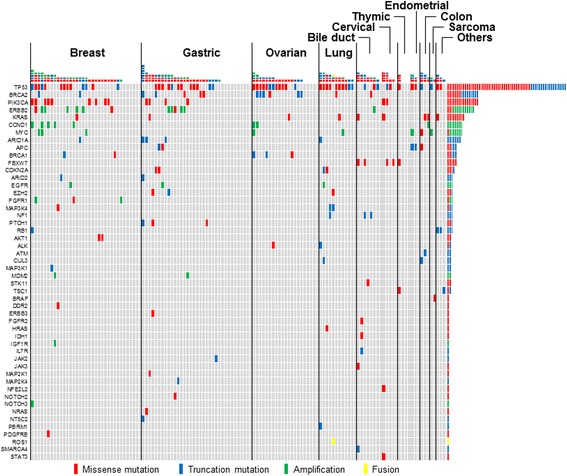
Summary of identified genomic alterations. The mutations, amplifications and fusions observed and reported in the study
Entry into phase I clinical trials
By June 2015, 29 (22%) of the 131 patients who underwent the sequencing test had entered phase I trials. Therefore, the primary endpoint, the inclusion of 25% patients in phase I trials, was not met. Seventeen different regimens were used in the 29 patients who entered phase I trials. Eleven (8% of the 131 patients) entered phase I trials of targeted therapies that matched their genomic alterations (Table 1). Forty-two of 59 patients who had actionable alterations could not enter accessible phase I trials because of ongoing standard treatment (18 patients), disease progression (18 patients), the patients’ wishes (4 patients), or no available matched phase I trials that targeted their actionable alterations (2 patients).
Table 1.
Characteristic of patient entering into Phase I with matched therapy
| No. | Cancer type | Molecular alteration | Matched therapy | Response | Progression free survival (month) |
|---|---|---|---|---|---|
| 1 | Bile duct | FGFR2 | FGFR inhibitor | PD | 0.95 |
| 2 | Cervical |
PIK3CA
E542K |
PI3K inhibitor | NE | 2.53 |
| 3 | Liver | TSC1 | mTOR inhibitor | NE | 0.82 |
| 4 | Breast | BRCA1 | PARP inhibitor | PR | 8.09 |
| 5 | Peritoneal | BRCA1 | PARP inhibitor | SD | 7.17 |
| 6 | Cervical |
PIK3CA
E542K |
AKT inhibitor | PD | 0.72 |
| 7 | Breast |
PIK3CA
H1047R |
PI3K inhibitor | PR | 6.18 |
| 8 | Breast |
PIK3CA
E545K |
PI3K inhibitor | SD | 2.80 |
| 9 | Breast |
PIK3CA
E545V |
PI3K inhibitor | SD | 5.72 |
| 10 | Breast |
AKT1
E17K |
AKT inhibitor | PR | 14.1 |
| 11 | Breast | BRCA1 | PARP inhibitor | SD | 5.53 |
PD progression disease, SD stable disease, PR partial response, NE not evaluable
Matched therapies and response
Seven different regimens were used in the 11 patients who received matched therapy. Two patients could not be assessed for antitumor activity. We evaluated response in the 9 assessable patients. Of them, 3 showed response and 4 had stable disease (Table 1). Partial response (PR) was seen in 3 patients, all responders had breast cancer. Case 1 was an ER-positive breast cancer patient with a PIK3CA H1047R mutation. She was treated with 5 kinds of hormonal therapy and 3 kinds of chemotherapy containing anthracycline and capecitabine after first relapse, and then received PI3K inhibitor treatment. PR began 60 days after first administration and lasted for 16 months. Case 2 was an ER-positive breast cancer patient with an AKT1 E17K mutation. She was treated with 6 kinds of hormonal therapy and 3 kinds of chemotherapy containing anthracycline and taxane, and then received AKT inhibitor treatment. PR began 60 days after first administration and lasted for 27 months. Case 3 was a triple-negative breast cancer patient with a BRCA1 truncation mutation. She experienced her first relapse after adjuvant chemotherapy containing anthracycline and taxane and then received combination therapy with a PARP inhibitor and eribulin. PR began 60 days after first administration and lasted for 6 months. The response and disease control rates were 33 and 78%, respectively. In contrast, the 18 patients who received non-matched therapy had response and disease control rates of only 6 and 56%, respectively. Moreover, we assessed progression free survival (PFS) (Table 1). Median PFS of the patients with matched therapy was longer than those with non-matched therapy (5.5 months, 95% CI; 2.1 to 9.0 vs. 1.9 months, 95% CI; 0.5 to 3.2) (Additional file 6: Figure S1).
Several trials, including SAFIR01 [NCT01414933] [7], NCI-MATCH [NCT02465060], MOSCATO01 and −02 trials [NCT01566019], SHIVA [NCT01771458] [16], PROFILER [NCT01774409], the EORTC SPECTA initiatives, are carried out to investigate the feasibility and utility of NGS-based screening. However, it is controversial for phase I trials, especially.
We demonstrate the feasibility of in-house, gene panel–based NGS screening for entry into phase I clinical trials for anti-cancer drugs. One of the distinctive features of this study is the customized assay design. Considering the flexibility of target genes, we adopted a custom gene panel consisting of 90 genes for mutations and amplifications and 10 genes for fusions (Additional file 7: Table S4). The analytical accuracy of this in-house system was validated. The second distinctive feature of this study is the use of formalin-fixed paraffin-embedded (FFPE) samples, which are easily available in clinical practice. The use of FFPE samples for sequencing creates the opportunity to characterize cancer-relevant genes even in cases where tissue retrieval is difficult. The quality of FFPE samples was related to fixation time and storage duration. To ensure stable sequencing, we changed the DNA amounts used for library preparation in response to the quality of the extracted DNA (Additional file 8: Table S2).
Genomic analysis was performed in 72% of the enrolled patients, and enabled matching of therapy in 8% of the patients in whom sequencing was performed. Twenty-two percent of the analyzed patients entered phase I trials after the sequencing test, although the primary endpoint was not met. This result was affected by patients’ performance status and the numbers of accessible phase I trials. However, it was feasible for the candidate patients to entry to phase I trials based on sequencing results. Moreover, genomic analyses led to PR in 33% and disease control in 78% of the patients who received matched therapy. The success rate of receiving matched therapy was consistent with other report [17]. On average, phase I trials show response rates between 5 and 10% [18–21]. In addition, in this study the response and disease control rates of genomic alteration–matched therapies were higher than those of non-matched therapies (33% versus 6%, and 78% versus 56%, respectively). Median PFS of the patients with matched therapy was longer than those with non-matched therapy (5.5 months, 95% CI; 2.1 to 9.0 vs. 1.9 months, 95% CI; 0.5 to 3.2). These results suggest the clinical utility of the sequencing test. The value of the sequencing test should increase if more predictive markers are defined or more novel targeted therapies are developed.
“Actionable genomic alterations” are a moving target. The evidence level of these alterations will probably change in the coming years as experimental agents move through the developmental pipeline. Many of the tumors that we tested harbored more than one potentially actionable alteration, but few treatment algorithms existed to stratify treatment options for these cases. In our original gene panel, target genes can be flexibly changed responding to the needs of the study (customized panel).
The current study involved 9 tumor types. The ratio of patients who were able to receive matched therapy was higher in breast cancer patients than those with other cancer types, and objective responses were observed only in breast cancer patients. The high efficacy in this population might be due to the higher frequency of driver mutations in breast cancer than in other tumor types (Additional file 9: Table S3). Moreover, it might be helpful that breast cancer is less aggressive and the tumor tissue is easy to access. Given our results, it is likely that the clinical utility of molecular prescreening differs among tumor types, and organ-specific screening might be useful.
Regarding future directions, we first need to determine the utility of small, organ-specific gene panels compared with the present pan-cancer gene panel. Second, we need to reconsider the timing of sequencing tests, such as perioperatively or at first recurrence. Third, to improve the accessibility of target drugs we need to construct a global social networking system that will allow patients to enter clinical trials.
Conclusions
This report showed that the NGS-based molecular screening was feasible in clinical setting and would be potentially useful for selecting adequate patients for entry into clinical studies. Since most recent phase I trials have tried to identify early signals regarding the efficacy of targeted agents, there is a strong rationale for proposing molecular selection for patients eligible for these studies.
Acknowledgments
The authors acknowledge Yutaka Fujiwara, Shunsuke Kondo, Shigehisa Kitano, Akihiko Shimomura, Tatsunori Shimoi, Hidenori Mizugaki, Emi Kubo, Keiko Goto, Nobuyoshi Hiraoka, Koji Tsuta, Akihiko Yoshida, Masayuki Yoshida, Hirokazu Taniguchi, Chikako Shimizu, Kan Yonemori, Harukaze Yamamoto, Makoto Kodaira, Chigusa Morizane, Yasuhide Yamada, Tetuya Hamaguchi, Yoshitaka Honma, and Atsuo Takashima for their support with the clinical trial. We also thank Sachiyo Mitani, Shizuka Shinohara, and Erika Arakawa for the sequencing analysis, Isao Kurosaka for bioinformatics analysis, and Suga Yamagami and Nao Nakamura for helping with the data collection and manuscript revisions.
Funding
This study was supported by National Cancer Center Research and Development Funds (T.K.) (24-A-1 and 27-A-1), and Grant-in-aid from the Ministry of Health, Labour and Welfare, Japan for Accelerating Regulatory Science Initiative (YF). The National Cancer Center Biobank was supported by National Cancer Center Research and Development Funds (23-A-1 and 26-A-1).
Authors’ contributions
KT, YT, HI, TK and NY conceived and designed the experiments. HI, TK, HY, TK, KT, NY and YT performed the experiments. HI, TK, YF, TK and YT analyzed the data. MK performed bioinformatic analyses. YT, HI, MK and KT wrote the manuscript. HI, TK, HY, TK, SI, AO, NY and KT contributed reagents, material and analytical tools. All authors have read and approved the final manuscript.
Competing interests
N.Y. reports research grants from Quintiles Transnational Japan K.K., Astellas Pharma Inc., Chugai Pharmaceutical Co., Ltd, Eisai Co., Ltd, Taiho Pharmaceutical Co., Ltd, Bristol Myers Squibb, Novartis Pharma K.K., Daiichi-Sankyo Co., Ltd, Boehringer Ingelheim and Kyowa-Hakko Kirin Co., Ltd. Y.F. reports grants and other from Japan Agency for Medical Research and Development, grants and other from The Ministry of Health Labor and Welfare, Japan, during the conduct of the study; other from Astra Zeneca KK, other from Eisai Co., Ltd, other from Daiichi Sankyo Co., Ltd, grants from Taiho Pharmaceutical Co., Ltd, grants from Takeda Pharmaceutical Co., Ltd, grants and other from Chugai Pharmaceutical Co., Ltd, grants and other from Eli Lilly Japan KK, grants from Nippon Kayaku Co., Ltd, other from Yakult Honsha Co., Ltd, outside the submitted work. K.T. reports grants from Pfizer, Astra Zeneca K.K., MSD K.K. and Novartis Pharma K.K. The other authors have no conflicts of interest to declare.
Consent for publication
Written informed consent was obtained from the patients for publication of this letter.
Ethics approval and consent to participate
This study was performed in agreement with the Declaration of Helsinki, and approved by the Ethical Committee at National Cancer Center Hospital, Tokyo (JAPAN).
Additional files
Supplementary Methods. (DOCX 17 kb)
List of patients registered into the study. (XLSX 21 kb)
List of identified mutations after SNP elimination. (XLSX 27 kb)
List of identified amplifications. (XLSX 10 kb)
List of actionable genomic alterations. (XLSX 10 kb)
Progression free survival of matched and non-matched therapy. (TIF 111 kb)
Gene list of NCC oncopanel ver. 2. (XLSX 10 kb)
DNA quantities used for sequencing library preparation. (XLSX 9 kb)
Actionable genomic alterations in histological tumor types. (XLSX 10 kb)
Contributor Information
Yuko Tanabe, Email: ytanabe@toranomon.gr.jp.
Hitoshi Ichikawa, Email: hichikaw@ncc.go.jp.
Takashi Kohno, Email: tkkohno@ncc.go.jp.
Hiroshi Yoshida, Email: hiroyosh@ncc.go.jp.
Takashi Kubo, Email: tkubo@ncc.go.jp.
Mamoru Kato, Email: mamkato@ncc.go.jp.
Satoru Iwasa, Email: siwasa@ncc.go.jp.
Atsushi Ochiai, Email: aochiai@east.ncc.go.jp.
Noboru Yamamoto, Email: nbryamam@ncc.go.jp.
Yasuhiro Fujiwara, Email: yfujiwar@ncc.go.jp.
Kenji Tamura, Phone: +81-3-3542-2511, Email: ketamura@ncc.go.jp.
References
- 1.Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, et al. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle. 2009;8(9):1352–8. doi: 10.4161/cc.8.9.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 3.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–29. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 4.Lieu C, Heymach J, Overman M, Tran H, Kopetz S. Beyond VEGF: inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin Cancer Res. 2011;17(19):6130–9. doi: 10.1158/1078-0432.CCR-11-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25(3):552–63. doi: 10.1093/annonc/mdt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welcsh PL, Owens KN, King MC. Insights into the functions of BRCA1 and BRCA2. Trends Genet. 2000;16(2):69–74. doi: 10.1016/S0168-9525(99)01930-7. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.1000 genomes browsers. http://www.1000genomes.org. Accessed 17 Oct 2016.
- 13.NHLBI Exome Sequencing Project (ESP). Exome Variant Server. http://evs.gs.washington.edu/EVS/.
- 14.Human Genetic Variation. http://www.genome.med.kyoto-u.ac.jp/SnpDB.
- 15.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Le Tourneau C, Delord JP, Gonçalves A, Gavoille C, Dubot C, Isambert N, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–34. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 17.André F, Bachelot F, Commo F, Campone M, Arnedos M, Dieras V, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER) Lancet Oncol. 2014;15(3):267–74. doi: 10.1016/S1470-2045(13)70611-9. [DOI] [PubMed] [Google Scholar]
- 18.Estey E, Hoth D, Simon R, Marsoni S, Leyland-Jones B, Wittes R. Therapeutic response in phase I trials of antineoplastic agents. Cancer Treat Rep. 1986;70(9):1105–15. Accessed 17 Oct 2016. [PubMed]
- 19.Horstmann E, McCabe M, Grochow L, Yamamoto S, Rubinstein L, Budd T, et al. Risks and benefits of phase 1 oncology trials, 1991 through 2002. N Engl J Med. 2005;352(9):895–904. Accessed 17 Oct 2016. [DOI] [PubMed]
- 20.Smith TL, Lee JJ, Kantarjian H, Legha SS, Raber MN. Design and results of phase I cancer clinical trials: three-year experience at M.D. Anderson Cancer Center. J Clin Oncol. 1996;14(1):287–95. Accessed 17 Oct 2016. [DOI] [PubMed]
- 21.Sekine I, Yamamoto N, Kunitoh H, Ohe Y, Tamura T, Kodama T, et al. Relationship between objective responses in phase I trials and potential efficacy of non-specific cytotoxic investigational new drugs. Ann Oncol. 2002;13(8):1300–6. doi: 10.1093/annonc/mdf202. [DOI] [PubMed] [Google Scholar]