

Abstract
The utilization of substoichiometric amounts of commercially available nickel(II) triflate as an activator in the reagent-controlled glycosylation reaction for the stereoselective construction of biologically relevant targets containing 1,2-cis-2-amino glycosidic linkages is reported. This straightforward and accessible methodology is mild, operationally simple and safe through catalytic activation by readily available Ni(OTf)2 in comparison to systems employing our previously in-house prepared Ni(4-F-PhCN)4(OTf)2. We anticipate that the bench-stable and inexpensive Ni(OTf)2, coupled with little to no extra laboratory training to set up the glycosylation reaction and no requirement of specialized equipment, should make this methodology be readily adopted by non-carbohydrate specialists. This report further highlights the efficacy of Ni(OTf)2 to prepare several bioactive motifs, such as blood type A-type V and VI antigens, heparin sulfate disaccharide repeating unit, aminooxy glycosides, and α-GalNAc-Serine conjugate, which cannot be achieved in high yield and α-selectivity utilizing in-house prepared Ni(4-F-PhCN)4(OTf)2 catalyst. The newly-developed protocol eliminates the need for the synthesis of Ni(4-F-PhCN)4(OTf)2 and is scalable and reproducible. Furthermore, computational simulations in combination with 1H NMR studies analyzed the effects of various solvents on the intramolecular hydrogen bonding network of tumor-associated mucin Fmoc-protected GalNAc-threonine amino acid antigen derivative, verifying discrepancies found that were previously unreported.
Graphical abstract

1. Introduction
Since Arthur Michael reported the first chemical glycosylation in 1879,1 the field of synthetic glycosciences has been continually striving to match nature’s ability to perform regio- and stereoselective glycosylations of complex oligosaccharides and glycoconjugates.2 It is well known that preparation of 1,2-trans-glycosides can be conveniently accessed in high selectivity through anchimeric assistance by a neighboring C(2)-ester protecting group.3 Unfortunately, no such facile technique exists for general formation of its 1,2-cis counterpart. The 1,2-cis glycosides, such as α-glucosides and β-mannosides, are present in a multitude of bioactive oligosaccharides and aim for their stereoselective construction pushes the forefront of chemical glycosylation reactions.4
In particular, 1,2-cis-2-aminoglycosides are ubiquitous in a range of naturally occurring oligosaccharides and glycoconjugates that play a role in many biological functions and human disease processes. Obtaining an adequate supply of these well-defined carbohydrate structures from natural sources can be challenging due to them being found in low concentrations and heterogeneous forms. Chemical glycosylation is one of the most reliable and efficient routes to obtain sufficient quantities of these desired bioactive molecules. For many current synthetic strategies, the stereochemical outcome of 1,2-cis-2-aminoglycoside formation is difficult to predict. Most of the current methodologies rely on the nature of the substrate’s protecting groups to control selectivity during formation of glycosidic bonds. In addition, glycosylation scenarios often require stoichiometric amounts of activating agents to sufficiently activate donors, resulting in excessive waste materials. Some of these activating agents can be air- and moisture-sensitive and must be used under anhydrous and low-temperature conditions, especially if glycosyl donors or acceptors incorporate acid-labile protecting groups. Amongst these methodologies, the two major categories utilize the non-assisting C(2)-azido or C(2,3)-oxazolidinone donors which rely on the substrate’s structural features and protecting groups to invoke a higher ratio of 1,2-cis-2-amino selectivity.2a,d,g,5
Recently, we have illustrated that under mild activation conditions C(2)-N-substituted benzylidenamino trihaloacetimidate donors could be highly stereoselective in the construction of many traditionally difficult 1,2-cis-2-aminoglycosides 2 (Scheme 1). We hypothesize that this is being achieved through a potentially reagent controlled pathway by activation with a substoichiometric amount of nickel.6 Evolution of this methodology over the years has seen the switch to N-phenyl trifluoroacetimidate donor 3 (both α- and β-anomers now capable of activation) as well as utilizing the inductive effects of the N-ortho-trifluoromethylbenzylidiene group which were able to provide increases in stability and ease of use and preparation of the donors, while maintaining the high α-selectivity of the desired glycoside products 4 (Scheme 1b).
Scheme 1.
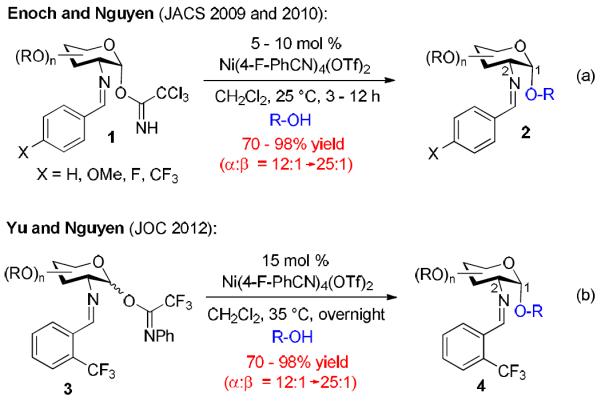
Evolution of the Nguyen nickel-catalyzed 1,2-cis-2-aminoglycosylation.
However, this system still relied on the non-commercially available, Ni(4-F-PhCN)4(OTf)2 catalyst which requires in-house production (Scheme 2a). This complexation reaction requires a prolonged reaction time (72 h), has variable yield (10-76%), and unless thoroughly dried, discrepancies in the selectivity can arise. During preliminary mechanistic elucidation,7 it was hypothesized that ligation by the para-fluorobenzonitrile was deemed unnecessary from that during the activation process both the nitrogen containing imidate and benzylidiene could potentially serve as temporary ligands to properly modulate the electronic nature of the nickel catalyst. In an effort to expand the scope of the potential users of this methodology to specialists and non-specialist alike, we have developed a glycosylation method utilizing Ni(OTf)2, which is readily available or can be quickly derived from commercially available NiCl2 and AgOTf (Scheme 2b). Herein, we report a straightforward and accessible methodology into the construction of 1,2-cis-aminoglycosides that is operationally simple and mild through the catalytic activation by a commercially available nickel catalyst, allowing for little to no extra laboratory training. The efficient access and simple operation of the readily available and inexpensive nickel triflate allows for a wider range of researchers to synthesize a variety of 1,2-cis-2-aminosugars in excellent yield and defined arrangement and addresses many of the significant synthetic limitations previously associated with the use of in-house prepared Ni(4-F-PhCN)4(OTf)2.
Scheme 2.

Simplification of nickel catalyst to expand the scope of potential users to non-specialists.
Furthermore, we envision that the use of the early transition-metal nickel in catalytic amount along with being at near room temperature will be very conducive with the synthesis of biological constructs as well as with the principles of green chemistry. As opposed to other late transition-metal activators of imidates [Pd(II), In(III), Au(I), Au(III), Ag(I), Sn(III)],8 nickel could be considered more biologically benign in such a substoichiometric amount in forming biological targets. For instance, nickel is incorporated into metalloenzymes that control the carbon cycle,9 as well as it is used for purification of peptides by ligation to histidine tags for affinity columns.10 Simple extract of nickel can be done through methods such as chelation by EDTA or by sorbent resins.
2. Results and discussion
2.1 Identification of Bench-Stable and Commercially Available Metal Catalysts
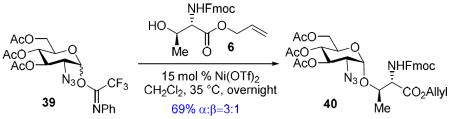
In recent years, we have demonstrated that Ni(4-F-PhCN)4(OTf)2 catalyst effectively promoted the α-coupling of acceptors with donors. However, due to unreproducible preparation and extensive manipulations of Ni(4-F-PhCN)4(OTf)2 prior to being used in the coupling event, many challenging 1,2-cis-2-aminoglycosides cannot be achieved in reproducible yield and α-selectivity. To address this unmet challenge, our goal is streamline and simplify the catalyst and procedure as much as possible without decreasing the reaction efficacy. We hypothesize that a simple and commercially available Ni(OTf)2 could be a catalyst of choice since both imidate and benzylidene nitrogens on glycosyl donor could serve as temporary ligands to modulate its reactivity. To validate our proposed hypothesis, we examined the formation of the simple glycoside 7.11 Since it was not known how the unligated version, Ni(OTf)2, would affect the yield and especially the α-stereoselectivity, a direct comparison of the glycosylation of Fmoc-protected threonine 6 with C(2)-N-ortho-(trifluoromethyl)benzylidene glucosamine donor 5 mediated by 15 mol% of both the ligated and unligated nickel(II) catalyst was carried out (entry 1 vs entry 2). We were delighted to find that both synthetically prepared ligated Ni(4-F-PhCN)4(OTf)2 and readily available unligated Ni(OTf)2 catalysts produced the desired coupled product 7 in similar yield, 66% (entry 1) and 69% (entry 2), and importantly the high α-selectivity remained unchanged (α:β = 9:1). These results validated our hypothesis. The ligated and unligated catalysts were both produced in situ by stirring their corresponding nickel chloride precursors along with silver triflate (2 equiv. relative to NiCl2) in CH2Cl2 for 30 min prior to the coupling event (Scheme 2). To determine the robustness of the in situ generation of Ni(OTf)2, the stirring of the NiCl2/AgOTf mixture was extended from 30 min to 24 h and then subjected to the same reaction conditions with donor 5 and acceptor 6. This in situ catalyst proceeded to form 7 with no change in either selectivity or yield (61%, α:β = 8:1). Since it has been previously shown that AgOTf is capable of activating acetimidate donors,8h,j to further demonstrate that Ni(OTf)2 is the actual activating agent required for the glycosylation to occur not the initial catalyst precursors, NiCl2 and AgOTf, two control reactions were performed (entries 3 and 4). Accordingly, treatment of donor 5 and amino acid residue 6 with NiCl2 and AgOTf individually provided the desired product 7 in very poor yield and selectivity (entries 3 and 4). It was not until a stoichiometric amount of AgOTf (entry 5) was utilized that the donor 5 was fully consumed after 16 h; however, a significant drop in α-selectivity (α:β = 9:1 → 2:1) was observed (entry 5). These control experiments exemplify the need for the nickel triflate catalyst in achieving α-selective coupling reaction. The unligated Ni(OTf)2 was also examined in the coupling of acceptor 6 with C(2)-azido version of donor 5, providing the desired coupling product with lower anomeric selectivity (α:β = 9:1 → 3:1), which corresponds to previous findings.12 The active catalyst, Ni(OTf)2, is commercially available from several different sources and air-stable, allowing it to be handled without the use of advanced inert gas techniques. When used in the coupling of 6 with 5, Ni(OTf)2 was able to promote formation of 1,2-cis-2-aminoglycoside 7 in similar yield and α-selectivity (entry 6) to the in situ version (entry 2). This further simplified the reaction to just a one-pot operationally simple technique. It is noted that discrepancies did arise in the yield when different batches of commercially available Ni(OTf)2 were used for this reaction and will be discussed later (vide infra, Table 2).
Table 2.
Discrepancies in yield based on variations in commercially acquired nickel triflate.
| Entry | Vendor | Lot # | Catalyst Color | Yield (%)a | α:βb |
|---|---|---|---|---|---|
| 1 | Strem | 24039600 | blue | 67 | 10:1 |
| 2 | Strem | 24039600 | green | 54 | 8:1 |
| 3 | Strem | 25453100 | tan | 16 | 7:1 |
Isolated by chromatography.
Calculated based on 1H NMR.
Several other commercially available metal triflate salts were also tested for facilitation of the desired α-aminoglycoside 7 (Table 1, entries 7–11). Previously, several of these catalysts were tried with the C(2)-N-para-(methoxy)benzylidene donor;13 however, due to the labile nature of the para-(methoxy)benzylidene group the glycosylation reactions only proceeded in low yield. Taking advantage of the switch to the more robust ortho-(trifluoromethyl)benzylidene donor, we hypothesize that these metal triflate catalysts could enable the coupling reaction. Although Cu(OTf)2 (entry 7) was able to proceed smoothly to provide the coupling product 7 in comparable yield (60%) and α-selectivity (α:β = 8:1), it did not provide the products with reproducible yield and selectivity in comparison to Ni(OTf)2.14 The use of higher valent and more Lewis acidic metals, including Fe(OTf)3, In(OTf)3, and Zn(OTf)2 (entries 9–11), resulted in lower yield of 7. These metals were also deliquescent, difficult to handle, and required handling in a glove box.
Table 1.
Initial catalyst screening in the streamline of metal-catalyzed 1,2-cis-2-amino glycosides.
| Entry | Catalyst | Mol % | Yield (%)b | α:βc |
|---|---|---|---|---|
| 1 | Ni(4-F-PhCN)4CI2/AgOTf | 15 | 66 | 9:1 |
| 2 | NiCI2/AgOTf | 15 | 69 | 9:1 |
| 3 | NiCI2 | 15 | 0 | 0 |
| 4 | AgOTf | 30 | 16 | 4:1 |
| 5 | AgOTf | 100 | 66 | 2:1 |
| 6d | Ni(OTf)2 | 15 | 67 | 10:1 |
| 7 | Cu(OTf)2 | 15 | 60 | 8:1 |
| 8 | Zn(OTf)2 | 15 | 45 | 7:1 |
| 9 | Fe(OTf)3 | 15 | 47 | 7:1 |
| 10e | Au(OTf)3 | 15 | 70 | 7:1 |
| 11 | In(OTf)3 | 15 | 53 | 8:1 |
All reactions were conducted with 0.08 mmol of glycosyl donor 5.
Isolated by chromatography.
Calculated based on 1H NMR.
For additonal details see Table 2.
Made in situ with 15 mol % AuCl3 and 30 mol % AgOTf.
Furthermore, through the screening process it was found that the glycosylation was tolerant of small amounts moisture and required the use of 4Å MS only during times of high humidity, to help suppress formation of the anomeric hydrolyzed product. Due to concerns with the stability of the C(2)-N-benzylidene group on glycosyl donors, we established that buffering silica gel with 1% triethylamine in the mobile phase prevents the potential removal of the benzylidene group. The system was then applied to the purification of the 1,2-cis-2-amino glycoside products and also allowed trivial separation of their α- and β-anomers. This standard purification procedure is not, however, suitable for isolation of 7 because the threonine Fmoc protecting group was partially removed. This obstacle was solved by addition of toluene as a co-solvent. We hypothesized that due to the higher boiling point of toluene (bp = 110 °C), triethylamine (88 °C) was removed first before it could reach concentration levels high enough to cleave the Fmoc group (see the SI for details).
As noted above, it was determined that utilization of commercially available Ni(OTf)2 resulted in inconsistencies in the yield when different bottles were used (Table 2). These variances could arise from batch-to-batch variation and purity of Ni(OTf)2. As illustrated in Table 2, when three different bottles of Ni(OTf)2 were used in the coupling of acceptor 6 with donor 5 the yield of the 1,2-cis-2-aminoglycoside 7 varied from 16 - 67% with the highest yield coming when Ni(OTf)2 was a blue color upon opening (67%, entry 1). In comparison, if the catalyst was a tan color there was a drastic decrease in the yield (16%, entry 3). In stark contrast, the in situ generation of Ni(OTf)2, from commercially available NiCl2 and AgOTf, resulted in far more reproducible and consistent results. Therefore, in situ generated Ni(OTf)2 became the catalyst of choice for the construction of several highly desired saccharide motifs containing 1,2-cis-2-amino glycosidic bonds.
2.2 Substrate Scope
The goal is to highlight the mild and operationally simple utility of available unligated Ni(OTf)2 for the construction of a number of biologically relevant 1,2-cis-2-aminoglycosides which could not be achieved previously in high yield and α-stereoselectivity with use of the synthetically-prepared ligated Ni(4-F-PhCN)4(OTf)2. For this purpose, only triacetyl protected glucosamine and galactosamine donors were utilized. Complete and systematic understanding of how various protecting groups affect the selectivity and yield is currently under investigation and will be reported in due course. In addition to glucosamine and galactosamine substrates, the utility and limitations of Ni(OTf)2 are currently being explored with a wide range of biologically relevant glycosyl donors such as fucosamine,15 quinovosamine,15cd,16 and 4-amino-galactosamine.15d,17
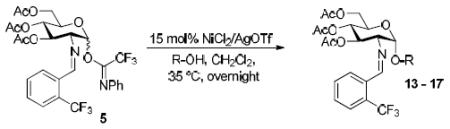
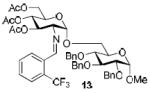
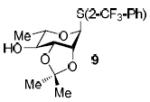
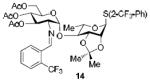
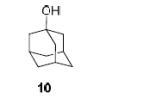
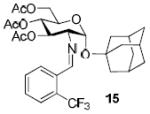
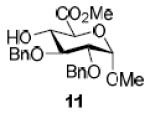
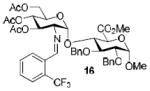
To demonstrate the utility of Ni(OTf)2 being able to efficiently promote glycosylations, primary and secondary carbohydrate acceptors 8 and 9 as well as the traditionally troublesome tertiary 1-adamantanol 10 (Table 3, entries 1-3) were examined with N-phenyl trifluoroacetimidate donor 5. The simple and unligated Ni(OTf)2 was found to be effective, providing the desired 1,2-cis-2-aminoglycosides 13-15 (entries 1-3) in high yield (85 - 93%) and with excellent α-stereoselectivity (α:β = 11:1 - α-only). These results are comparable to what was accomplished with use of the Ni(4-F-PhCN)4OTf2 catalyst.6b,c Next, we examined the efficacy of Ni(OTf)2 to mediate stereoselective formation of GlcNα(1→4)GlcA disaccharides 16 and 17 (entries 4 and 5), which are an important subunit of the anticoagulant heparin oligosaccharides2a and could not be prepared in high yield and α-selectivity with use of in-housed prepared Ni(4-F-PhCN)4(OTf)2. Accordingly, coupling of armed glucuronic acid acceptor 11 and disarmed acceptor 12 with glucosamine donor 5 to provide α-exclusive disaccharides 16 and 17, respectively, in moderate to good yield. One of the important features of the Ni(OTf)2-catalyzed chemistry is that the scope can be expanded to glucuronic acid thioglycoside acceptor 12 with no aglycon thiol transfer, allowing for subsequent iterative couplings with no anomeric protection group exchange. Due to the low nucleophilic nature of 12, the donor to acceptor ratio was switched to the acceptor as the limiting reagent in order to suppress the formation of the undesired elimination product.
Table 3.
Coupling of various glycosyl acceptors to C(2)-N-benzylidene substituted glucosamine donor 5.
| Entry | Acceptors | Products | Yieldd (α:βe) |
|---|---|---|---|
| 1a |
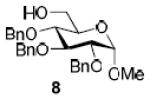
|
|
85%, 15:1 |
| 2b |
|
|
93%, α-only |
| 3a |
|
|
90%, 11:1 |
| 4a |
|
|
76%, α-only 71%, α-onlyf |
| 5c |
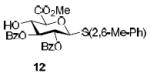
|
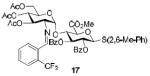
|
58%, α-only |
Donor/Acceptor = 1:1.2.
Donor/Acceptor = 1:2.
Donor/Acceptor = 2:1.
Isolated by chromatography.
Calculated based on 1H NMR.
Conducted with 15 mol % commercially available Ni(OTf)2 for 16 h.
Importantly, Ni(OTf)2 was successfully employed to catalyze the construction of two different complex blood group tetrasaccharide antigens 21 and 22 (Scheme 3), overcoming the limitations previously associated with use of in-house prepared Ni(4-F-PhCN)4(OTf)2 to generate these antigen motifs. Accordingly, glycosylation of the group H type-VI antigen trisaccharide acceptor 19 (1 equiv.) to N-substituted benzylidene galactosamine donor 18 (2 equiv.) provided the fully protected A type-VI antigen 21 in 63% yield and with α-only selectivity (Scheme 3a). Likewise, coupling of the H type-V antigen trisaccharide acceptor 20 with donor 18 in the presence of 15 mol % NiOTf2 resulted in a 59% yield of the protected tetrasaccharide blood group A type-V antigen 22 with complete α-selectivity (Scheme 3b). Similar yield and selectivity were achieved when 3 equivalents of the galactosamine azido donor and moisture-sensitive TMSOTf activating reagent were used in the coupling event.18
Scheme 3.

Ni(OTf)2-mediated formation of blood group tetrasaccharide antigens.
One of the major roadblocks in glycosciences is the lack of ability to obtain large quantities of well-defined oligosaccharide structures, from natural sources and/or synthesis, which is necessary to perform biological testing. One target of interest which has been prioritized in the top five cancer antigens is MUC1.19 This antigen is on the apical surface of epithelial cells; however, when a cell becomes cancerous MUC1 is highly overexpressed and aberrantly glycosylated. These new glycans are much smaller than that of non-tumor associated MUC1 glycans, typically shortened to just a simple α-GalNAc-Thr/Ser conjugate known as TN antigen. Due to the fact that this is exclusively expressed in carcinomas and found nowhere else, it has been seen to have potential as a biomarker or as a vaccine against several different types of cancer. Thus, the scalability of its production is of utmost importance.2d,20 Safety could become a major issue during the scale-up process with C(2)-azido donors. Synthesis of these donors requires the use of the TfN3 and when not handled properly can be highly explosive.21 Although we have illustrated that use of 15 mol % Ni(4-F-PhCN)4OTf2 was effective at safely providing the threonine-containing TN antigen derivative 23 in high yield and α-anomeric selectivity (Scheme 4a),6f the major drawback is to prepare Ni(4-F-PhCN)4OTf2 in reproducible and large quantities. Thus, our goal is to investigate the ability of Ni(OTf)2 to provide large quantities of 23 in a comparable yield and selectivity to that of Ni(4-F-PhCN)4OTf2. Accordingly, the reaction between galactosamine-derived donor 18 and Fmoc-protected threonine amino acid 6 was first trialed on the small scale (0.08 mmol) by both in situ generated and commercially available version of Ni(OTf)2 (Schemes 4b and 4c). These nickel catalysts were able to provide the desired aminoglycoside 23 in similar yield to that of the ligated version, Ni(4-F-PhCN)4OTf2 (Scheme 4a), although a slight drop in selectivity was observed (α-only → α:β =10:1 - 12:1). The reaction again proceeded smoothly when the scale was quadrupled to 0.32 mmol (Scheme 4d). These results demonstrate three important points: (1) the commercially available Ni(OTf)2 catalyst is much more reliable than the in-house prepared Ni(4-F-PhCN)4OTf2 counterpart for generating large quantities of 23; (2) the mild and operational simple Ni(OTf)2-catalyzed glycosylation procedure can be adopted by both specialist and nonspecialists; and (3) the C(2)-N-benzylideneamino donor is much more α-selective than the commonly used C(2)-azido donor for generating 1,2-cis-2-aminoglycoside. For instance, TMSOTf-mediated coupling with threonine acceptor 6 with the C(2)-azido galactose trichloroacetimidate donor provided the desired product in 55% yield with poor selectivity (α:β = 2:1) even with diethyl ether to serve as putative nucleophile.22 The threonine-containing compound 24 could be subsequently prepared according to literature6f or by our improved procedure (see Scheme 6).23 The synthesis of compound 24 would allow us to spectroscopically observe its structural variation in different deuterated solvents (vide infra, Fig.1).
Scheme 4.

Comparison studies: In-house prepared Ni(4-F-PhCN)4 (OTf)2 vs. readily available Ni(OTf)2
Scheme 6.

Scalable synthesis of TN antigen bearing serine amino acid.
Fig. 1.

The 1H NMR of GalNAc-Thr 24 in three different deuterated solvents (CDCl3, MeOD-d4, and DMSO-d6).
2.3 Synthetic Applications
Over the years there have been several elegant methodologies to integrate TN antigen into biological systems by means other than in peptide sequences. The major strategy involves conjugation of α-aminooxy glycoside 28 (Scheme 5) to an aldehyde or ketone unit on a scaffold. We question if Ni(OTf)2 could be utilized to promote high yielding and α-selective formation of 28, which could not be achieved in high yield and α-selectivity with use of in-house prepared Ni(4-F-PhCN)4OTf2 catalyst. Accordingly, synthesis of this glycoside was attempted under nickel-catalyzed conditions by the glycosylation of NHS ester 25 (2 equiv.) with galactosamine donor 18 (Scheme 5). The coupling proceeded smoothly to provide the desired α-aminooxy analogue 26 in 63% yield.24 Previous introduction of this type of α-aminooxy derivatives, utilizing C(2)-azido glycosyl donor, required 4 equivalents of NHS ester 25 to out compete the glycosylation by the more nucleophilic N-succinimide following activation of a thioglycoside with NIS.25 As well, the use of C(2)-azido halides resulted in 67% yield however with low selectivity (α:β = 3:1); α-only selectivity has been previously reported through the use of an expensive phase transfer catalyst and a difficult to fashion β-chloride.26 Subsequent conversion of 26 into N-acetylated 27 (81%) was achieved by removal of the benzylidene group followed by acetylation. Hydrolysis of the known compound 27 would reveal the free α-aminoxy glycoside 28.25,26 Andreana and coworkers were able to conjugate 28 to the modified version of immunostimulant PS A1 to produce 29 (Scheme 5) for use as potential vaccine.26e On other the hand, Bertozzi and coworkers conjugated 28 to a polymer scaffold containing pendant ketone to generate 30 (Scheme 5) for use to mimic the MUC1 mucin of epithelial cells.27
Scheme 5.

Ni(OTf)2-catalyzed formation of α-aminooxy glycoside.
The α-GalNAc-Serine derivative of mucin TN antigen is not as conformationally locked as its threonine counterpart.28 This freedom of rotation allows it to achieve stronger binding to potential lectins.28 Gram-scale synthesis of this potent cancer biomarker could allow for an effective therapeutic. Unfortunately, this freedom makes construction of the α-GalNAc-Ser compound more challenging, and typically lowering of the α-selectivity is often observed.29 Under Ni(4-F-PhCN)4OTf2-catalyzed conditions, coupling of serine residue 31 with galactosamine donor 18 provided the desired glycosyl amino acid 32 (Scheme 6) in unreliable yield and α-selectivity. To illustrate the reproducibility and scalability of the commercially available Ni(OTf)2 catalyst, coupling of 31 with 18 was first carried out in a small-scale to provide the desired glycosyl amino acid 32 in 68% yield with α:β = 7:1 (Scheme 6a). We then proceeded to scale up to one gram of donor 18 which produced 0.975 g of 32 in 76% yield and with α:β = 7:1 (Scheme 6b), validating the efficacy of Ni(OTf)2 for use in reproducible and large scale preparation. The aminoglycoside 32 was then subjected to traditional acidic conditions to remove the N-benzylidene group. This was first attempted with the in situ production of HCl from acetyl chloride in methanol, the optimal conditions used for the removal of the benzylidene group of glycosyl threonine amino acid 23 (Scheme 4).6f Unfortunately, these previous conditions are not suitable for glycosyl serine amino acid 32 because the use of methanol resulted in a transesterification reaction converting an allyl protected acid to a methyl protected carboxylic acid. This methyl ester byproduct came in a 1:1 ratio along with the desired allyl protected intermediate. To suppress transesterification, the bulkier 2-propanol (5 equiv.) was then investigated in combination with acetyl chloride (2.2 equiv.). The N-benzylidene group of 32 was cleanly removed under new conditions (Scheme 6). Without purifying the resulting amine salt intermediate was then dissolved in pyridine followed by addition of excess acetic anhydride (Scheme 6), and the corresponding N-acetylated 33 was produced in 70% over two steps. Removal of the allyl protecting group was done within 1 h using 10 mol % Pd(PPh3)4 with N-methyl aniline as the allyl scavenger.30 This resulted in the solid-phase-peptide synthesizer compatible 35 near quantitatively. Finally, serine-containing TN antigen 34 was obtained as the result of global hydrolysis of 33 (Scheme 6).
2.4 1H NMR Analysis of Threonine- and Serine-Containing Aminoglycosides:
Interestingly, when a 1H NMR analysis of 35 was conducted in CDCl3 it appeared that a nearly 1:1 mixture of two compounds were observed. This observation was far more distinct on the threonine-containing compound 24 (Fig. 1). At first, we speculated that the desired aminoglycoside 24 was epimerized. However, without the presence of a strong base it would be unlikely to happen under these conditions and has never been mentioned in the literature. On the other hand, when 24 was spectroscopically analyzed in either DMSO-d6 or MeOD the peaks merged into one compound (Fig. 1). The spectrum of 24 was also compared to that of known spectra of each GalNAc-Thr epimer. The chemical shifts of 24 in DMSO-d6 matched that of the GalNAc bearing the desired, natural l-threonine amino acid.31 The discrepancy between CDCl3 and DMSO-d6 spectra suggests the involvement of hydrogen bonding. We hypothesize that a 1:1 mixture of 24 observed in the 1H NMR spectrum is the result of two major hydrogen bonding conformations not being able to interconvert in CDCl3. This type of intramolecular hydrogen bonding is disrupted when 24 was spectroscopically analyzed in DMSO-d6 or MeOD, explaining why only one product was observed in 1H NMR analysis (Fig. 1).
To further confirm our hypothesis, a variable temperature from 25 to 45 °C of GalNAc-Thr compound 24 in CDCl3 was conducted (Fig. 2). As the temperature rises, the peaks slowly become broad and less distinct and begin to coalesce with the known compound peaks as the rate of exchange increases, including that of the anomeric hydrogen peak at 5.03 ppm and the methyl peak at around 1.25 ppm. This result suggests that there is actually only one pure compound. Unfortunately, complete coalescence could not be achieved due to low boiling point of chloroform. To the best of our knowledge, this phenomena has never been mentioned in the literature on this substrate.31,32 We believe that this transparent analysis would minimize confusion when non-specialists try to use one of the aforementioned methods for the construction of Fmoc-protected GalNAc-Ser/Thr compounds 24 and 35. These compounds are versatile building blocks required for the solid-phase peptide synthesis of tumor associated mucin-type glycopeptides.
Fig. 2.

Variable temperature 1H NMR experiment of GalNAc-Thr 24 in CDCl3 in which the temperature was raised from 25 °C to 45 °C.
2.5 Computational Analysis of Threonine-Containing Aminoglycosides in Various Solvents
To confirm that hydrogen bonding played important role in the aforementioned 1H NMR results (Fig. 1 and 2), we turned to computational analysis of GalNAc-Thr 24 in three solvent systems (chloroform, dimethyl sulfoxide, and methanol) to simulate the intramolecular movement of the hydrogen bond network over time. These solvents were chosen in particular due to their range of relative polarities from medium to very high. Upon initial mapping it was found that there were five major potential intramolecular hydrogen bonds that could occur between several donor (D-H) and acceptor (A) atoms on 24 (Fig. 3). We present the results of the interaction between each solvent system and the key hydrogen bonding (intra- and intermolecular) donor-acceptor pairs while comparing and contrasting the differences in the dihedral angle distribution of O1-C-C-NH of the threonine component of compound 24.
Fig. 3.

GalNAc-Thr molecule 24 with all the possible intramolecular hydrogen bonds. The Roman numerals show the numbering of the five H-bonds that we calculated in this molecule. The arrows indicate the hydrogen bonds with the arrow head pointing from the donor (D-H) to acceptor (A) (Oxygen = Red, Nitrogen = Blue, Carbon = Cyan, Hydrogen = Gray).
Overall, the analysis of the intramolecular hydrogen bonds as an evolution of time in these three solvents found that only three hydrogen bonds were of key significance. Specifically, hydrogen bonds between the C(2)-NH group of GalNAc sugar unit and the carboxylic acid oxygen of threonine (I, Fig. 3), the carboxylic acid and the carbonyl oxygen of the N-Fmoc protecting group on threonine (II, Fig. 3), and the C(2)-NH group and the carbonyl oxygen of the C(3)-acetyl group of GalNAc unit (III, Fig. 3). These hydrogen bonds (I, II, and III) were determined to be of significance based on the standard distances between the donor (D-H) and acceptor (A) atoms for strong and medium strength hydrogen bonds, established by Jeffrey and coworkers, being 2.2 - 2.5 and 2.5 - 3.2 Å, respectively.33 Although hydrogen bonds IV and V were observed in the simulation (Fig. 3), their greater distances (> 5 Å) were not relevant to this study. To determine how the solvent would interact/interfere with these hydrogen bonds of the solute, the distances between these donors and acceptors were simulated over 400 ns and plotted (Fig. 4). As chloroform is a relatively non-polar solvent, GalNAc-Thr compound 24 prefers intramolecular hydrogen bonding because there is no possibility of hydrogen bonds formed between the sugar and chloroform. Over time these three hydrogen bonds did not deviate significantly. In particular, H-bond II maintains a near distance of less than 3.2 Å. The result suggests that hydrogen bonding was strong and conformationally locked the molecule in CHCl3 (Fig. 4a). When the simulation was attempted in the more polar solvents, methanol and DMSO (Figs. 4b and 4c), which is in stark contrast to the results shown in t chloroform (Fig. 4a). Although all three of the same hydrogen bonds were observed, there was a greater fluctuation of the distances between the donor and acceptor atoms caused by competing intermolecular bonding with the solvent (Figs. 4b and 4c). An analysis of the solvation geometry for the hydrogen bonding in both DMSO and methanol illustrates that compound 24 is in hydrogen bonding with the solvent for >99% of time (See S1 for a representative snapshot of the solvated GalNAc-Thr by methanol).
Fig. 4.

Time vs. distance plot for GalNAc-Thr 24 in all three solvent systems, a) CHCl3, b) DMSO, and c) MeOH. Refer to Fig. 3 for the H-bond donor/acceptor pairs.
Normalized population distributions of the hydrogen bond distance vs time plots (Fig. 4) further confirmed that over time the probability of strong intramolecular hydrogen bonding occurring in chloroform is much larger than that of both methanol and DMSO (Fig. 5). Having weak intramolecular hydrogen bonds would lower the barrier of rotation around the bonds allowing for fast interconversion between conformations. This could explain why only one set of peaks in the 1H NMR spectrum was observed for MeOD-d4 and DMSO-d6, whereas in CDCl3 the strong hydrogen bonding locked into two separate conformations with no interconversion. Thus, the two blocked conformations explains why two sets of 1H NMR peaks were observed in CDCl3. Dihedral angle analysis for O1-C-C-NH of threonine that connects GalNAc to the Fmoc moiety is also in agreement with this statement (see the Supporting Information). In DMSO and methanol, dihedral angle of O1-C-C-NH freely shifts between three populations centered around −50, 80 and 170 degrees. On the other hand, in chloroform only two populations were centered around 80 and 170 degrees (Fig. S2). When thoroughly analyzing the population distributions of GalNAc-Thr 24 in chloroform, we hypothesize since there are two very distinct and similar populated regions (2.5 −3.2 Å and 4.2-5 Å) that hydrogen bond III plays a role in these two different conformations. Theoretically, there could be intramolecular hydrogen bonding competition for the C(2)-N-acetamide hydrogen by both the carbonyl of the free carboxylic acid and also the carbonyl of the C(3)-acetyl group.
Fig. 5.

Hydrogen bond distance population analysis for the three major hydrogen bonds of GalNAc-Thr 24 over 400 nanoseconds in all three solvent systems. Refer to Fig. 3 for the H-bond donor/acceptor pairs.
3. Conclusion
In summary, we have developed an operationally-simple and efficient approach to the synthesis of 1,2-cis-2-aminoglycosides in very high α-selectivity through the activation of N-phenyltrifluoroacetimidates by bench-stable and substoichiometric amounts of inexpensive and commercially available Ni(OTf)2. This simple protocol is water-tolerant, does not require specialized equipment, and is compatible with a wide variety of protecting groups. Importantly, we demonstrated that the Ni(OTf)2-mediated 1,2-cis-2-aminoglycosylation methodology is a straightforward approach to effortlessly and safely construct a number of highly α-selective saccharide targets, such as blood type A-type V and VI antigens, heparin sulfate disaccharide repeating unit, aminooxy glycosides, and GalNAc-Serine conjugate, which could not be easily achieved with use of in-house prepared Ni(4-F-PhCN)4(OTf)2. Moreover, under these nickel conditions the scale-up production of these targets transitioned smoothly. This methodology provides increased access to these targets as it requires minimal training and simple laboratory equipment so that potential users could be both specialist and non-specialists alike. Finally, discrepancies seen by 1H NMR in various solvents were computationally addressed as a way to solidify the purity and structure of the product by analyzing the solvent effect on intramolecular hydrogen bonding of the Fmoc-protected GalNAc Serine/Threonine compounds, one of the most important tumor-associated mucin antigens.
4. Experimental section
4.1 General methods
All reactions were performed in dried flasks fitted with septa under a positive pressure of nitrogen atmosphere unless otherwise noted. Organic solutions were concentrated using a Buchi rotary evaporator below 40 °C at 25 torr. Analytical thin-layer chromatography (TLC) was routinely utilized to monitor the progress of the reactions and performed using pre-coated glass plates with 230-400 mesh silica gel impregnated with a fluorescent indicator (250 nm). Visualization was achieved using UV light, iodine, or ceric ammonium molybdate stain. Flash column chromatography was performed using 40-63 μm silica gel (SiliaFlash® F60 from Silicycle). Dry solvents were obtained from a SG Waters solvent system utilizing activated alumina columns under an argon pressure. Anhydrous nickel(II) chloride, silver triflate, and nickel(II) triflate were purchased from Strem Chemicals Inc. and Sigma Aldrich Co. All other metal triflates, solvents, and commercial reagents were used as received from Sigma Aldrich, Alfa Aesar, Acros Organics, TCI and Combi-Blocks, unless otherwise noted. All new compounds were analyzed by NMR spectroscopy and High Resolution Mass spectrometry. All 1H NMR spectra were recorded on either Bruker 400 or 500 MHz spectrometers. All 13C NMR spectra were recorded on either Bruker 100 or 125 MHz NMR spectrometer. Chemical shifts are expressed in parts per million (δ scale) downfield from tetramethylsilane and are referenced to the residual proton in the NMR solvent (CDCl3: δ 7.27 ppm, δ 77.16 ppm; DMSO-d6: δ 2.50 ppm, δ 39.52 ppm; D2O: δ 4.79 ppm). Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and bs = broad singlet), integration, and coupling constant in hertz (Hz). Infrared (IR) spectra were reported in cm−1. High resolution (ESI) mass spectrometry was performed at the University of Iowa. Preparation of donors 5 and 18 and allyl-protected threonine and serine residues 6 and 31 as well as global hydrolysis were done according to our previous report.8f
4.2 General Glycosylation of Threonine/Serine Amino Acids Mediated by In Situ Generated Ni(OTf)2
4.2.1. Fmoc-protected threonine-(O-allyl) 3,4,6-tri-O-acetyl-2-deoxy-2-O-trifluoromethylbenzylideneamino-α-d-glucopyranoside (7)
First, a stock solution of active Ni(OTf)2 catalyst was prepared by charging a dried 10 mL reaction flask A with NiCl2 (4.5 mg, 0.036 mmol, 45 mol %) and AgOTf (18.3 mg, 0.071 mol, 90 mol %) under nitrogen. A reaction flask A was wrapped in aluminum foil since AgOTf is light-sensitive, and 1 mL of dry CH2Cl2 was then added under flowing nitrogen and quickly capped to prevent evaporation. This mixture was allowed to stir at room temperature for 30 min. Into a different dried 10 mL reaction flask B, d-glucososamine N-phenyl trifluoroacetimidate donor 5 (50 mg, 0.079 mmol, 1 equiv.) and threonine 6 (36.6 mg, 0.095 mmol, 1.2 equiv.) were charged under nitrogen and then dissolved in 0.7 mL of dry CH2Cl2. After the catalyst mixture in reaction flask A had been stirring for 30 min, a 1 mL plastic syringe fitted with a 20 gauge needle was inserted into the flask and to make sure there is high turbidity in the solution was extracted into the syringe serval times before finally withdrawing the desired amount [0.33 mL containing 15 mol % of the active Ni(OTf)2 catalyst]. This solution was then transferred into a reaction flask B under nitrogen. The resulting mixture was stirred under nitrogen at 35 °C overnight while covered in an aluminum foil tent. Reaction was monitored using TLC (5/1 toluene/acetonitrile, Rf = 0.25). Once complete, 0.5 mL of toluene was added to the reaction and then concentrated at room temperature until CH2Cl2 was completely removed. The remaining toluene containing the reaction mixture was then loaded directly on to a silica gel column and purified by flash chromatography (10 g of silica, 1/2in ID X 12in column, 5/1(180 mL)→3/1(200 mL)→2/1(200 mL) hexanes/ethyl acetate + 1% triethylamine). AXer purificaZon, the fractions containing the product were combined into a 250 mL round bottom flask, diluted with 50 mL toluene,7 and then concentrated using a Buchi rotary evaporator at room temperature until the solvent level reaches the previously marked 50 mL volume (at this point, all Et3N was removed). Once this solvent level was achieved, the solution was concentrated at 50 °C to provide the desired 1,2-cis-aminoglycoside 7 as a semi solid (44.9 mg, 69%, α:β = 9:1). 7α: 1H NMR (400 MHz, CDCl3): δ 8.65 (q, J = 2.4 Hz, 1H), 8.39 – 8.34 (m, 1H), 7.77 (d, J = 7.6 Hz, 2H), 7.66 – 7.61 (m, 3H), 7.46 – 7.42 (m, 2H), 7.39 (q, J = 6.9 Hz, 3H), 7.32 – 7.27 (m, 2H), 6.31 (d, J = 8.9 Hz, 1H), 5.77 – 5.59 (m, 2H), 5.18 – 5.08 (m, 3H), 4.99 (d, J = 3.5 Hz, 1H, H-1), 4.54 – 4.25 (m, 11H), 4.16 (d, J = 11.6 Hz, 1H), 3.64 (dd, J = 10.2, 3.5 Hz, 1H), 2.12 (s, 3H), 2.06 (s, 3H), 1.89 (s, 3H), 1.44 (d, J = 6.4 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 170.78, 170.28, 170.01, 170.00, 162.02, 157.01, 144.06, 143.89, 141.38, 141.34, 133.02, 132.39, 131.34, 131.06, 129.56, 129.29, 129.16, 128.85, 128.35, 127.81, 127.25, 125.80, 125.32, 122.85, 120.07, 120.05, 119.13, 99.61, 75.89, 72.91, 70.71, 68.94, 68.44, 67.71, 66.20, 62.34, 58.94, 47.30, 20.88, 20.84, 20.56, 19.50; HRMS (ESI): calc. for C42H43N2O12F3 (M + Na)+: 847.2666; found 847.2669. 7β: 1H NMR (400 MHz, CDCl3): δ 8.63 (q, J = 2.5 Hz, 1H), 8.10 (d, J = 7.5 Hz, 1H), 7.76 (d, J = 7.6 Hz, 2H), 7.70 (d, J = 7.4 Hz, 1H), 7.56 (dt, J = 15.7, 7.3 Hz, 4H), 7.39 (t, J = 7.4 Hz, 2H), 7.30 – 7.22 (m, 2H), 6.00 – 5.86 (m, 1H), 5.63 (d, J = 9.2 Hz, 1H), 5.46 (t, J = 9.6 Hz, 1H), 5.36 (dd, J = 17.2, 1.3 Hz, 1H), 5.26 (dd, J = 10.4, 1.1 Hz, 1H), 5.14 (t, J = 9.8 Hz, 1H), 4.82 (d, J = 7.8 Hz, 1H, H-1), 4.74 – 4.58 (m, 2H), 4.47 (qd, J = 6.2, 2.6 Hz, 1H), 4.42 – 4.30 (m, 4H), 4.23 (t, J = 7.3 Hz, 1H), 4.15 (dd, J = 12.2, 2.3 Hz, 1H), 3.81 (ddd, J = 10.0, 4.4, 2.4 Hz, 1H), 3.44 (dd, J = 9.8, 7.8 Hz, 1H), 2.10 (s, 3H), 2.06 (s, 3H), 1.94 (s, 3H), 1.19 (d, J = 6.4 Hz, 3H); 13C NMR (126 MHz, CDCl3): δ 170.61, 169.84, 169.80, 169.70, 161.64, 156.68, 143.93, 143.79, 141.26, 133.62, 132.22, 131.71, 130.69, 129.20 (q, 2JC-F = 31.0 Hz), 128.48, 128.21, 127.66, 127.02, 125.64 (q, 3JC-F = 5.6 Hz), 125.12, 125.10, 119.92, 118.55, 99.61, 74.94, 73.99, 73.02, 71.96, 68.38, 67.38, 66.10, 62.06, 58.66, 47.15, 20.68, 20.64, 20.39, 17.14; HRMS (ESI): calc. for C42 H43N2O12F3 (M + Na)+ : 847.2666; found 847.2671.
4.3. General Glycosylation of Threonine/Serine Amino Acids Mediated by Commercially Available Ni(OTf)2
4.3.1. Fmoc-protected threonine-(O-allyl) 3,4,6-tri-O-acetyl-2-deoxy-2-O-trifluoromethylbenzylideneamino-α-d-glucopyranoside (7)
To a dried 10 mL reaction flask was charged with d-glucososamine N-phenyl trifluoroacetimidate donor 5 (50 mg, 0.079 mmol, 1 equiv.), threonine 6 (36.6 mg, 0.095 mmol, 1.2 equiv.), and NiOTf2 (4.25 mg, .0119 mmol, 15 mol %) under nitrogen, and the mixture was then dissolved in 1 mL of dry CH2Cl2. The resulting solution was stirred under nitrogen at 35 °C (16 h) overnight. Reaction was monitored by TLC (5/1 toluene/acetonitrile, Rf = 0.25). Once complete, 0.5 mL of toluene was added to the reaction and then concentrated at room temperature until CH2Cl2 removed. The remaining toluene containing the crude product was then loaded directly on to a silica gel column and purified by flash chromatography (10 g of silica, 1/2in ID X 12in column, 5/1(180 mL)→3/1(200 mL)→2/1(200 mL) hexanes/ethyl acetate + 1% triethylamine). AXer purificaZon, the fractions containing the product were combined into a 250 mL round bottom flask and diluted with 50 mL toluene. The round bottom was then concentrated via a Buchi rotary evaporator at room temperature until the solvent level reaches the previously marked 50 mL volume. Once this level is achieved, the solution was concentrated at 50 °C to provide the desired glycosyl amino acid 7 as a semi solid (43.6 mg, 67%, α:β = 10:1).
Supplementary Material
Research Highlights.
Commercial Ni(OTf)2 is an effective catalyst to form α-glycosidic bonds.
Comparison of Ni(OTf)2 and in-house prepared Ni(4-F-PhCN)4(OTf)2 is discussed.
Ni(OTf)2 prepared several bioactive motifs difficult under traditional methods.
Investigatory studies of solvent effects on Tn-antigen hydrogen bonding network.
Acknowledgement
We would like to thank Dr. Ravi Loki, Dr. Fei Yu, Alisa Fairweather (graduate student), and Greg Jenson (undergraduate student) for verifying the reproducibility of these glycosylation reactions. Dr. Fei Yu and Jiayi Li (undergraduate student) for constructing some of the glycosyl acceptors. Professor Todd Lowry at the University of Alberta for providing tetrasaccharides 19 and 20. This work is supported the National Institutes of Health (R01 GM098285).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Michael A. Am. Chem. J. 1879;1:305. [Google Scholar]
- 2 (a).Petitou M, van Boeckel CAA. Angew. Chem., Int. Ed. 2004;43:3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]; (b) Echardt KJ. Nat. Prod. 1983;46:544–550. doi: 10.1021/np50028a020. [DOI] [PubMed] [Google Scholar]; (c) Magnet S, Blanchard JS. Chem. Rev. 2005;105:477–497. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]; (d) Danishefsky SJ, Allen JR. Angew. Chem., Int. Ed. 2000;39:836–837. doi: 10.1002/(sici)1521-3773(20000303)39:5<836::aid-anie836>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]; (e) Marcaurelle LA, Bertozzi CR. Glycobiology. 2002;12:69R–77R. [PubMed] [Google Scholar]; (f) Ferguson MAJ, Williams AF. Annu. Rev. Biochem. 1988;57:285–320. doi: 10.1146/annurev.bi.57.070188.001441. [DOI] [PubMed] [Google Scholar]; (g) Bongat AFG, Demchenko AV. Carbohydr. Res. 2007;342:374–406. doi: 10.1016/j.carres.2006.10.021. [DOI] [PubMed] [Google Scholar]; (h) Bertozzi CR, Kiessling LL. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]; (i) Galonic DP, Gin DY. Nature. 2007;446:1000–1007. doi: 10.1038/nature05813. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Seeberger PH, Werz DB. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]; (k) Nicolaou KC, Mitchell HJ. Angew. Chem., Int. Ed. 2001;40:1576–1624. [PubMed] [Google Scholar]
- 3 (a).Goodman L. Adv. Carbohydr. Chem. Biochem. 1967;22:109–175. doi: 10.1016/s0096-5332(08)60152-6. [DOI] [PubMed] [Google Scholar]; (b) Nukada T, Berces A, Zgierski MZ, Whitfield DM. J. Am. Chem. Soc. 1998;120:13291–13295. [Google Scholar]
- 4 (a).Nigudkar SS, Demchenko AV. Chem. Sci. 2015;6:2687. doi: 10.1039/c5sc00280j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McKay MJ, Nguyen HM. ACS Catal. 2012;2:1563–1595. doi: 10.1021/cs3002513. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu X, Schmidt RR. Angew. Chem., Int. Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 5 (a).Manabe S. Methods Enzymol. 2010;478:413–435. doi: 10.1016/S0076-6879(10)78020-4. [DOI] [PubMed] [Google Scholar]; (b) Hakomori S. Annual Rev. Immunol. 1984;2:103–126. doi: 10.1146/annurev.iy.02.040184.000535. [DOI] [PubMed] [Google Scholar]; (c) Banoub J, Boullanger P, Lafont D. Chem. Rev. 1992;92:1167–1195. [Google Scholar]; (d) Kerns RJ, Wei P. ACS Sym. Ser. 2006;932:205–236. [Google Scholar]; (e) Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Paulsen H, Kolar C, Stenzel W. Chem. Ber-Recl. 1978;111:2358–2369. [Google Scholar]; (g) Paulsen H, Stenzel W. Chem. Ber-Recl. 1978;111:2348–2357. [Google Scholar]; (h) Lemieux RU, Ratcliffe RM. Can. J. Chem. 1979;57:1244–1251. [Google Scholar]; (i) Manabe S, Satoh H, Hutter J, Lüthi HP, Laino T, Ito Y. Chem. Eur. J. 2014;20:124–132. doi: 10.1002/chem.201303474. [DOI] [PubMed] [Google Scholar]; (j) Park J, Kawatkar S, Kim J-H, Boons G-J. Org. Lett. 2007;9:1959–1962. doi: 10.1021/ol070513b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6 (a).Mensah EA, Nguyen HM. J. Am. Chem. Soc. 2009;131:8778–8780. doi: 10.1021/ja903123b. [DOI] [PubMed] [Google Scholar]; (b) Mensah EA, Yu F, Nguyen HM. J. Am. Chem. Soc. 2010;132:14288–14302. doi: 10.1021/ja106682m. [DOI] [PubMed] [Google Scholar]; (c) Yu F, Nguyen HM. J. Org. Chem. 2012;77:7330–7343. doi: 10.1021/jo301050q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McConnell MS, Mensah EA, Nguyen HM. Carbohydr. Res. 2013;381:146–152. doi: 10.1016/j.carres.2013.09.006. [DOI] [PubMed] [Google Scholar]; (e) McConnell MS, Yu F, Nguyen HM. Chem. Comm. 2013;49:4313–4315. doi: 10.1039/c2cc35823a. [DOI] [PubMed] [Google Scholar]; (f) Yu F, McConnell MS, Nguyen HM. Org. Lett. 2015;17:2018–2021. doi: 10.1021/acs.orglett.5b00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The mechanistic pathway is currently being investigated.
- 8 (a).Yang J, Cooper-Vanosdell C, Mensah EA, Nguyen HM. J. Org. Chem. 2008;73:794–800. doi: 10.1021/jo702436p. [DOI] [PubMed] [Google Scholar]; (b) McKay MJ, Naab BD, Mercer GJ, Nguyen HM. J. Org. Chem. 2009;74:4705–4711. doi: 10.1021/jo9002807. [DOI] [PubMed] [Google Scholar]; (c) Mensah EA, Azzarelli JM, Nguyen HM. J. Org. Chem. 2009;74:1650–1657. doi: 10.1021/jo802468p. [DOI] [PubMed] [Google Scholar]; (d) Goetze S, Fitzner R, Kunz H. Synlett. 2009;20:3346–3348. [Google Scholar]; (e) Mattson AL, Michel AK, Cloninger MJ. Carbohydr. Res. 2012;347:142–146. doi: 10.1016/j.carres.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Peng P, Schmidt RR. J. Am. Chem. Soc. 2015;137:12653–12659. doi: 10.1021/jacs.5b07895. [DOI] [PubMed] [Google Scholar]; (g) Bartek J, Müller R, Kosma P. Carbohydr. Res. 1998;308:259–273. doi: 10.1016/s0008-6215(98)00082-2. [DOI] [PubMed] [Google Scholar]; (h) Douglas SP, Whitfield DM, Krepinsky JJ. J. Carbohydr. Chem. 1993;12:131–136. [Google Scholar]; (i) Wei G, Gu G, Du Y. J. Carbohydr. Chem. 2003;22:385–393. [Google Scholar]; (j) Roy R, Palanivel AK, Mallick A, Vankar YD. Eur. J. Org. Chem. 2015;18:4000–4005. [Google Scholar]
- 9.Boer JL, Mulrooney SB, Hausinger RP. Arch. Biochem. Biophys. 2014;544:142–152. doi: 10.1016/j.abb.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hochuli E, Bannwarth W, Döbeli H, Gentz R, Stüber D. Nature Biotechnol. 1988;6:1321–1325. [Google Scholar]
- 11.We have established that 15 mol % is the standard amount to be effective in this glycosylation reaction.
-
12.
- 13.Mensah EA. Ph.D. Dissertation. University of Iowa; Iowa City, IA: 2011. Palladium and Nickel Catalyzed Stereoselective Formation of Glycosides. [Google Scholar]
- 14.The mechanism of Ni(OTf)2 catalyzed glycosylation is under investigation. In addition, the utility of these catalysts is currently being explored with other coupling partners and will be reported in due course.
- 15 (a).Beczała A, Duda KA, Skurnik M, Holst O. Carbohydr. Res. 2012;359:97–101. doi: 10.1016/j.carres.2012.06.002. [DOI] [PubMed] [Google Scholar]; (b) Jones C. Carbohydr. Res. 2005;340:1097–1106. doi: 10.1016/j.carres.2005.02.001. [DOI] [PubMed] [Google Scholar]; (c) Pieretti G, Puopolo G, Carillo S, Zoina A, Lanzeaa R, Parrilli M, Evidente A, Corsaro MM. Carbohydr. Res. 2011;346:2705–2709. doi: 10.1016/j.carres.2011.09.027. [DOI] [PubMed] [Google Scholar]; (d) Pieretti G, Corsaro MM, Lanzetta R, Parrilli M, Canals R, Merino S, Tomas JM. Eur. J. Org. Chem. 2008;2008:3149–3155. [Google Scholar]; (e) King JD, Mulrooney EF, Vinogradov E, Kneidinger B, Mead K, Lam JS. J. Bacteriol. 2008;190:1671–1679. doi: 10.1128/JB.01708-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sanapala SR, Kulkarni SS. J. Am. Chem. Soc. 2016;138:4938–4947. doi: 10.1021/jacs.6b01823. [DOI] [PubMed] [Google Scholar]
- 16 (a).Reddy GP, Hayat U, Abeygunawardana C, Fox C, Wright AC, Maneval DR, Bush CA, Morris JG. J. Bacteriol. 1992;174:2620–2630. doi: 10.1128/jb.174.8.2620-2630.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kondakova AN, Kolodziejska K, Zych K, Senchenkova SN, Shashkov AS, Knirel YA, Sidorczyk Z. Carbohydr. Res. 2003;338:1999–2004. doi: 10.1016/s0008-6215(03)00327-6. [DOI] [PubMed] [Google Scholar]; (c) Reddy GP, Hayat U, Xu Q, Reddy KV, Wang Y, Chiu K-W, Morris JG, Bush CA. Eur. J. Biochem. 1998;255:279–288. doi: 10.1046/j.1432-1327.1998.2550279.x. [DOI] [PubMed] [Google Scholar]; (d) Shashkov AS, Senchenkova SN, Nazarenko EL, Zubkov VA, Gorshkova NM, Knirel YA, Gorshkova RP. Carbohydr. Res. 1997;303:333–338. doi: 10.1016/s0008-6215(97)00165-1. [DOI] [PubMed] [Google Scholar]
- 17 (a).Hunter SW, Fujiwara T, Murphy RC, Brennan PJ. J. Biol. Chem. 1984;259:9729–9734. [PubMed] [Google Scholar]; (b) Yoshimura J, Sato KI, Singh RB. Chem. Lett. 1985;14:69–70. [Google Scholar]; (c) Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Chem. Rev. 2005;105:739–758. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 18.Meloncelli PJ, Lowary TL. Aust. J. Chem. 2009;62:558–574. [Google Scholar]
- 19.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM. Clin. Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20 (a).Ju T, Otto VI, Cummings RD. Angew. Chem. Int. Ed. 2011;50:1770–1791S. doi: 10.1002/anie.201002313. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hakomori S. Adv. Cancer Res. 1989;52:257–331. doi: 10.1016/s0065-230x(08)60215-8. [DOI] [PubMed] [Google Scholar]; (c) Singhal A, Fohn M, Hakomori S. Cancer Res. 1991;51:1406–1411. [PubMed] [Google Scholar]; (d) Singhal A, Hakomori S. Bioessays. 1990;12:223–230. doi: 10.1002/bies.950120506. [DOI] [PubMed] [Google Scholar]; (e) Henningsson CM, Selvaraj S, MacLean GD, Suresh MR, Noujaim AA, Longenecker BM. Cancer Immunology, Immunotherapy : CII. 1987;25:231–241. doi: 10.1007/BF00199152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Samuel J, Noujaim AA, MacLean GD, Suresh MR, Longenecker BM. Cancer Res. 1990;50:4801–4808. [PubMed] [Google Scholar]; (g) Xu YF, Sette A, Sidney J, Gendler SJ, Franco A. Immunol. Cell. Biol. 2005;83:440–448. doi: 10.1111/j.1440-1711.2005.01347.x. [DOI] [PubMed] [Google Scholar]
- 21.Nyffeler PT, Liang C-H, Koeller KM, Wong C-H. J. Am. Chem. Soc. 2002;124:10773–10778. doi: 10.1021/ja0264605. [DOI] [PubMed] [Google Scholar]
- 22.Payne RJ, Ficht S, Tang S, Brik A, Yang Y-Y, Case DA, Wong C-H. J. Am. Chem. Soc. 2007;129:13527–13536. doi: 10.1021/ja073653p. [DOI] [PubMed] [Google Scholar]
- 23.Threonine containing 24 was prepared for spectroscopic analysis in yields comparable to that of the serine counterpart.
- 24.Overall yield and selectivity of this reaction is α:β = 7:1, 73%. Due to highly varied Rf purification of the β-anomer requires a drastic switch to a far more polar mobile phase.
- 25.Bourgault JP, Trabbic KR, Shi M, Andreana PR. Org. Biomol. Chem. 2014;12:1699–1702. doi: 10.1039/c4ob00128a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26 (a).Cao S, Tropper FD, Roy R. Tetrahedron. 1995;51:6679–6686. [Google Scholar]; (b) Marcaurelle LA, Shin Y, Goon S, Bertozzi CR. Org. Lett. 2001;3:3691–3694. doi: 10.1021/ol0166247. [DOI] [PubMed] [Google Scholar]; (c) Marcaurelle LA, Rodriguez EC, Bertozzi CR. Tetrahedron Lett. 1998;39:8417–8420. [Google Scholar]; (d) Rodriguez EC, Marcaurelle LA, Bertozzi CR. J. Org. Chem. 1998;63:7134–7135. doi: 10.1021/jo981351n. [DOI] [PubMed] [Google Scholar]; (e) De Silva RA, Wang Q, Chidley T, Appulage DK, Andreana PR. J. Am. Chem. Soc. 2009;131:9622–9623. doi: 10.1021/ja902607a. [DOI] [PubMed] [Google Scholar]
- 27.Rabuka D, Parthasarathy R, Lee GS, Chen X, Groves JT, Bertozzi CR. J. Am. Chem. Soc. 2007;129:5462–5471. doi: 10.1021/ja067819i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28 (a).Coelho H, Matsushita T, Artigas G, Hinou H, Cañada FJ, Lo-Man R, Leclerc C, Cabrita EJ, nez-Barbero JJ, Nishimura S-I, Garcia-Martín F, Marcelo F. J. Am. Chem. Soc. 2015;137:12438–12441. doi: 10.1021/jacs.5b06787. [DOI] [PubMed] [Google Scholar]; (b) MarZnez-Saez N., Castro-Lopez, J., Valero-Gonzalez J, Madariaga D, Companon I, Somovilla VJ, Salvado M, Asensio JL, Jimenez-Barbero J, Avenoza A, Busto JH, Bernardes GJL, Peregrina JM, Hurtado-Guerrero R, Corzana F. Angew. Chem. Int. Ed. 2015;54:9830–9834. doi: 10.1002/anie.201502813. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sames D, Chen X-T, Danishefsky SJ. Nature. 1997;389:587–591. doi: 10.1038/39292. [DOI] [PubMed] [Google Scholar]; (d) Bielfeldt T, Peters S, Meldal M, Bock K, Paulsen H. Angew. Chem. Int. Ed. 1992;31:857–859. [Google Scholar]
- 29.Graham ME, Stone RS, Robinson PJ, Payne RJ. Org. Biomol. Chem. 2012;10:2545–2551. doi: 10.1039/c2ob07139h. [DOI] [PubMed] [Google Scholar]
- 30 (a).Heggemann C, Budke C, Schomburg B, Majer Z, Wißbrock M, Koop T, Sewald N. Amino Acids. 2010;38:213–222. doi: 10.1007/s00726-008-0229-0. [DOI] [PubMed] [Google Scholar]; (b) Nagel L, Budke C, Dreyer A, Koop T, Sewald N. Beilstein J. Org. Chem. 2012;8:1657–1667. doi: 10.3762/bjoc.8.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31 (a).Buba AE, Lowe H, Kunz H. Eur. J. Org. Chem. 2015:5764–5774. [Google Scholar]; (b) H., Paulsen H, Adermann K. Liebigs Ann. Chem. 1989;8:751–769. [Google Scholar]
- 32.Jeffrey GA. An Introduction to Hydrogen Bonding. Oxford University Press; Oxford: 1997. [Google Scholar]

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.