

Abstract
Myocardial infarction results in scar tissue that cannot actively contribute to heart mechanical function and frequently causes lethal arrhythmias. The healing response after infarction involves inflammation, biochemical signaling, changes in cellular phenotype, activity, and organization, and alterations in electrical conduction due to variations in cell and tissue geometry and alterations in protein expression, organization, and function – particularly in membrane channels. The intensive research focus on regeneration of myocardial tissues has, as of yet, only met with modest success, with no near-term prospect of improving standard-of-care for patients with heart disease. An alternative concept for novel therapeutic approach is the rejuvenation of cardiac electrical and mechanical properties through the modification of scar tissue. Several peptide therapeutics, locally applied genetic therapies, or delivery of genetically modified cells have shown promise in improving the characteristics of the fibrous scar and post-myocardial infarction prognosis in experimental models. This review highlights several factors that contribute to arrhythmogenesis in scar formation and how these might be targeted to regenerate some of the electrical and mechanical function of the post-MI scar.
Keywords: myocardial infarction, regeneration, fibrosis, structure, tissue properties
1. Introduction
Myocardial infarction (MI) results in scar formation to repair and replace myocardium lost to ischemic insult. While the scar has mechanical assignments in preventing the heart wall from rupturing, scar tissue does not have the same structure or function as native myocardium. The fibrous scar that forms is unable to contract rhythmically and is not an efficient conductor of electrical signals - two key functions of myocardial tissue. The presence of scar places elevated stress on the heart to fulfill its contractile role, further affecting cardiac performance.
Taking a cue from the neonatal heart and hearts of non-mammals [1–3], many recent studies have been aimed at regenerating myocytes lost to MI [4,5], or reprogramming mature adult non-muscle cells to become myocytes in an attempt to restore cardiac function [6–8]. Recent advances in this area include strategies that enhance fibroblast reprogramming rates, at least in cultured cells [9], and the identification of small molecules that prompt conversion of fibroblasts into myocytes [10]. Though conceptually exciting, these novel findings have, as yet, not demonstrated levels of efficacy in preclinical models or early-stage clinical trials that would justify translation to the next level. Thus far in vivo, such approaches have yielded low numbers of regenerated muscle cells of immature phenotype, and have proven more difficult to elicit from human cells than in model species. Emerging, and perhaps more immediately tractable, approaches to mitigating the loss of myocardial tissue to disease in humans are therapeutic strategies aimed at re-engineering scar properties.
While many factors affect the characteristics of the healed MI, targeted alterations in electrical or mechanical function of the scar and its adjacent infarct border zone (IBZ), may be achievable and could lead to improvements in cardiac performance in hearts damaged by disease [11,12]. However, the ability to therapeutically modify cardiac scar tissue is still limited by a lack of knowledge on what constitutes benefit. Achieving healed infarct that performs similarly to intact cardiac muscle is not possible, though attaining a useful intermediate tissue, in which some biomechanical and/or electrical conduction properties are restored may be on the horizon. This review focuses on regenerating cardiac function lost to disease through manipulation of scar and IBZ tissues. Perfect healing may not be possible, but clinicians could eventually be able to give “Mother Nature an assist”, inhibiting or slowing the presently unstoppable progression toward heart failure faced by many patients.
2. Changes in the structure and function of the infarcted heart
2.1. Formation of a (mostly) non-electrically conducting, non-contractile scar
Prolonged cardiac ischemia is the cause of tissue death in MI [13], with cells dying within minutes to hours of the onset of ischemia [13]. The ischemic tissue then undergoes a healing and remodeling response in the following days and months. Since adult human myocardium has limited to no ability to regenerate, a fibrous scar that cannot perform as normal myocardium replaces lost cardiac muscle (Fig. 1A). The scar does serve mechanical roles - showing some level of compliance during active pumping, as well as preventing the heart wall from rupturing [14]. Interestingly, increased numbers of myofibroblasts in the scar have been associated with less dilation in mouse models of MI [14]. However, fibrotic tissue cannot rhythmically contract, nor is it an efficient conductor of electrical signals, both critical functions of myocardium. Since the majority of patients now survive MI, these alterations result in arrhythmia generation, left ventricular (LV) dysfunction, and often, eventual decline into heart failure and death.
Figure 1.
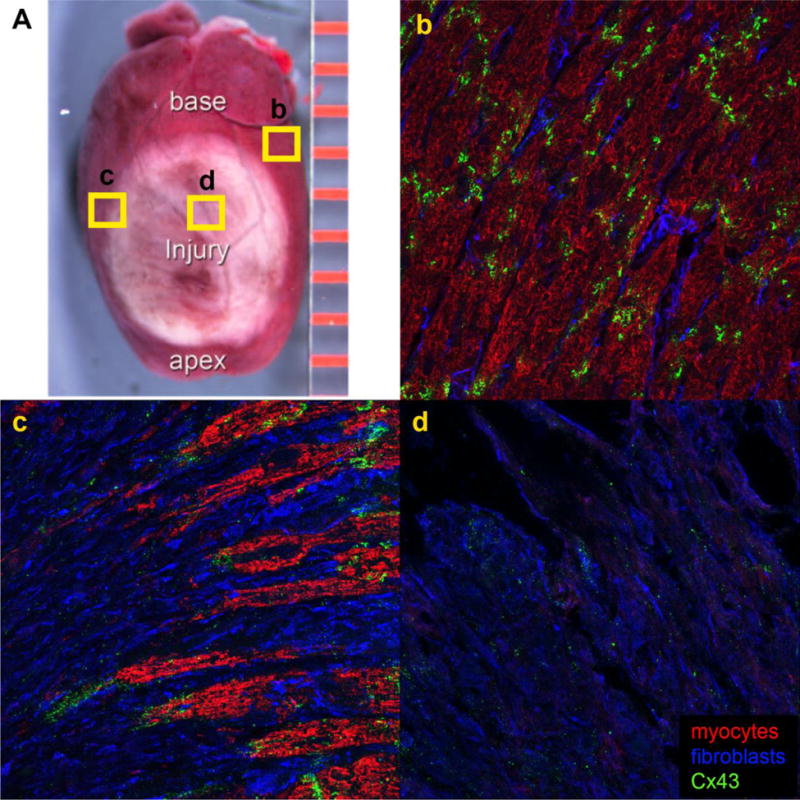
Structural changes in healed infarct scar. A) A scar differentiates in the ischemic portion of the heart. b) Cx43 (green) is localized to the IDs in remote myocardium. c) Fibroblasts (blue) interrupt normal myocyte (red) connections in the IBZ adjacent to the scar and Cx43 reorganizes away from the IDs. Cx43 is often found between myocytes and fibroblasts. d) Punctate Cx43 is found within the scar.
In the ischemic portion of the heart, myocytes die, and cardiac repair begins with inflammatory cells infiltrating the injury within the first day after MI [15,16]. This inflammatory response clears the infarct region of dead cells and debris and is thought to be a necessary step in healing of the infarct. Reperfusion and other clinical interventions can significantly alter the course of healing and the characteristics of the healed infarct [17–20]. Inflammation is significantly increased and accelerated in reperfused tissue (which results from the common clinical intervention coronary angioplasty), though reperfusion improves tissue repair because of this increased inflammatory response [21–25]. On the other hand, delayed resolution of inflammation leads to LV enlargement and adverse remodeling in mouse MI models [26–28]. Resident macrophages appear to have important roles in the chronic phase after myocardial infarction [29], while knockout or depletion of macrophages results in impaired healing [30,31]. Additionally, several inflammatory molecules contribute to the healing process after MI and are implicated in post-MI arrhythmogenesis, including interleukin-1 (IL-1) and transforming growth factor-β (TGF-β) [32]. This being said, factors such as TGF-β play a number of contrasting roles in myocardial healing [33,34]. Thus, although modulation of inflammation represents an attractive strategy for development of new treatments for ischemic heart disease, cell and molecular targets and timing may prove critical to optimizing outcomes from novel approaches to therapy.
Together with macrophages and endothelial cells, fibroblasts are major cellular contributors to cardiac granulation tissue formation and infarct scar remodeling [35–39]. Mesenchymal populations responding to the MI come mainly from resident fibroblasts [40], which in turn largely have epicardial/mesothelial origins [41,42]. Fibroblasts recruited into the wound assume an activated status, differentiating into myofibroblasts - exhibiting among other characteristics elevated levels of collagen secretion and cytoskeletal differentiation, as well as an increased ability to generate mechanical force [43–46].
Remodeling of the scar is a balance between extracellular matrix (ECM) degradation and generation. The rapid degradation of ECM occurs via the action of matrix metalloproteinases (MMPs) and collagenases, released either by damaged cells within the myocardium, or by immune cells that have infiltrated the infarct [47–49]. The fibroblasts infiltrating the ischemic area construct a new network of collagen-rich granulation tissue, changing the normal fibrous ECM skeleton of myocardial tissue within the first week. Remodeling of the infarct is time-dependent and a healed infarction may not differentiate substantively until several weeks after the MI, though ongoing scar maturation may continue for months and years, and in some cases never reaches an equilibrium state [13]. As the heart is less able to efficiently pump due to the loss of myocytes during the scar maturation process, the heart begins to dilate and remaining myocytes hypertrophy [13].
Cardiac injury leads to an initial abrupt decline in cardiac performance, with changes occurring within minutes of the onset of ischemia. This is well demonstrated in isolated heart models, where acute MI can be shown to rapidly impair both force development and contraction [50]. As the infarct heals, the newly generated fibrotic tissues remodel – a process involving extension, expansion, and re-organization of its cellular and acellular components. This not only results in changes to the size and geometry of the infarcted region, but also modifies the myocardium’s ability to contract and transfer electrical signals. Ultimately, functional myocardium is replaced by non-contractile and non-conductive scar. The next section details why this is a problem.
2.2. Electrophysiological remodeling of the scar and border zone after MI
Transmission of electrical signals through the myocardium is held to be mediated by GJs, which are preferentially localized at intercalated discs (IDs) between adjacent myocytes (Fig. 1b). There is also growing evidence for an ephaptic contribution to cardiac conduction [51,52]. Ephaptic conduction of electrical impulses has been proposed to occur via transients in extracellular ions in the ID that prompt activation of adjacent myocytes – albeit that the mechanism appears to be localized in the perinexal region of cell membrane surrounding GJs. GJs are comprised of connexin proteins, the most common of which in the mammalian ventricle is connexin 43 (Cx43) [53]. Post-MI, there is a loss in both the density of GJs and in gap junctional intercellular communication (GJIC) in the ischemic myocardium compared to the remote regions [54,55]. Further, a hallmark of the IBZ is the reorganization of Cx43 from IDs at zones of end-to-end contact between myocytes to non-ID lateral membranes (Fig. 1c) [56–59]. Decreased electrical coupling during acute MI has been correlated with dephosphorylation and reorganization of Cx43, as well as translocation from the cell surface to intracellular pools [60,61]. While the reduction in Cx43 is likely a protective mechanism (GJs are implicated in the spread of injury signals in skin [62], neural tissues [63], and heart [64]), Cx43 lateralization interrupts normal electrical conduction patterns in the heart, and may contribute to arrhythmias [10–13].
Bordering the infarct area is the IBZ, which is a few cell layer-thick region of transitional tissue between the scar and adjacent viable myocardium that has survived infarction. The changes in tissue electrophysiology, cell organization, collagen deposition, and inefficient removal of toxic biochemical products, among other factors, can contribute to the generation of lethal arrhythmias in the IBZ [65–67]. Previous studies demonstrate action potential (AP) slowing through the IBZ [68,69], though propagation of activation increases in the scar, possibly due to the presence of surviving islands of myocytes [70]. Passive conduction via fibroblasts may also contribute to within-scar increases in AP conduction speed [70,71].
In the population of Cx43 that remains at the ID in an ischemic setting, there is an increase in phosphorylation at serine (S) 368 [72] and S373 [73]. Additionally, ischemic preconditioning has been shown to augment protein kinase C-ε–mediated Cx43 phosphorylation of S262 and S368, and prevent Cx43 lateralization following an ischemic insult [74]. Further, Akt-mediated phosphorylation of S373 is elevated in hypoxia. Though increasing GJ size and GJIC, S373 phosphorylation is thought to precede internalization of Cx43 [73]. Phosphorylation events in Cx43 are known to regulate channel effects like unitary conductance, so connexin phosphorylation, in addition to connexin downregulation, may be protective mechanisms against the spread of injury effects into otherwise undamaged tissues surrounding the initial ischemic field.
Surviving myocytes in the IBZ have reduced Na+ [75,76] and repolarizing K+ currents that can lead to prolonged action potential duration (APD) [77,78], and altered intracellular calcium handling, which may result in decreases in cellular excitability and triggered activity [79]. The change in repolarization results in altered and variable resting membrane potential [77,80–82]. Expression of sarcoplasmic reticulum (SR) calcium ATPase (SERCA) is also reduced, as is the activity of the sodium/calcium exchanger [83] - changes that are associated with LV dysfunction. Lower levels of SERCA2a may contribute to alterations in intracellular calcium that are linked to variability in APD and the onset of arrhythmias [84,85]. While reentrant arrhythmias are the most common after MI, triggered and automatic arrhythmias may also occur.
Cx43 is increased in activated fibroblasts in the scar and IBZ [86–88], enhancing the possibility for myocytes and fibroblasts to be electrically coupled. Additionally, infarcted fibroblasts show more hyperpolarized membrane potentials, increased outward currents [89], and higher membrane resistance compared to fibroblasts isolated from normal hearts [90].
Non-muscle cells migrating to the MI from the blood circulation may also affect cardiac electrophysiological properties [40,91,92]. Bone marrow derived cells (BMCs) are mobilized by injury signals and recruited to the infarct from the peripheral circulation. Since the first report in 2001 [93], BMCs have been extensively studied for their utility in cardiac repair, though there have been varying accounts of the numbers of these cells recruited following MI [40,91,94,95]. Some clinical trials of exogenously applied BMCs post-MI indicate modest benefit from paracrine factors [96] and exosome release [97,98]. However, studies of large patient cohorts in meta-analyses have raised questions as to whether experimental cell therapies of this type will provide significant benefit beyond existing standard-of-care, in terms of favorable clinical events or improvement in metrics of LV function [99,100].
2.3. Changes in cellular organization and interactions
The changes in electrophysiological characteristics of scar tissue may be related to sub-acute and longer-term remodeling that occurs following MI. While nearly all myocytes in the ischemic area die, numerous myocytes at the infarct periphery in the IBZ survive. Structural remodeling in the IBZ involves an interspersion of myocytes with fibroblasts that have migrated into, and/or proliferated in the wound, and the ECM that these mesenchymal cells produce - leading to separations between strands of surviving myocardial cells and enhanced opportunity for electrical connection between myocytes and fibroblasts (Fig. 1c) [65,87,101,102]. Fibroblasts have higher membrane resistance and capacitance than myocytes, potentially making them better sustainers of electrical signals when paired to each other, or to myocytes [103]. However, fibroblasts also have slightly depolarized resting membrane potentials compared to myocytes [104–106]. Electrical coupling between fibroblasts and myocytes in diseased tissue thus may result in myocardial cell depolarization, increasing the risk of conduction disturbance and arrhythmia - especially with the potential for numerous heterocellular contacts of this type to occur over the extent of the IBZ [107].
In addition to reorganization of cells and increased deposition of ECM proteins post-MI, one of the alterations in fibroblasts in the scar is an increase in connexin expression (Fig. 1d) [86–88], which enhances the capability of fibroblasts to be paired with myocytes via GJs [87]. Myocyte-fibroblast coupling is relatively rare in the uninjured myocardium in homeostasis and has only been definitively demonstrated in sinoatrial tissue [108]. However, immunolabeling studies suggest that GJ-based heterocellular contacts may accrete in the IBZ (Fig. 1c) [87,88,109]. While intercellular communication of myocardial cells is well-established, as electrical impulse spread through the myocardium relies primarily on the passage of ions through GJs [110,111], the presence and activity of heterocellular GJ coupling is not well understood [103].
In an elegant optical mapping study of infarction in rabbit, AP propagated into post-MI scar tissue, indicating a possible role for myocyte-fibroblast interconnection in sustaining electrical signals [70]. The AP measured within the scar demonstrated profiles similar to those of cardiac fibroblasts connected to myocytes [104,106]. Additionally, fibroblasts in the infarct scar were shown to lack the calcium transients occurring in myocytes [68], suggesting that these cells passively conduct electrical signals through the scar. However, alterations in cell organization and communication may also contribute to the arrhythmogenic propensity of the IBZ [112].
Myocyte-fibroblast coupling is well established in vitro, and is reinforced by the fact that fibroblasts removed from native myocardium upregulate their expression of connexin proteins [113–116]. Heterocellular interactions mediated by GJs are also increased in disease [88], as fibroblast phenotype changes in response to injury, making interactions more likely [117–119]. The overall effect of increased intercellular contact in the context of other changes to the post-MI environment is currently unknown. Changes in myocyte-fibroblast coupling may nonetheless be responsible for key aspects of long-term and pathologic changes to conduction at the IBZ.
Tunneling nanotubes – thin, extended membrane-bound projections – are a further type of myocyte-fibroblast interaction known to occur within cardiac tissues [120]. The function of these ephemeral structures is presently unknown. Interestingly, there are reports of Cx43 enrichment in tunneling nanotubes in a number of cell types [121,122], though the existence of punctate Cx43 labeling of nanotubes, suggestive of GJ formation, was not found between myocardial cells and fibroblasts isolated from uninjured neonatal ventricles [120]. The presence of tunneling nanotubes between myocytes and fibroblasts within the IBZ, and the extent to which such structures in the injured heart express connexins, seem pertinent questions for ongoing study.
2.4. Mechanical properties and infarct size
Infarct size is a major predictor of outcome in MI patients [123]. Diabetic and dysglycemic individuals tend to have larger infarcts and higher post-MI mortality [124]. This may be related to GJ coupling in the cardiac injury, as insulin administration was found to decrease GJIC and increase phosphorylation of Cx43 at S368 [64], which is associated with lower channel conductance. Large infarcts result in extensive cardiac remodeling, including changes in gross morphology, histology, and molecular function and interactions [125]. Within a few days of infarction, fibroblasts begin to deposit ECM, and collagen content increases significantly in the first month [67,126]. Scar stiffness is widely held to increase with escalating collagen levels [126,127], though some studies suggest that even whilst the amount of collagenous tissue deposition continues to increase over time, stiffness peaks a few weeks after infarction and then declines [128,129]. This may be accounted for by the differences in collagen content and MMP profiles in rodents versus large animals.
MI often results in systolic and diastolic dysfunction and can predispose patients to arrhythmias because of the extensive remodeling that occurs [130]. The remodeling process is typically ongoing; with continued dilation, cardiac performance deteriorates over time [131], leading to heart failure and the eventual death of the patient.
3. Experimental approaches to improving myocardial function
The intention of a number of emerging therapies is to modify the properties of the infarct scar, preventing adverse remodeling or functional events, and to mitigate the progression to heart failure. These therapies are directed at restoring several important heart characteristics that are not usually attributes of cardiac scar tissue.
3.1. Amelioration of electrical properties
The scar and IBZ have been the object of many therapies aimed at ameliorating the electrophysiological medium of these tissues. While traditional pharmacological solutions are limited by their global action, resulting in many off-target effects, several directed therapeutic approaches can be taken to alter electrical transfer in the scar and reduce the propensity for arrhythmias. If the border zone and scar tissue could efficiently propagate electrical signals instead of acting as a structure promoting reentrant arrhythmia generation via disruption of uniform conduction, a path to abating the effects and incidence of lethal arrhythmias could be provided.
3.1.1. Connexins
Several experimental treatments that attempt to modify electrical communication post-MI have focused on the connexins. Timing is critical in therapies aimed at increasing connexin expression, as these proteins are known to be involved in the spread of injury signals [62,132]. Cx40 and Cx43 have been used to speed conduction in the pig atria [133,134]. Upregulation of these two connexins enhanced propagation in the atria, as well as prevented atrial fibrillation and lessened arrhythmia susceptibility. Cx43 was found to improve the electrical substrate of the ventricular scar, as Cx43-expressing cells engrafted into murine infarct were shown to prevent arrhythmogenesis post-MI [11]. Following Cx43 gene transfer via intracoronary adenoviral delivery to infarcts in pig, hearts experienced fewer inducible arrhythmias and faster CV through the border zone, results which were correlated with a doubling of Cx43 expression in the IBZ [135]. Interestingly, the ratios of ID-localized Cx43 and phosphorylated Cx43 were similar in transduced infarcts versus controls. This suggests that heightened Cx43 levels in the IBZ may be sufficiently therapeutic to reduce arrhythmias post-MI.
Low pH in infarcted tissue during ischemia can cause Cx43 GJ channels to close [136]. In contrast, Cx32 remains open during low pH [137]. However, when Cx32 was ectopically expressed in an attempt to increase CV through the border zone, though GJIC was increased, no effect on arrhythmia incidence was found [138]. Importantly, Cx32 expression was found to significantly increase infarct size [138], highlighting a long-established role of connexins in injury spread [139]. Cx43 can increase CV in the IBZ of a healed infarct [135], but Cx32 added at the time of injury leads to an increase in the extent of injured myocardial tissue [138]. This provides a cautionary example that timing and channel properties of connexin isoforms are essential considerations in any therapeutic strategy involving manipulation of these proteins.
Connexins have also been modulated by microRNAs to improve scar characteristics. Wang et al. noticed that overexpression of microRNA-1 (miR-1) increased arrhythmogenesis in the infarcted heart [140]. Amplified miR-1 expression was associated with slowed conduction spread and depolarized membrane potential, likely caused by reduced expression of Cx43 and inward rectifying potassium channel 2.1 (Kir2.1) [140]. Using antagomirs to block miR-1 activity in a rat model of infarction relieved arrhythmogenesis.
Several Cx43 mimetic peptides have shown potential for novel therapeutic approach to improve prognosis after experimental MI. αCT1, a mimic of the Cx43 carboxyl-terminus, was shown to reduce arrhythmia incidence and severity in a murine cryo-injury model of infarction [141]. This likely occurred because of an enhanced CV through the IBZ, and a tendency for Cx43 to be maintained at IDs. The opening of hemichannels, which are localized to the perinexus at the edges of GJ plaques [51,142,143], is thought to contribute to the phenomenon of connexin-mediated injury spread following MI, with an accompanying efflux of ATP and other pro-inflammatory molecules and influx of toxic levels of ions [143–145]. Gap 26/27, mimetics of extracellular loop domains of Cx43, inhibited electrical connection via GJs and hemichannel activity [146], and diminished infarct size in a rat MI model [144]. Gap19, an intracellular loop mimetic that reportedly blocks Cx43 hemichannels, but not GJs formed by this isoform [143], modestly decreased infarct size in a murine model of MI [143]. Gap19 and Gap 26/27 thus appear to have cardioprotective effects, though no study to date has examined the effects of these peptides on CV post-injury or probed their ability to inhibit arrhythmias.
3.1.2. Myocyte-fibroblast coupling
While myocytes are widely thought to be the prime cellular medium for improving the propagation of cardiac APs, the question of whether non-myocytes could be targets for therapeutically improving conduction through the IBZ and scar remains open. Fibroblasts in the normal heart do express connexins [147,148], albeit at low levels, and dye transfer studies in rabbit sinoatrial node have shown that fibroblasts can functionally couple to myocytes [108]. After MI, activated fibroblasts upregulate expression of Cx43 [86–88], potentially further enhancing myocyte-fibroblast interconnections. Optical mapping studies of experimental MI in rabbit show that, even after chemical ablation of surviving myocytes in the infarct, the remaining cells within the scar can propagate electrical signals [70]. Additionally, a recent report of conduction between myocytes and non-myocytes in the IBZ was demonstrated by a fluorescent voltage-reporting protein under the Wilm’s Tumor 1 (WT1) promoter [149]. In light of these studies, instead of ensuring the scar is non-conducting, it may be beneficial to patients in some instances to augment propagation of electrical signals through the scar. This would not regenerate the contractile attributes of fibrotic tissues in the heart, but it could recapitulate aspects of electrical transfer and reduce propensity for conduction disturbance and arrhythmia.
3.1.3. Ion channels
The skeletal muscle voltage-gated sodium channel isoform 4a (SCN4a) has been used in attempts to speed up CV through the border zone into the infarct scar [150]. Lau et al. observed that the skeletal muscle sodium channel isoform was more active at the depolarized membrane potentials found in the damaged myocytes of the IBZ than was the cardiac sodium channel, SCN5a. Adenoviral transduction of SCN4a into the epicardial border zone reduced arrhythmia inducibility and increased CV, with no change in infarct size in a canine infarct model [150]. In a simulation model of Nav1.5 redistribution in IBZ myocytes, Na+ channel blockade with class I antiarrhythmic drugs tended to result in conduction block and reentrant arrhythmia generation at the border zone [151]. Additionally, it was found that Nav1.5 expression is reduced in the IBZ of mouse hearts, and that the transcription factor FoxO1 negatively regulates Nav1.5 α- and β3-subunits and sodium current density [152], indicating that FoxO1 could be an interesting target for modulating sodium channel expression in the IBZ.
Variability in APD is linked to the onset of arrhythmias [84], and one of the mechanisms that may be responsible for this is variation in intracellular calcium handling that can result, in part, by a reduction in SERCA2a [85]. When SERCA2a was transfected into rat hearts 16 weeks post-MI, the hearts had fewer spontaneous premature ventricular contractions and spontaneous or induced non-sustained VT episodes compared to controls. There were fewer spontaneous SR calcium release events, reduced total SR calcium leak, and fewer triggered arrhythmias [153]. Similar observations were made in a sheep model, with enhanced contractility and stroke volume, and smaller and fewer apoptotic myocytes in the IBZ [154]. As SERCA2a gene therapy can improve the mechanical pump capacity of the heart, Phase I/Phase II clinical trials have been completed [155,156]. However, heart failure patients in the Phase IIb CUPID 2 trial received no benefit from treatment [157].
Since potassium channel alterations can lead to prolonged APD, they have been the subject of several studies. A dominant negative potassium channel mutation in which a glycine (G) was mutated to an S at position 628 (KCNH2-G628S) increased APD by eliminating a key myocyte repolarization current. Localized in vivo gene transfer of this mutant channel to the VT site prolonged refractory period, extending the reentrant wavelength and preventing arrhythmias [158]. Similarly, voltage-gated potassium channel 1.3 (Kv1.3)-expressing fibroblasts engrafted into rat heart led to a prolonged local refractory period, but had no effect on the regions of the heart not receiving cells [159]. This effect was maintained at slower pacing cycles in pig hearts with fibroblasts expressing Kv1.3 and Kir2.1 [159]. These genetically engineered non-myocytes were reported to be able to couple with myocytes, as well as being associated with reduced levels of cardiac automaticity and prolonged refractoriness. Additionally, semaphorin 3a is known to decrease sympathetic neural remodeling after MI, and may be involved in electrical remodeling at the IBZ, as levels of potassium channels were restored by adenoviral delivery of the signaling molecule in a rat model of MI [160].
3.2. Targeting scar structure
Resolution of the inflammatory phase of healing after infarction and the proper timing of reparative fibrosis are critical for healing of the scar. An overactive reparative phase of healing can lead to collagen accumulation outside the ischemic area, contributing to interstitial fibrosis in the IBZ. Modifying the inflammatory response could have an impact on both electrophysiological and mechanical properties by altering the proteins directly involved in electrogenesis [161–163], the tissue architecture that supports efficient activation spread within the scar and IBZ [161], and the extent of fibrosis contributing to tissue stiffness.
3.2.1. Altering inflammation and the fibrotic response
One aspect of the healing process that can affect the amount of fibrous ECM deposition is the inflammatory response. Delayed resolution of inflammation leads to LV dilation and adverse remodeling in mouse MI models [26–28]. Consistent with this, knockout or depletion of macrophages results in impaired healing [30,31] and inflammation may be required for regeneration in the zebrafish heart [164]. This suggests the necessity of balancing the inflammatory response for normal healing of the infarct scar.
Non-steroidal anti-inflammatories should NOT be used in MI patients, as they have been shown to worsen clinical outcomes [165]. On the other hand, the literature on the ability of steroidal anti-inflammatories to reduce fibrosis is controversial, with some studies showing decreased mortality and no effect on risk of rupture [166], while others demonstrated detrimental effects, including an increased risk of ventricular rupture [167].
A number of established clinical interventions appear to modulate fibrosis in an “off-target” manner, including angiotensin converting enzyme inhibitors [168], angiotensin receptor blockers [169], and statins [170–174], which have been shown to reduce fibrosis and inhibit detrimental myocardial remodeling. One study suggests that the anti-fibrotic effect of statins is mediated by inhibiting TGF-β-induced differentiation of fibroblasts to myofibroblasts [175].
Modulation of neurohormonal factors can also affect cardiac fibrosis. Endothelin is known to increase collagen deposition by heart fibroblasts [176–179], and endothelin antagonism has been shown to have cardioprotective effects [180,181]. However, clinical trials to reduce LV remodeling have demonstrated that endothelin antagonism is mostly ineffective, though it does not adversely affect remodeling [182–184]. Inhibiting the angiotensin II pathway by administration of losartan was reported to decrease interstitial fibrosis, increase CV, and raise GJ conductance in infarcted hearts [185,186]. Losartan mode-of-action is thought to include the deactivation of MMPs [187–189].
Several studies directed at inhibiting specific inflammatory molecules or the conversion of fibroblasts to myofibroblasts have been completed. Studies in mouse and human using IL-1 inhibition attenuated LV remodeling and preserved cardiac function, while allowing normal infarct healing [190–193]. Inhibition of the TGFβ pathway can prevent the conversion of fibroblasts to myofibroblasts [194], and has been shown to reduce fibrosis and progression to heart failure in a mouse model of pressure overload [195].
Remodeling of the infarct area after injury is a balance between ECM degradation by enzymes and ECM synthesis by fibroblasts. Even though a number of reports have demonstrated that MMPs play a role in LV myocardial remodeling after MI [49,196], preventing excess degradation of collagen by MMPs has produced controversial results in preclinical studies. The overall collagen area of the MI scar was found to change very little with broad spectrum MMP inhibition [197–199] and changes in collagen content were modest and mostly not significant in targeted reduction or transgenic knockouts of MMP-1, -3, -9, -12, and MT1-MMP [200–203]. However, broad spectrum MMP inhibition did limit LV dilation [197,199], reduce myocyte hypertrophy in the non-infarcted regions of myocardium [198], and decrease tissue and chamber compliance [198,199], suggesting a beneficial effect on cardiac structure.
Although much progress has been made in understanding the contribution of cardiac fibroblasts to fibrosis, cell lineage analyses suggest differences in fibroblast response to ischemic versus hypertrophic diseases [40,92,204], as well as differences in fibroblast response in atrium versus ventricle [34,205–208]. These studies emphasize the importance of examining potential remedies in appropriate disease models.
3.2.2. Altering scar structure and ultrastructure
The ultimate long-term goal of regenerative therapy is to replace MI scar with new myocytes. Unfortunately, experiments performed thus far in vivo suggest low efficiency of myocyte regeneration and immature myocyte phenotypes, which frequently display lateralized GJs, immature calcium handling, slower conduction velocity, and residual pacemaking currents, all of which can contribute to aberrant AP propagation and arrhythmia [209–211]. In line with this, there has been a shift in interest among researchers from a sole emphasis on the prospect of myocardial regeneration to the paracrine effects of experimental cellular therapies – raising interesting unanswered questions as to whether the target for such paracrine effect is cardiac muscle or scar. To date, studies of regenerative and direct reprogramming strategies have focused on myocardial properties, paying less attention to effects on scar electrical and mechanical properties, CV, arrhythmogenesis, and/or cardioprotection.
At the border zone, interstitial fibrosis leads to separation of myocytes, interfering with normal electrical connections. Because of the variation in angle of myocardial sheetlets from the surface of the epicardium through to the endocardium [212,213], fibrosis at the border of the scar may be nearly parallel, nearly perpendicular (or some intermediate orientation) to surviving myocytes [214]. This results in a heterogeneous tissue, which is a well-known predictor of arrhythmia risk [215,216]. Few experimental studies have examined how electrical cues propagate in a 3D heterogeneous tissue. This being noted, an image-based modeling study of the IBZ demonstrated that heterogeneous IBZ composition is correlated with tortuous communication pathways and conduction block [214]. Understanding the geometry of cells and tissue that contribute to conduction in this context may be important for modifying scar configuration and ultrastructure post-MI.
Infarct size is a critical predictor of outcome in MI patients [123,125]. Weakened ventricular function [217], adverse LV remodeling [218], and heart failure [219] are more likely to result from larger infarcts. Revascularization of infarct tissue has been posited as the first step in reducing infarct size and has indeed found to be associated with extended survival of MI patients [220]. There is some evidence that infarcts may compact spontaneously [221]. Other groups have also addressed the potential of the Wnt pathway in minimizing cardiac scars [222–224]. Mechanisms of cardioprotection may also lead to smaller infarcts. Gap 19/26/27, which are known to inhibit hemichannel activity, reduced infarct size in rat and murine models of MI [143,144]. Additionally, though an unlikely remedy to precede an MI, ischemic preconditioning confers some level of cardioprotection, even when performed on extremities [225].
Another aspect of scar ultrastructure that can shed some light on arrhythmia mechanism is the cellular composition of the IBZ. Mathematical models have demonstrated that when myocyte-fibroblast coupling via GJs and ionic current remodeling is considered in the IBZ, activated fibroblasts at intermediate densities prompt AP shortening and elevated arrhythmia propensity. By contrast, at high density, fibroblasts cause resting depolarization and block propagation, protecting against arrhythmias [226]. While this study did not take into account the organization of cells in the IBZ, it does suggest that varying the cellular profile could be critical for arrhythmia prevention.
In simulation models of AF, fibroblast proliferation, but not collagen accumulation alone, was suggested to be responsible for disturbances to electrical activity [227]. Additionally, spatial variations in accumulation of fibrosis may affect electrical propagation in the heart, and patient-specific modeling of this fibrosis may allow identification of areas-at-risk for developing arrhythmias [228]. Rotors in AF were trapped by fiber discontinuities, and fibrosis or scarring of sufficient size could terminate this [229]. Patient-specific distribution of fibrosis, thus, may be critical in causing and maintaining electrical abnormalities [230].
Few studies have focused on changing the patterns of cellular contact in the IBZ, though 2D studies have shown that the geometry of cell interactions may play a role in arrhythmia generation [231,232].
3.2.3. Altering the mechanical properties of the infarct
Mechanical properties of healed infarct are critical, as the scar must be stiff enough to prevent rupture or stretching that leads to poor force generation, but not so stiff that filling during diastole is limited. The scar must also maintain its mural thickness over time to avoid LV dilation.
Early computer modeling work, that treated infarcts as isotropic tissue, suggested that increasing stiffness of the infarct resulted in confounding changes to cardiac LV metrics [233,234]. In a finite-element model in dog heart, Holmes and co-workers determined that isotropic stiffening of the scar did not alter stroke volume, but longitudinal stiffening, though maintaining some level of compliance in the circumferential direction, led to increases in stroke volume [233]. A follow-up study in dogs confirmed this finding by mechanically reinforcing the infarct in the longitudinal direction [12]. While these studies demonstrated a key proof-of-concept for the field, a therapeutic strategy aimed at generating a longitudinally stiff and (somewhat) circumferentially compliant scar requires further development. In an attempt to better understand scar anisotropy, this group generated a mathematical model that shed light on the mechanical cues that influence collagen alignment [235], though this model does not as yet take into account biochemical effects on this process.
Several studies have examined the effect of polymer injection to control mechanical characteristics of the scar [236–238]. Polymer injection into the scar seems to only provide benefit to cardiac mechanics if a stiff polymer is used [237]. These experimental results were reflected by a finite element modeling study that suggested injections into the IBZ at multiple locations might be the most beneficial, while injections into the scar produced mixed results on cardiac pressure-volume relationships [234].
Another approach to increasing stiffness of infarct scar is to amplify collagen content, either by inhibiting MMPs (as reviewed in section 3.2.1) or boosting tissue inhibitors of metalloproteinases (TIMPs) [239–241]. Hydrogel or adenoviral delivery of TIMPs to infarcts led to reduced MMP activity and LV remodeling in a porcine model [239] and greater fibrillar collagen deposition and LV ejection fraction in mice [241].
Though we still don’t fully understand the ideal mechanical properties of a healing or healed infarct scar, many experimental and modeling studies have provided a wealth of information. Hearts with small scars that are longitudinally stiff seem to have the best prognosis.
4. Conclusion
The MI scar and IBZ present significant clinical challenges, as these tissues cannot contribute actively to the contractile ability of the heart, do not efficiently pass electrical signals, and are associated with life-threatening arrhythmias. Advances have been made, including, in a few instances, the progression of promising early-stage therapies to clinical testing. However, the attributes of the healing infarct that are the most amenable to decreasing the likelihood of adverse outcomes are not well understood. Further research is required to gain insight into the basis of variation in scar mechanical and electrical properties between individuals, and how patient-to-patient differences in fibrotic tissue organization and function ultimately contribute to lethal arrhythmias and heart failure. It is also notable that extant preclinical and early clinical-stage studies have mostly examined the effects of therapies based on manipulation of a single factor. In the long term, it is envisaged that improvements beyond the current standard-of-care, as well as mitigation of off-target effects, may require the orchestration of effects on several facets of scar and IBZ microenvironment during the healing process. This being said, that the electrical and mechanical properties of diseased hearts may be partially restored by targeting cardiac scar tissue is a proposition that requires serious consideration.
Table 1.
Therapeutic targets for regeneration of electrical and mechanical properties of MI scar tissue.
| Target | Targeting strategy | Stage | Findings l | Mode | References |
|---|---|---|---|---|---|
| Connexin 40, 43 | Adenovirus; engraftment of cells expressing Cx43 | Preclinical | Enhanced atrial conduction, prevented atrial fibrillation, reduced arrhythmia susceptibility, faster CV through ventricular border zone | Pig, mouse | [11,133–135] |
| Connexin 43 | Mimeticpeptides: αCT1, Gap19/26/27 | Preclinical | Reduce arrhythmia incidence and severity, increased CV through IBZ, Cx43 maintained at IDs; Decreased infarct size; inhibit electrical coupling and/or hemichannel activity | Mouse, rat | [141,143,144,146] |
| Connexin 32 | Adenovirus | Preclinical | Increased GJIC, no change in arrhythmia incidence, increased infarct size | Dog | [138] |
| miR-1 | Antagomir | Preclinical | Decreased arrhythmogenesis | Rat | [140] |
| Cardiac sodium channel 4a (SCN4a) | Adenovirus | Preclinical | Reduced arrhythmia inducibility, increased CV through border zone | Dog | [150] |
| Sarcoplasmic reticulum calcium-ATPase 2a (SERCA2a) | Adeno-associated virus | Preclinical | Fewer spontaneous premature ventricular contractions and spontaneous or induced non-sustained VT episodes, fewer spontaneous SR calcium release events, reduced total SR calcium leak, fewer triggered arrhythmias | Rat, Sheep | [153] |
| Sarcoplasmic reticulum calcium-ATPase 2a (SERCA2a) | Adeno-associated virus | Clinical, Phase II | Decreased incidence of ventricular arrhythmias and triggers, improved arrhythmogenic substrate | Human | [155,156] |
| KCNH2-G628S | Adenovirus | Preclinical | Increased APD; prolonged refractory period, extending the reentrant wavelength and preventing arrhythmias | Pig | [158] |
| Potassium channel (Kv1.3, Kir2.1) | Engraftment of Kv1.3- and Kir2.1-expressing fibroblasts | Preclinical | Prolonged local refractory period, engrafted cells coupled with myocytes leading to reduced automaticity and prolonged refractoriness | Rat, Pig | [159] |
| Semaphorin 3a | Adenovirus | Preclinical | Increased levels of potassium channels at the border zone | Rat | [160] |
| Endothelin | ET-1 antagonists | Preclinical, Clinical | Cardioprotective effects; no improvements in cardiac remodeling in clinical trials, but also no adverse effects | Rat, Human | [180–184] |
| Angiotensin II pathway | Ang-II inhibition | Preclinical, Clinical | Decreased interstitial fibrosis, increased CV, and increased GJ conductance | Hamster, Human | [185,186] |
| Interleukin-1 | IL-1 inhibition | Preclinical, Clinical | Attenuated LV remodeling, preserved cardiac function, allowed normal infarct healing | Mouse, Human | [190–193] |
| TGF-β pathway | TGF-β inhibition | Preclinical | Reduced fibrosis and progression to heart failure in a model of pressure overload | Mouse | [195] |
| MMP | Broad spectrum MMP inhibition; targeted MMP inhibition | Preclinical | Overall collagen area of MI scar changes very little with broad spectrum inhibition, but did limit LV dilation, reduced myocyte hypertrophy in non-infarcted regions, decreased tissue and chamber compliance; No significant changes in collagen content in targeted reduction or transgenic knockouts of MMP-1, -3, -9, -12, and MT1-MMP | Mouse, Rat, Pig | [197–203] |
| Wnt pathway | Wnt pathway transgenics or interfering peptides | Preclinical | Reduced MMP level and leukocyte infiltration, decreased scar size, improved cardiac function, reduced fibrosis, reduced infarct expansion and development of heart failure | Mouse | [222–224] |
| Fibroblast density | NA | Modeling | At high density, fibroblasts cause resting depolarization and block propagation, protecting against arrhythmias | In silico | [226] |
| Scar stiffness | Scar anisotropy | Modeling, Preclinical | Increasing stiffness of isotropic infarct tissue does not alter LV pump function of the heart, stiffening scar in longitudinal direction (or longitudinal mechanical restraint) increases pump function | In silico, Dog | [12,233,234] |
| Scar stiffness | Intra-scar polymer injection | Preclinical, Modeling | Maintained fractional shortening and wall thickness; Stiff polymers improve cardiac mechanics; Injections into IBZ at multiple locations is most beneficial, while injections into scar produce mixed results on cardiac pressure-volume relationships | Rat, Sheep, In silico | [234,236–238] |
| Increase scar stiffness by increasing collagen content | Inhibiting MMPs or increasing TIMPs by hydrogel or adenoviral delivery | Preclinical | Reduced MMP activity and LV remodeling, increased fibrillary collagen content and LV ejection fraction | Mouse, Pig | [239–241] |
Highlights.
The adult heart cannot regenerate, so myocardial infarction (MI) results in a scar.
Significant electrical and mechanical remodeling occurs after MI, resulting in arrhythmias.
Modifying scar properties is a viable strategy for improving post-MI prognosis.
Proteins involved in conduction and fibrous tissue structure are primary targets for altering scar properties.
Further study is required to understand how modifying scar tissue affects heart function.
Acknowledgments
ELO acknowledges support by the National Institutes of Health.
RGG acknowledges support by the National Institutes of Health, the Center for Innovative Technology and the Virginia Biosciences Health Research Corporation.
Abbreviations
- αCT1
alpha-carboxyl-terminal peptide 1
- AP
action potential
- APD
action potential duration
- BMCs
bone marrow derived cells
- CV
conduction velocity
- Cx43
connexin 43
- ECM
extracellular matrix
- GJ
gap junction
- GJIC
gap junction intercellular communication
- IBZ
injury border zone
- ID
intercalated disc
- Kir2.1
inward rectifying potassium current 2.1
- KCNH2
voltage gated potassium channel
- Kv1.3
voltage gated potassium channel 1.3
- LV
left ventricular
- MI
Myocardial infarction
- miR-1
microRNA-1
- MMP
matrix metalloproteinase
- SERCA
sarcoplasmic reticulum calcium ATPase
- SCN4a
voltage gated sodium channel isoform 4a
- SCN5a
voltage gated sodium channel isoform 5a
- SR
sarcoplasmic reticulum
- TIMP
tissue inhibitor of metalloproteinase
- TGF-β
transforming growth factor-β
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
ELO: none. RGG is a member of the scientific advisory board of FirstString Research Inc and holds modest ownership in this company (<5%).
Contributor Information
Emily L. Ongstad, Email: ongstad@vt.edu.
Robert G. Gourdie, Email: gourdier@vtc.vt.edu.
References
- 1.Porrello ER, Mahmoud AI, Simpson E, Hill Ja, Richardson Ja, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poss KD. Heart Regeneration in Zebrafish. Science (80-) 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 3.Fang Y, Gupta V, Karra R, Holdway JE, Kikuchi K, Poss KD. Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc Natl Acad Sci. 2013;110:13416–13421. doi: 10.1073/pnas.1309810110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. doi: 10.1016/S0140-6736(12)60195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yacoub MH, Terrovitis J. CADUCEUS, SCIPIO, ALCADIA: Cell therapy trials using cardiac-derived cells for patients with post myocardial infarction LV dysfunction, still evolving. Glob Cardiol Sci Pract. 2013;2013:3. doi: 10.5339/gcsp.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, et al. Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrée B, Zweigerdt R. Directing Cardiomyogenic Differentiation and Transdifferentiation By Ectopic Gene Expression - Direct Transition Or Reprogramming Detour? Curr Gene Ther. 2016;16:14–20. doi: 10.2174/1566523216666160104141522. http://www.ncbi.nlm.nih.gov/pubmed/26725881. [DOI] [PubMed] [Google Scholar]
- 9.Zhou H, Dickson ME, Kim MS, Bassel-Duby R, Olson EN. Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc Natl Acad Sci U S A. 2015;112:11864–9. doi: 10.1073/pnas.1516237112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao N, Huang Y, Zheng J, Spencer CI, Zhang Y, Fu JD, et al. Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science. 2016 doi: 10.1126/science.aaf1502. [DOI] [PubMed] [Google Scholar]
- 11.Roell W, Lewalter T, Sasse P, Tallini YN, Choi BR, Breitbach M, et al. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature. 2007;450:819–824. doi: 10.1038/nature06321. [DOI] [PubMed] [Google Scholar]
- 12.Fomovsky GM, Clark SA, Parker KM, Ailawadi G, Holmes JW. Anisotropic reinforcement of acute anteroapical infarcts improves pump function. Circ Heart Fail. 2012;5:515–22. doi: 10.1161/CIRCHEARTFAILURE.111.965731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33:2551–2567. doi: 10.1093/eurheartj/ehs184. [DOI] [PubMed] [Google Scholar]
- 14.van den Borne SWM, van de Schans VAM, Strzelecka AE, Vervoort-Peters HTM, Lijnen PM, Cleutjens JPM, et al. Mouse strain determines the outcome of wound healing after myocardial infarction. Cardiovasc Res. 2009;84:273–282. doi: 10.1093/cvr/cvp207. [DOI] [PubMed] [Google Scholar]
- 15.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–77. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frangogiannis NG. Regulation of the Inflammatory Response in Cardiac Repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hochman JS, Choo H. Limitation of myocardial infarct expansion by reperfusion independent of myocardial salvage. Circulation. 1987;75:299–306. doi: 10.1161/01.cir.75.1.299. http://www.ncbi.nlm.nih.gov/pubmed/3791612. [DOI] [PubMed] [Google Scholar]
- 18.Horie H, Takahashi M, Minai K, Izumi M, Takaoka A, Nozawa M, et al. Long-term beneficial effect of late reperfusion for acute anterior myocardial infarction with percutaneous transluminal coronary angioplasty. Circulation. 1998;98:2377–82. doi: 10.1161/01.cir.98.22.2377. http://www.ncbi.nlm.nih.gov/pubmed/9832481. [DOI] [PubMed] [Google Scholar]
- 19.Abbate A, Bussani R, Biondi-Zoccai GGL, Rossiello R, Silvestri F, Baldi F, et al. Persistent infarct-related artery occlusion is associated with an increased myocardial apoptosis at postmortem examination in humans late after an acute myocardial infarction. Circulation. 2002;106:1051–4. doi: 10.1161/01.cir.0000030936.97158.c4. http://www.ncbi.nlm.nih.gov/pubmed/12196327. [DOI] [PubMed] [Google Scholar]
- 20.Sadanandan S, Buller C, Menon V, Dzavik V, Terrin M, Thompson B, et al. The late open artery hypothesis—A decade later. Am Heart J. 2001;142:411–421. doi: 10.1067/mhj.2001.117774. [DOI] [PubMed] [Google Scholar]
- 21.Richard V, Murry CE, Reimer KA. Healing of Myocardial Infarcts in Dogs: Effects of Late Reperfusion. Circulation. 1995;92:1891–1901. doi: 10.1161/01.CIR.92.7.1891. [DOI] [PubMed] [Google Scholar]
- 22.Reimer KA, Vander Heide RS, Richard VJ. Reperfusion in acute myocardial infarction: effect of timing and modulating factors in experimental models. Am J Cardiol. 1993;72:13G–21G. doi: 10.1016/0002-9149(93)90102-i. http://www.ncbi.nlm.nih.gov/pubmed/8279349. [DOI] [PubMed] [Google Scholar]
- 23.Jugdutt BI. Effect of reperfusion on ventricular mass, topography, and function during healing of anterior infarction. Am J Physiol. 1997;272:H1205–11. doi: 10.1152/ajpheart.1997.272.3.H1205. http://www.ncbi.nlm.nih.gov/pubmed/9087594. [DOI] [PubMed] [Google Scholar]
- 24.Solomon A, B MD, Gersh MD. THE OPEN-ARTERY HYPOTHESIS. Annu Rev Med. 1998;49:63–76. doi: 10.1146/annurev.med.49.1.63. [DOI] [PubMed] [Google Scholar]
- 25.Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, et al. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–808. doi: 10.4049/jimmunol.165.5.2798. http://www.ncbi.nlm.nih.gov/pubmed/10946312. [DOI] [PubMed] [Google Scholar]
- 26.Dobaczewski M, de Haan JJ, Frangogiannis N. the extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J Cardiovasc Transl Res. 2012;5:837–847. doi: 10.1016/j.biotechadv.2011.08.021.Secreted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cochain C, Auvynet C, Poupel L, Vilar J, Dumeau E, Richart A, et al. The Chemokine Decoy Receptor D6 Prevents Excessive Inflammation and Adverse Ventricular Remodeling After Myocardial Infarction. Arterioscler Thromb Vasc Biol. 2012;32:2206–2213. doi: 10.1161/ATVBAHA.112.254409. [DOI] [PubMed] [Google Scholar]
- 28.Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Brás LE, et al. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol. 2015;185:198–208. doi: 10.1016/j.ijcard.2015.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, et al. Differential Contribution of Monocytes to Heart Macrophages in Steady-State and After Myocardial Infarction. Circ Res. 2014;115:284–295. doi: 10.1161/CIRCRESAHA.115.303567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJA. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–29. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ben-Mordechai T, Holbova R, Landa-Rouben N, Harel-Adar T, Feinberg MS, Abd Elrahman I, et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. 2013;62:1890–901. doi: 10.1016/j.jacc.2013.07.057. [DOI] [PubMed] [Google Scholar]
- 32.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–28. http://www.ncbi.nlm.nih.gov/pubmed/7856752. [PMC free article] [PubMed] [Google Scholar]
- 33.Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J Mol Cell Cardiol. 2000;32:187–95. doi: 10.1006/jmcc.1999.1065. [DOI] [PubMed] [Google Scholar]
- 34.Nakajima H, Nakajima HO, Salcher O, Dittiè AS, Dembowsky K, Jing S, et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000;86:571–9. doi: 10.1161/01.res.86.5.571. http://www.ncbi.nlm.nih.gov/pubmed/10720419. [DOI] [PubMed] [Google Scholar]
- 35.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frangogiannis NG. Pathophysiology of Myocardial Infarction. Compr Physiol. 2015;5:1841–75. doi: 10.1002/cphy.c150006. [DOI] [PubMed] [Google Scholar]
- 37.Deb A, Ubil E. Cardiac fibroblast in development and wound healing. J Mol Cell Cardiol. 2014;70:47–55. doi: 10.1016/j.yjmcc.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldsmith EC, Bradshaw AD, Zile MR, Spinale FG. Myocardial fibroblast-matrix interactions and potential therapeutic targets. J Mol Cell Cardiol. 2014;70:92–9. doi: 10.1016/j.yjmcc.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gourdie RG, Dimmeler S, Kohl P. Novel Therapeutic Strategies Targeting Fibroblasts and Fibrosis in Heart Disease. Nat Rev Drug Discov. 2016 doi: 10.1038/nrd.2016.89. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruiz-Villalba A, Simón AM, Pogontke C, Castillo MI, Abizanda G, Pelacho B, et al. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J Am Coll Cardiol. 2015;65:2057–2066. doi: 10.1016/j.jacc.2015.03.520. [DOI] [PubMed] [Google Scholar]
- 41.Mikawa T, Gourdie RG. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev Biol. 1996;174:221–232. doi: 10.1006/dbio.1996.0068. [DOI] [PubMed] [Google Scholar]
- 42.Dettman RW, Denetclaw W, Ordahl CP, Bristow J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev Biol. 1998;193:169–81. doi: 10.1006/dbio.1997.8801. [DOI] [PubMed] [Google Scholar]
- 43.van Putten S, Shafieyan Y, Hinz B. Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol. 2015 doi: 10.1016/j.yjmcc.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 44.Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol. 2014;70:9–18. doi: 10.1016/j.yjmcc.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leask A. Getting to the Heart of the Matter. Circ Res. 2015;116:1269–1276. doi: 10.1161/CIRCRESAHA.116.305381. [DOI] [PubMed] [Google Scholar]
- 46.van den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–7. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 47.Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, et al. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation. 2001;103:2181–7. doi: 10.1161/01.cir.103.17.2181. http://www.ncbi.nlm.nih.gov/pubmed/11331260. [DOI] [PubMed] [Google Scholar]
- 48.Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, Schulz R. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation. 2000;101:1833–9. doi: 10.1161/01.cir.101.15.1833. http://www.ncbi.nlm.nih.gov/pubmed/10769285. [DOI] [PubMed] [Google Scholar]
- 49.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 50.Stefanon I, Valero-Muñoz M, Fernandes AA, Ribeiro RF, Rodríguez C, Miana M, et al. Left and right ventricle late remodeling following myocardial infarction in rats. PLoS One. 2013;8:e64986. doi: 10.1371/journal.pone.0064986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhett JM, Ongstad EL, Jourdan J, Gourdie RG. Cx43 associates with Na(v)1.5 in the cardiomyocyte perinexus. J Membr Biol. 2012;245:411–22. doi: 10.1007/s00232-012-9465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG, Poelzing S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflügers Arch - Eur J Physiol. 2015;467:2093–2105. doi: 10.1007/s00424-014-1675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gourdie RG, Severs NJ, Green CR, Rothery S, Germroth P, Thompson RP. The spatial distribution and relative abundance of gap-junctional connexin40 and connexin43 correlate to functional properties of components of the cardiac atrioventricular conduction system. J Cell Sci. 1993;105:985–991. doi: 10.1242/jcs.105.4.985. [DOI] [PubMed] [Google Scholar]
- 54.Kieval RS, Spear JF, Moore EN. Gap junctional conductance in ventricular myocyte pairs isolated from postischemic rabbit myocardium. Circ Res. 1992;71:127–36. doi: 10.1161/01.res.71.1.127. http://www.ncbi.nlm.nih.gov/pubmed/1606660. [DOI] [PubMed] [Google Scholar]
- 55.Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–75. doi: 10.1161/01.cir.88.3.864. http://www.ncbi.nlm.nih.gov/pubmed/8394786. [DOI] [PubMed] [Google Scholar]
- 56.Green CR, Severs NJ. Robert Feulgen Prize Lecture. Distribution and role of gap junctions in normal myocardium and human ischaemic heart disease. Histochemistry. 1993;99:105–20. doi: 10.1007/BF00571871. http://www.ncbi.nlm.nih.gov/pubmed/8478212. [DOI] [PubMed] [Google Scholar]
- 57.Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- 58.Severs NJ, Dupont E, Coppen SR, Halliday D, Inett E, Baylis D, et al. Remodelling of gap junctions and connexin expression in heart disease. Biochim Biophys Acta - Biomembr. 2004;1662:138–148. doi: 10.1016/j.bbamem.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 59.Bernus O, Zemlin CW, Zaritsky RM, Mironov SF, Pertsov AM. Alternating conduction in the ischaemic border zone as precursor of reentrant arrhythmias: A simulation study. Europace. 2005;7 doi: 10.1016/j.eupc.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 60.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–62. doi: 10.1161/01.res.87.8.656. http://www.ncbi.nlm.nih.gov/pubmed/11029400. [DOI] [PubMed] [Google Scholar]
- 61.Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547–52. doi: 10.1161/01.cir.101.5.547. http://www.ncbi.nlm.nih.gov/pubmed/10662753. [DOI] [PubMed] [Google Scholar]
- 62.Lin JHC, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, et al. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- 63.Davidson JO, Green CR, Bennet L, Nicholson LFB, Danesh-Meyer H, O’Carroll SJ, et al. A key role for connexin hemichannels in spreading ischemic brain injury. Curr Drug Targets. 2013;14:36–46. doi: 10.2174/138945013804806479. http://www.ncbi.nlm.nih.gov/pubmed/23170795. [DOI] [PubMed] [Google Scholar]
- 64.Palatinus JA, Gourdie RG. Diabetes Increases Cryoinjury Size with Associated Effects on Cx43 Gap Junction Function and Phosphorylation in the Mouse Heart. J Diabetes Res. 2016;2016:1–9. doi: 10.1155/2016/8789617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ursell PC, Gardner PI, Albala A, Fenoglio JJ, Wit AL. Structural and electrophysiological changes in the epicardial border zone of canine myocardial infarcts during infarct healing. Circ Res. 1985;56:436–51. doi: 10.1161/01.RES.56.3.436. [DOI] [PubMed] [Google Scholar]
- 66.Driesen RB, Verheyen FK, Dijkstra P, Thoné F, Cleutjens JP, Lenders MH, et al. Structural remodelling of cardiomyocytes in the border zone of infarcted rabbit heart. Mol Cell Biochem. 2007;302:225–232. doi: 10.1007/s11010-007-9445-2. [DOI] [PubMed] [Google Scholar]
- 67.Jugdutt BI. Ventricular Remodeling After Infarction and the Extracellular Collagen Matrix: When Is Enough Enough? Circulation. 2003;108:1395–1403. doi: 10.1161/01.CIR.0000085658.98621.49. [DOI] [PubMed] [Google Scholar]
- 68.Saba S, Mathier MA, Mehdi H, Liu T, Choi BR, London B, et al. Dual-dye optical mapping after myocardial infarction: does the site of ventricular stimulation alter the properties of electrical propagation? J Cardiovasc Electrophysiol. 2008;19:197–202. doi: 10.1111/j.1540-8167.2007.00998.x. [DOI] [PubMed] [Google Scholar]
- 69.Cabo C, Yao J, Boyden Pa, Chen S, Hussain W, Duffy HS, et al. Heterogeneous gap junction remodeling in reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc Res. 2006;72:241–249. doi: 10.1016/j.cardiores.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 70.Walker NL, Burton FL, Kettlewell S, Smith GL, Cobbe SM. Mapping of epicardial activation in a rabbit model of chronic myocardial infarction: Response to atrial, endocardial and epicardial pacing. J Cardiovasc Electrophysiol. 2007;18:862–868. doi: 10.1111/j.1540-8167.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- 71.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 72.Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ Res. 2006;98:1498–1505. doi: 10.1161/01.RES.0000227572.45891.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunn CA, Lampe PD. Injury-triggered Akt phosphorylation of Cx43: a ZO-1-driven molecular switch that regulates gap junction size. J Cell Sci. 2014;127:455–464. doi: 10.1242/jcs.142497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Srisakuldee W, Jeyaraman MM, Nickel BE, Tanguy S, Jiang ZS, Kardami E. Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc Res. 2009;83:672–681. doi: 10.1093/cvr/cvp142. [DOI] [PubMed] [Google Scholar]
- 75.Baba S. Remodeling in Cells From Different Regions of the Reentrant Circuit During Ventricular Tachycardia. Circulation. 2005;112:2386–2396. doi: 10.1161/CIRCULATIONAHA.105.534784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dun W, Lowe JS, Wright P, Hund TJ, Mohler PJ, Boyden PA. Ankyrin-G Participates in INa Remodeling in Myocytes from the Border Zones of Infarcted Canine Heart. PLoS One. 2013;8:e78087. doi: 10.1371/journal.pone.0078087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lue WM, Boyden PA. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation. 1992;85:1175–88. doi: 10.1161/01.cir.85.3.1175. http://www.ncbi.nlm.nih.gov/pubmed/1371431. [DOI] [PubMed] [Google Scholar]
- 78.Pinto JM, Boyden PA. Electrical remodeling in ischemia and infarction. Cardiovasc Res. 1999;42:284–97. doi: 10.1016/s0008-6363(99)00013-9. http://www.ncbi.nlm.nih.gov/pubmed/10533567. [DOI] [PubMed] [Google Scholar]
- 79.Pu J, Robinson RB, Boyden PA. Abnormalities in Ca(i) handling in myocytes that survive in the infarcted heart are not just due to alterations in repolarization. J Mol Cell Cardiol. 2000;32:1509–23. doi: 10.1006/jmcc.2000.1184. [DOI] [PubMed] [Google Scholar]
- 80.Dun W. Diverse phenotypes of outward currents in cells that have survived in the 5-day-infarcted heart. AJP Hear Circ Physiol. 2005;289:H667–H673. doi: 10.1152/ajpheart.00180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang M, Cabo C, Yao J, Boyden PA, Tseng G. Delayed rectifier K currents have reduced amplitudes and altered kinetics in myocytes from infarcted canine ventricle. Cardiovasc Res. 2000;48:34–43. doi: 10.1016/s0008-6363(00)00159-0. http://www.ncbi.nlm.nih.gov/pubmed/11033106. [DOI] [PubMed] [Google Scholar]
- 82.Pu J, Boyden PA. Alterations of Na+ Currents in Myocytes From Epicardial Border Zone of the Infarcted Heart: A Possible Ionic Mechanism for Reduced Excitability and Postrepolarization Refractoriness. Circ Res. 1997;81:110–119. doi: 10.1161/01.RES.81.1.110. [DOI] [PubMed] [Google Scholar]
- 83.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 84.Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med. 1994;330:235–41. doi: 10.1056/NEJM199401273300402. [DOI] [PubMed] [Google Scholar]
- 85.Iijima K, Geshi E, Nomizo A, Arata Y, Katagiri T. Alterations in Sarcoplasmic Reticulum and Angiotensin II Type 1 Receptor Gene Expression After Myocardial Infarction in Rats. Jpn Circ J. 1998;62:449–454. doi: 10.1253/jcj.62.449. [DOI] [PubMed] [Google Scholar]
- 86.Vasquez C, Benamer N, Morley GE. The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts. J Cardiovasc Pharmacol. 2011;57:380–388. doi: 10.1097/FJC.0b013e31820cda19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Y, Kanter EM, Yamada KA. Remodeling of cardiac fibroblasts following myocardial infarction results in increased gap junction intercellular communication. Cardiovasc Pathol. 2010;19:e233–e240. doi: 10.1016/j.carpath.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Camelliti P, Devlin GP, Matthews KG, Kohl P, Green CR. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc Res. 2004;62:415–425. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 89.Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ Res. 2010;107:1011–1020. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kiseleva I, Kamkin A, Pylaev A, Kondratjev D, Leiterer KP, Theres H, et al. Electrophysiological properties of mechanosensitive atrial fibroblasts from chronic infarcted rat heart. J Mol Cell Cardiol. 1998;30:1083–1093. doi: 10.1006/jmcc.1998.0673. [DOI] [PubMed] [Google Scholar]
- 91.Visconti RP, Markwald RR. Recruitment of new cells into the postnatal heart: potential modification of phenotype by periostin. Ann N Y Acad Sci. 2006;1080:19–33. doi: 10.1196/annals.1380.003. [DOI] [PubMed] [Google Scholar]
- 92.Crawford JR, Haudek SB, Cieslik KA, Trial J, Entman ML. Origin of developmental precursors dictates the pathophysiologic role of cardiac fibroblasts. J Cardiovasc Transl Res. 2012;5:749–59. doi: 10.1007/s12265-012-9402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–5. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 94.Möllmann H, Nef HM, Kostin S, von Kalle C, Pilz I, Weber M, et al. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc Res. 2006;71:661–671. doi: 10.1016/j.cardiores.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 95.van Amerongen M, Bou-Gharis G, Popa E, van Ark J, Petersen A, van Dam G, et al. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J Pathol. 2008;214:377–386. doi: 10.1002/path.2281. [DOI] [PubMed] [Google Scholar]
- 96.Abdel-Latif A, Bolli R, Tleyjeh IM, Montori VM, Perin EC, Hornung CA, et al. Adult bone marrow-derived cells for cardiac repair: a systematic review and meta-analysis. Arch Intern Med. 2007;167:989–97. doi: 10.1001/archinte.167.10.989. [DOI] [PubMed] [Google Scholar]
- 97.Lai RC, Arslan F, Lee MM, Sze NSK, Choo A, Chen TS, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4:214–222. doi: 10.1016/j.scr.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 98.Ibrahim AGE, Cheng K, Marbán E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports. 2014;2:606–19. doi: 10.1016/j.stemcr.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nowbar AN, Mielewczik M, Karavassilis M, Dehbi HM, Shun-Shin MJ, Jones S, et al. Discrepancies in autologous bone marrow stem cell trials and enhancement of ejection fraction (DAMASCENE): weighted regression and meta-analysis. BMJ. 2014;348:g2688–g2688. doi: 10.1136/bmj.g2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gyongyosi M, Wojakowski W, Lemarchand P, Lunde K, Tendera M, Bartunek J, et al. Meta-Analysis of Cell-based CaRdiac stUdiEs (ACCRUE) in Patients With Acute Myocardial Infarction Based on Individual Patient Data. Circ Res. 2015;116:1346–1360. doi: 10.1161/CIRCRESAHA.116.304346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.C. Vasquez, P. Mohandas, K.L. Louie, N. Benamer, A.C. Bapat, G.E. Morley, 12, 100, (n.d.) 1–5.
- 102.Goldsmith EC, Bradshaw AD, Zile MR, Spinale FG. Myocardial fibroblast-matrix interactions and potential therapeutic targets. J Mol Cell Cardiol. 2014;29:92–99. doi: 10.1016/j.biotechadv.2011.08.021.Secreted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kohl P, Gourdie RG. Fibroblast-myocyte electrotonic coupling: Does it occur in native cardiac tissue? J Mol Cell Cardiol. 2014;70:37–46. doi: 10.1016/j.yjmcc.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kohl P, Kamkin AG, Kiseleva IS, Noble D. Mechanosensitive fibroblasts in the sinoatrial node region of rat heart: Interaction with cardiomyocytes and possible role. Exp Physiol. 1994;79:943–956. doi: 10.1113/expphysiol.1994.sp003819. [DOI] [PubMed] [Google Scholar]
- 105.Kamkin A, Kiseleva I, Wagner KD, Pylaev A, Leiterer KP, Theres H, et al. A possible role for atrial fibroblasts in postinfarction bradycardia. Am J Physiol Heart Circ Physiol. 2002;282:H842–H849. doi: 10.1152/ajpheart.00240.2001. [DOI] [PubMed] [Google Scholar]
- 106.Rook MB, van Ginneken ACG, de Jonge B, el Aoumari A, Gros D, Jongsma HJ. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am J Physiol - Cell Physiol. 1992;263:C959–C977. doi: 10.1152/ajpcell.1992.263.5.C959. [DOI] [PubMed] [Google Scholar]
- 107.Nguyen TP, Xie Y, Garfinkel A, Qu Z, Weiss JN. Arrhythmogenic consequences of myofibroblast-myocyte coupling. Cardiovasc Res. 2012;93:242–251. doi: 10.1093/cvr/cvr292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: Structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res. 2004;94:828–835. doi: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]
- 109.Xie Y, Garfinkel A, Camelliti P, Kohl P, Weiss JN, Qu Z. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: A computational study. Hear Rhythm. 2009;6:1641–1649. doi: 10.1016/j.hrthm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lo CW. Role of gap junctions in cardiac conduction and development: insights from the connexin knockout mice. Circ Res. 2000;87:346–348. doi: 10.1161/01.RES.87.5.346. [DOI] [PubMed] [Google Scholar]
- 111.Gros DB, Jongsma HJ. Connexins in mammalian heart function. BioEssays. 1996;18:719–730. doi: 10.1002/bies.950180907. [DOI] [PubMed] [Google Scholar]
- 112.El-Sherif N, Mehra R, Gough WB, Zeiler RH. Ventricular activation patterns of spontaneous and induced ventricular rhythms in canine one-day-old myocardial infarction. Evidence for focal and reentrant mechanisms. Circ Res. 1982;51:152–166. doi: 10.1161/01.RES.51.2.152. [DOI] [PubMed] [Google Scholar]
- 113.Asazuma-Nakamura Y, Dai P, Harada Y, Jiang Y, Hamaoka K, Takamatsu T. Cx43 contributes to TGF-β signaling to regulate differentiation of cardiac fibroblasts into myofibroblasts. Exp Cell Res. 2009;315:1190–1199. doi: 10.1016/j.yexcr.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 114.Mark GE, Strasser FF. Pacemaker activity and mitosis in cultures of newborn rat heart ventricle cells. Exp Cell Res. 1966;44:217–233. doi: 10.1016/0014-4827(66)90427-7. [DOI] [PubMed] [Google Scholar]
- 115.Hyde A, Blondel B, Matter A, Cheneval JP, Filloux B, Girardier L. Homo- and heterocellular junctions in cell cultures: An electrophysiological and morphological study. Prog Brain Res. 1969;31:283–311. doi: 10.1016/S0079-6123(08)63247-1. [DOI] [PubMed] [Google Scholar]
- 116.Goshima K. Formation of nexuses and electrotonic transmission between myocardial and FL cells in monolayer culture. Exp Cell Res. 1970;63:124–130. doi: 10.1016/0014-4827(70)90339-3. [DOI] [PubMed] [Google Scholar]
- 117.Santiago JJJ, Dangerfield AL, Rattan SG, Bathe KL, Cunnington RH, Raizman JE, et al. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. 2010;239:1573–1584. doi: 10.1002/dvdy.22280. [DOI] [PubMed] [Google Scholar]
- 118.Rohr S. Myofibroblasts in diseased hearts: New players in cardiac arrhythmias? Hear Rhythm. 2009;6:848–856. doi: 10.1016/j.hrthm.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 119.Masur SK, Dewal HS, Dinh TT, Erenburg I, Petridou S. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc Natl Acad Sci U S A. 1996;93:4219–4223. doi: 10.1073/pnas.93.9.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.He K, Shi X, Zhang X, Dang S, Ma X, Liu F, et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc Res. 2011;92:39–47. doi: 10.1093/cvr/cvr189. [DOI] [PubMed] [Google Scholar]
- 121.Wang X, Gerdes HH. Long-distance electrical coupling via tunneling nanotubes. Biochim Biophys Acta - Biomembr. 2012;1818:2082–2086. doi: 10.1016/j.bbamem.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 122.Wang X, Veruki ML, Bukoreshtliev NV, Hartveit E, Gerdes HH. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc Natl Acad Sci U S A. 2010;107:17194–17199. doi: 10.1073/pnas.1006785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McKay RG, Pfeffer MA, Pasternak RC, Markis JE, Come PC, Nakao S, et al. Left ventricular remodeling after myocardial infarction: a corollary to infarct expansion. Circulation. 1986;74:693–702. doi: 10.1161/01.CIR.74.4.693. [DOI] [PubMed] [Google Scholar]
- 124.Mather AN, Crean A, Abidin N, Worthy G, Ball SG, Plein S, et al. Relationship of dysglycemia to acute myocardial infarct size and cardiovascular outcome as determined by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2010;12:61. doi: 10.1186/1532-429X-12-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pfeffer JM, Pfeffer Ma, Fletcher PJ, Braunwald E. Progressive ventricular remodeling in rat with myocardial infarction. Am J Physiol. 1991;260:H1406–14. doi: 10.1016/j.bbadis.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 126.Fomovsky GM, Holmes JW. Evolution of scar structure, mechanics, and ventricular function after myocardial infarction in the rat. AJP Hear Circ Physiol. 2010;298:H221–H228. doi: 10.1152/ajpheart.00495.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng. 2005;7:223–53. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]
- 128.Gupta KB, Ratcliffe MB, Fallert MA, Edmunds LH, Bogen DK. Changes in passive mechanical stiffness of myocardial tissue with aneurysm formation. Circulation. 1994;89:2315–26. doi: 10.1161/01.cir.89.5.2315. http://www.ncbi.nlm.nih.gov/pubmed/8181158. [DOI] [PubMed] [Google Scholar]
- 129.McGarvey JR, Mojsejenko D, Dorsey SM, Nikou A, Burdick JA, Gorman JH, et al. Temporal Changes in Infarct Material Properties: An In Vivo Assessment Using Magnetic Resonance Imaging and Finite Element Simulations. Ann Thorac Surg. 2015;100:582–9. doi: 10.1016/j.athoracsur.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gaudron P, Kugler I, Hu K, Bauer W, Eilles C, Ertl G. Time course of cardiac structural, functional and electrical changes in asymptomatic patients after myocardial infarction: Their inter-relation and prognostic impact. J Am Coll Cardiol. 2001;38:33–40. doi: 10.1016/S0735-1097(01)01319-5. [DOI] [PubMed] [Google Scholar]
- 131.Holmes JW, Yamashita H, Waldman LK, Covell JW. Scar remodeling and transmural deformation after infarction in the pig. Circulation. 1994;90:411–420. doi: 10.1161/01.CIR.90.1.411. [DOI] [PubMed] [Google Scholar]
- 132.Kanno S, Kovacs A, Yamada KA, Saffitz JE. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J Am Coll Cardiol. 2003;41:681–686. doi: 10.1016/S0735-1097(02)02893-0. [DOI] [PubMed] [Google Scholar]
- 133.Logeart D, Hatem SN, Rücker-Martin C, Chossat N, Névo N, Haddada H, et al. Highly efficient adenovirus-mediated gene transfer to cardiac myocytes after single-pass coronary delivery. Hum Gene Ther. 2000;11:1015–22. doi: 10.1089/10430340050015329. [DOI] [PubMed] [Google Scholar]
- 134.Bikou O, Thomas D, Trappe K, Lugenbiel P, Kelemen K, Koch M, et al. Connexin 43 gene therapy prevents persistent atrial fibrillation in a porcine model. Cardiovasc Res. 2011;92:218–25. doi: 10.1093/cvr/cvr209. [DOI] [PubMed] [Google Scholar]
- 135.Greener ID, Sasano T, Wan X, Igarashi T, Strom M, Rosenbaum DS, et al. Connexin43 Gene Transfer Reduces Ventricular Tachycardia Susceptibility After Myocardial Infarction. J Am Coll Cardiol. 2012;60:1103–1110. doi: 10.1016/j.jacc.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ek-Vitorín JF, Calero G, Morley GE, Coombs W, Taffet SM, Delmar M. PH regulation of connexin43: molecular analysis of the gating particle. Biophys J. 1996;71:1273–84. doi: 10.1016/S0006-3495(96)79328-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stergiopoulos K, Alvarado JL, Mastroianni M, Ek-Vitorin JF, Taffet SM, Delmar M. Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ Res. 1999;84:1144–55. doi: 10.1161/01.res.84.10.1144. http://www.ncbi.nlm.nih.gov/pubmed/10347089. [DOI] [PubMed] [Google Scholar]
- 138.Boink GJJ, Lau DH, Shlapakova IN, Sosunov EA, Anyukhovsky EP, Driessen HE, et al. SkM1 and Cx32 improve conduction in canine myocardial infarcts yet only SkM1 is antiarrhythmic. Cardiovasc Res. 2012;94:450–459. doi: 10.1093/cvr/cvs107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.García-Dorado D, Rodríguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc Res. 2004;61:386–401. doi: 10.1016/j.cardiores.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 140.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–91. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 141.O’Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res. 2011;108:704–715. doi: 10.1161/CIRCRESAHA.110.235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rhett JM, Veeraraghavan R, Poelzing S, Gourdie RG. The perinexus: Sign-post on the path to a new model of cardiac conduction? Trends Cardiovasc Med. 2013;23:222–228. doi: 10.1016/j.tcm.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang N, De Vuyst E, Ponsaerts R, Boengler K, Palacios-Prado N, Wauman J, et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2013;108:309. doi: 10.1007/s00395-012-0309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hawat G, Hélie P, Baroudi G. Single intravenous low-dose injections of connexin 43 mimetic peptides protect ischemic heart in vivo against myocardial infarction. J Mol Cell Cardiol. 2012;53:559–566. doi: 10.1016/j.yjmcc.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 145.Sáez JC, Schalper KA, Retamal MA, Orellana JA, Shoji KF, Bennett MVL. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp Cell Res. 2010;316:2377–89. doi: 10.1016/j.yexcr.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 146.Desplantez T, Verma V, Leybaert L, Evans WH, Weingart R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol Res. 2012;65:546–552. doi: 10.1016/j.phrs.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 147.Zhang Y, Kanter EM, Laing JG, Aprhys C, Johns DC, Kardami E, et al. Connexin43 expression levels influence intercellular coupling and cell proliferation of native murine cardiac fibroblasts. Cell Commun Adhes. 2008;15:289–303. doi: 10.1080/15419060802198736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Louault C, Benamer N, Faivre JF, Potreau D, Bescond J. Implication of connexins 40 and 43 in functional coupling between mouse cardiac fibroblasts in primary culture. Biochim Biophys Acta - Biomembr. 2008;1778:2097–2104. doi: 10.1016/j.bbamem.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 149.Quinn TA, Camelliti P, Siedlecka U, Poggioli T, Loew LM, Knopfel T, et al. Abstract 11749: Cell-specific expression of voltage-sensitive protein confirms cardiac myocyte to non-myocyte electrotonic coupling in healed murine infarct border tissue. Circulation. 2014;130:A11749. http://circ.ahajournals.org/cgi/content/long/130/Suppl_2/A11749 (accessed September 29, 2015) [Google Scholar]
- 150.Lau DH, Clausen C, Sosunov EA, Shlapakova IN, Anyukhovsky EP, Danilo P, et al. Epicardial border zone overexpression of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation. 2009;119:19–27. doi: 10.1161/CIRCULATIONAHA.108.809301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tsumoto K, Ashihara T, Haraguchi R, Nakazawa K, Kurachi Y. Ischemia-Related Subcellular Redistribution of Sodium Channels Enhances the Proarrhythmic Effect of Class I Antiarrhythmic Drugs: A Simulation Study. PLoS One. 2014;9:e109271. doi: 10.1371/journal.pone.0109271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cai B, Wang N, Mao W, You T, Lu Y, Li X, et al. Deletion of FoxO1 leads to shortening of QRS by increasing Na+ channel activity through enhanced expression of both cardiac NaV1.5 and β3 subunit. J Mol Cell Cardiol. 2014;74:297–306. doi: 10.1016/j.yjmcc.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lyon AR, Bannister ML, Collins T, Pearce E, Sepehripour AH, Dubb SS, et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ Arrhythm Electrophysiol. 2011;4:362–72. doi: 10.1161/CIRCEP.110.961615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fargnoli AS, Katz MG, Yarnall C, Isidro A, Petrov M, Steuerwald N, et al. Cardiac Surgical Delivery of the Sarcoplasmic Reticulum Calcium ATPase Rescues Myocytes in Ischemic Heart Failure. Ann Thorac Surg. 2013;96:586–595. doi: 10.1016/j.athoracsur.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, et al. Long-Term Effects of AAV1/SERCA2a Gene Transfer in Patients With Severe Heart FailureNovelty and Significance. Circ Res. 2014;114:101–108. doi: 10.1161/CIRCRESAHA.113.302421. [DOI] [PubMed] [Google Scholar]
- 156.AP-H de P.C. Corporation AAV1-CMV-Serca2a GENe Therapy Trial in Heart Failure (AGENT-HF) Clin [Internet] Bethesda Natl Libr Med (US) 2015 https://clinicaltrials.gov/ct2/show/NCT01966887 accessed January 3, 2016.
- 157.Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–1186. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 158.Sasano T, McDonald AD, Kikuchi K, Donahue JK. Molecular ablation of ventricular tachycardia after myocardial infarction. Nat Med. 2006;12:1256–1258. doi: 10.1038/nm1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yankelson L, Feld Y, Bressler-Stramer T, Itzhaki I, Huber I, Gepstein A, et al. Cell therapy for modification of the myocardial electrophysiological substrate. Circulation. 2008;117:720–731. doi: 10.1161/CIRCULATIONAHA.106.671776. [DOI] [PubMed] [Google Scholar]
- 160.Wen H, Jiang H, Li L, Xie P, Li J, Lu Z, et al. Semaphorin 3A attenuates electrical remodeling at infarct border zones in rats after myocardial infarction. Tohoku J Exp Med. 2011;225:51–7. doi: 10.1620/tjem.225.51. http://www.ncbi.nlm.nih.gov/pubmed/21869591. [DOI] [PubMed] [Google Scholar]
- 161.Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431–488. doi: 10.1152/physrev.00025.2003. [DOI] [PubMed] [Google Scholar]
- 162.Lindsey ML. Matrix Metalloproteinase-7 Affects Connexin-43 Levels, Electrical Conduction, and Survival After Myocardial Infarction. Circulation. 2006;113:2919–2928. doi: 10.1161/CIRCULATIONAHA.106.612960. [DOI] [PubMed] [Google Scholar]
- 163.Wu X, Huang W, Luo G, Alain LA. Hypoxia induces connexin 43 dysregulation by modulating matrix metalloproteinases via MAPK signaling. Mol Cell Biochem. 2013;384:155–62. doi: 10.1007/s11010-013-1793-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lafontant PJ, Burns AR, Grivas Ja, Lesch Ma, Lala TD, Reuter SP, et al. The Giant Danio (D. Aequipinnatus) as A Model of Cardiac Remodeling and Regeneration. Anat Rec Adv Integr Anat Evol Biol. 2012;295:234–248. doi: 10.1002/ar.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.O’Gara PT, Kushner FG, Ascheim DD, Casey DE, Chung MK, de Lemos JA, et al. 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction. J Am Coll Cardiol. 2013;61:e78–e140. doi: 10.1016/j.jacc.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 166.Hammerman H, Kloner RA, Schoen FJ, Brown EJ, Hale S, Braunwald E. Indomethacin-induced scar thinning after experimental myocardial infarction. Circulation. 1983;67:1290–5. doi: 10.1161/01.cir.67.6.1290. http://www.ncbi.nlm.nih.gov/pubmed/6851023. [DOI] [PubMed] [Google Scholar]
- 167.Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am J Cardiol. 2003;91:1055–9. doi: 10.1016/s0002-9149(03)00148-6. http://www.ncbi.nlm.nih.gov/pubmed/12714146. [DOI] [PubMed] [Google Scholar]
- 168.Watanabe R, Ogawa M, Suzuki J, Hirata Y, Nagai R, Isobe M. A Comparison Between Imidapril and Ramipril on Attenuation of Ventricular Remodeling After Myocardial Infarction. J Cardiovasc Pharmacol. 2012;59:323–330. doi: 10.1097/FJC.0b013e3182422c1a. [DOI] [PubMed] [Google Scholar]
- 169.Palaniyappan A, Uwiera RRE, Idikio H, Jugdutt BI. Comparison of vasopeptidase inhibitor omapatrilat and angiotensin receptor blocker candesartan on extracellular matrix, myeloperoxidase, cytokines, and ventricular remodeling during healing after reperfused myocardial infarction. Mol Cell Biochem. 2009;321:9–22. doi: 10.1007/s11010-008-9905-3. [DOI] [PubMed] [Google Scholar]
- 170.Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–24. doi: 10.1161/hc2801.094031. http://www.ncbi.nlm.nih.gov/pubmed/11457751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bauersachs J, Galuppo P, Fraccarollo D, Christ M, Ertl G. Improvement of left ventricular remodeling and function by hydroxymethylglutaryl coenzyme a reductase inhibition with cerivastatin in rats with heart failure after myocardial infarction. Circulation. 2001;104:982–5. doi: 10.1161/hc3401.095946. http://www.ncbi.nlm.nih.gov/pubmed/11524389. [DOI] [PubMed] [Google Scholar]
- 172.Hasegawa H, Yamamoto R, Takano H, Mizukami M, Asakawa M, Nagai T, et al. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors prevent the development of cardiac hypertrophy and heart failure in rats. J Mol Cell Cardiol. 2003;35:953–60. doi: 10.1016/s0022-2828(03)00180-9. http://www.ncbi.nlm.nih.gov/pubmed/12878482. [DOI] [PubMed] [Google Scholar]
- 173.Abulhul E, McDonald K, Martos R, Phelan D, Spiers JP, Hennessy M, et al. Long-Term Statin Therapy in Patients With Systolic Heart Failure and Normal Cholesterol: Effects on Elevated Serum Markers of Collagen Turnover, Inflammation, and B-Type Natriuretic Peptide. Clin Ther. 2012;34:91–100. doi: 10.1016/j.clinthera.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 174.Elnakish MT, Hassona MDH, Alhaj MA, Moldovan L, Janssen PML, Khan M, et al. Rac-Induced Left Ventricular Dilation in Thyroxin-Treated ZmRacD Transgenic Mice: Role of Cardiomyocyte Apoptosis and Myocardial Fibrosis. PLoS One. 2012;7:e42500. doi: 10.1371/journal.pone.0042500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shiroshita-Takeshita A, Brundel BJJM, Burstein B, Leung TK, Mitamura H, Ogawa S, et al. Effects of simvastatin on the development of the atrial fibrillation substrate in dogs with congestive heart failure. Cardiovasc Res. 2007;74:75–84. doi: 10.1016/j.cardiores.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 176.Adiarto S, Heiden S, Vignon-Zellweger N, Nakayama K, Yagi K, Yanagisawa M, et al. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci. 2012;91:651–7. doi: 10.1016/j.lfs.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 177.Ammarguellat F, Larouche I, Schiffrin EL. Myocardial Fibrosis in DOCA-Salt Hypertensive Rats: Effect of Endothelin ETA Receptor Antagonism. Circulation. 2001;103:319–324. doi: 10.1161/01.CIR.103.2.319. [DOI] [PubMed] [Google Scholar]
- 178.Guarda E, Katwa LC, Myers PR, Tyagi SC, Weber KT. Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardiovasc Res. 1993;27:2130–4. doi: 10.1093/cvr/27.12.2130. http://www.ncbi.nlm.nih.gov/pubmed/8313419. [DOI] [PubMed] [Google Scholar]
- 179.Piacentini L, Gray M, Honbo NY, Chentoufi J, Bergman M, Karliner JS. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol. 2000;32:565–76. doi: 10.1006/jmcc.2000.1109. [DOI] [PubMed] [Google Scholar]
- 180.Singh AD, Amit S, Kumar OS, Rajan M, Mukesh N. Cardioprotective effects of bosentan, a mixed endothelin type A and B receptor antagonist, during myocardial ischaemia and reperfusion in rats. Basic Clin Pharmacol Toxicol. 2006;98:604–10. doi: 10.1111/j.1742-7843.2006.pto_405.x. [DOI] [PubMed] [Google Scholar]
- 181.Gupta SK, Saxena A, Singh U, Arya DS. Bosentan, the mixed ETA-ETB endothelin receptor antagonist, attenuated oxidative stress after experimental myocardial ischemia and reperfusion. Mol Cell Biochem. 2005;275:67–74. doi: 10.1007/s11010-005-1999-2. http://www.ncbi.nlm.nih.gov/pubmed/16335785. [DOI] [PubMed] [Google Scholar]
- 182.Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:347–354. doi: 10.1016/S0140-6736(04)16723-8. [DOI] [PubMed] [Google Scholar]
- 183.Prasad SK, Dargie HJ, Smith GC, Barlow MM, Grothues F, Groenning BA, et al. Comparison of the dual receptor endothelin antagonist enrasentan with enalapril in asymptomatic left ventricular systolic dysfunction: a cardiovascular magnetic resonance study. Heart. 2006;92:798–803. doi: 10.1136/hrt.2004.049734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rodríguez-Pascual F, Busnadiego O, González-Santamaría J. The profibrotic role of endothelin-1: Is the door still open for the treatment of fibrotic diseases? Life Sci. 2014;118:156–164. doi: 10.1016/j.lfs.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 185.De Mello WC, Specht P. Chronic blockade of angiotensin II AT1-receptors increased cell-to-cell communication, reduced fibrosis and improved impulse propagation in the failing heart. J Renin Angiotensin Aldosterone Syst. 2006;7:201–5. doi: 10.3317/jraas.2006.038. [DOI] [PubMed] [Google Scholar]
- 186.Shibasaki Y, Nishiue T, Masaki H, Tamura K, Matsumoto N, Mori Y, et al. Impact of the angiotensin II receptor antagonist, losartan, on myocardial fibrosis in patients with end-stage renal disease: assessment by ultrasonic integrated backscatter and biochemical markers. Hypertens Res. 2005;28:787–95. doi: 10.1291/hypres.28.787. [DOI] [PubMed] [Google Scholar]
- 187.Coker ML, Jolly JR, Joffs C, Etoh T, Holder JR, Bond BR, et al. Matrix metalloproteinase expression and activity in isolated myocytes after neurohormonal stimulation. Am J Physiol Heart Circ Physiol. 2001;281:H543–51. doi: 10.1152/ajpheart.2001.281.2.H543. http://www.ncbi.nlm.nih.gov/pubmed/11454555. [DOI] [PubMed] [Google Scholar]
- 188.Rouet-Benzineb P, Gontero B, Dreyfus P, Lafuma C. Angiotensin II induces nuclear factor- kappa B activation in cultured neonatal rat cardiomyocytes through protein kinase C signaling pathway. J Mol Cell Cardiol. 2000;32:1767–78. doi: 10.1006/jmcc.2000.1211. [DOI] [PubMed] [Google Scholar]
- 189.Wang TL, Yang YH, Chang H, Hung CR. Angiotensin II signals mechanical stretch-induced cardiac matrix metalloproteinase expression via JAK-STAT pathway. J Mol Cell Cardiol. 2004;37:785–94. doi: 10.1016/j.yjmcc.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 190.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GGL, Van Tassell BW, Robati R, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010;105:1371–1377.e1. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 191.Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A, et al. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail. 2010;12:319–22. doi: 10.1093/eurjhf/hfq017. [DOI] [PubMed] [Google Scholar]
- 192.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci. 2011;108:19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Van Tassell BW, Varma A, Salloum FN, Das A, Seropian IM, Toldo S, et al. Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J Cardiovasc Pharmacol. 2010;55:117–22. doi: 10.1097/FJC.0b013e3181c87e53. [DOI] [PubMed] [Google Scholar]
- 194.Cucoranu I. NAD(P)H Oxidase 4 Mediates Transforming Growth Factor- 1-Induced Differentiation of Cardiac Fibroblasts Into Myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 195.Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, et al. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–2312. doi: 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mukherjee R, Mingoia JT, Bruce JA, Austin JS, Stroud RE, Escobar GP, et al. Selective spatiotemporal induction of matrix metalloproteinase-2 and matrix metalloproteinase-9 transcription after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H2216–28. doi: 10.1152/ajpheart.01343.2005. [DOI] [PubMed] [Google Scholar]
- 197.Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez-Anaya A, et al. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999;99:3063–70. doi: 10.1161/01.cir.99.23.3063. http://www.ncbi.nlm.nih.gov/pubmed/10368126. [DOI] [PubMed] [Google Scholar]
- 198.Villarreal FJ, Griffin M, Omens J, Dillmann W, Nguyen J, Covell J. Early short-term treatment with doxycycline modulates postinfarction left ventricular remodeling. Circulation. 2003;108:1487–92. doi: 10.1161/01.CIR.0000089090.05757.34. [DOI] [PubMed] [Google Scholar]
- 199.Yarbrough WM, Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Escobar GP, et al. Matrix metalloproteinase inhibition modifies left ventricular remodeling after myocardial infarction in pigs. J Thorac Cardiovasc Surg. 2003;125:602–610. doi: 10.1067/mtc.2003.197. [DOI] [PubMed] [Google Scholar]
- 200.King MK, Coker ML, Goldberg A, McElmurray JH, Gunasinghe HR, Mukherjee R, et al. Selective matrix metalloproteinase inhibition with developing heart failure: effects on left ventricular function and structure. Circ Res. 2003;92:177–85. doi: 10.1161/01.res.0000052312.41419.55. http://www.ncbi.nlm.nih.gov/pubmed/12574145. [DOI] [PubMed] [Google Scholar]
- 201.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–42. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 202.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Koenig GC, Rowe RG, Day SM, Sabeh F, Atkinson JJ, Cooke KR, et al. MT1-MMP–Dependent Remodeling of Cardiac Extracellular Matrix Structure and Function Following Myocardial Infarction. Am J Pathol. 2012;180:1863–1878. doi: 10.1016/j.ajpath.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cieslik Ka, Trial J, Crawford JR, Taffet GE, Entman ML. Adverse fibrosis in the aging heart depends on signaling between myeloid and mesenchymal cells; role of inflammatory fibroblasts. J Mol Cell Cardiol. 2014;70:56–63. doi: 10.1016/j.yjmcc.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Burstein B, Libby E, Calderone A, Nattel S. Differential behaviors of atrial versus ventricular fibroblasts: A potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation. 2008;117:1630–1641. doi: 10.1161/CIRCULATIONAHA.107.748053. [DOI] [PubMed] [Google Scholar]
- 206.Kamkin A, Kiseleva I, Isenberg G. Activation and inactivation of a non-selective cation conductance by local mechanical deformation of acutely isolated cardiac fibroblasts. Cardiovasc Res. 2003;57:793–803. doi: 10.1016/S0008-6363(02)00775-7. [DOI] [PubMed] [Google Scholar]
- 207.Chatelier A, Mercier A, Tremblier B, Thériault O, Moubarak M, Benamer N, et al. A distinct de novo expression of Na v 1.5 sodium channels in human atrial fibroblasts differentiated into myofibroblasts. J Physiol. 2012;590:4307–4319. doi: 10.1113/jphysiol.2012.233593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Verheule S, Sato T, Everett T, Engle SK, Otten D, Rubart-von der Lohe M, et al. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res. 2004;94:1458–65. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chong JJH, Yang X, Don CW, Minami E, Liu YW, Weyers JJ, et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273–277. doi: 10.1038/nature13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dolnikov K, Shilkrut M, Zeevi-Levin N, Gerecht-Nir S, Amit M, Danon A, et al. Functional properties of human embryonic stem cell-derived cardiomyocytes: intracellular Ca2+ handling and the role of sarcoplasmic reticulum in the contraction. Stem Cells. 2006;24:236–45. doi: 10.1634/stemcells.2005-0036. [DOI] [PubMed] [Google Scholar]
- 211.Liu J, Fu JD, Siu CW, Li RA. Functional sarcoplasmic reticulum for calcium handling of human embryonic stem cell-derived cardiomyocytes: insights for driven maturation. Stem Cells. 2007;25:3038–44. doi: 10.1634/stemcells.2007-0549. [DOI] [PubMed] [Google Scholar]
- 212.LeGrice I, Pope A, Smaill B. The architecture of the heart: myocyte organization and the cardiac extracellular matrix. In: Villarreal FJ, editor. Interstitial Fibros Hear Fail V 253 Dev Cardiovasc Med. Springer; New York: 2005. pp. 3–21. [DOI] [Google Scholar]
- 213.Hales PW, Schneider JE, Burton RA, Wright BJ, Bollensdorff C, Kohl P. Histo-anatomical structure of the living isolated rat heart in two contraction states assessed by diffusion tensor MRI. Prog Biophys Mol Biol. 2012;110:319–330. doi: 10.1016/j.pbiomolbio.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rutherford SL, Trew ML, Sands GB, Legrice IJ, Smaill BH. High-resolution 3-dimensional reconstruction of the infarct border zone: Impact of structural remodeling on electrical activation. Circ Res. 2012;111:301–311. doi: 10.1161/CIRCRESAHA.111.260943. [DOI] [PubMed] [Google Scholar]
- 215.Dillon SM, Allessie MA, Ursell PC, Wit AL. Influences of anisotropic tissue structure on reentrant circuits in the epicardial border zone of subacute canine infarcts. Circ Res. 1988;63:182–206. doi: 10.1161/01.res.63.1.182. http://www.ncbi.nlm.nih.gov/pubmed/3383375. [DOI] [PubMed] [Google Scholar]
- 216.Arenal A, Hernandez J, Perez-David E, Rubio-Guivernau JL, Ledesma-Carbayo MJ, Fernandez-Aviles F. Do the spatial characteristics of myocardial scar tissue determine the risk of ventricular arrhythmias? Cardiovasc Res. 2012;94:324–332. doi: 10.1093/cvr/cvs113. [DOI] [PubMed] [Google Scholar]
- 217.Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, et al. Myocardial infarct size and ventricular function in rats. Circ Res. 1979;44:503–512. doi: 10.1161/01.RES.44.4.503. [DOI] [PubMed] [Google Scholar]
- 218.Masci P, Ganame J, Francone M, Desmet W, Lorenzoni V, Iacucci I, et al. Relationship between location and size of myocardial infarction and their reciprocal influences on post-infarction left ventricular remodeling. J Cardiovasc Magn Reson. 2011;13:P84. doi: 10.1186/1532-429X-13-S1-P84. [DOI] [PubMed] [Google Scholar]
- 219.Zimmer HG, Gerdes AM, Lortet S, Mall G. Changes in heart function and cardiac cell size in rats with chronic myocardial infarction. J Mol Cell Cardiol. 1990;22:1231–43. doi: 10.1016/0022-2828(90)90060-f. http://www.ncbi.nlm.nih.gov/pubmed/2149393. [DOI] [PubMed] [Google Scholar]
- 220.U.S. National Library of Medicine. NHLBI health topics: Heart attack. PubMed Heal. 2014 http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0062989/
- 221.Richardson WJ, Holmes JW. Why Is Infarct Expansion Such an Elusive Therapeutic Target? J Cardiovasc Transl Res. 2015;8:421–30. doi: 10.1007/s12265-015-9652-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Barandon L, Couffinhal T, Ezan J, Dufourcq P, Costet P, Alzieu P, et al. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation. 2003;108:2282–9. doi: 10.1161/01.CIR.0000093186.22847.4C. [DOI] [PubMed] [Google Scholar]
- 223.Kobayashi K, Luo M, Zhang Y, Wilkes DC, Ge G, Grieskamp T, et al. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat Cell Biol. 2009;11:46–55. doi: 10.1038/ncb1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Laeremans H, Hackeng TM, van Zandvoort MAMJ, Thijssen VLJL, Janssen BJA, Ottenheijm HCJ, et al. Blocking of frizzled signaling with a homologous peptide fragment of wnt3a/wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation. 2011;124:1626–35. doi: 10.1161/CIRCULATIONAHA.110.976969. [DOI] [PubMed] [Google Scholar]
- 225.Heusch G, Bøtker HE, Przyklenk K, Redington A, Yellon D. Remote ischemic conditioning. J Am Coll Cardiol. 2015;65:177–95. doi: 10.1016/j.jacc.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.McDowell KS, Arevalo HJ, Maleckar MM, Trayanova Na. Susceptibility to arrhythmia in the infarcted heart depends on myofibroblast density. Biophys J. 2011;101:1307–1315. doi: 10.1016/j.bpj.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ashihara T, Haraguchi R, Nakazawa K, Namba T, Ikeda T, Nakazawa Y, et al. The Role of Fibroblasts in Complex Fractionated Electrograms During Persistent/Permanent Atrial Fibrillation: Implications for Electrogram-Based Catheter Ablation. Circ Res. 2012;110:275–284. doi: 10.1161/CIRCRESAHA.111.255026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.McDowell KS, Vadakkumpadan F, Blake R, Blauer J, Plank G, MacLeod RS, et al. Methodology for patient-specific modeling of atrial fibrosis as a substrate for atrial fibrillation. J Electrocardiol. 2012;45:640–645. doi: 10.1016/j.jelectrocard.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gonzales MJ, Vincent KP, Rappel WJ, Narayan SM, McCulloch AD. Structural contributions to fibrillatory rotors in a patient-derived computational model of the atria. Europace. 2014;16:iv3–iv10. doi: 10.1093/europace/euu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.McDowell KS, Zahid S, Vadakkumpadan F, Blauer J, MacLeod RS, Trayanova NA. Virtual Electrophysiological Study of Atrial Fibrillation in Fibrotic Remodeling. PLoS One. 2015;10:e0117110. doi: 10.1371/journal.pone.0117110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Fast VG, Kléber G. Microscopic conduction in cultured strands of neonatal rat heart cells measured with voltage-sensitive dyes. Circ Res. 1993;73:914–925. doi: 10.1161/01.RES.73.5.914. [DOI] [PubMed] [Google Scholar]
- 232.Fast VG, Kléber AG. Cardiac tissue geometry as a determinant of unidirectional conduction block: assessment of microscopic excitation spread by optical mapping in patterned cell cultures and in a computer model. Cardiovasc Res. 1995;29:697–707. http://www.ncbi.nlm.nih.gov/pubmed/7606760. [PubMed] [Google Scholar]
- 233.Fomovsky GM, Macadangdang JR, Ailawadi G, Holmes JW. Model-based design of mechanical therapies for myocardial infarction. J Cardiovasc Transl Res. 2011;4:82–91. doi: 10.1007/s12265-010-9241-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wall ST, Walker JC, Healy KE, Ratcliffe MB, Guccione JM. Theoretical impact of the injection of material into the myocardium: a finite element model simulation. Circulation. 2006;114:2627–35. doi: 10.1161/CIRCULATIONAHA.106.657270. [DOI] [PubMed] [Google Scholar]
- 235.Rouillard AD, Holmes JW. Mechanical regulation of fibroblast migration and collagen remodelling in healing myocardial infarcts. J Physiol. 2012;590:4585–4602. doi: 10.1113/jphysiol.2012.229484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Christman KL, Fok HH, Sievers RE, Fang Q, Lee RJ. Fibrin Glue Alone and Skeletal Myoblasts in a Fibrin Scaffold Preserve Cardiac Function after Myocardial Infarction. Tissue Eng. 2004;10:403–409. doi: 10.1089/107632704323061762. [DOI] [PubMed] [Google Scholar]
- 237.Ifkovits JL, Tous E, Minakawa M, Morita M, Robb JD, Koomalsingh KJ, et al. Injectable hydrogel properties influence infarct expansion and extent of postinfarction left ventricular remodeling in an ovine model. Proc Natl Acad Sci. 2010;107:11507–11512. doi: 10.1073/pnas.1004097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Rane AA, Christman KL. Biomaterials for the Treatment of Myocardial Infarction. J Am Coll Cardiol. 2011;58:2615–2629. doi: 10.1016/j.jacc.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 239.Purcell BP, Lobb D, Charati MB, Dorsey SM, Wade RJ, Zellars KN, et al. Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat Mater. 2014;13:653–661. doi: 10.1038/nmat3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Eckhouse SR, Purcell BP, McGarvey JR, Lobb D, Logdon CB, Doviak H, et al. Local Hydrogel Release of Recombinant TIMP-3 Attenuates Adverse Left Ventricular Remodeling After Experimental Myocardial Infarction. Sci Transl Med. 2014;6:223ra21–223ra21. doi: 10.1126/scitranslmed.3007244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zavadzkas JA, Stroud RE, Bouges S, Mukherjee R, Jones JR, Patel RK, et al. Targeted overexpression of tissue inhibitor of matrix metalloproteinase-4 modifies post-myocardial infarction remodeling in mice. Circ Res. 2014;114:1435–45. doi: 10.1161/CIRCRESAHA.114.303634. [DOI] [PMC free article] [PubMed] [Google Scholar]