

Abstract
Objective:
We describe 2 additional patients with early-onset epilepsy with a de novo FGF12 mutation.
Methods:
Whole-exome sequencing was performed in 2 unrelated patients with early-onset epilepsy and their unaffected parents. Genetic variants were assessed by comparative trio analysis. Clinical evolution, EEG, and neuroimaging are described. The phenotype and response to treatment was reviewed and compared to affected siblings in the original report.
Results:
We identified the same FGF12 de novo mutation reported previously (c.G155A, p.R52H) in 2 additional patients with early-onset epilepsy. Similar to the original brothers described, both presented with tonic seizures in the first month of life. In the first patient, seizures responded to sodium channel blockers and her development was normal at 11 months. Patient 2 is a 15-year-old girl with treatment-resistant focal epilepsy, moderate intellectual disability, and autism. Carbamazepine (sodium channel blocker) was tried later in her course but not continued due to an allergic reaction.
Conclusions:
The identification of a recurrent de novo mutation in 2 additional unrelated probands with early-onset epilepsy supports the role of FGF12 p.R52H in disease pathogenesis. Affected carriers presented with similar early clinical phenotypes; however, this report expands the phenotype associated with this mutation which contrasts with the progressive course and early mortality of the siblings in the original report.
High-throughput sequencing technology, including whole-exome sequencing (WES), has revolutionized gene discovery in epilepsy. It has also demonstrated the significant genetic heterogeneity of epilepsy and the importance of de novo mutations in the epileptic encephalopathies,1 which are the most severe forms of epilepsy. Recently, WES in a family quintet with 2 affected siblings with early-onset epileptic encephalopathy (EOEE) and cerebellar atrophy identified fibroblast growth factor 12 (FGF12, also known as FHF1) as a putative gene responsible.2 A de novo gain-of-function variant (p.R52H) was found in 2 affected siblings with lethal disease. FGF12 encodes a cytosolic protein that interacts with neuronal sodium channels and increases their voltage-dependent fast inactivation.3 Here, we performed WES-trio analysis and identified the same de novo missense mutation in 2 patients with severe neonatal-onset epilepsy and variable clinical outcomes.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was approved by site-specific Institutional Review Boards, and informed consent was obtained from all individuals.
Genetic analysis.
Peripheral blood samples were collected from the proband and her parents. Genomic DNA was extracted from peripheral blood lymphocytes following standard protocols. Patient 1 exonic regions were captured using the Ion AmpliSeq Exome Kit (57.7 Mb), and WES was performed on the Ion Proton System according to manufacturers' recommendations (Life Technologies, Carlsbad, CA). Candidate variants were validated by Sanger sequencing as previously described.4 Primer sequences and PCR conditions are available on request. Patient 2 was identified through GeneMatcher (genematcher.org/).5 WES and data analysis were performed as part of a trio-based clinical sequencing study at HudsonAlpha. Exome capture was completed using Nimblegen SeqCap EZ Exome v3 (Roche, Basel, Switzerland), and sequencing was conducted on the Illumina HiSeq 2000 (Illumina, San Diego, CA). Sanger confirmation of variants, including confirmation of absence from both biological parents, was performed by a College of American Pathologists/Clinical Laboratory Improvement Amendments–certified or Diagnostic Accreditation Program–accredited diagnostic laboratory.
RESULTS
Family trio analysis identified the same FGF12 missense mutation in both patients, identical to the variant reported previously.2 The mutation is a de novo heterozygous G>A transition at position 155 of the complementary DNA (NM_004113) which results in a p.R52H missense substitution. Sanger sequencing was performed in both trios and confirmed that the mutation was only present in the affected probands. No other pathogenic mutations in known epilepsy genes were present.
Clinical phenotypes.
Patient 1 is a developmentally normal 11-month-old baby, born at term after an uncomplicated pregnancy. She presented on day 3 of life with a 2-minute seizure involving extension of all limbs, head, and eye deviation to the right side and apnea with desaturation. On day 3, the interictal EEG showed multifocal spikes. During continuous video-EEG monitoring on day 8, multiple independent left and right frontal seizures were recorded. Seizures occurred from sleep, and 1 typical seizure with corresponding EEG is shown in the video at Neurology.org/ng and in the figure. Seizures continued despite treatment with phenobarbital, levetiracetam, topiramate, and a midazolam infusion. She required intubation and ventilation for 3 days. Her seizures stopped on day 20 of age after the addition of phenytoin. She had 1–3 seizures per month on phenytoin and topiramate. Carbamazepine was added to topiramate at 6 weeks of age, and she has been seizure free since 5 months of age on carbamazepine (30 mg/kg/d). Her initial neurologic examination showed mild hypotonia which later resolved. Her development remains normal. Her head circumference remains at the 85th percentile, and her EEG performed at 10 months of age was normal. Extensive neurometabolic investigations and a head MRI performed at day 4 of life were unremarkable.
Figure. Patient 1: Ictal EEG.

Seizure began with arousal from sleep and extension of all 4 limbs followed by clonic jerking of the right arm and bradycardia (see video). Ictal EEG demonstrated (A) generalized attenuation of EEG activity at seizure onset, followed by rhythmic spikes in left central area; (B) spread to right central area; (C) spread to bilateral central frontal parietal regions; and (D) postictal suppression.
Patient 2 is a 15-year-old girl, born at term following an uncomplicated pregnancy and delivery. She presented on day 2 of life with tonic seizures characterized by generalized stiffening which evolved to intermittent stiffening and twitching of the limbs. On day 11, her EEG demonstrated suppression of the background and multifocal spikes. Seizures did not respond to phenobarbital, clonazepam, topiramate, and lamotrigine. At 4 years of age, she developed focal seizures with impaired awareness which could evolve to bilateral tonic-clonic seizures. The interictal EEG showed mild background slowing and left frontotemporal spikes. Carbamazepine was tried at 4 years of age; however, it was stopped immediately following an allergic reaction. Rufinamide was added to lamotrigine at the age of 8 years and at most recent follow-up, the patient has been seizure free for 1 year. Developmental delay was noted by 1 year of age, and her development regressed at times of increased seizure frequency. She currently has moderate intellectual disability and autism. MRI of the brain at 2 weeks and 2 years of age was normal except for an incidental Chiari I malformation.
DISCUSSION
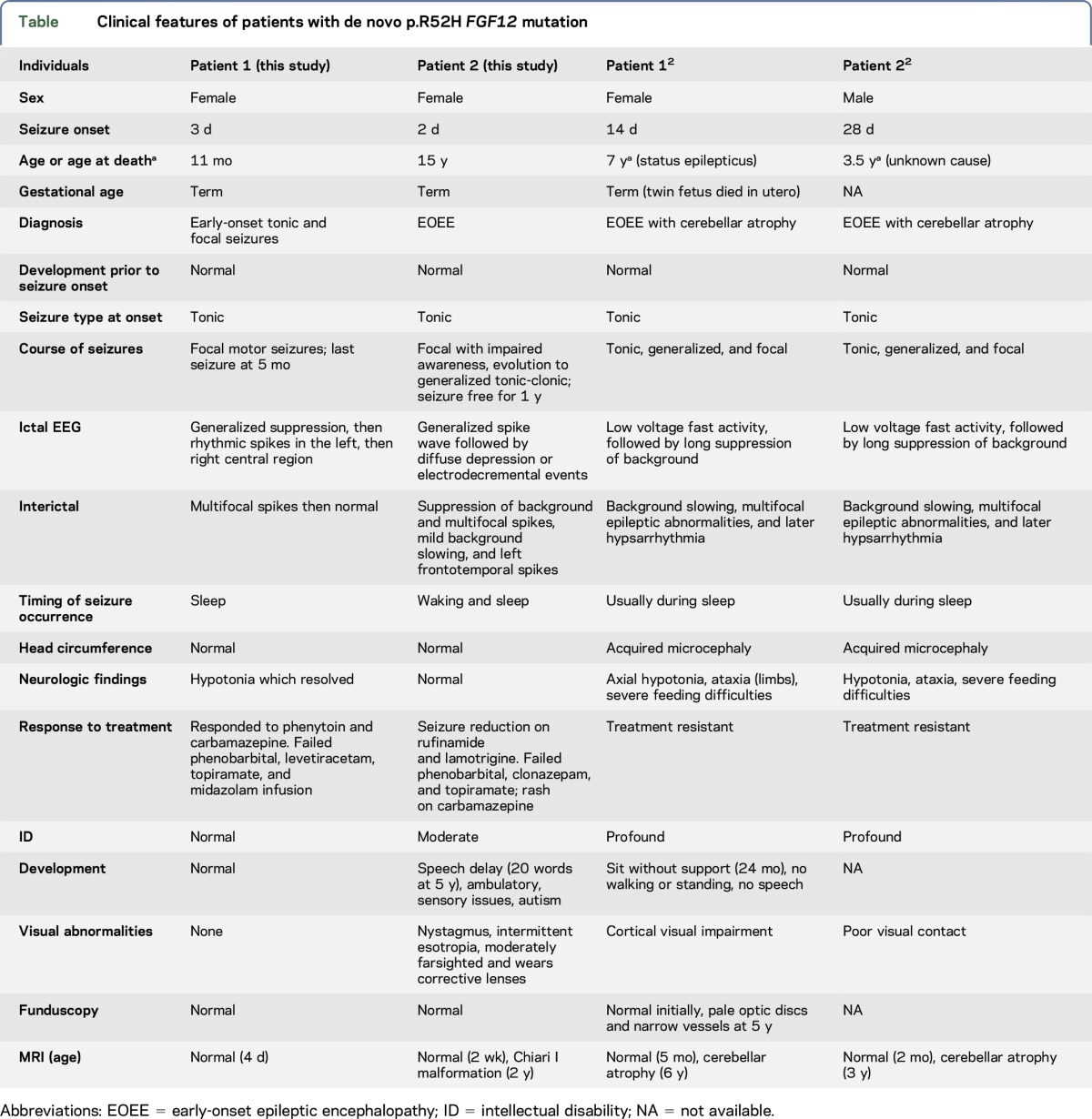
In this report, we describe 2 additional patients with a de novo p.R52H mutation in FGF12 but with milder phenotypes. The same variant observed in our patients was recently described in 2 siblings with a lethal disorder characterized by EOEE and cerebellar atrophy.2 All 4 affected individuals presented with early-onset tonic seizures and most had seizures from sleep (table). Patient 1 does not have an epileptic encephalopathy given the cessation of seizures, normal development, and normal EEG. However, patient 2 and the original reported clinical features of the siblings2 are consistent with an epileptic encephalopathy. In all cases, seizures were refractory to multiple antiseizure medications. However, patient 1 had a good response to sodium channel blockers and has been seizure free since 5 months of age. In patient 2, carbamazepine resulted in an allergic reaction and phenytoin was not tried. Patient 2 is currently seizure free on a combination of lamotrigine and rufinamide, which is possibly due to the effects of these drugs on the sodium channel. The current neurologic examination and development are normal for patient 1. This contrasts with the report of the siblings, which indicates a neurodegenerative course associated with early death. The clinical course of patient 2 also is not as severe as that of the siblings; however, her seizures were refractory to treatment except for the past year, and she did have developmental regression associated with increased seizure activity. She also has intellectual disability and autism. In the original report,2 progressive cerebellar atrophy was also a prominent feature of the disease phenotype which differs from the normal MRI seen in patient 1 and the incidental Chiari I malformation identified on MRI in patient 2. It is difficult to accurately compare MRI findings given the young age of patient 1; however, MRI of patient 2 showed no sign of cerebellar atrophy at age 2. The milder phenotype with normal clinical outcome seen in patient 1 may be influenced by the early seizure control. However, other genetic and/or environmental factors may also be playing a role. Although the young age of patient 1 limits a clear impression of the natural history of this disorder, there is a striking clinical difference between patient 1 normal clinical course to date and moderate to severe course of patient 2 and the lethal outcome of the previously described siblings. Early seizure control in patient 1 suggests possible important treatment implications for this genetic disorder which could affect outcome.
Table.
Clinical features of patients with de novo p.R52H FGF12 mutation
During the preparation of this manuscript, we became aware of 3 additional patients with an epileptic encephalopathy carrying the same FGF12 de novo mutation.6 The identification of a recurrent de novo mutation in 6 unrelated patients all presenting with early-onset epilepsy strongly supports the involvement of FGF12 in disease pathogenesis.7,8 Moreover, the identification of additional patients carrying the same FGF12 variant broadens the phenotypic spectrum associated with this gene.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the parents and children involved. Clinical research was performed as part of the EPGEN program on the genetics of refractory seizure disorders at Children's Hospital and the University of British Columbia. They thank the broader EPGEN team of health care professionals (including Cyrus Boelman, Corneliu Bolbocean, Tara Candido, Patrice Eydoux, William Gibson, Gabriella Horvath, Clara van Karnebeek, Graham Sinclair, Tamsin Tarling, Katelin N. Townsend, and Suzanne Vercauteren) and to the Division of Genetics & Genomics, Department of Pathology & Laboratory Medicine, BC Children's Hospital for providing clinical confirmation of identified mutations. They thank members of the HudsonAlpha CSER team, especially Shirley Simmons, Kelly East, Whitley Kelley, Kevin Bowling, Susan Hiatt, Candice Finnila, David Gray, and Michelle Amaral, for help with sequencing data analysis. They appreciate the support of the Canada Excellence Research Chair (M.J.F.) and the Alva Foundation (M.D.).
GLOSSARY
- EOEE
early-onset epileptic encephalopathy
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Dr. Ilaria Guella: conception and design of the study; analysis and interpretation of data; and drafting the manuscript. Dr. Linda Huh: clinical assessment and data acquisition for patient 1. Ms. Marna B. McKenzie: execution of the study; acquisition and analysis of genetic data for patient 1; and drafting the manuscript. Mr. Eric B. Toyota: data acquisition. Dr. E. Martina Bebin: recruitment and clinical characterization of patient 2 and collection of biological samples. Dr. Michelle L. Thompson: acquisition and analysis of genetic data for patient 2. Dr. Gregory M. Cooper: design of the study for patient 2 and guided implementation across all components. Mr. Daniel M. Evans: bioinformatics and data analysis. Dr. Sarah Buerki: data acquisition and revising manuscript. Ms. Shelin Adam: review of literature. Dr. Margot I. Van Allen and Dr. Tanya N. Nelson: review of literature and revising manuscript. Dr. Mary B. Connolly: review of patient 1 clinically; revising manuscript; and obtaining funding. Dr. Matthew J. Farrer: conception and design of the study; obtaining funding; and revising manuscript. Dr. Michelle Demos: conception and design of the study; review of patient 1 clinically; obtaining funding; and revising manuscript.
STUDY FUNDING
Supported by Canada Excellence Research Chair (M.J.F.), the Alva Foundation, and US National Human Genome Research Institute (NHGRI) (UM1HG007301) (G.M.C.).
DISCLOSURE
Dr. Ilaria Guella reports no disclosures. Dr. Linda Huh has served on the scientific advisory boards of the British Columbia Epilepsy Society and Matthew's Friends (nonprofit). Ms. Marna B. McKenzie and Mr. Eric B. Toyota report no disclosures. Dr. E. Martina Bebin has served on scientific advisory boards for Novartis, GW Pharmaceuticals, FDA Orphan Drug Program, and NIH; has received gifts from Novartis and GW Pharmaceuticals; has received travel funding/speaker honoraria from GW Pharmaceuticals; has served on the editorial board of Pediatric Neurology; has been a consultant for and served on the speakers' bureau of GW Pharmaceuticals; and has received research support from NIH. Dr. Michelle L. Thompson reports no disclosures. Dr. Gregory M. Cooper has served on the editorial board of PLoS Genetics Genome Research and has received research support from NIH. Mr. Daniel M. Evans, Dr. Sarah E. Buerki, Ms. Shelin Adam, and Dr. Margot I. Van Allen report no disclosures. Dr. Tanya N. Nelson has received research support from the BCCH Foundation and Genome BC. Dr. Mary B. Connolly has served on the editorial board of the Canadian Journal of Neurological Sciences and has served on speakers' bureaus for Esai and Biocodex. Dr. Matthew J. Farrer has served on the scientific advisory boards of the Michael J. Fox Foundation, Parkinson's Society Canada, EURAC, and Parkinson's UK; has served on the editorial boards of Neurobiology of Disease and Parkinsonism & Related Disorders; holds the following patents: (1) International Publication Number WO 2006/045392 A2; (2) International Publication Number WO 2006/068492 A1; (3) US Patent Number 7,544,786; and (4) Norwegian patent 323175—provisionally filed in 2004–2005; has received research support from the Canadian Federal Government (CERC, CFI, and CIHR) and the Cunhill Foundation; and has received royalty payments from Lundbeck Inc. and Merck for Lrrk2 mouse models (2016), and from Isis Pharma. for SNCA mouse model (2015). Dr. Michelle Demos has received research support from the Rare Disease Foundation and the Alva Foundation. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siekierska A, Isrie M, Liu Y, et al. Gain-of-function FHF1 mutation causes early-onset epileptic encephalopathy with cerebellar atrophy. Neurology 2016;86:2162–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldfarb M, Schoorlemmer J, Williams A, et al. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron 2007;55:449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steele JC, Guella I, Szu-Tu C, et al. Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol 2015;77:458–468. [DOI] [PubMed] [Google Scholar]
- 5.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015;36:928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Mehmadi S, Splitt M, Venkateswaran R, et al. The FHF1 (FGF12) epileptic encephalopathy. Neurol Genet 2016. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014;508:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.