

Abstract
Prognosis of primary refractory and relapsed pediatric B-lineage acute lymphoblastic leukemia (ALL) is very poor. Relapse rates significantly correlate with persistent minimal residual disease (MRD). In MRD, favorable effector-target ratios prevail and thus this situation might be optimally suited for immunotherapy with antibodies recruiting immunological effector cells. We here report on the generation, preclinical characterization and first clinical application in B-lineage ALL of an Fc-optimized CD19 antibody. This third-generation antibody (4G7SDIE) mediated enhanced antibody-dependent cellular cytotoxicity (ADCC) against leukemic blasts with effector cells from healthy volunteers and B-lineage ALL patients. The antibody was produced in a university-owned production unit and was applied on a compassionate use basis to 14 pediatric patients with refractory and relapsed B-lineage ALL at the stage of MRD. In 10/14 patients, MRD was reduced by ≥ 1 log or below the patient-individual detection limit, and 5/14 patients have achieved ongoing complete molecular remission with a median leukemia-free survival of 428 days. Two additional patients died in complete molecular remission due to complications not related to antibody therapy. Besides profound in vivo B-cell depletion, side effects were negligible. A clinical phase 1/2 study to further assess the therapeutic activity of 4G7SDIE is in preparation.
Introduction
Pediatric B-lineage acute lymphoblastic leukemia (ALL) is the most common malignancy in childhood. Although well-designed clinical trials have led to excellent survival rates, patients with refractory or relapsed disease remain a therapeutic challenge.1,2,3 Persistent or increasing minimal residual disease (MRD) has already been shown to be an important adverse prognostic factor for those patients within chemotherapy protocols. Prior to allogeneic hematopoietic stem cell transplantation (HSCT), low or undetectable levels of MRD appear to improve clinical outcome and several attempts are currently evaluated to eradicate or reduce persistent MRD by additional chemotherapy cycles.4,5,6,7,8 Moreover, rising MRD levels after HSCT have recently been suggested to predict hematological relapse.9 In general, relapse after HSCT is associated with a poor prognosis although in some cases, retransplantation can be a curative option.10,11,12 In these cases, however, induction chemotherapy is often associated with high toxicity and limited efficiency, due to the development of multidrug resistance.13 Therefore, the implementation of new and efficient treatment strategies with low toxicity is urgently needed.14,15
The CD20-specific chimeric monoclonal antibody rituximab was the first antibody demonstrating convincing activity against human cancer and has considerably improved the treatment of B-cell-derived lymphomas. Currently the Fc-engineered CD20 antibody obinutuzumab (GA101) is being evaluated in large clinical trials (e.g., NCT01300247) and has demonstrated superior therapeutic activity against CLL.16 In particular, the antibody has achieved a higher response rate in relapsed CLL patients compared to rituximab.16,17 It is particularly noteworthy that the percentage of patients achieving deep remissions was markedly increased in the group treated with the Fc-optimized antibody.17
However, only a minority of pediatric B-lineage ALLs uniformly express CD20.18,19,20 This restricts the potential use of CD20 targeting antibodies in these patients. In contrast, CD19 is consistently expressed on childhood B-lineage ALL blasts.18,21 Two CD19 antibodies are already used in clinical trials.18,22,23,24 These antibodies are Fc-optimized either by genetic modification of the glycosylation pattern (MEDI-551) or by the exchange of defined amino acids in the CH2 domain of the human Fc-part of IgG1 (MOR00208).25,26 Both antibodies have been demonstrated to exert markedly enhanced ADCC activity compared to the “second generation” non-Fc-optimized counterparts. MEDI-551, lacking fucosylation in the Fc-part, is being evaluated in a clinical trial of adults with relapsed and refractory B-cell malignancies (NCT00983619). The CD19 antibody MOR00208, a humanized version of the 4G7-antibody, was Fc-optimized by two amino acid modifications (S239D and I332E, SDIE modification).22,23 MOR00208 has demonstrated some efficacy in relapsed CLL27 and is currently evaluated in a clinical trial with adult B-ALL patients (NCT01685021).
Since, in MRD, a favorable ratio of leukemic target cells and FcR-expressing effector cells prevails, we reasoned that this situation might be particularly suited for treatment with Fc-optimized antibodies.
Here, we report on the generation, preclinical characterization, and clinical application of a chimerized SDIE-modified CD19 antibody, termed 4G7SDIE. The antibody was produced in pharmaceutical quality and quantity in a university-owned production unit and applied to B-lineage ALL patients with persistent MRD on a compassionate need basis. The aim was to significantly reduce the MRD load during the course of treatment with low toxicity prior to or after HSCT or both, with the intention to minimize relapse rates in this high-risk situation.
Results
In vitro activity of 4G7SDIE
Generation of the chimerized Fc-optimized CD19 antibody 4G7SDIE and the chimerized IgG1 counterpart χ4G7 have been described previously.28 For this study, 4G7SDIE was produced in pharmaceutical quality in a university-owned production unit.
CD19 expression, on primary B-lineage ALL blasts, was analyzed quantitatively on 23 samples, including 5 samples of 4G7SDIE-treated patients (Figure 1a). CD19 was expressed on all B-lineage ALL blasts with a mean of 1.5 × 104 molecules per cell (range 4.5 × 103 to 2.4 × 104). In Supplementary Figure S2a, half-maximal target cell lysis of leukemic blasts with peripheral blood mononuclear cell (PBMC) of three healthy volunteers (HV) is depicted. EC50 was 65 ng/ml (± 55) and saturating concentrations were reached below 1 µg/ml. In CD107a-degranulation assays, a significantly increased CD107a, TNFα-, and IFNγ-expression was observed on NK cells when incubated with target cells and Fc-optimized antibody 4G7SDIE compared to target cells alone (Supplementary Figure S2b). The chimerized antibody χ4G7 induced neither an increase in CD107a-, nor in intracellular TNFα- and IFNγ-expression. In real-time cytotoxicity assays (xCELLigence) with an adherent CD19-expressing cell line (MCF-7-CD19) and PBMC of 12 different healthy volunteers (HV), no ADCC was induced by χ4G7 whereas 4G7SDIE mediated strong ADCC (Figure 1b). Specific lysis of primary B-lineage ALL blasts of five different patients by PBMC of one HV was increased by 4G7SDIE in the presence of autologous serum but heterogeneous between different patient samples (Figure 1c). That a correlation of CD19-expression on leukemic blasts and 4G7SDIE-mediated lysis nevertheless exists is demonstrated in Figure 1d where MCF7 cells transfected with different amounts of CD19 were used as ADCC targets.
Figure 1.

In vitro characterization of 4G7SDIE and B-lineage acute lymphoblastic leukemia (ALL) blasts. (a) B-lineage ALL blasts (n = 18 patients (black), n = 5 4G7SDIE-treated patients (hatched)) were incubated with murine 4G7 (5 µg/ml), washed and bound antibody was detected by indirect immunofluorescence staining and flow cytometry. CD19 molecules/cell were calculated by comparison with calibrated beads (QIFIKIT). (b) A MCF-7-CD19-transfectant expressing 1.9 × 104 CD19 molecules/cell was incubated with PBMC (n = 12 HVs) and medium, 1 µg/ml of 4G7SDIE or 1 µg/ml χ4G7 and analyzed in xCELLigence assays. (c) B-lineage ALL blasts (n = 5) were incubated with PBMC of one HV (E:T 20:1), autologous serum, medium, and 1 µg/ml 4G7SDIE, respectively. Specific lysis was measured in 2 h-EuTDA assays. (d) ADCC with PBMC (E:T 20:1) from 3 HVs was assessed in xCELLigence assays with 4G7SDIE (2.5 µg/ml) and MCF-7-CD19-transfectants expressing the indicated levels of CD19 molecules/cell. ADCC after 10 hours was depicted and correlation was assessed using Spearman's rank correlation coefficient.
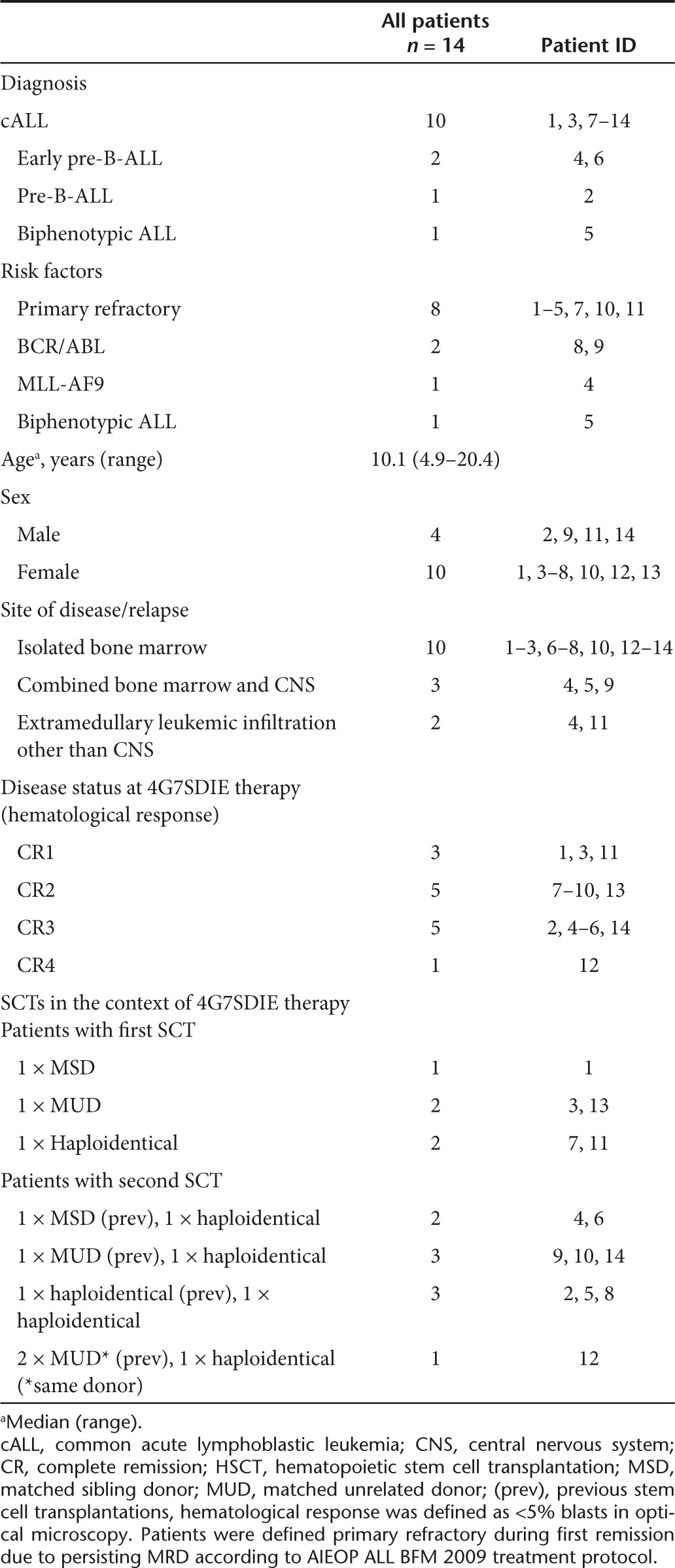
Patient characteristics
A cohort of 14 MRD-positive patients, suffering from primary refractory and/or relapsed CD19-positive B-lineage ALL, at very high risk of relapse were treated with 4G7SDIE to reduce or eradicate residual leukemic cells and prevent recurrence. Only three patients were treated in complete remission 1 (CR1), five patients in CR2 and six patients in ≥ CR3. Patient characteristics are listed in Table 1. Besides refractory leukemia and/or leukemia recurrence, adverse prognostic factors were early post-HSCT relapse, unfavorable genetic rearrangements including BCR/ABL and MLL-AF9, combined bone marrow and central nervous system relapse (#4, #5, #9) as well as extramedullary leukemic manifestation other than central nervous system (#4, #11).
Table 1. Patient characteristics.
Treatment
3/14 patients (# 1, #3, #11) received 4G7SDIE in CR1, 11/14 patients in ≥ CR2. 5/14 patients (#1, #3, #7, #11, #13) received the first 4G7STabDIE application in the autologous immune state prior to allogeneic HSCT, and 9/14 patients received the first 4G7SDIE infusion in the allogeneic immune state post-HSCT (Table 2). 11/14 patients received the 4G7SDIE mAb in the course of treatment before and after the corresponding allogeneic HSCT. Two patients (#2 and #10) and 1 patient (#13) received 4G7SDIE only post- or pre-allotransplant, respectively (Table 2, Supplementary Table S2). Patients received 1 to 40 applications at 5 to 60 mg/m2, every 2 or 4 weeks with exceptions. Prior to the corresponding HSCT, a median number of 2 (range: 1–18) infusions were applied with low toxicity. Post-HSCT, a rather long-lasting application schedule with a median number of 10 infusions (range: 2–41) was applied to reduce the MRD load and to prevent relapse with a maximum duration of mAb infusions up to 2 years (1st year: every 2 weeks; 2nd year: every 4 weeks). To reduce the MRD load 1 to 6, 4G7SDIE mAb infusions were necessary at 5–60 mg/m2 (Table 2, Figure 4).
Table 2. Overview and outcome.

All patients received myeloablative conditioning regimens for allogeneic HSCT. 5/14 patients received first HSCT and 9/14 patients ≥ 2nd HSCT. Donor source was diverse and comprised matched unrelated, matched related, and mismatched haploidentical-related donors (Table 1). 12/14 patients did not receive any other anti-leukemic treatment during 4G7SDIE therapy. 2/14 patients (#8, #9) with Ph+ leukemia were treated with tyrosine kinase inhibitors for several months before, during and after 4G7SDIE application.
Monitoring of 4G7SDIE-treated patients
Cytolysis of pediatric CD19+ pre-B ALL cell line MHH-cALL-4 by PBMC of 5 4G7SDIE-treated patients was assessed in 2 hours cytotoxicity assays (Figure 2a). Either serum of HVs, saturating concentrations of 4G7SDIE (1 µg/ml) diluted in serum of HVs or autologous patient serum taken 1 hour after antibody infusion, were used for the tests. The 4G7SDIE serum concentrations 1 hour after 4G7SDIE infusion were ≥ 10 µg/ml (Supplementary Table S1). In three out of five patient PBMC samples a markedly enhanced cytolysis was observed with 4G7SDIE in serum of HVs as well as with patient serum after 4G7SDIE infusion. Notably, low 4G7SDIE-mediated cytolysis was observed for patients #13 and #14, who did not respond to 4G7SDIE treatment.
Figure 2.

Cytotoxic activity of PBMC from 4G7SDIE-treated patients, serum half-life of 4G7SDIE and antigenic shift of CD19. (a) Cell line MHH-CALL-4 was incubated with PBMC of five patients (E:T 20:1) and serum of HVs with 1 µg/ml 4G7SDIE or autologous patient serum and analyzed in 2 h-EuTDA assays. Coincubation with patient serum significantly increased cytolysis of MHH-CALL-4, n = 5, P = 0.0239 (one-way ANOVA). PBMC were taken prior to indicated antibody infusion cycle, autologous serum was taken 1 h after antibody infusion. (b) Primary B-lineage acute lymphoblastic leukemia blasts from five 4G7SDIE-treated patients were incubated with indicated concentrations of 4G7SDIE. After 24 h cells were washed, incubated with 4G7SDIE (2 µg/ml) and remaining CD19 surface expression on CD10+ and or CD34+ blasts was analyzed by indirect immunofluorescence and flow cytometry. Relative surface expression was calculated, based on the mean fluorescence intensity of 4G7SDIE-stained cells without preincubation. (c) Serum samples of two patients and defined concentrations of 4G7SDIE diluted in autologous serum, taken prior to first antibody treatment, were incubated with NALM-16 cells. Cells were washed and 4G7SDIE was detected by indirect immunofluorescence staining and flow cytometry. Serum titer of 4G7SDIE was calculated with the generated standard curve and t1/2 was calculated. (d) CD19+ and CD20+ B cell depletion and recovery in patient #2. B cell counts in whole blood were determined over course of treatment by routine flow cytometry. First day of 4G7SDIE was day +22 after HSCT, last day was day +740 after hematopoietic stem cell transplantation.
The serum half-life of 4G7SDIE in the first treatment cycle post-HSCT was for patient #1 43 hours and for patient #7 20 hours (Figure 2c). Patient #1 received 40 mg/m2 4G7SDIE at a MRD level of 5 × 10−3, patient #7 20 mg/m2 at an MRD level of 1.1 × 10−2. Serum titer of patient #2 was monitored for 1 year (Supplementary Figure S1) and high-serum titers were sustained over the course of treatment. In five out of five analyzed patients, no human anti-chimeric antibodies were detectable 30 days after the 4G7SDIE treatment was initiated.
Downmodulation of the target antigen is a commonly observed phenomenon during antibody therapy.29 CD19 shift was evaluated on primary B-lineage ALL blasts of five 4G7SDIE-treated patients (Figure 2b) and reached 90 to 40% of initial CD19 expression after 24 hours. Antigenic shift was not significantly increased by longer exposure to 4G7SDIE (data not shown).
In order to evaluate 4G7SDIE-mediated cytolysis of autologous leukemic blasts, cryopreserved blasts of patient #2 were analyzed in cytotoxicity assays with PBMC of patient #2 at different time-points after haploidentical HSCT (Figure 3a). Cytolysis with medium, 4G7SDIE (1 µg/ml) or autologous serum, containing saturating concentrations of 4G7SDIE ≥ 1 µg/ml (Supplementary Figure S1), were assessed. 4G7SDIE-mediated cytolysis by 4G7SDIE as well as cytolysis in presence of autologous serum was markedly enhanced in the early post-HSCT phase, but decreased to moderate cytolysis at day +54 after HSCT. Notably, early after HSCT NK cell frequencies were correspondingly higher and declined over time (Figure 3b).
Figure 3.

Cytotoxic activity of patient PBMC over course of 4G7SDIE treatment against autologous blasts. (a) Primary B-lineage acute lymphoblastic leukemia blasts from patient #2 were incubated with autologous PBMC of the patient (PBMC: target ratio 20:1) with or without 1 µg/ml 4G7SDIE and with autologous serum. PBMC were taken prior to antibody infusion, autologous serum was taken 1 hour after antibody infusion. Specific lysis was measured in DELFIA 2 h-EuTDA cytotoxicity assays. 4G7SDIE infusions are indicated by arrows. 1 µg/ml 4G7SDIE significantly increased cytolysis, n = 6, P = 0.02 (one-way ANOVA). (b) B cell counts (CD19+ or CD20+) and NK cell counts (CD56+CD16+) in whole blood were determined over course of treatment by routine flow cytometry.
During 4G7SDIE treatment, a profound B-cell depletion and prevention of reconstitution was observed. While CD56+CD16+ NK cells of patient #2 reconstituted as expected, neither CD19+ nor CD20+ B-cells were detectable over the course of treatment (Figure 3b and Supplementary Figure S1). After discontinuation of 4G7SDIE therapy, B cells rapidly recovered to a normal level (Figure 2d).
Reduction of MRD by the Fc-optimized CD19 antibody 4G7SDIE
In 10/14 patients (#1 to #10) a significant reduction of MRD was observed. The best response over the course of treatment was illustrated in Figure 4. Either a reduction of MRD ≥ 1 log was documented (#4, #6, #7, #8) or MRD was reduced below the patient-individual detection limit (#1, #2, #3, #5, #9, #10). Out of 12 patients receiving 4G7SDIE prior to HSCT, 7 responded. Application of 4G7SDIE post-HSCT resulted in a response in 3/13 patients. As mentioned above, patient #8 and #9 had received tyrosine kinase inhibitor treatment in parallel. The immune system at response was autologous (prior to first HSCT) in 2/10, haploidentical in 4/10, matched unrelated in 2/10 and matched related in 2/10 responding patients (Table 2).
Figure 4.

Minimal residual disease (MRD) reduction after 4G7SDIE treatment. MRD was assessed by multiparameter flow cytometry and detection of individual clonal immunoglobulin and TCR gene rearrangements or bcr/abl translocation fusion genes by real-time quantitative PCR. The origin of the y-axis indicates the individual MRD-detection limit. Best response for each eligible patient #P1-10 is shown. The nonresponding patients #P11-14 are spared.
Clinical outcome
Taken together, 6/14 patients are alive in enduring molecular remission with a median follow up of 543 days postcorresponding HSCT (range: 208 to 1,137 days) and with a median follow up of 720 days post-first 4G7SDIE (range: 264 to 1,115 days). The Kaplan-Meier survival estimation of event-free survival probability after corresponding HSCT and post-first 4G7SDIE is 39% (median follow up 221 days, Figure 5a) and 40% (median follow up 324 days, Figure 5b), respectively. 7/14 patients died. Five patients died from leukemia relapse (#3, #4, #7, #10, #14). Patient #5 died from fludarabine associated neurotoxicity 72 days post-HSCT and patient #8 from late post-HSCT infectious complication, Gram-negative sepsis 347 days post-HSCT. Both patients were in complete molecular remission when they died. Patient #11 is alive (day +149) but experienced relapse 72 days post-HSCT.
Figure 5.

Event-free survival (EFS) after antibody treatment. Kaplan-Meier estimates for EFS probability were calculated from the date of first 4G7SDIE administration (a) or the date of last hematopoietic stem cell transplantation (b) to the date of relapse, other events or the date of last follow-up in continuous complete remission.
Side effects and toxicity
Side-effect profile of 4G7SDIE infusion was very low (Table 3). Intermittent increased temperature and fever, headache, and nausea were observed. Neither gastrointestinal nor neurotoxic events were documented.
Table 3. Adverse events during the course of CD19 treatment.
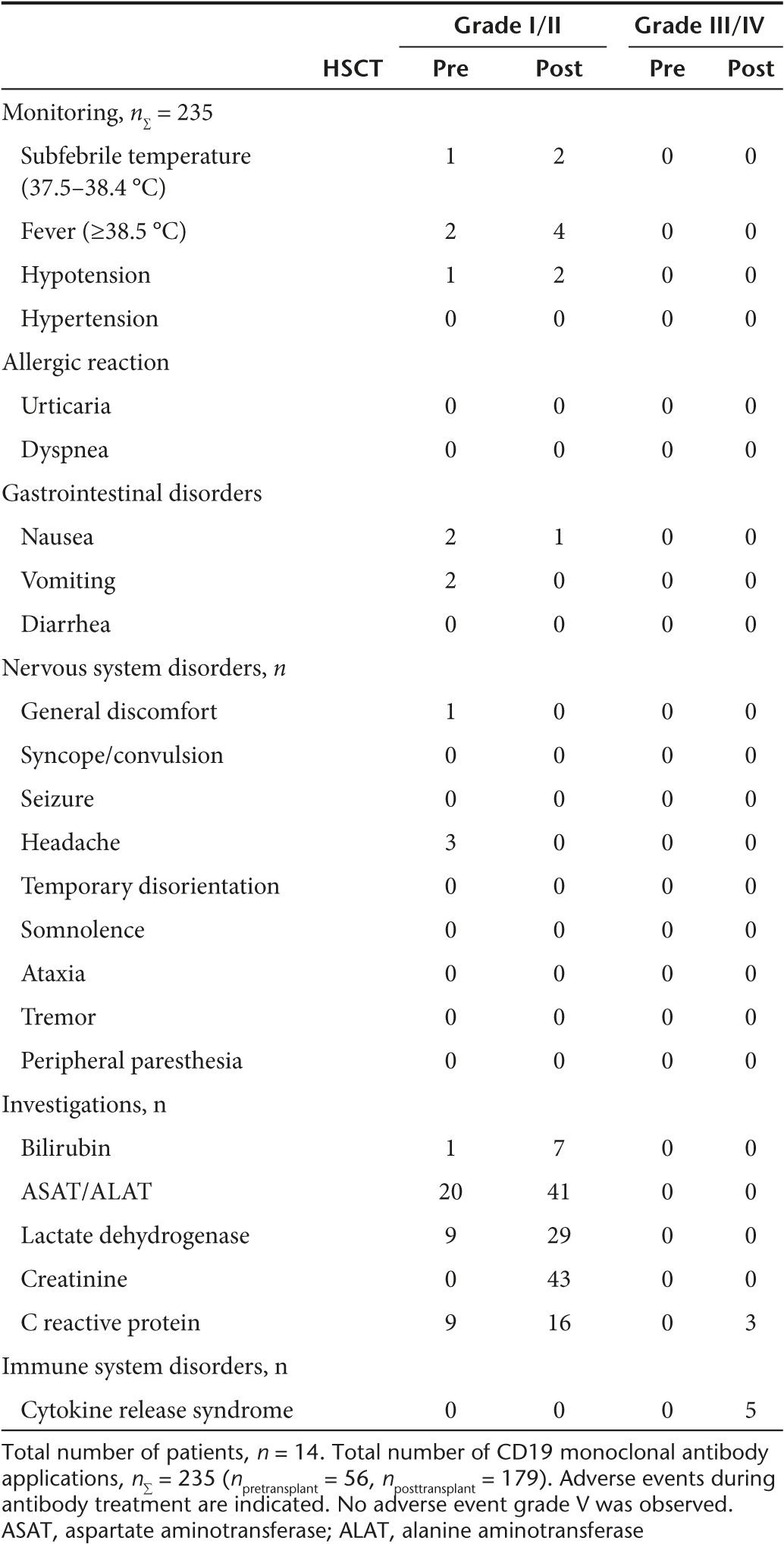
In only five antibody infusions out of 235 (patients #3, #7, #11, and #12), severe side effects occurred. Symptoms of cytokine release syndrome were observed including high fever, reduced general condition, tachycardia, and hypotension. Symptoms were transient and could be controlled after fluid substitution. In each of these patients, increasing leukemic load was detected in subsequent bone marrow aspirates. Thus, the amount of available CD19-positive target cells seemed to determine the severity level of side effects associated with 4G7SDIE application. In all patients, a profound B-cell depletion was observed and B-cell reconstitution after HSCT was suppressed completely until 4G7SDIE application was terminated as exemplarily shown for patient #2 in Figure 2d. After 4G7SDIE therapy was discontinued, B-cells recovered rapidly in this patient (Figure 2d). To prevent infections, human immunoglobulin was substituted above 500 mg/deciliter in all patients over the course of treatment. Neither induction of graft-versus-host disease nor 4G7SDIE-related death was observed.
Discussion
Persistent MRD or rising MRD levels after successful HSCT have been shown to increase the relapse risk in pediatric patients with B-lineage ALL and new treatment strategies capable of eradicating MRD at low side effects are needed. Thus our off-the-shelf approach may be complementary to other CD19 targeting strategies like blinatumomab and CAR-T cells. In our view, Fc-optimized third-generation antibodies with enhanced ADCC activity are particularly suited to cope with this task.
Here, we report on the generation and preclinical characterization of the Fc-optimized CD19 antibody 4G7SDIE as well as the first clinical application in pediatric MRD+ B-lineage ALL. Initially, expression of CD19 was confirmed in all analyzed B-lineage ALL samples, verifying CD19 as a suitable target antigen. The SDIE-modified version of CD19 antibody 4G7 exerted dramatically enhanced ADCC activity against cultured cell lines as well as leukemic blasts. Maximal cytolysis was reached at concentrations below 1 µg/ml.
Treatment with 4G7SDIE reduced MRD load in 10 of 14 patients by at least 1 log in 4 and below detection limit in 6 patients. This might have contributed to the favorable event-free survival (39%) in our extremely high-risk patients, characterized by persistent MRD positivity as well as additional adverse factors, that are, ≥ CR2 (in 11 patients), ≥ 2nd HSCT (in 9 patients) and unfavorable rearrangements in 3 patients.
Remarkable findings were that MRD-reduction ≥ 1 log by Fc-optimized CD19-mAb can occur prior to as well as after HSCT and sometimes several infusions may be necessary to induce response. Further, response was seen in the autologous setting pretransplant as well as in the post-transplant situation irrespective of donor origin. These observations broaden the applicability of the Fc-optimized CD19 antibody to patients at different stages of treatment. However, we propose that during the early post-transplant phase, when NK cell numbers are particularly high (NK cell wave), the treatment with 4G7SDIE, might be especially promising due to favorable effector-to-target ratios.30,31 Patient #2 provides some evidence for this hypothesis. This patient's ADCC activity was extensively monitored during and after treatment. It was found to be significantly increased by 4G7SDIE but decreased over the course of treatment most likely due to a corresponding decline of NK cell numbers.
With PBMC of three out of five analyzed patients, a high 4G7SDIE-mediated ADCC was observed in cytotoxicity assays. Interestingly, the 2 patients with a low ADCC activity did not respond to 4G7SDIE treatment. In general, several effector-cell and target-cell based factors may influence the extent of ADCC. In our experiments with different HVs, a wide variability of cytolysis was observed against the same B-lineage ALL targets; and different blasts were heterogeneously lysed by effector cells of a single HV. A possible target-cell based factor might be the extent of CD19 expression. Albeit no correlation with CD19 expression and 4G7SDIE-mediated lysis was observed in the case of B-lineage ALL blasts, a significant positive correlation was shown in a model with CD19 transfectants with the same cellular background. Thus, the CD19-surface expression level might be of critical value to determine the outcome of 4G7SDIE-treatment and the correlation of target antigen expression and ADCC might underlie a logarithmic correlation as it has been demonstrated for cetuximab in lung cancer cell lines.32
Antigen modulation is a frequently observed phenomenon and may have been the reason for limited therapeutic efficacy of CD20 antibody rituximab.33,34 CD19 antigen loss has been described after treatment with CD19 chimeric-antigen receptor modified T cells (CD19-CARs) and CD19xCD3 bispecific antibody blinatumomab treatment.35,36,37,38,39 In the 4G7SDIE-treated patients, no CD19 antigen loss has been observed in relapsed patients (data not shown). However, 4G7SDIE modulated the surface expression of CD19 on leukemic blasts of treated patients in vitro. The observed shift was heterogeneous, ranging from 10 to 60% after 24 hours. Although no correlation with the clinical response could be found in our very few patients, the modulation or loss of CD19 may hamper the therapeutic efficacy of 4G7SDIE.
The serum half-life of 4G7SDIE ranged from 20 to 43 hours. This might be due to variable amounts of antigen present not only on leukemic but also on normal B cells.40 In any case, biweekly administration of 20 mg/m2 was sufficient to constantly maintain saturating 4G7SDIE concentrations in the blood (≥ 1 µg/ml).
Due to the expression of the antigen on normal B cells, a profound B-cell depletion was expected and in fact observed in all patients treated with 4G7SDIE. In patients with X-linked agammaglobulinemia and those receiving rituximab, an increased risk to develop severe infections, opportunistic infections, tuberculosis, sepsis, and reactivation of hepatitis B has been described.41,42 Thus, four weekly immunoglobulin substitution was carried out during 4G7SDIE treatment and may have prevented severe infections in our patients. The immune reconstitution of T cells and NK cells was not impaired and no neurotoxicity, gastrointestinal toxicity, acute allergic reaction, induction of graft versus host disease, and treatment-related death was observed under 4G7SDIE treatment. Due to the very mild side effects (mainly fever), long-term preemptive treatment may be reasonable also in patients in CR with no molecular detectable MRD as current technology is unable to detect MRD below 1 × 10−7, which can correspond to a significant residual tumor load of 1 × 107 cells.43 Other CD19-based immunotherapies recruiting T-cells like blinatumomab, a bispecific CD19xCD3-antibody and CD19-CARs yield significant response rates even in patients with massive leukemia burden; on the other hand, with these reagents frequently occurring severe adverse events have to be accepted like life-threatening cytokine release syndrome and seizures.36,37,44
Thus, the antibody format described here, may be preferably used (i) in a MRD situation, (ii) in patients who may not be able to tolerate severe side effects, and (iii) as prophylactic treatment in patients without detectable MRD but at high risk for leukemic relapse.
Taken together, academia should participate in the development and process of treatment innovations. We demonstrated that MRD can be eradicated or reduced by the Fc-optimized CD19 antibody 4G7SDIE in high-risk refractory or relapsed B-lineage ALL patients. Further investigation of 4G7SDIE within a clinical trial is required to confirm its efficacy to reduce the risk of relapse and to improve long-term survival.
Materials and Methods
Experimental design. G7SDIE was generated and produced in pharmaceutical quality in a university-owned production unit for compassionate use treatment. Preclinical characterization of 4G7SDIE was performed with cell lines, cryopreserved B-lineage ALL samples, and PBMC of healthy volunteers (HVs) and B-lineage ALL patients after obtaining written informed consent of the donors or their representatives in law. Sample acquisition and performed experiments were approved by the Institutional Review Board.
A cohort of 14 MRD-positive B-lineage ALL patients with chemorefractory persistent MRD was offered a treatment using the 4G7SDIE-mAb on a compassionate-use basis at the University Children's Hospital Tübingen (12 patients) or at the University Children's Hospital Düsseldorf (2 patients) between August 2011 and September 2014. Patients disease status and response was defined according to AIEOP ALL BFM 2009 treatment protocol. The data presented is not a clinical trial. All patients were registered at the local authorities responsible for the University Children's Hospital Tübingen or Düsseldorf according to German codes and regulations. The treatment was conducted in accordance with the provisions of the Declaration of Helsinki. All representatives in law of the patients gave informed consent before the treatment with 4G7SDIE was started. All experiments with patient samples were approved by the Institutional Review Board. Experiments and treatment were not performed blinded. The aim of the 4G7SDIE treatment was to significantly reduce MRD (≥ 1 log) with low toxicity prior to HSCT or post-transplant in order to minimize the risk of leukemia recurrence.
Production and purification of 4G7SDIE. Initial chimerization and Fc-optimization of χ4G7 and 4G7SDIE, respectively, was described previously28 as well as production in pharmaceutical quality and quantity.45
Primary cells and cell lines. PBMC and primary leukemic blasts, isolated by density gradient centrifugation using Biocol Separating Solution (Biochrom, Berlin, Germany) and MHH-CALL-4 (ACC 337, DSMZ, Braunschweig, Germany) and NALM-16 (ACC 680) were cultured in RPMI 1640 (Biochrom), MCF-7 cells (ACC 115), and the MCF-7-CD19 transfectant were kept in EMEM (Lonza, Basel, Switzerland). All media were supplemented with 10 to 20% fetal calf serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 1 mmol/l sodium pyruvate and 2 mmol/l L-glutamine (all reagents Biochrom). Generation of MCF-7-CD19 transfectant has been described previously.45
Application schedule of 4G7SDIE. Patients received 4G7SDIE on an individual compassionate-use basis (not within a clinical trial) as intravenous infusion (3 hours) at a dose of 5 to 60 mg/m2/day for 1 to 3 subsequent days. Treatment was started prior to HSCT and/or post-transplant. The evolved treatment schedule was 20 mg/m2/day every 2 weeks in the first year post-transplant and every 4 weeks in the second year post-transplant.
Monitoring during antibody infusion and assessment of toxicity. Clinical surveillance parameters were assessed in all patients during antibody infusion. Post-transplant 4G7SDIE treatment was performed on an outpatient basis. Side effects were assessed according to CTCA v.4 criteria. Standard laboratory parameters (differential blood count, liver enzymes, bilirubin, pancreatic enzymes, kidney retention values, albumin, immunoglobulins, coagulation parameters, C-reactive protein, and electrolytes) were assessed during treatment.
Monitoring of bone marrow and MRD detection. MRD level of all patients was assessed prior to and after 4G7SDIE administration by multi-parameter flow cytometry46 as well as detection of individual clonal immunoglobulin and T-cell receptor gene rearrangements or bcr/abl translocation fusion genes by real-time quantitative PCR in certified laboratories in Frankfurt and Kiel, Germany. Data were analyzed according to the guidelines of the EuroMRD consortium.47 Response to the mAb treatment was defined a significant MRD reduction of ≥ 1 log change of leukemic load during the compassionate-use treatment.
Antibodies and flow cytometry. CD20-FITC, CD22-PE, CD19-APC, CD19-PE, CD3-FITC, CD56/CD16-PE, CD34-APC, CD45-PerCp, CD10-PE, CD19-FITC, unconjugated CD19 (4G7)-, and isotype control antibodies were purchased from BD Biosciences (Heidelberg, Germany). CD34-APC from Biolegend (San Diego, CA) and CD10-PerCp from Exbio (Prague, Czech Republic). For indirect immunofluorescence, PE-conjugated goat-anti-human F(ab')2 fragments (Jackson ImmunoResearch, West Grove, PA) were used. For multiparameter analysis, a combination of direct and indirect immunofluorescence was used by adding direct labeled antibodies to the final staining step. All antibodies were incubated with cells for either 10 minutes at room temperature or 30 minutes at 4 °C. Quantitative analyses were performed with QIFIKIT (Dako, Hamburg, Germany) according to the manufacturers' instructions. Cells were analyzed with a FACSCalibur or a LSRII (BD Biosciences).
Cytotoxicity and CD107a-degranulation assays. The cytolytic activity of PBMC from HVs and patients was analyzed in a 2 h-DELFIA EuTDA cytotoxicity assay (PerkinElmer, Waltham, MA) according to the manufacturers recommendations and as described previously.45,48 Real-time cytotoxicity assays were performed with an xCELLigence RTCA SP instrument (ACEA Biosciences, San Diego, CA) as described previously.45 CD107a-degranulation assays were performed as described previously.45
Antigenic shift assay. Antigenic shift assays were performed as described previously.28 Briefly, primary leukemic blasts were incubated with various 4G7SDIE concentrations in supplemented RPMI 1640 at 37 °C. After 24 hours, cells were washed and incubated with 2 µg/ml of 4G7SDIE for 30 minutes at 4 °C. Detection of residual CD19 molecules was performed by staining with PE-conjugated goat-anti-human F(ab')2 fragments and flow cytometric analysis. Relative surface expression was calculated, defining the mean fluorescence intensity preincubated with no 4G7SDIE as 100%.
Serum titer assay. NALM-16 cells were incubated with diluted serum samples for 30 minutes at 4 °C. Cells were washed and bound 4G7SDIE was stained with PE-conjugated goat-anti-human F(ab')2 fragments. Standard curve was generated by diluting defined 4G7SDIE concentrations in serum taken prior to antibody treatment. Cells were analyzed by flow cytometry.
Graphical and statistical analysis. Flow cytometry data were analyzed with FlowJo V10 software (Tree Star, Ashland, OR). xCELLigence data were analyzed with RTCA Software 1.2 (ACEA Biosciences). Other analyses were performed using GraphPad Prism software V5.04 (GraphPad Software, La Jolla, CA). Statistical significance was accepted at P < 0.05 and is indicated by * (P < 0.05), ** (P < 0.01), *** (P < 0.001), and **** (P < 0.0001).
SUPPLEMENTARY MATERIAL Table S1. Patient characteristics at time-point of 2 h-EuTDA assays with MHH-cALL-4 Table S2. Chemotherapy and CD19 mAb treatment. Figure S1. 4G7SDIE serum levels of patient #2. Figure S2. In vitro characterization of 4G7SDIE and B-lineage ALL blasts.
Acknowledgments
The authors acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG), CRC685 Immunotherapy, the Bundesministerium für Bildung und Forschung (BMBF iVac ALL, BMBF GO-Bio 0315096 / 0316070), the Reinhold Beitlich Stiftung, the Deutsche José Carreras Leukämie-Stiftung, the Stefan Morsch Stiftung, the Förderverein für krebskranke Kinder Tübingen, the German Cancer Consortium (DKTK) and the German Cancer Research Center (DKFZ) Heidelberg, Germany. The authors thank Annie Babarin-Dorner for technical assistance and Peter-Michael Weber for editing the layout of figures. The authors declare no conflict of interest.
The work was done at the University Children's Hospital and in the Department of Immunology of the University of Tübingen, Germany.
Supplementary Material
References
- Smith, MA, Seibel, NL, Altekruse, SF, Ries, LA, Melbert, DL, O'Leary, M et al. (2010). Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28: 2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, LM and Gore, L (2014). Blinatumomab, a Bi-Specific Anti-CD19/CD3 BiTE(®) Antibody for the Treatment of Acute Lymphoblastic Leukemia: Perspectives and Current Pediatric Applications. Front Oncol 4: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möricke, A, Reiter, A, Zimmermann, M, Gadner, H, Stanulla, M, Dördelmann, M et al.; German-Austrian-Swiss ALL-BFM Study Group. (2008). Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111: 4477–4489. [DOI] [PubMed] [Google Scholar]
- Bader, P, Kreyenberg, H, Henze, GH, Eckert, C, Reising, M, Willasch, A et al.; ALL-REZ BFM Study Group. (2009). Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J Clin Oncol 27: 377–384. [DOI] [PubMed] [Google Scholar]
- Lankester, AC, Bierings, MB, van Wering, ER, Wijkhuijs, AJ, de Weger, RA, Wijnen, JT et al. (2010). Preemptive alloimmune intervention in high-risk pediatric acute lymphoblastic leukemia patients guided by minimal residual disease level before stem cell transplantation. Leukemia 24: 1462–1469. [DOI] [PubMed] [Google Scholar]
- Sramkova, L, Muzikova, K, Fronkova, E, Krejci, O, Sedlacek, P, Formankova, R et al. (2007). Detectable minimal residual disease before allogeneic hematopoietic stem cell transplantation predicts extremely poor prognosis in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 48: 93–100. [DOI] [PubMed] [Google Scholar]
- Eckert, C, von Stackelberg, A, Seeger, K, Groeneveld, TW, Peters, C, Klingebiel, T et al. (2013). Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia - long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 49: 1346–1355. [DOI] [PubMed] [Google Scholar]
- Eckert, C, Henze, G, Seeger, K, Hagedorn, N, Mann, G, Panzer-Grümayer, R et al. (2013). Use of allogeneic hematopoietic stem-cell transplantation based on minimal residual disease response improves outcomes for children with relapsed acute lymphoblastic leukemia in the intermediate-risk group. J Clin Oncol 31: 2736–2742. [DOI] [PubMed] [Google Scholar]
- Bader, P, Kreyenberg, H, von Stackelberg, A, Eckert, C, Salzmann-Manrique, E, Meisel, R et al. (2015). Monitoring of minimal residual disease after allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia allows for the identification of impending relapse: results of the ALL-BFM-SCT 2003 trial. J Clin Oncol 33: 1275–1284. [DOI] [PubMed] [Google Scholar]
- Kato, M, Horikoshi, Y, Okamoto, Y, Takahashi, Y, Hasegawa, D, Koh, K et al. (2012). Second allogeneic hematopoietic SCT for relapsed ALL in children. Bone Marrow Transplant 47: 1307–1311. [DOI] [PubMed] [Google Scholar]
- Lang, P, Teltschik, HM, Feuchtinger, T, Müller, I, Pfeiffer, M, Schumm, M et al. (2014). Transplantation of CD3/CD19 depleted allografts from haploidentical family donors in paediatric leukaemia. Br J Haematol 165: 688–698. [DOI] [PubMed] [Google Scholar]
- Muñoz, A, Badell, I, Olivé, T, Verdeguer, A, Gómez, P and Bureo, E; Spanish Working Party for Bone Marrow Transplantation in Children (GETMON) (2002). Second allogeneic hematopoietic stem cell transplantation in hematologic malignancies in children: long-term results of a multicenter study of the Spanish Working Party for Bone Marrow Transplantation in Children (GETMON). Haematologica 87: 331–332. [PubMed] [Google Scholar]
- Bosi, A, Laszlo, D, Labopin, M, Reffeirs, J, Michallet, M, Gluckman, E et al.; Acute Leukemia Working Party of the European Blood and Marrow Transplant Group. (2001). Second allogeneic bone marrow transplantation in acute leukemia: results of a survey by the European Cooperative Group for Blood and Marrow Transplantation. J Clin Oncol 19: 3675–3684. [DOI] [PubMed] [Google Scholar]
- Pui, CH and Jeha, S (2007). New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat Rev Drug Discov 6: 149–165. [DOI] [PubMed] [Google Scholar]
- Orentas, RJ, Lee, DW and Mackall, C (2012). Immunotherapy targets in pediatric cancer. Front Oncol 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patz, M, Isaeva, P, Forcob, N, Müller, B, Frenzel, LP, Wendtner, CM et al. (2011). Comparison of the in vitro effects of the anti-CD20 antibodies rituximab and GA101 on chronic lymphocytic leukaemia cells. Br J Haematol 152: 295–306. [DOI] [PubMed] [Google Scholar]
- Cartron, G, de Guibert, S, Dilhuydy, MS, Morschhauser, F, Leblond, V, Dupuis, J et al. (2014). Obinutuzumab (GA101) in relapsed/refractory chronic lymphocytic leukemia: final data from the phase ½ GAUGUIN study. Blood 124: 2196–2202. [DOI] [PubMed] [Google Scholar]
- Lang, P, Barbin, K, Feuchtinger, T, Greil, J, Peipp, M, Zunino, SJ et al. (2004). Chimeric CD19 antibody mediates cytotoxic activity against leukemic blasts with effector cells from pediatric patients who received T-cell-depleted allografts. Blood 103: 3982–3985. [DOI] [PubMed] [Google Scholar]
- Jeha, S, Behm, F, Pei, D, Sandlund, JT, Ribeiro, RC, Razzouk, BI et al. (2006). Prognostic significance of CD20 expression in childhood B-cell precursor acute lymphoblastic leukemia. Blood 108: 3302–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworzak, MN, Schumich, A, Printz, D, Pötschger, U, Husak, Z, Attarbaschi, A et al. (2008). CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood 112: 3982–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, O (2012). CD19 as an attractive target for antibody-based therapy. MAbs 4: 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, HM, Bernett, MJ, Pong, E, Peipp, M, Karki, S, Chu, SY et al. (2008). Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res 68: 8049–8057. [DOI] [PubMed] [Google Scholar]
- Kellner, C, Zhukovsky, EA, Pötzke, A, Brüggemann, M, Schrauder, A, Schrappe, M et al. (2013). The Fc-engineered CD19 antibody MOR208 (XmAb5574) induces natural killer cell-mediated lysis of acute lymphoblastic leukemia cells from pediatric and adult patients. Leukemia 27: 1595–1598. [DOI] [PubMed] [Google Scholar]
- Matlawska-Wasowska, K, Ward, E, Stevens, S, Wang, Y, Herbst, R, Winter, SS et al. (2013). Macrophage and NK-mediated killing of precursor-B acute lymphoblastic leukemia cells targeted with a-fucosylated anti-CD19 humanized antibodies. Leukemia 27: 1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel, UJ, Schlegel, P and Lang, P (2013). Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front Immunol 4: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, A, Wurch, T, Bailly, C and Corvaia, N (2010). Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 10: 345–352. [DOI] [PubMed] [Google Scholar]
- Woyach, JA, Awan, F, Flinn, IW, Berdeja, JG, Wiley, E, Mansoor, S et al. (2014). A phase 1 trial of the Fc-engineered CD19 antibody XmAb5574 (MOR00208) demonstrates safety and preliminary efficacy in relapsed CLL. Blood 124: 3553–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, M, Große-Hovest, L, Nübling, T, Pyż, E, Bamberg, ML, Aulwurm, S et al. (2012). Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 26: 1228–1237. [DOI] [PubMed] [Google Scholar]
- Feldman, EJ, Brandwein, J, Stone, R, Kalaycio, M, Moore, J, O'Connor, J et al. (2005). Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J Clin Oncol 23: 4110–4116. [DOI] [PubMed] [Google Scholar]
- Foley, B, Felices, M, Cichocki, F, Cooley, S, Verneris, MR and Miller, JS (2014). The biology of NK cells and their receptors affects clinical outcomes after hematopoietic cell transplantation (HCT). Immunol Rev 258: 45–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handgretinger, R, Chen, X, Pfeiffer, M, Schumm, M, Mueller, I, Feuchtinger, T et al. (2008). Cellular immune reconstitution after haploidentical transplantation in children. Biol Blood Marrow Transplant 14(1 Suppl 1): 59–65. [DOI] [PubMed] [Google Scholar]
- Kurai, J, Chikumi, H, Hashimoto, K, Yamaguchi, K, Yamasaki, A, Sako, T et al. (2007). Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res 13: 1552–1561. [DOI] [PubMed] [Google Scholar]
- Beers, SA, French, RR, Chan, HT, Lim, SH, Jarrett, TC, Vidal, RM et al. (2010). Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115: 5191–5201. [DOI] [PubMed] [Google Scholar]
- Duman, BB, Sahin, B, Ergin, M and Guvenc, B (2012). Loss of CD20 antigen expression after rituximab therapy of CD20 positive B cell lymphoma (diffuse large B cell extranodal marginal zone lymphoma combination): a case report and review of the literature. Med Oncol 29: 1223–1226. [DOI] [PubMed] [Google Scholar]
- Mackall, CL, Merchant, MS and Fry, TJ (2014). Immune-based therapies for childhood cancer. Nat Rev Clin Oncol 11: 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp, SA, Kalos, M, Barrett, D, Aplenc, R, Porter, DL, Rheingold, SR et al. (2013). Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, DW, Kochenderfer, JN, Stetler-Stevenson, M, Cui, YK, Delbrook, C, Feldman, SA et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp, MS, Gökbuget, N, Zugmaier, G, Degenhard, E, Goebeler, ME, Klinger, M et al. (2012). Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood 120: 5185–5187. [DOI] [PubMed] [Google Scholar]
- Maude, SL, Frey, N, Shaw, PA, Aplenc, R, Barrett, DM, Bunin, NJ et al. (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371: 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keizer, RJ, Huitema, AD, Schellens, JH and Beijnen, JH (2010). Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 49: 493–507. [DOI] [PubMed] [Google Scholar]
- Quartier, P, Debré, M, De Blic, J, de Sauverzac, R, Sayegh, N, Jabado, N et al. (1999). Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: a retrospective survey of 31 patients. J Pediatr 134: 589–596. [DOI] [PubMed] [Google Scholar]
- Petropoulou, AD, Porcher, R, Peffault de Latour, R, Xhaard, A, Weisdorf, D, Ribaud, P et al. (2012). Increased infection rate after preemptive rituximab treatment for Epstein-Barr virus reactivation after allogeneic hematopoietic stem-cell transplantation. Transplantation 94: 879–883. [DOI] [PubMed] [Google Scholar]
- Campana, D (2010). Minimal residual disease in acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2010: 7–12. [DOI] [PubMed] [Google Scholar]
- Schlegel, P, Lang, P, Zugmaier, G, Ebinger, M, Kreyenberg, H, Witte, KE et al. (2014). Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica 99: 1212–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel, UJ, Vogt, F, Grosse-Hovest, L, Jung, G, Handgretinger, R and Lang, P (2014). γδ T cell-mediated antibody-dependent cellular cytotoxicity with CD19 antibodies assessed by an impedance-based label-free real-time cytotoxicity assay. Front Immunol 5: 618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerst, G, Kreyenberg, H, Roth, C, Well, C, Dietz, K, Coustan-Smith, E et al. (2005). Concurrent detection of minimal residual disease (MRD) in childhood acute lymphoblastic leukaemia by flow cytometry and real-time PCR. Br J Haematol 128: 774–782. [DOI] [PubMed] [Google Scholar]
- van der Velden, VH, Cazzaniga, G, Schrauder, A, Hancock, J, Bader, P, Panzer-Grumayer, ER et al.; European Study Group on MRD detection in ALL (ESG-MRD-ALL). (2007). Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia 21: 604–611. [DOI] [PubMed] [Google Scholar]
- Lang, P, Pfeiffer, M, Handgretinger, R, Schumm, M, Demirdelen, B, Stanojevic, S et al. (2002). Clinical scale isolation of T cell-depleted CD56+ donor lymphocytes in children. Bone Marrow Transplant 29: 497–502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.