

Abstract
Several studies over the last few decades have shown that antibiotic resistance mechanisms frequently confer a fitness cost and that these costs can be genetically ameliorated by intra- or extragenic second-site mutations, often without loss of resistance. Another, much less studied potential mechanism by which the fitness cost of antibiotic resistance could be reduced is via a regulatory response where the deleterious effect of the resistance mechanism is lowered by a physiological alteration that buffers the mutational effect. In mycobacteria, resistance to the clinically used tuberactinomycin antibiotic capreomycin involves loss-of-function mutations in rRNA methylase TlyA or point mutations in 16S rRNA (in particular the A1408G mutation). Both of these alterations result in resistance by reducing drug binding to the ribosome. Here we show that alterations of tlyA gene expression affect both antibiotic drug susceptibility and fitness cost of drug resistance. In particular, we demonstrate that the common resistance mutation A1408G is accompanied by a physiological change that involves increased expression of the tlyA gene. This gene encodes an enzyme that methylates neighboring 16S rRNA position C1409, and as a result of increased TlyA expression the fitness cost of the A1408G mutation is significantly reduced. Our findings suggest that in mycobacteria, a nonmutational mechanism (i.e., gene regulatory) can restore fitness to genetically resistant bacteria. Our results also point to a new and clinically relevant treatment strategy to combat evolution of resistance in multidrug-resistant tuberculosis. Thus, by utilizing antagonistic antibiotic interactions, resistance evolution could be reduced.
Keywords: Mycobacterium tuberculosis, antibiotic resistance, fitness, compensation, rRNA methylation, tlyA
INTRODUCTION
One of the key factors determining the success of an antibiotic-resistant bacterial clone in a clinical setting is the impact of the resistance mechanism on the fitness of the bacteria. Several studies have shown that resistance typically is associated with a reduction in fitness (Andersson and Levin 1999; Andersson and Hughes 2010), even though a few exceptions exist (Bottger et al. 1998; Sander et al. 2002), and that resistant mutants generally grow slower and have reduced virulence and survival when compared to their susceptible siblings. Studies of streptomycin resistance in Mycobacterium tuberculosis were the first to combine data on the frequency of molecular resistance determinants in clinical isolates in vivo with in vitro experimental data on the genetics and costs associated with resistance. In these clinical isolates, an inverse correlation was observed between the frequency of a resistance mutation and its fitness cost, i.e., the most costly mutations were also the most rare among clinical isolates (Bottger et al. 1998; Sander et al. 2002).
In M. tuberculosis, the etiologic agent of tuberculosis, drug resistance is exclusively due to chromosomal alterations that affect either the drug target itself or bacterial enzymes modifying the drug (for review, see Bottger and Springer 2008). Injectable antibiotics such as the aminoglycosides kanamycin and amikacin and the tuberactinomycins capreomycin and viomycin are key second-line drugs for the treatment of multidrug-resistant tuberculosis (MDR TB) disease (for review, see Falzon et al. 2011). These antibiotics inhibit protein synthesis by binding to the small ribosomal subunit (Shcherbakov et al. 2010; Stanley et al. 2010). As all four drugs share the same molecular target and bind to a similar location in the ribosome, cross-resistance is frequently observed (McClatchy et al. 1977; Maus et al. 2005a; Feuerriegel et al. 2009; Jugheli et al. 2009; Akbergenov et al. 2011; Sirgel et al. 2012). Resistance to the tuberactinomycin antibiotics capreomycin and viomycin involves two mechanisms, both of which result in reduced binding of the antibiotics to the 30S subunit of the ribosome: loss-of-function mutations in rRNA methylase TlyA (Maus et al. 2005b; Monshupanee et al. 2012) and point mutations in 16S rRNA (Akbergenov et al. 2011), in particular at position 1408. While loss-of-function mutations in rRNA methylase TlyA only affect tuberactinomycins but not aminoglycosides (Maus et al. 2005b), point mutations in 16S rRNA in general confer cross-resistance to both tuberactinomycins and aminoglycosides (Sirgel et al. 2012; Reeves et al. 2015).
Fitness costs conferred by resistance mutations can be reduced by compensatory evolution, where second-site mutations reduce or completely ameliorate the cost, often without loss of resistance (Andersson and Levin 1999; Andersson and Hughes 2010). Such genetically based compensatory evolution has been demonstrated with various combinations of bacteria and resistance mechanisms in vitro, in animal models and in humans and often occurs rapidly and with high efficiency. It is expected that compensation will result in stabilization of the resistance in the bacterial population (Andersson and Levin 1999; Andersson and Hughes 2010). In M. tuberculosis, secondary-site compensatory mutations have been demonstrated in clinical isolates resistant to aminoglycosides. Thus, specific point mutations in rRNA that confer aminoglycoside resistance can be compensated by mutations that restore rRNA secondary structure (Shcherbakov et al. 2010). Similarly, there is circumstantial evidence for compensatory mutations restoring fitness for isoniazid, and rifampicin resistant mutants (Sherman et al. 1996; Comas et al. 2012; Meftahi et al. 2016).
Another potential mechanism of compensation could be by alterations in gene regulation where the initial cost of the resistance mechanism is reduced by a physiological alteration. Such mechanisms would generally be much more difficult to identify since the physiological response might be very rapid and not involve any genetic changes. One rare example illustrating such a physiological response was provided by the studies of Olivares et al. (2014), who showed that the costs of overexpressing the multidrug efflux pump MexEF-OprN (a proton/substrate antiporter system) in Pseudomonas aeruginosa can be compensated by an unknown mechanism that causes a metabolic rewiring to increase aerobic respiration and reduce the deleterious effects of an increased proton influx and acidification due to the overexpressed efflux pump.
In this study, we show that mycobacterial adaptation to antibiotic exposure and mutational fitness cost involves alteration of tlyA gene expression. In particular, we demonstrate that the substantial fitness cost associated with the common rRNA resistance mutation A1408G can be reduced by up-regulation of the tlyA rRNA methylase. The TlyA methylase modifies the neighboring position 1409, reducing the cost of the A1408G mutation while concomitantly also reducing the resistance level.
RESULTS
Mutant ribosomes with the A1408G mutation in 16S rRNA are more resistant in cell-free in vitro translation than in vivo
During analysis of M. smegmatis mutants resistant to capreomycin and viomycin, we observed an unexpected discrepancy in drug susceptibility for the wild type versus the A1408G mutant when comparing whole-cell MIC determinations with drug susceptibility as determined in cell-free ribosomal translation assays. In a standard MIC assay, the A1408G mutant bacteria showed an eight- to 16-fold increase in resistance for capreomycin when compared to the susceptible wild type bacteria (Table 1). However, when resistance was analyzed in an in vitro translation system based on cellular extracts from susceptible wild type and resistant A1408G mutant bacteria, there was a 100-fold difference in the level of resistance (Table 2; Supplemental Fig. 1). Thus, for both capreomycin and viomycin the relative resistance was approximately 10-fold higher when analyzed at the ribosomal target when compared to the MIC determined using intact cells. In contrast, for the tlyA deletion mutant no such discrepancy in tuberactinomycin susceptibility between whole-cell MIC and ribosomal IC50 was observed. As a control, drug susceptibility to the aminoglycoside amikacin was determined, and for both MIC and ribosomal translation assays the A1408G mutation increased resistance >1000-fold, and as expected, inactivation of tlyA did not affect drug susceptibility. Significantly, a similar fourfold increase in MIC levels for capreomycin conferred by the A1408G mutation was also observed in clinical M. tuberculosis isolates (Table 3). An implication of these findings is that in the living cell the tuberactinomycin resistance levels are reduced by an unknown mechanism.
TABLE 1.
Drug susceptibility (MIC, µg/mL) of recombinant M. smegmatis strains
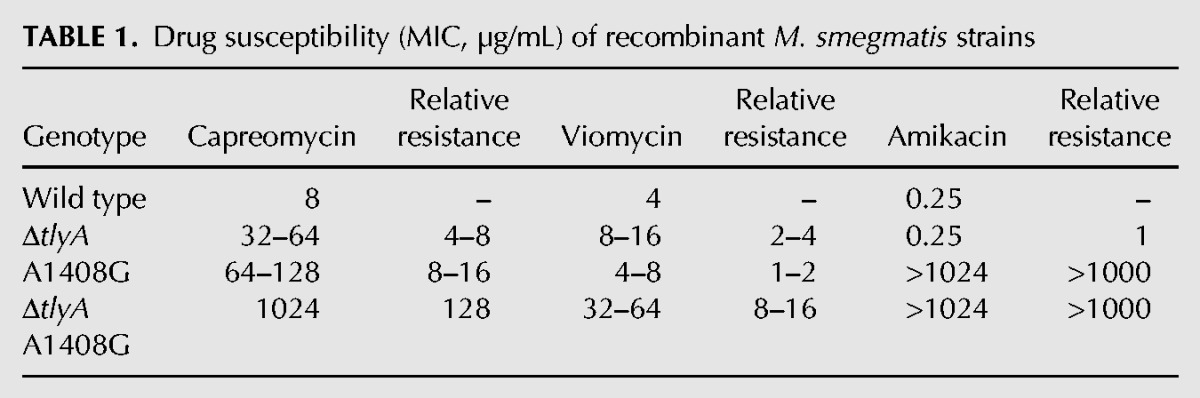
TABLE 2.
Drug susceptibility (IC50, µM) of M. smegmatis ribosomes
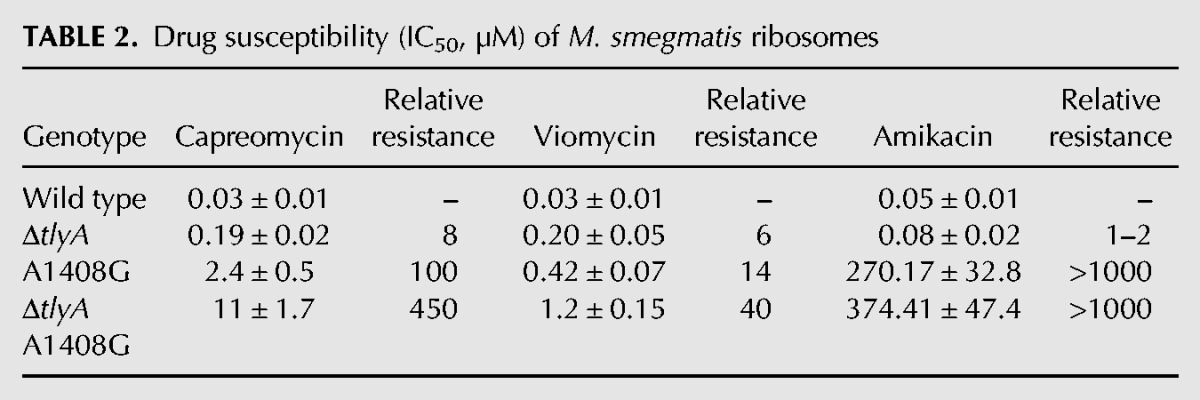
TABLE 3.
Drug susceptibility (MIC, µg/mL) of M. tuberculosis clinical isolates (wild type and A1408G mutant)a

In response to capreomycin, cells down-regulate TlyA-mediated methylation of 16S and 23S rRNA resulting in decreased drug susceptibility
Previous studies showed that the TlyA methylase modifies position 1409 in 16S rRNA and position 1920 in 23S rRNA and that inactivation of TlyA-dependent methylation confers resistance to capreomycin and viomycin (Johansen et al. 2006). Furthermore, as reported below, we observed that incubation with capreomycin during bacterial growth resulted in a reduced post-transcriptional modification of rRNA at TlyA-dependent sites (1409 in 16S and 1920 in 23S). Based on these observations and the discrepancy between MIC values in whole cells (eight- to 16-fold increase in A1408G mutant compared to wild type) and susceptibility in cell-free translation (100-fold increase in A1408G mutant compared to wild type), we hypothesized that alterations in TlyA-dependent rRNA methylation modulated drug susceptibility. Thus, in the wild type, the presence of drug should result in a down-regulation of TlyA-mediated methylation and cause an increased resistance. Conversely, the A1408G mutation in the absence of drug should result in an increased TlyA-expression and a concomitant reduction in resistance. A prediction from this reasoning is that inactivation of the tlyA gene should have the same effect as pregrowth in the presence of capreomycin.
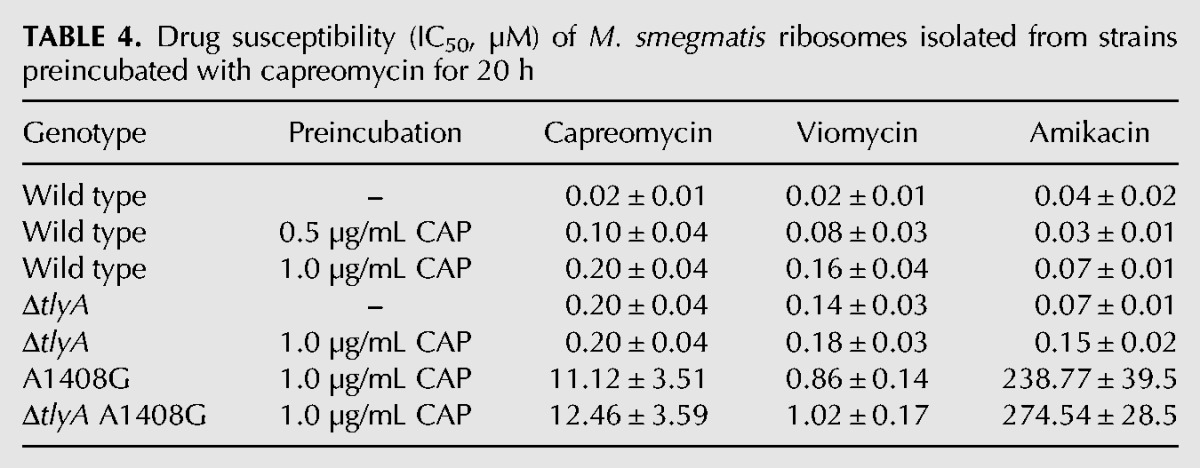
To test this idea, we first compared the drug susceptibility of ribosomes prepared from wild-type cells pregrown in the presence of capreomycin and corresponding tlyA-inactivated mutants. We preincubated whole cells with capreomycin and then prepared ribosomes to measure activity in cell-free translation and level of rRNA methylation. Preincubation with drug resulted in increased resistance as well as reduced post-transcriptional modification of residues 1409 in 16S rRNA and 1920 in 23S rRNA. Thus, preincubation of wild type cells with capreomycin resulted in a dose-dependent decrease of tuberactinomycin susceptibility in wild type M. smegmatis, as determined in cell-free ribosomal translation assays, and a 20 h preincubation with 1 µg/mL capreomycin resulted in IC50 values similar to the ΔtlyA mutant. Similarly, preincubation with capreomycin increased tuberactinomycin resistance of the A1408G mutant as assessed in cell-free translation assays. Thus, a 20 h preincubation of A1408G mutants with 1 µg/mL capreomycin resulted in IC50 values similar to that of the A1408G ΔtlyA double mutant. As predicted, preincubation with capreomycin did not affect tuberactinomycin resistance in the ΔtlyA mutant nor in the A1408G ΔtlyA double mutant (Table 4, compare with Table 2; see Supplemental Figs. 2, 3).
TABLE 4.
Drug susceptibility (IC50, µM) of M. smegmatis ribosomes isolated from strains preincubated with capreomycin for 20 h
The increased resistance in cell-free translation was associated with a reduced post-transcriptional modification of residues 1409 in 16S rRNA and 1920 in 23S rRNA (Fig. 1). These experiments demonstrated that in the presence of low capreomycin levels, TlyA-dependent rRNA modification was reduced, and as a result, ribosomal resistance was increased. In summary, inactivation of the tlyA gene had the same effect as pregrowth in the presence of capreomycin.
FIGURE 1.

Capreomycin affects post-transcriptional methylation in 16S rRNA and 23S rRNA. Primer extension analysis of Cm1409*, helix 44 (A) and Cm1920*, helix 69 (B) in the rRNA of M. smegmatis wild type and M. smegmatis ΔtlyA mutant. The strains were grown in liquid culture for 20 h. Wild type strains were grown without drug and in the presence of 0.5 and 1 µg/mL capreomycin. The size marker indicates positions from A1408 to C1412 and A1919 to C1925, respectively. Densitometric analysis of position Cm1409 (C) and Cm1920 (D).
Increased TlyA expression reduces resistance in the A1408G mutant and concomitantly increases fitness
A second prediction of the hypothesis that TlyA-dependent rRNA methylation modulates resistance in the A1408G mutant is that TlyA expression is increased in the presence of the A1408G mutation and results in reduced resistance. We performed two experiments to test this prediction. First, we examined whether TlyA levels were increased in the A1408G mutant. Results shown in Figure 2A demonstrate that levels of tlyA mRNA were increased approximately sevenfold in the A1408G M. smegmatis mutants when compared to the wild type. In addition, and to examine whether our findings observed in M. smegmatis also apply to the pathogen M. tuberculosis, we studied tlyA gene expression in the laboratory strain M. tuberculosis H37Rv. Spontaneous A1408G mutants were obtained by selective plating. Compared to the wild type strain, A1408G mutants showed increased levels of tlyA mRNA (Fig. 2B). Finally, we studied clinical isolates of M. tuberculosis. Compared to fully drug susceptible clinical isolates, aminoglycoside-resistant clinical isolates of M. tuberculosis with the corresponding A1408G mutation on average showed a fourfold increase in levels of tlyA mRNA (Fig. 2C). As expected for a regulatory, nonmutational compensatory mechanism, the levels of tlyA mRNA expression in clinical A1408G M. tuberculosis isolates varied significantly both between different isolates and within a single isolate (see Fig. 2C, inset). To corroborate the conclusion that a nonmutational mechanism is involved in up-regulation of tlyA mRNA levels in the A1408G mutant, we sequenced the tlyA promoter region (position 1915029–1915526) and the 5′ upstream region of tlyA (position 1917769–1917939) in all M. tuberculosis strains investigated (H37Rv in vitro selected A1408G mutants, clinical wild type and A1408G isolates); no mutations were found.
FIGURE 2.
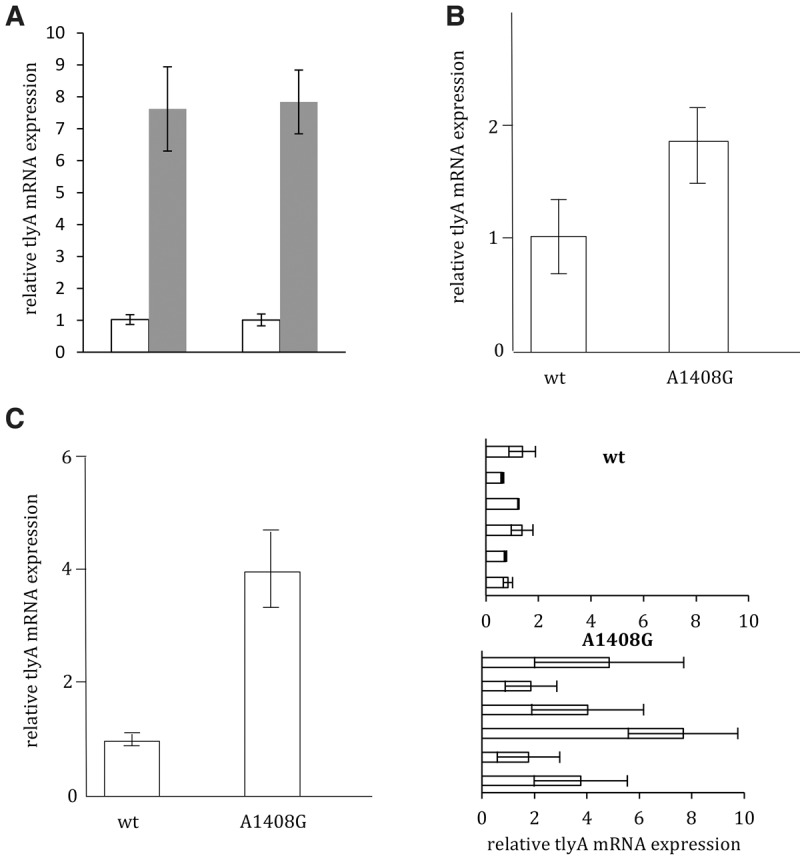
tlyA mRNA levels measured by quantitative PCR. (A) TlyA mRNA levels in M. smegmatis; wild type (white bar) and A1408G (gray bar). 16S rRNA (left) and EF-Tu (right) were used as internal controls. The average expression levels of three wild-type strains and three A1408G strains are given, SDs are indicated, P < 0.01. (B) TlyA mRNA expression in laboratory strain M. tuberculosis H37Rv ATCC27294. Averaged expression levels of triplicate measurements (three wild type clones, five A1408G mutants), SDs are indicated, P < 0.01. (C) TlyA mRNA expression in M. tuberculosis wild-type versus A1408G clinical isolates. Averaged expression levels of triplicate measurements (six clinical isolates of each M. tuberculosis wild type and M. tuberculosis A1408G). Mean and SD are given, P < 0.001. (Inset) TlyA mRNA expression at the level of individual clinical isolates. Mean and SD are given, P < 0.01.
As a second test, we determined the effect of preincubation with capreomycin on MIC levels in whole cells. As expected, pregrowth in the presence of capreomycin did not affect MIC levels in the ΔtlyA mutant. In contrast, and as predicted on the basis of increased tlyA mRNA levels, preincubation increased the MICs of capreomycin and viomycin in the A1408G mutant to the same extent as inactivation of the tlyA gene (Table 5).
TABLE 5.
Drug susceptibility (MIC, µg/mL) of M. smegmatis ΔtlyA and A1408G mutant strains preincubated with 1 µg/mL capreomycin for 20 h

We finally studied whether inactivation of the tlyA gene affected the fitness cost of the A1408G mutation in addition to the increase in resistance. Measurements of fitness as growth rate in vitro showed that loss of TlyA function comes without any measurable fitness cost whereas the A1408G mutation carried a fitness cost of 5%. In the presence of a loss of function of tlyA, the A1408G mutation confers a fitness cost of 10% per generation (Fig. 3), demonstrating that a functional TlyA methylase significantly reduces the fitness cost of the A1408G mutation. Overall, these results suggest that in the A1408G mutant, in the absence of drug, the TlyA enzyme is up-regulated, resulting in sustained rRNA methylation and partial amelioration of the fitness cost of the A1408G resistance mutation.
FIGURE 3.
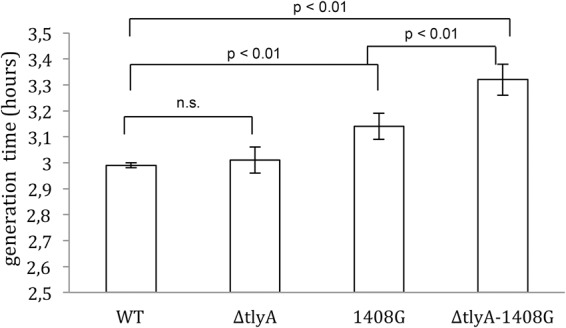
Generation times. M. smegmatis wild type; ΔtlyA mutant; A1408G mutant; and ΔtlyA, A1408G double mutant. Average generation times and SDs are indicated. The average generation times were as follows: wild type 2.99 ± 0.01, ΔtlyA 3.01 ± 0.07, A1408G 3.14 ± 0.05, ΔtlyA, A1408G 3.32 ± 0.06. Each strain was measured in triplicate; P-values are indicated.
DISCUSSION
Essentially all types of organisms can respond and adapt to altered environmental conditions by various regulatory and physiological changes. Similarly, changes in gene expression and cell physiology can occur in response to mutational alterations, and there are numerous examples of how mutations might cause alterations in gene expression (Felix and Barkoulas 2015; Ho and Zhang 2016; Naseeb et al. 2016). However, whether these changes are adaptive and homeostatic in the sense that they act to buffer the negative effect of the mutation and thereby restore fitness, or whether they are only downstream pleiotropic effects of no adaptive value is more difficult to say. The reason for this is that to distinguish between these two (not mutually exclusive) possibilities one has to examine how the mutation affects fitness both in the presence and absence of the observed change in gene expression, which might be difficult to achieve experimentally.
Here we report on a compensatory effect that involves a gene regulatory response that dampens the fitness cost of a mutation that confers resistance to tuberactinomycins, second-line drugs for the treatment of MDR-TB. Our results suggest that the fitness cost of the A1408G mutation, a mutation that confers cross-resistance to aminoglycosides and tuberactinomycins, can be reduced by an increased expression of the tlyA gene, and associated sustained methylation of rRNA. How the A1408G mutation confers the increase in tlyA gene expression is at present unclear, but the compensatory mechanism involves a physiological alteration that increases fitness and buffers the mutational effect at the expense of reduced resistance. Thus, our data show that expression of the rRNA methylase TlyA is up-regulated when the A1408G mutation is present, thereby partly restoring fitness (the fitness cost measured as growth rate was reduced from 10% to 5%). This increase in fitness is importantly associated with a reduced resistance to capreomycin, presumably as a result of the increased methylation of rRNA.
The observation that increased fitness is associated with reduced capreomycin resistance is particularly relevant with regard to treatment of tuberculosis. Thus, previous work has formulated ideas for rationally selecting drug combinations that limit resistance evolution based on antagonistic antibiotic interactions where resistance to one drug increases susceptibility to another, so-called collateral sensitivity (Michel et al. 2008; Munck et al. 2014; Baym et al. 2016). Our findings add to this framework and suggest a new option to combat the clinically highly relevant problem of MDR-TB. MDR treatment is frequently complicated by resistance development as per the A1408G mutation (Shcherbakov et al. 2010). As the A1408G alteration confers cross-resistance to capreomycin, the corresponding mutants are resistant to both aminoglycosides and tuberactinomycins (Maus et al. 2005a; Feuerriegel et al. 2009; Jugheli et al. 2009; Akbergenov et al. 2011; Sirgel et al. 2012). The A1408G mutants observed in clinical M. tuberculosis isolates are mostly, if not all, a result of treatment with aminoglycosides, whereas resistance mutations selected during treatment with capreomycin mainly affect tlyA. Thus, resistance evolution via the A1408G mutation could be reduced by first using capreomycin rather than kanamycin or amikacin in MDR TB treatment. If capreomycin resistance develops, mainly due to nonsense mutations in tlyA, it can subsequently be replaced by an aminoglycoside as tlyA nonsense mutations do not affect susceptibility to kanamycin or amikacin. However, the loss of TlyA function would significantly increase the fitness cost of the A1408G mutation impeding its fixation in the population. In this context, it is relevant to note that a double A1408G tlyA mutant has to our knowledge never been observed in clinical isolates and it is conceivable that the substantial fitness cost of the double mutation precludes it from becoming fixed in the population. We searched public genome databases (EMBL, GMTV) for M. tuberculosis isolates with the A1408G mutation. We extracted genomic data from a total of 291 M. tuberculosis A1408G mutant strains (220 genomes EMBL database, 71 genomes GMTV database); none of these strains carried a mutation in the tlyA gene.
In conclusion, our results demonstrate how nonmutational compensation, i.e., a regulatory change in gene expression of the tlyA gene, can reduce the deleterious effects of a common resistance mutation and how these effects potentially can be utilized to optimize treatment regimes. It is likely that such nongenetic compensatory mechanisms are more common than previously thought and that an understanding of the specific mechanisms can be used to identify regulatory responses that are amenable to modulation by altered treatment regimes and/or inhibition by drugs.
MATERIALS AND METHODS
Antibiotics
Capreomycin sulfate and amikacin sulfate were obtained from Sigma (catalog no. C4142 and A2324, respectively); viomycin sulfate was obtained from Tocris Bioscience (catalog no. 3787).
MIC assay
Drug susceptibility testing of M. smegmatis was done in by broth microdilution in triplicates using twofold dilution series of antibiotics, as described previously (Hobbie et al. 2007). The MIC was defined as the drug concentration at which growth of the cultures was completely inhibited after 72 h incubation of 37°C. Drug susceptibility of M. tuberculosis was assessed using the MGIT 960 instrumentation equipped with the TBeXiST software (Becton Dickinson) as described previously (Springer et al. 2009).
Bacterial strains and DNA techniques
The strains and plasmids used in this study are given in Supplemental Tables 1, 2, and 3. Clinical strains of Mycobacterium tuberculosis were obtained from the Diagnostic Department, Institute of Medical Microbiology, University of Zurich. The single rRNA allelic Mycobacterium smegmatis ΔrrnB strain (SZ380) was obtained by unmarked deletion mutagenesis (Hobbie et al. 2006, 2007) and used for all genetic constructions. Strains SZ459-461 are derivatives of SZ380 with a mutated rrn operon (A1408G) obtained by site-directed mutagenesis (Shcherbakov et al. 2010). M. smegmatis ΔrrnA ΔrrnB attB::int-hyg-rrnB (SZ633) was used for tlyA gene inactivation. In this strain, the two endogenous rrn operons have been deleted by unmarked deletion mutagenesis, and synthesis of rrn is provided by an integrative plasmid which carries the complete rrnB operon under control of its own promoter (Hobbie et al. 2007). A tlyA knockout mutant was generated by homologous recombination using the gene targeting vector pZ566 followed by selection on sucrose and kanamycin. Vector pZ566 was constructed by ligating the left and right chromosomal regions of tlyA (obtained by PCR) into a suicide vector carrying sacB and aph. Successful gene deletion was controlled by Southern blot analysis. M. smegmatis ΔtlyA ΔrrnA ΔrrnB attB::int-Gm-rrnB (SZ907-909) was generated by plasmid exchange using the rrn complementing vector pH144 and selection with gentamicin. M. smegmatis ΔtlyA ΔrrnA ΔrrnB attB::int-hyg-rrnB (SZ910-912) was obtained by plasmid exchange in strain SZ907 using rrn complementing vector pH150 and selection with hygromycin. M. smegmatis ΔtlyA ΔrrnA ΔrrnB attB::int-hyg-rrnB A1408G (SZ913-915) was generated from strain SZ907 by plasmid exchange using rrn complementing vector pH187 and selection with hygromycin. Vector pH187 carries a 16S rRNA A1408G mutation and was obtained from vector pH150 by PCR-mediated mutagenesis.
Determination of generation time
Growth experiments were done in triplicates in 200 mL Erlenmeyer flasks in a total volume of 50 mL of LB-Tween 0.05%. Freshly, homogeneously grown cells were picked from an agar plate, resuspended thoroughly in 1 mL of LB-Tween and diluted for inoculation, resulting in an A600 of 0.01. The measurements were started after overnight incubation at an A600 of approximately 0.1. The A600 was measured every 2.5 h for 12 h and generation times were calculated.
Isolation and purification of ribosomes
Ribosomes were purified from bacterial cell pellets by sucrose gradient (10% to 40%) centrifugation as described previously (Beringer et al. 2005; Bruell et al. 2008). Ribosome concentrations of 70S were determined by absorption measurements on the basis of 23 pmol ribosomes per A260 unit.
Cell-free luciferase translation assays
Purified 70S ribosomes and in vitro-transcribed firefly luciferase mRNA were used in translation reactions. Firefly luciferase mRNA was produced using T7 RNA polymerase in vitro. A typical translation reaction mixture contained 0.21 µM 70S ribosomes, 450 ng mRNA, 20% (v/v) M. smegmatis S100 extract, 200 µM amino acid mixture, 0.3 µL 50× protease inhibitor coctail (Roche) and Ribo Lock RNase inhibitor (12 units; Promega), 0.4 mg/ml tRNAs and 40% (v/v) commercial S30 premix without amino acids (Promega). 1.5 µL antibiotics (final reaction concentrations between 0.01 and 1000 µM) or water were added to the mixture. The total volume of the reaction mixture was 15 µL. The reaction mixture was incubated at 37°C for 30 min and stopped on ice. Finally, 75 µL luciferase assay substrate (Promega) was added and the bioluminescence measured in a luminometer (Flx800; Bio-Tek Instruments). The IC50 is defined as drug concentration required to inhibit synthesis of active luciferase to 50%.
Ribosomal RNA methylation
rRNA methylations at 16S rRNA position C1409 (h44) and at 23S rRNA position C1920 (h69) were determined by primer extension analysis. Ribosomal RNA was isolated from purified ribosomes obtained by sucrose gradient centrifugation using TRIzol reagent (Life Technologies). Ribosomal RNA concentrations were determined by absorption measurements at A260 nm. Primers 5′-GGTGTTACCGACTTTCATG-3′ for h44 and 5′-GGAATTTCGCTACC TTAG-3′ for h69 were ordered from Microsynth and radiolabeled with γ 32-ATP (Hartman Analytics, catalog no. FP-501) using the following reaction: 2 µL T4-polynucleotide kinase (10 U/µL; Fermentas), 2 µL 10× buffer A PNK (Fermentas), 1.5 µL (0.37 MBq/µl) γ 32-ATP, 2 µL 10 mM primer and 12.5 µL water. The reaction mixture was incubated at 37°C for 30 min followed by column purification using Quick spin columns (Sephadex G-25, fine, Roche).
For primer annealing, 5 µL radiolabeled primer, 2 µL rRNA (0.5 µg/µL) and 7.5 µL water were heated up at 90°C for 30 sec, followed by 15 min incubation at room temperature. For primer extension reactions, the following components were added and incubated at 50°C for 1 h: 0.5 µL AMV reverse transcriptase (20 U/µL; Fermentas), 4 µL 5× AMV reverse transcriptase buffer (Fermentas), and 1 µL dNTP mixture (10 mM dATP, 10 mM dCTP, 10 mM dTTP, 0.1 µM dGTP). After the primer extension reaction, samples were purified with the Quick spin columns (Sephadex G-25, fine, Roche). Subsequently, the samples were heated at 95°C for 5 min in formamide sample buffer (95% formamide, 5% saturated bromphenol blue solution) and run on polyacrylamide gels (20% acylamide/bis-acrylamide 19:1, 8 M urea) using TBE running buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 8.0). Gels were dried and exposed to phosphor imaging screens (Kodak SO230). The screens were scanned with fluorescent image analyzer (Fujifilm FLA-5100). The intensity of bands was quantified using Aida Image Analyser software.
Determination of mRNA expression
M. smegmatis strains were grown in liquid culture (LB-Tween) and harvested in logarithmic growth at an OD600 0.2–0.4. M. tuberculosis clinical isolates were grown in liquid culture (7H9 OHDC) and harvested in logarithmic growth at an OD600 0.3–0.35. RNA was extracted using TRIzol extraction (Life Technologies) and reverse transcribed using the High Capacity RNA-to-cDNA kit (Applied Biosystems) according to the manufacturer's instructions. For quantitative PCR, the Quantitect SYBR Green PCR kit (Qiagen) was used together with the 7500 Fast Real-Time PCR System (Applied Biosystems). Primers for M. smegmatis were:
tlyA: forward 5′-GTGTCAGATCGCGAGGTTCT-3′, reverse 5′-GGAGGATTCACCGAGGTTCT-3′;
16S rRNA: forward 5′-GCGTTGTTCGTGAAAACTCA-3′, reverse 5′-CCTCCTGATATCTGCGCATT-3′;
EF-Tu: forward 5′-GACAAGTTCCCCGATTTGA 3′, reverse 5′-GTGTGCGTAGTGACGCTTGT-3′.
Primers for M. tuberculosis were:
tlyA: forward 5′-GTTCATCTCGTTGGCTACC-3′, reverse 5′-CTTCACCAGTGGAACGATA-3′;
16S rRNA: forward 5′-GCAGATATCAGGAGGAACA-3′, reverse 5′-GGACATCCAGGGTATCTAATC-3′.
For relative quantification of tlyA gene expression, 16S rRNA and EF-Tu served as reference. CT-values were calculated according to Pfaffl (Pfaffl 2001). Three biological replicates were run in triplicates each and means and SDs were calculated. Every biological replicate contained a negative control without reverse transcription. Relative tlyA mRNA expression was calculated by relating the individual expression level to the average values of wt isolates.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the University of Zurich (EB) and the Swedish Research Council (DIA). We thank Dr. Peter Sander and Michael Dal Molin for providing the A1408G mutant of M. tuberculosis H37Rv, and Tanja Janusic, Claudia Ritter, and Nora Köhler for technical assistance.
Author contributions: E.C.B. conceived the study and P.F., R.A., Y.T., and R.J. performed the experiments. All authors analyzed the data and wrote the manuscript.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.057257.116.
REFERENCES
- Akbergenov R, Shcherbakov D, Matt T, Duscha S, Meyer M, Wilson DN, Bottger EC. 2011. Molecular basis for the selectivity of antituberculosis compounds capreomycin and viomycin. Antimicrob Agents Chemother 55: 4712–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8: 260–271. [DOI] [PubMed] [Google Scholar]
- Andersson DI, Levin BR. 1999. The biological cost of antibiotic resistance. Curr Opin Microbiol 2: 489–493. [DOI] [PubMed] [Google Scholar]
- Baym M, Stone LK, Kishony R. 2016. Multidrug evolutionary strategies to reverse antibiotic resistance. Science 351: aad3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringer M, Bruell C, Xiong L, Pfister P, Bieling P, Katunin VI, Mankin AS, Bottger EC, Rodnina MV. 2005. Essential mechanisms in the catalysis of peptide bond formation on the ribosome. J Biol Chem 280: 36065–36072. [DOI] [PubMed] [Google Scholar]
- Bottger EC, Springer B. 2008. Tuberculosis: drug resistance, fitness, and strategies for global control. Eur J Pediatr 167: 141–148. [DOI] [PubMed] [Google Scholar]
- Bottger EC, Springer B, Pletschette M, Sander P. 1998. Fitness of antibiotic-resistant microorganisms and compensatory mutations. Nat Med 4: 1343–1344. [DOI] [PubMed] [Google Scholar]
- Bruell CM, Eichholz C, Kubarenko A, Post V, Katunin VI, Hobbie SN, Rodnina MV, Bottger EC. 2008. Conservation of bacterial protein synthesis machinery: initiation and elongation in Mycobacterium smegmatis. Biochemistry 47: 8828–8839. [DOI] [PubMed] [Google Scholar]
- Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. 2012. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet 44: 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzon D, Jaramillo E, Schunemann HJ, Arentz M, Bauer M, Bayona J, Blanc L, Caminero JA, Daley CL, Duncombe C, et al. 2011. WHO guidelines for the programmatic management of drug-resistant tuberculosis: 2011 update. Eur Respir J 38: 516–528. [DOI] [PubMed] [Google Scholar]
- Felix MA, Barkoulas M. 2015. Pervasive robustness in biological systems. Nat Rev Genet 16: 483–496. [DOI] [PubMed] [Google Scholar]
- Feuerriegel S, Cox HS, Zarkua N, Karimovich HA, Braker K, Rusch-Gerdes S, Niemann S. 2009. Sequence analyses of just four genes to detect extensively drug-resistant Mycobacterium tuberculosis strains in multidrug-resistant tuberculosis patients undergoing treatment. Antimicrob Agents Chemother 53: 3353–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WC, Zhang J. 2016. Adaptive genetic robustness of Escherichia coli metabolic fluxes. Mol Biol Evol 33: 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbie SN, Bruell C, Kalapala S, Akshay S, Schmidt S, Pfister P, Bottger EC. 2006. A genetic model to investigate drug-target interactions at the ribosomal decoding site. Biochimie 88: 1033–1043. [DOI] [PubMed] [Google Scholar]
- Hobbie SN, Kalapala SK, Akshay S, Bruell C, Schmidt S, Dabow S, Vasella A, Sander P, Bottger EC. 2007. Engineering the rRNA decoding site of eukaryotic cytosolic ribosomes in bacteria. Nucleic Acids Res 35: 6086–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen SK, Maus CE, Plikaytis BB, Douthwaite S. 2006. Capreomycin binds across the ribosomal subunit interface using tlyA-encoded 2′-O-methylations in 16S and 23S rRNAs. Mol Cell 23: 173–182. [DOI] [PubMed] [Google Scholar]
- Jugheli L, Bzekalava N, de Rijk P, Fissette K, Portaels F, Rigouts L. 2009. High level of cross-resistance between kanamycin, amikacin, and capreomycin among Mycobacterium tuberculosis isolates from Georgia and a close relation with mutations in the rrs gene. Antimicrob Agents Chemother 53: 5064–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus CE, Plikaytis BB, Shinnick TM. 2005a. Molecular analysis of cross-resistance to capreomycin, kanamycin, amikacin, and viomycin in Mycobacterium tuberculosis. Antimicrob Agents Chemother 49: 3192–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus CE, Plikaytis BB, Shinnick TM. 2005b. Mutation of tlyA confers capreomycin resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 49: 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchy JK, Kanes W, Davidson PT, Moulding TS. 1977. Cross-resistance in M. tuberculosis to kanamycin, capreomycin and viomycin. Tubercle 58: 29–34. [DOI] [PubMed] [Google Scholar]
- Meftahi N, Namouchi A, Mhenni B, Brandis G, Hughes D, Mardassi H. 2016. Evidence for the critical role of a secondary site rpoB mutation in the compensatory evolution and successful transmission of an MDR tuberculosis outbreak strain. J Antimicrob Chemother 71: 324–332. [DOI] [PubMed] [Google Scholar]
- Michel JB, Yeh PJ, Chait R, Moellering RC Jr, Kishony R. 2008. Drug interactions modulate the potential for evolution of resistance. Proc Natl Acad Sci 105: 14918–14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monshupanee T, Johansen SK, Dahlberg AE, Douthwaite S. 2012. Capreomycin susceptibility is increased by TlyA-directed 2′-O-methylation on both ribosomal subunits. Mol Microbiol 85: 1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munck C, Gumpert HK, Wallin AI, Wang HH, Sommer MO. 2014. Prediction of resistance development against drug combinations by collateral responses to component drugs. Sci Transl Med 6: 262ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseeb S, Carter Z, Minnis D, Donaldson I, Zeef L, Delneri D. 2016. Widespread impact of chromosomal inversions on gene expression uncovers robustness via phenotypic buffering. Mol Biol Evol 33: 1679–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares J, Alvarez-Ortega C, Martinez JL. 2014. Metabolic compensation of fitness costs associated with overexpression of the multidrug efflux pump MexEF-OprN in Pseudomonas aeruginosa. Antimicrob Agents Chemother 58: 3904–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AZ, Campbell PJ, Willby MJ, Posey JE. 2015. Disparities in capreomycin resistance levels associated with the rrs A1401G mutation in clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 59: 444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander P, Springer B, Prammananan T, Sturmfels A, Kappler M, Pletschette M, Bottger EC. 2002. Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob Agents Chemother 46: 1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakov D, Akbergenov R, Matt T, Sander P, Andersson DI, Bottger EC. 2010. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol Microbiol 77: 830–840. [DOI] [PubMed] [Google Scholar]
- Sherman DR, Mdluli K, Hickey MJ, Arain TM, Morris SL, Barry CE III, Stover CK. 1996. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272: 1641–1643. [DOI] [PubMed] [Google Scholar]
- Sirgel FA, Tait M, Warren RM, Streicher EM, Bottger EC, van Helden PD, Gey van Pittius NC, Coetzee G, Hoosain EY, Chabula-Nxiweni M, et al. 2012. Mutations in the rrs A1401G gene and phenotypic resistance to amikacin and capreomycin in Mycobacterium tuberculosis. Microb Drug Resist 18: 193–197. [DOI] [PubMed] [Google Scholar]
- Springer B, Lucke K, Calligaris-Maibach R, Ritter C, Bottger EC. 2009. Quantitative drug susceptibility testing of Mycobacterium tuberculosis by use of MGIT 960 and EpiCenter instrumentation. J Clin Microbiol 47: 1773–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley RE, Blaha G, Grodzicki RL, Strickler MD, Steitz TA. 2010. The structures of the anti-tuberculosis antibiotics viomycin and capreomycin bound to the 70S ribosome. Nat Struct Mol Biol 17: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.