

Abstract
In Pseudomonas putida, the Hfq and Crc proteins regulate the expression of many genes in response to nutritional and environmental cues, by binding to mRNAs that bear specific target motifs and inhibiting their translation. The effect of these two proteins is antagonized by the CrcZ and CrcY small RNAs (sRNAs), the levels of which vary greatly according to growth conditions. The crcZ and crcY genes are transcribed from promoters PcrcZ and PcrcY, respectively, a process that relies on the CbrB transcriptional activator and the RpoN σ factor. Here we show that crcZ can also be transcribed from the promoter of the immediate upstream gene, cbrB, a weak constitutive promoter. The cbrB-crcZ transcript was processed to render a sRNA very similar in size to the CrcZ produced from promoter PcrcZ. The processed sRNA, termed CrcZ*, was able to antagonize Hfq/Crc because, when provided in trans, it relieved the deregulated Hfq/Crc-dependent hyperrepressing phenotype of a ΔcrcZΔcrcY strain. CrcZ* may help in attaining basal levels of CrcZ/CrcZ* that are sufficient to protect the cell from an excessive Hfq/Crc-dependent repression. Since a functional sRNA can be produced from PcrcZ, an inducible strong promoter, or by cleavage of the cbrB-crcZ mRNA, crcZ can be considered a 3′-untranslated region of the cbrB-crcZ mRNA. In the absence of Hfq, the processed form of CrcZ was not observed. In addition, we show that Crc and Hfq increase CrcZ stability, which supports the idea that these proteins can form a complex with CrcZ and protect it from degradation by RNases.
Keywords: global regulation of gene expression, catabolite repression, small RNAs, RNA processing, bacteria
INTRODUCTION
Small RNAs (sRNAs) are key elements in controlling the expression of bacterial genomes. They act post-transcriptionally, usually to inhibit translation of a given mRNA, although some can also activate translation or bind to and regulate the activity of specific proteins (Frohlich and Vogel 2009; Bobrovskyy and Vanderpool 2013; Wagner and Romby 2015). In addition to controlling the expression of specific genes, their influence on diverse transcriptional regulators allows them to modulate important regulatory networks (Romby et al. 2006; Waters and Storz 2009; Storz et al. 2011; Mandin and Guillier 2013). sRNAs are particularly useful for controlling rapid responses to environmental or physiological signals (Wassarman 2002; Bobrovskyy and Vanderpool 2013). For a metabolically versatile bacterium that thrives in a constantly changing environment, a rapid response can be especially important to allow it to profit from transitory nutrients.
Bacteria of the genus Pseudomonas are a clear example in this respect. They are metabolically and physiologically very flexible and have a considerable adaptive capacity that allows them to thrive in many different environments, even in extreme conditions (Silby et al. 2011; Wu et al. 2011; Moreno and Rojo 2014). Several recently described sRNAs have key roles in controlling many aspects of Pseudomonas physiology (Sharma and Storz 2011; Sonnleitner and Haas 2011; Gómez-Lozano et al. 2015). One of these, CrcZ, participates in a complex regulatory network that modulates the expression of many genes involved in nutrient assimilation, thus optimizing metabolism and improving growth (Sonnleitner et al. 2009; Moreno et al. 2012). CrcZ is found in all Pseudomonas for which the genome sequence is available. Many Pseudomonas species contain, in addition to CrcZ, other very similar sRNAs that are functionally redundant, such as CrcY in Pseudomonas putida (Moreno et al. 2012) or CrcX in Pseudomonas syringae (Filiatrault et al. 2013). CrcZ and its sRNA homologs regulate gene expression in association with the Hfq and Crc proteins.
Hfq is a hexameric RNA-binding protein that can recognize specific targets in RNAs and has a central role in post-transcriptional gene regulation. In Escherichia coli, it can facilitate the annealing of sRNAs to specific mRNAs to modulate translation, influence RNA decay, and enable the assembly of some ribonucleoprotein complexes (for recent reviews, see Vogel and Luisi 2011; Sobrero and Valverde 2012; De Lay et al. 2013; Wagner and Romby 2015). In Pseudomonas, Hfq is a global regulator that influences diverse features such as growth, metabolism, virulence, motility, quorum sensing, and tolerance to stress (Sonnleitner et al. 2006; Sonnleitner and Blasi 2014; Arce-Rodriguez et al. 2015; Madhushani et al. 2015; Moreno et al. 2015).
The Crc protein cooperates with Hfq to form stable complexes with RNAs bearing an A-rich motif with the consensus sequence AAnAAnAA. Although Crc was initially thought to bind RNA on its own (Moreno et al. 2009; Sonnleitner et al. 2009), recent results indicate that this is not the case, but rather that Hfq is the protein that initially recognizes the A-rich motif (Sonnleitner and Blasi 2014). The Crc protein appears to stabilize the Hfq–RNA complex and forms a tripartite Hfq–RNA–Crc complex (Madhushani et al. 2015; Moreno et al. 2015). Several mRNAs that specify proteins involved in the uptake and metabolism of carbon sources, or in other physiological processes, bear A-rich Hfq targets that overlap the translation initiation site; Hfq/Crc binding to these targets inhibits translation initiation (Sonnleitner and Blasi 2014; Madhushani et al. 2015; Moreno et al. 2015).
When cells grow in nutritionally rich conditions, a situation that elicits a carbon catabolite repression (CCR) response, the repressive effect of Hfq and Crc is strong. In contrast, when the preferred nutrient-rich compounds are consumed or are lacking, the effect of Hfq/Crc is low or undetectable. Hfq and Crc levels are similar in distinct growth conditions (Moreno et al. 2015). CrcZ levels (and those of homologous sRNAs such as CrcY) vary greatly in response to nutritional cues, however, and are inversely proportional to the strength of the inhibitory effect of Hfq and Crc proteins. CrcZ and its homologous sRNAs have several A-rich motifs and can bind Hfq/Crc in vitro. It was thus suggested that these sRNAs counteract Hfq/Crc repression by sequestering one or both proteins, making them unavailable to act on mRNAs with A-rich motifs. CrcZ/CrcY levels are low in nutritionally rich conditions, which would allow Hfq/Crc proteins to repress target mRNAs. When nutrients are scarce or render low energetic return, CrcZ/CrcY levels increase and could then sequester Hfq/Crc proteins, impeding their action on target mRNAs (Sonnleitner et al. 2009; Moreno et al. 2012; Filiatrault et al. 2013; Valentini et al. 2014).
The role of the Hfq/Crc/CrcZ-CrcY regulatory system in controlling gene expression, and the underlying molecular mechanisms, have been studied in some detail in Pseudomonas putida, a ubiquitous soil bacterium that has become an important model system in biotechnology (Poblete-Castro et al. 2012; Nikel et al. 2014). The P. putida crcZ and crcY genes are transcribed from promoters PcrcZ and PcrcY, respectively, which require the CbrB transcriptional activator and the RNA polymerase form bound to the RpoN alternative σ factor (Moreno et al. 2012; García-Mauriño et al. 2013). The activity of these promoters varies considerably, depending on the nutritional conditions. The crcY gene can also be transcribed from an unknown promoter upstream of PcrcY. The resulting transcript is processed to render a sRNA similar in size to that of CrcY (García-Mauriño et al. 2013); the possible function of this processed transcript was not studied.
Here we show that crcZ is transcribed not only from PcrcZ, but from the promoter of the cbrB gene, located immediately upstream of crcZ. The cbrB-crcZ transcript is processed to render a sRNA very similar in size to the primary CrcZ produced from PcrcZ. This suggests that crcZ is a 3′-untranslated region (3′-UTR) of cbrB that can generate a sRNA either from PcrcZ or by cleavage of the cbrB-crcZ mRNA. We analyzed the ability of the processed form of CrcZ to control Hfq/Crc function in vivo. In addition, we show that Hfq and Crc protect the primary and processed CrcZ forms from cleavage by ribonucleases, thus increasing their stability. Our data support a model that explains how the Hfq/Crc/CrcZ regulatory system could react to nutritional signals.
RESULTS
Cells lacking RpoN contain substantial amounts of CrcZ sRNA
In agreement with reports that promoter PcrcZ requires the CbrB transcriptional activator and the RpoN-RNA polymerase (Moreno et al. 2012; García-Mauriño et al. 2013), the β-galactosidase activity derived from a plasmid-borne PcrcZ-lacZ transcriptional fusion was very low in P. putida cells that lacked the RpoN σ factor or the CbrB activator (Fig. 1A,B). Based on this finding, the amount of CrcZ in these mutant strains should be low or undetectable; nonetheless, real-time RT–PCR monitoring of crcZ transcript levels (or transcripts including crcZ sequences) showed that the abundance of crcZ transcripts in the RpoN-null strain was similar to that of the wild-type strain, both during exponential growth in LB and in the stationary phase of growth (Fig. 1C). The CbrB-null strain behaved as predicted, with very low crcZ transcript levels (Fig. 1C). These results suggest that, at least in the RpoN-null strain, crcZ can be transcribed from an RpoN-independent promoter. The DNA segment in the PcrcZ-lacZ transcriptional fusion, including the promoter PcrcZ, spanned from the end of cbrB to the start of crcZ (plasmid pPcrcZ; Fonseca et al. 2013). Since this fusion was inactive in the RpoN-null strain, crcZ transcription in this strain must derive from an RpoN-independent promoter located within cbrB or upstream of it. When sequenced, the chromosomal region spanning from the start of cbrB to crcZ in the RpoN-null strain was found to be identical to the same region in the wild-type strain. This rules out the presence of a new promoter in the RpoN-null strain that would explain the high crcZ transcript levels. This sequence identity also indicates that, in the wild-type strain, crcZ could be transcribed not only from PcrcZ, but also from an RpoN-independent promoter. The absence of crcZ transcripts in the MPO401 CbrB-null strain (García-Mauriño et al. 2013) is probably due to the deletion of almost the entire cbrB gene, and to the kanamycin-resistance gene that replaces the deleted cbrB sequences and includes a transcriptional terminator that can stop transcription originated in upstream regions.
FIGURE 1.
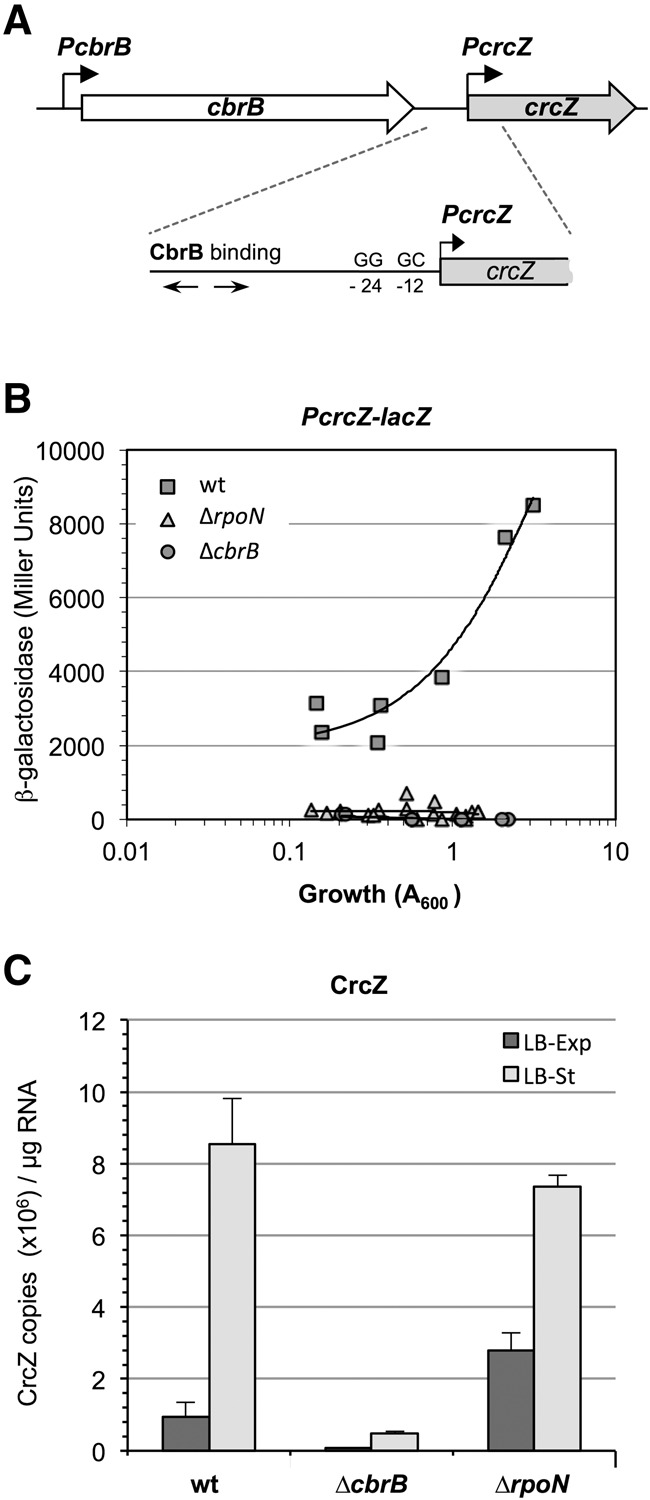
Influence of CbrB and RpoN on the activity of promoter PcrcZ and on CrcZ levels. (A) Scheme showing the location of promoter PcrcZ, the −12 and −24 consensus motifs for the RpoN σ factor, and the binding site for the CbrB transcriptional activator. (B) P. putida strains KT2442 (wt), MPO401 (cbrB::km derivative of KT2442, indicated as ΔcbrB), and KT2442rpoN::Ωkm (rpoN::Ωkm derivative of KT2442, indicated as ΔrpoN), transformed with plasmid pPcrcZ (includes a PcrcZ-lacZ transcriptional fusion, but lacks cbrB sequences), were cultured in LB medium, and β-galactosidase levels were measured at different times. The plot shows the values obtained as a function of cell growth (turbidity at 600 nm). (C) CrcZ levels in the above strains, cultured in LB medium and collected at mid-exponential phase (A600, 0.6; dark bars) or at the start of the stationary phase (turbidity, 2.2; light bars). RNA levels were determined by real-time RT-PCR; values shown are the mean ± SD for three independent assays.
Identification of promoters upstream of PcrcZ that can drive crcZ transcription
To assess whether crcZ is under the influence of a promoter located upstream of PcrcZ, we performed reverse transcription-PCR assays with total RNA from the wild-type and RpoN-null strains, and various primer pairs designed to detect transcripts that originate upstream of PcrcZ and span into crcZ. Results confirmed the presence of a transcript that includes crcZ and originates between the end of cbrB and PcrcZ, within cbrB, or upstream of cbrB (Fig. 2A).
FIGURE 2.

The crcZ gene can be transcribed from promoter PcbrB. (A) Identification of transcripts comprising crcZ that originate at or upstream of the promoter PcrcZ, detected by RT-PCR. Total RNA from the wild-type (KT2442) and RpoN-null (KT2442rpoN::Ωkm) strains cultured in LB medium was purified, transformed to cDNA with reverse transcriptase and a primer that hybridized within crcZ (Zrv; see figure); the cDNA was amplified by PCR using forward primers that hybridized up- (Zfw2) or downstream (Zfw1) from promoter PcrcZ, and the reverse Zrv primer that hybridized within crcZ. Control reactions to which RNase A was added, or in which the PCR was performed with genomic DNA, were included as controls. (M) DNA size ladder (in bp). (B) Transcriptional fusions of the cbrB gene, or of serial deletions affecting its 5′-region, to the lacZ reporter gene. The cbrB segments fused to lacZ were generated by PCR with forward primers FW1 to FW7, and with the reverse primer RV(7); note that only the segment generated with primer FW1 includes the PcbrB promoter and that none of the fusions includes the sequences corresponding to promoter PcrcZ or crcZ. DNA segment size is indicated. (C) β-Galactosidase activity displayed by P. putida KT2440 harboring plasmids that include the transcriptional fusions constructed in B, named according to the corresponding forward primer. A strain bearing the plasmid vector used to construct the fusions (pMP220) was included as control. Cells were cultured in LB medium, samples were withdrawn at various times, and β-galactosidase activity was determined using ONPG as the substrate; data shown are the mean ± SD for three independent assays. (D) Influence of RpoN on cbrB gene expression. The left panel shows the transcription of cbrB, as deduced from a cbrB-lacZ transcriptional fusion, in P. putida strains KT2442 (wt) and KT2442rpoN::Ωkm (ΔrpoN), transformed with plasmid pFW1 (contains the cbrB-lacZ transcriptional fusion; see panel B). Cells were cultured in LB medium; at mid-exponential phase (A600, 0.6), samples were taken and β-galactosidase activity was determined using ONPG as substrate. Data shown are the mean ± SD for three independent assays. The right panel shows the abundance of cbrB transcripts in P. putida strains KT2442 (wt) and KT2442rpoN::Ωkm (ΔrpoN), determined by real-time RT-PCR with RNA from cells cultured in LB medium and collected at mid-exponential phase (A600, 0.6). The primers used for the PCR reaction hybridized at the central region of cbrB. Values show the mean ± SD for three independent assays.
Two parallel approaches were used to locate this promoter. We first tested whether crcZ can be transcribed from the cbrB promoter, termed PcbrB (Amador 2011), or from an uncharacterized internal promoter within cbrB. The activity of a plasmid-borne transcriptional fusion of cbrB (including the PcbrB promoter) to lacZ was compared to that of a series of similar reporter fusions in which the 5′-end of cbrB had been trimmed progressively to eliminate PcbrB and downstream sequences (see Fig. 2B). Deletion of promoter PcbrB completely eliminated reporter gene transcription (Fig. 2C), indicating that cbrB has no internal promoters that could affect crcZ, and that the crcZ transcripts that originate upstream of PcrcZ might derive from promoter PcbrB.
Introduction of the cbrB-lacZ reporter fusion contained in plasmid pFW1 (see Fig. 2B,C) into the RpoN-null strain KT2442rpoN::Ωkm, showed that β-galactosidase levels were three times higher in the absence of RpoN than in its presence (Fig. 2D, left panel). Real-time RT-PCR assays confirmed that transcripts for cbrB were twofold more abundant in the RpoN-null than in the wild-type strain (Fig. 2D, right panel). PcbrB promoter activity can thus explain, at least in part, the presence of crcZ transcripts in the RpoN-null strain. This activity was nevertheless much lower than that of promoter PcrcZ in the same conditions (cf. Figs. 1B, 2C).
In an alternative approach, we used 5′ RACE to test for the possible presence of a crcZ promoter in the vicinity of PcrcZ (Fig. 3A). RNA preparations from the wild-type and RpoN-null strains growing exponentially in LB medium were treated with the TEX nuclease (terminator 5′-phosphate-dependent exonuclease), which digests RNAs that have a 5′ monophosphate, but is inactive on primary transcripts with a triphosphate at their 5′ end. In the wild-type strain, the 5′-end of most of the transcripts detected (64%) corresponded to an A residue located 2 nucleotides (nt) downstream from the start site mapped by Moreno et al. (2012) and 1 nt downstream from that reported by García-Mauriño et al. (2013), both of which were determined using a primer extension approach. In the RpoN-null strain, many fewer clones were isolated from the RACE assays, which suggests the absence of primary crcZ transcripts. In addition, the 5′-end indicated by the isolated clones mapped in most cases at positions 5–14 nt upstream of the +1 start site observed for CrcZ in the wild-type strain; 50% of these 5′-ends mapped at positions −5 and −6 (Fig. 3A). The heterogeneity of the 5′-ends suggests that they are processed transcripts that escaped the TEX treatment.
FIGURE 3.

Identification of the 5′ end of crcZ transcripts in wild-type and rpoN P. putida strains. Total RNA was obtained from P. putida KT2442 (wt) and KT2442rpoN::Ωkm (ΔrpoN) cultured in LB medium and the 5′ end of the crcZ transcripts were determined using 5′ RACE (A) or primer extension (B). (A) 5′ RACE assays were performed with RNA samples previously treated with terminator 5′-phosphate-dependent exonuclease (TEX). Fourteen independent clones were sequenced for each strain; the 5′ ends found for the wild type are indicated by black arrows, and those for the rpoN strain by gray arrows. The frequency with which each 5′ end was found is indicated in the lower panels (in %). (B) Primer extension reactions were performed with an end-labeled oligonucleotide and RNA samples, untreated or treated with TEX enzyme, as indicated. The extended cDNA products were analyzed by electrophoresis on a denaturing 6% urea–polyacrylamide gel, in parallel with a DNA sequence ladder (L). Numbers (right) indicate the position of the 5′ ends detected relative to the crcZ transcription start site (+1).
The 5′ end of crcZ transcripts was also analyzed using primer extension assays performed with an end-labeled oligonucleotide primer. When using TEX-treated RNA samples, we detected an extension product in the wild-type strain, which corresponded to a transcript originated at the transcription start site detected in the RACE assays (position +1; Fig. 3B). This extension product was barely visible in the RNA sample from the RpoN-null strain (Fig. 3B). In the absence of TEX treatment, most extension products in the RpoN-null strain samples were transcripts with 5′ ends matching positions −6, −9, −11, or −13. Within the accuracy limits of the primer extension assays, these 5′ ends coincide with those found using 5′ RACE assays. The extension products with 5′ ends in the −6 to −13 region were also detected in the TEX-untreated wild-type strain samples, although their abundance was much lower than in the RpoN-null strain and represented a minor fraction of overall crcZ transcripts (Fig. 3B). These results support the idea that promoter PcrcZ is inactive in the absence of RpoN and that additional promoters cannot be detected in this region. The transcripts detected in the RpoN-null strain are probably due to read-through transcription from cbrB and cleavage of the cbrB-crcZ transcripts.
The transcript originated at PcbrB can be processed to generate an sRNA similar to the primary CrcZ
When we used a probe for crcZ, Northern blot analysis of RNA samples from cells growing exponentially in LB medium showed that the RpoN-null strain bears a transcript that hybridizes to the probe and is very similar in size to CrcZ (Fig. 4). This transcript was barely detected in TEX-treated samples, which indicates that it is not a primary transcript, but derives from processing of a larger transcript that probably originated at the PcbrB promoter. This idea coincides with the results of the 5′ RACE and primer extension assays for the RpoN-null strain (Fig. 3), which suggest a processing site located 5–9 nt upstream of the promoter PcrcZ +1 start site. For simplicity, these processed transcripts of very similar size were collectively termed CrcZ*. In the wild-type strain, however, most crcZ transcripts were TEX-resistant, although quantitation of the bands in three independent assays indicated that transcript levels decreased by ∼20% after TEX treatment. This again suggests that processed CrcZ* transcripts are also present in the wild-type strain.
FIGURE 4.

Identification of CrcZ sRNA by Northern blot in strains KT2442 (wt), MPO401 (ΔcbrB), KT2442rpoN::Ωkm (ΔrpoN). Strain KT2440-Z, which has an inactivated crcZ::tet allele and lacks CrcZ, is included as control (indicated as ΔcrcZ). Total RNA was obtained from cells growing exponentially in LB medium (A600, 0.6). RNA samples were untreated or treated with TEX, resolved on a 6% polyacrylamide/7 M urea gel, transferred to a nitrocellulose membrane, and the presence of CrcZ and 5S rRNA determined by hybridization with specific probes. Lane M corresponds to an RNA size ladder.
Real-time RT-PCR assays performed with RNA from cells growing exponentially in LB medium showed that the number of transcripts that include crcZ sequences were much more abundant than those for cbrB (∼10-fold for the wild-type strain and 16-fold for the RpoN-null strain; compare data in Fig. 1C and Fig. 2D). This suggests that, after cleavage of the cbrB-crcZ transcript, the 5′-segment for cbrB is rapidly degraded, while the fragment corresponding to crcZ is more stable and accumulates, particularly in the RpoN-null strain.
In the CbrB-null strain MPO401, neither the primary nor the processed crcZ transcripts were detected (Fig. 4). Primary transcripts that originated at PcrcZ are predicted to be absent in this strain, since this promoter relies on the CbrB activator, which is lacking. The absence of processed crcZ transcripts is probably the consequence of the kanamycin-resistance determinant that interrupts the cbrB gene in this strain, and stops transcripts that originate at promoter PcbrB.
The processed form of CrcZ can antagonize Hfq/Crc function, complementing the hyperrepressing phenotype of a ΔcrcZΔcrcY double mutant
Our results showed that cells can bear two CrcZ variants, the primary CrcZ and the processed CrcZ*. The processed form is the predominant, if not the only variant in the RpoN-null strain, whereas most CrcZ in the wild-type strain corresponds to the primary transcript. Prediction of the secondary structure of CrcZ* and CrcZ, using the RNAfold software package (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi), suggested that the overall structure of these sRNAs is highly similar. Although there are some local modifications due to the extra nucleotides present at the CrcZ* 5′-end, they are small, and the stem–loops that include the Hfq binding sites remain unchanged (not shown). We thus assessed whether the processed CrcZ* variant is functional and can antagonize the repressing activity of Hfq/Crc proteins, which would open up a possible role for CrcZ* in the CbrB/Hfq/Crc regulatory system. We previously reported that a mutant P. putida strain bearing inactivated crcZ and crcY genes, and therefore lacking all forms of CrcZ and CrcY, is unable to grow in a minimal salts medium containing benzoate or citrate as the sole carbon source, and it has a markedly reduced growth rate when glucose, fumarate, or succinate are provided as the carbon source (Moreno et al. 2012). Since the wild-type strain can grow efficiently using any of these compounds, we proposed that lack of CrcZ and CrcY (and of its processed forms, CrcZ* and CrcY*) deregulates the Hfq/Crc system, leading to strong, constitutive Hfq/Crc-dependent repression of genes involved in the uptake and assimilation of these nonpreferred carbon sources (Moreno et al. 2012).
To test whether CrcZ* expression in trans complements the ΔcrcZΔcrcY strain growth defects, two plasmids were constructed. The first, p421-Pwt, bore a DNA segment that includes the cbrB and crcZ genes (expressed from their native promoters, PcbrB and PcrcZ) and can therefore generate CrcZ and CrcZ* (see Fig. 5A). The second plasmid, p421-Pmut, has a variant of this DNA fragment in which the GG and GC nucleotides at the consensus −24 and −12 regions of promoter PcrcZ were modified to CC and TT, respectively. This was predicted to inactivate promoter PcrcZ, such that plasmid p421-Pmut would generate the processed CrcZ* form but not the primary CrcZ transcript. To verify that the mutations inactivated PcrcZ, a DNA segment from p421-Pmut spanning from the end of cbrB to the promoter PcrcZ start site was PCR amplified and cloned into the pMP220 reporter plasmid, generating a transcriptional fusion to the lacZ indicator gene. The plasmid obtained, pPcrcZmut, was introduced into strain KT2440, and its ability to produce β-galactosidase was compared to that of plasmid pPcrcZ, which bears a wild-type PcrcZ promoter. As predicted, the mutant promoter was totally inactive (Fig. 5B).
FIGURE 5.

Plasmids designed to produce CrcZ and CrcZ*, or only CrcZ*. (A) Scheme of the insert in plasmids p421-Pwt and p421-Pmut. Plasmid p421-Pwt bears the cbrB and crcZ genes. Plasmid p421-Pmut contains cbrB and a mutant variant of PcrcZ in which the GG and GC nucleotides of the −24 and −12 regions, critical for recognition by RpoN-RNA polymerase, were modified (indicated in the blow-up). The RNAs predicted in each case are indicated. (B) P. putida strain KT2440 containing plasmid pPcrcZ (includes a PcrcZ-lacZ transcriptional fusion) or pPcrcZmut (includes a similar transcriptional fusion, with the mutant PcrcZmut promoter described in A), were cultured in LB medium and β-galactosidase levels measured at different times. The plot shows values obtained as a function of cell growth. (C) Northern blot identification of CrcZ/CrcZ* in strain KT2440-ZY containing plasmids pSEVA421 (control), p421-Pwt, or p421-Pmut. Total RNA from cells growing exponentially in LB medium was untreated or treated with TEX, resolved on a denaturing polyacrylamide/urea gel, transferred to a nitrocellulose membrane, and the presence of CrcZ and 5S rRNA determined by hybridization with specific probes. (D) The amount of CrcZ/CrcZ*, determined by real-time RT-PCR, in strains KT2440 and KT2440-ZY containing the plasmids is indicated in C; total RNA was purified from cells cultured in LB medium and collected at mid-exponential phase (A600 of 0.6; “Ex”) or in stationary phase (A600 of 2.2; “St”).
We introduced plasmids p421-Pwt and p421-Pmut into the ΔcrcZΔcrcY strain KT2440-ZY and analyzed their ability to produce CrcZ and/or CrcZ*. As control, the empty plasmid vector (pSEVA421) was also introduced into strain KT2440-ZY. Northern blot analysis showed that plasmid p421-Pwt allowed production of an sRNA that hybridizes to the CrcZ probe and whose abundance decreases little after TEX treatment (Fig. 5C). This suggests that most sRNA produced is primary CrcZ, although CrcZ* would also be present. In contrast, the sRNA species produced by plasmid p421-Pmut was completely degraded by TEX treatment, indicating that this plasmid generates CrcZ* but not primary CrcZ. Real-time RT-PCR indicated that, in cells growing exponentially in LB medium, the amount of CrcZ* in the mutant strain KT2440-ZY containing p421-Pmut was similar to that of CrcZ in the wild-type KT2440 strain (Fig. 5D); nonetheless, the amount of CrcZ produced from plasmid p421-Pwt in strain KT2440-ZY was twice as high.
We next analyzed the ability of plasmids p421-Pwt and p421-Pmut to restore the growth defect of strain KT2440-ZY in a minimal salts medium containing citrate, benzoate, glucose, or succinate as the sole carbon source. When using citrate or benzoate, the control strain KT2440-ZY(pSEVA421) could not grow even after 24 h in culture, while the wild-type strain KT2440(pSEVA421) grew efficiently (Fig. 6). The presence of plasmid p421-Pwt in the CrcZ/CrcY-null strain KT2440-ZY fully restored growth, with a growth rate similar to that of the wild-type KT2440 strain. This suggests that the CrcZ and CrcZ* sRNAs produced from plasmid p421-Pwt can complement the growth defect of strain KT2440-ZY when citrate or benzoate are the only carbon source available. Plasmid p421-Pmut, which generates CrcZ* but not the primary CrcZ (see Fig. 5C), provided partial complementation, since cells grew slowly and cultures eventually reached turbidity values similar to those of the wild-type strain after 24 h culture (Fig. 6). This suggests that CrcZ* can at least partially antagonize the inhibitory effect of Hfq/Crc. Most likely, complementation is only partial because the amount of CrcZ* produced from the weak PcbrB promoter is lower than that of CrcZ generated from the strong PcrcZ promoter.
FIGURE 6.

Growth of P. putida strains KT2440 (wild type) and KT2440-ZY (a ΔcrcZΔcrcY derivative of strain KT2440), containing plasmids pSEVA421 (vector with no insert), p421-Pwt (produces CrcZ and CrcZ*), or p421-Pmut (produces CrcZ*), in M9 minimal salts medium containing citrate, benzoate, glucose, or succinate as sole carbon source. Note discontinuity in the time scale after hour 7.
Lack of CrcZ and CrcY had a less severe effect when strain KT2440-ZY used glucose or succinate as the carbon source; cells grew slowly, although after 24 h, turbidity values were high (Fig. 6). Plasmid p421-Pmut could effectively complement the lack of CrcZ and CrcY, particularly when succinate was the carbon source (Fig. 6). Since this plasmid allows production of CrcZ*, but not of the primary CrcZ, its ability to restore the growth of strain KT2440-ZY on glucose or succinate further shows that the processed CrcZ* form can at least partially antagonize the repressive effect of the Hfq/Crc proteins, supporting that it is functional.
Since the amount of CrcZ* present in a wild-type strain is low, it can be asked whether it plays an active role in the modulation of Hfq/Crc-dependent catabolite repression when growth conditions change from a situation of high repression (for example, a complete medium) to one of low repression (a poor carbon source available). We therefore analyzed whether the amount of CrcZ* produced from promoter PcbrB is high enough to relieve the Hfq/Crc-dependent repression of the benA gene, which codes for the first enzyme of the benzoate assimilation pathway. Transcription of benA requires the BenR transcriptional activator. Earlier work showed that the Hfq/Crc system inhibits translation of benR mRNA, which bears an A-rich motif to which Hfq/Crc bind (Moreno and Rojo 2008; Hernández-Arranz et al. 2013). As a result, in cells growing exponentially in benzoate-containing LB medium, Hfq/Crc keep BenR levels below those needed for an efficient induction of the benA gene, exerting an indirect but strong inhibition of benA transcription (Morales et al. 2004; Moreno and Rojo 2008; Moreno et al. 2012; Hernández-Arranz et al. 2013).
The indirect Hfq/Crc-dependent inhibition of benA transcription can be monitored using P. putida strain PBA1, which bears a PbenA-lacZ transcriptional fusion in its chromosome, and its derivatives PBA1C (Crc-null) and PBAZY (CrcZ-null, CrcY-null; lacking all forms of these sRNAs) (Moreno and Rojo 2008; Moreno et al. 2012). Plasmids p421-Pwt and p421-Pmut were introduced into strain PBAZY, while the empty plasmid vector pSEVA421 was introduced into strains PBA1, PBAZY, and PBA1C, as controls. The ability of benzoate to induce transcription from promoter PbenA was followed in all strains by measuring β-galactosidase production in cells cultured in LB medium with 5 mM benzoate. β-Galactosidase expression in strain PBA1 (pSEVA421) was strongly inhibited during the exponential growth phase until the culture reached a turbidity of ∼1–1.5, when cells prepared to enter the stationary phase and Hfq/Crc-dependent repression was relieved (see Fig. 7), a process that coincides with a strong increase in CrcZ/CrcY levels (Moreno et al. 2012). Repression was not observed in the Crc-null strain, as predicted. In strain PBAZY, however, repression was strong and did not disappear after entry into stationary phase, as would be anticipated for a strain with a deregulated, constitutively active Hfq/Crc repression system. Introduction of plasmid p421-Pwt into strain PBAZY partially relieved repression, probably because the plasmid provides several copies of the cbrB and crcZ genes that give rise to increased CrcZ dosage (see Fig. 5D). The presence of p421-Pmut in strain PBAZY had no apparent effect. This plasmid allows production of CrcZ* but not of CrcZ. Since the results presented in Figure 6 had indicated that CrcZ* can at least partially antagonize the inhibitory effect of Hfq/Crc, the observation that it cannot relieve the Hfq/Crc-dependent inhibition of benA transcription suggests that the levels of CrcZ* produced from plasmid p421-Pmut are not high enough to counteract the Hfq/Crc-dependent inhibition of benA expression. In support of this idea, real-time RT-PCR assays (Fig. 5D) indicated that CrcZ* levels in a ΔcrcZΔcrcY strain containing plasmid p421-Pmut and growing exponentially in LB medium are about half those of CrcZ + CrcZ* generated from p421-Pwt, and very similar to those seen in the wild-type strain, levels that allow a strong Hfq/Crc-dependent repression. In addition, since plasmid p421-Pmut generates CrcZ* from promoter PcbrB, which is weak and constitutive, the amount of CrcZ* cannot increase in the stationary phase, and therefore Hfq/Crc-dependent repression of promoter PbenA is not relieved in the stationary phase, in contrast to what is observed when the primary CrcZ is expressed from the strong and inducible PcrcZ promoter. Altogether, the results presented suggest that the amounts of CrcZ* present in the cell are not sufficient to counteract the strong CCR effect that occurs in a complete medium, but can contribute to provide a basal protection from an excessive Hfq/Crc-dependent repression.
FIGURE 7.
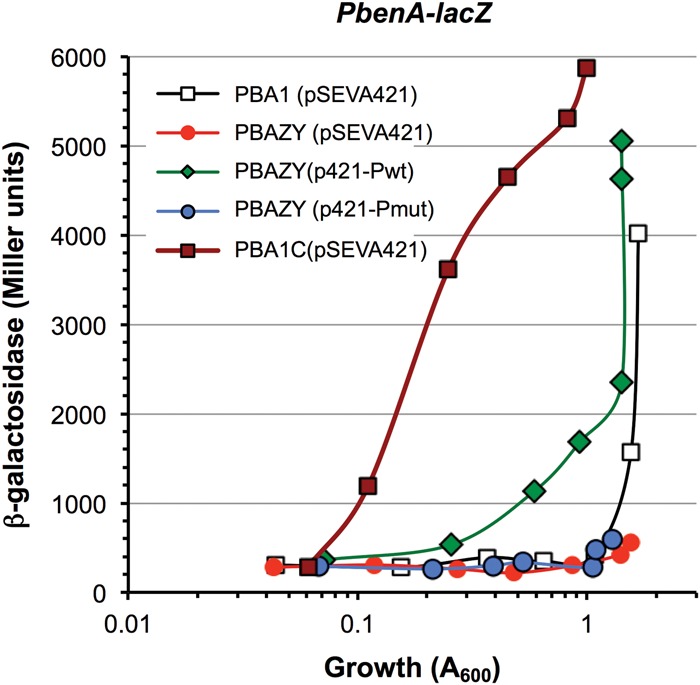
P. putida strains PBA1 (contains a PbenA-lacZ transcriptional fusion in its chromosome), PBA1C (a Crc-null derivative of strain PBA1), or PBAZY (a ΔcrcZΔcrcY derivative of strain PBA1), where indicated containing plasmids pSEVA421, p421-Pwt, or p421-Pmut, were cultured in LB medium with 5 mM benzoate, and β-galactosidase levels were measured at different times. The plot shows the values as a function of cell growth (turbidity at 600 nm). Exponential growth declined at a turbidity of ∼1.5, when cells prepared to enter the stationary growth phase.
Influence of Crc and Hfq proteins on CrcZ expression, stability, and processing
P. putida CrcZ levels decrease considerably following inactivation of the crc gene (García-Mauriño et al. 2013). This effect has been traced to a reduction in promoter PcrcZ activity when the Crc protein is absent. Although the underlying reasons are unclear, the authors proposed that Crc inhibits translation of an mRNA encoding a transcriptional repressor that inhibits PcrcZ activity. The effect of Hfq on CrcZ levels and PcrcZ activity was unknown. Northern blot analysis showed that lack of Hfq also led to a marked decrease in CrcZ levels (Fig. 8A), which was confirmed by real-time RT-PCR (not shown). The absence of Hfq also had a negative effect on promoter PcrcZ activity (Fig. 8B). The strong decrease in CrcZ when Crc or Hfq proteins are absent might nonetheless be due not only to reduced PcrcZ activity, but also to reduced sRNA stability, since the CrcZ sRNA has several A-rich motifs that are very similar to RNase E targets.
FIGURE 8.
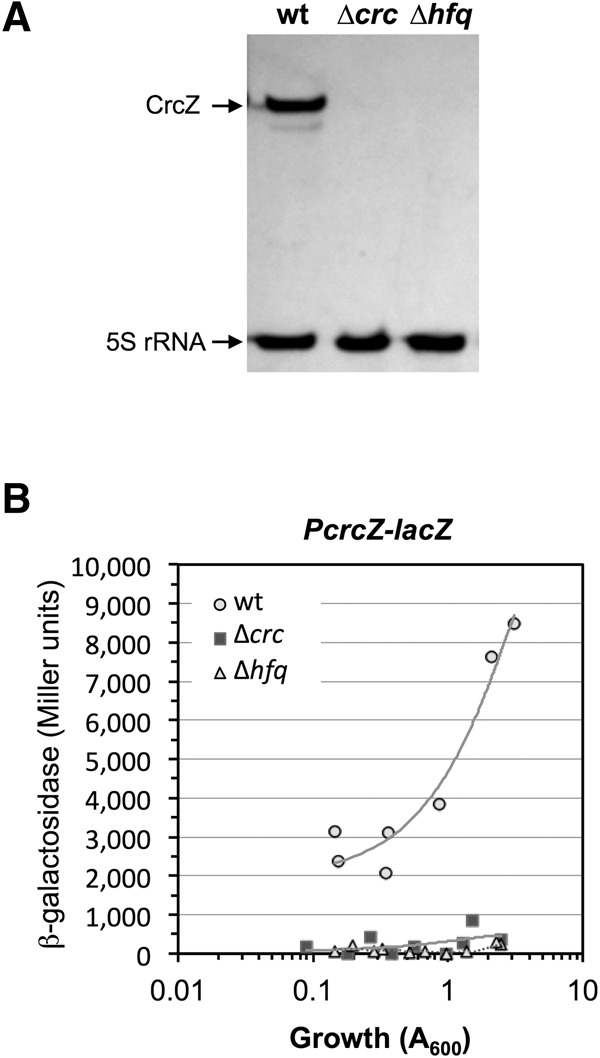
Amounts of CrcZ (A) and PcrcZ promoter activity (B) in P. putida strains KT2440 (wt), KTCRC (Δcrc), and KT2440Δhfq (Δhfq derivative of KT2440). (A) Total RNA was obtained from cells growing exponentially in LB medium (A600, 0.6), and CrcZ and 5S rRNA determined by Northern blot with specific probes. (B) The strains indicated above, transformed with plasmid pPcrcZ (which bears a PcrcZ-lacZ transcriptional fusion), were cultured in LB medium. Samples were taken at different times and β-galactosidase levels measured using ONPG as substrate. Two independent assays were performed; results from both of them are shown.
As Hfq/Crc binding to CrcZ might protect the sRNA from RNase degradation, which could affect CrcZ levels, we analyzed the influence of Crc and Hfq on CrcZ stability. Since CrcZ levels in cells lacking Crc or Hfq are essentially undetectable, we introduced plasmid p424-Z, which allows crcZ transcription from the heterologous IPTG-inducible Ptrc promoter, into P. putida strains KT2440Δhfq (Hfq-null) and KTCRC (Crc-null). As a control, the same plasmid was introduced into the CrcZ-null strain KT2440-Z, which is wild type for Crc and Hfq and in which all CrcZ must be generated from plasmid p424-Z. The three strains were cultured in LB medium with 1 mM IPTG (to induce crcZ transcription); cells were collected at mid-exponential phase, their RNA extracted, and CrcZ levels were analyzed by Northern blot and real-time RT-PCR. In the strain bearing wild-type crc and hfq alleles, CrcZ overproduction led to the appearance of two similarly sized bands that hybridized to the probe (Fig. 9A; lanes labeled “ΔcrcZ”). The larger band was TEX-resistant and probably corresponds to a primary, plasmid-produced crcZ transcript that contains 62 extra nucleotides at its 5′-end, which is the distance between the Ptrc promoter transcription start site and the first crcZ nucleotide. The smaller band was similar in size to the CrcZ sRNA in the wild-type KT2440 strain (Fig. 9A, labelled “wt”) and was selectively degraded by TEX. This smaller band probably corresponds to a processed transcript that has lost most or all of the 62-nt extra tail at the 5′ end. This processed transcript was not observed in the Hfq-null strain. This suggests that Hfq facilitates this processing or, alternatively, that Hfq protects CrcZ from degradation by RNases, protection that would be more important for the processed transcript than for the primary transcript. Although the processed crcZ transcript was detected in the Crc-null strain, it was somewhat smaller than in the wild-type strain (Fig. 9A), which suggests that Crc influences the structure of the RNA–Hfq complex that facilitates processing and favors a defined excision site. Real-time RT-PCR assays to quantify the combined abundance of primary and processed CrcZ forms showed that, when CrcZ was overproduced by the plasmid, lack of Crc or Hfq reduced CrcZ steady-state levels by 2.5- and 3.7-fold, respectively (Fig. 9B), which coincides with Northern blot assay results.
FIGURE 9.
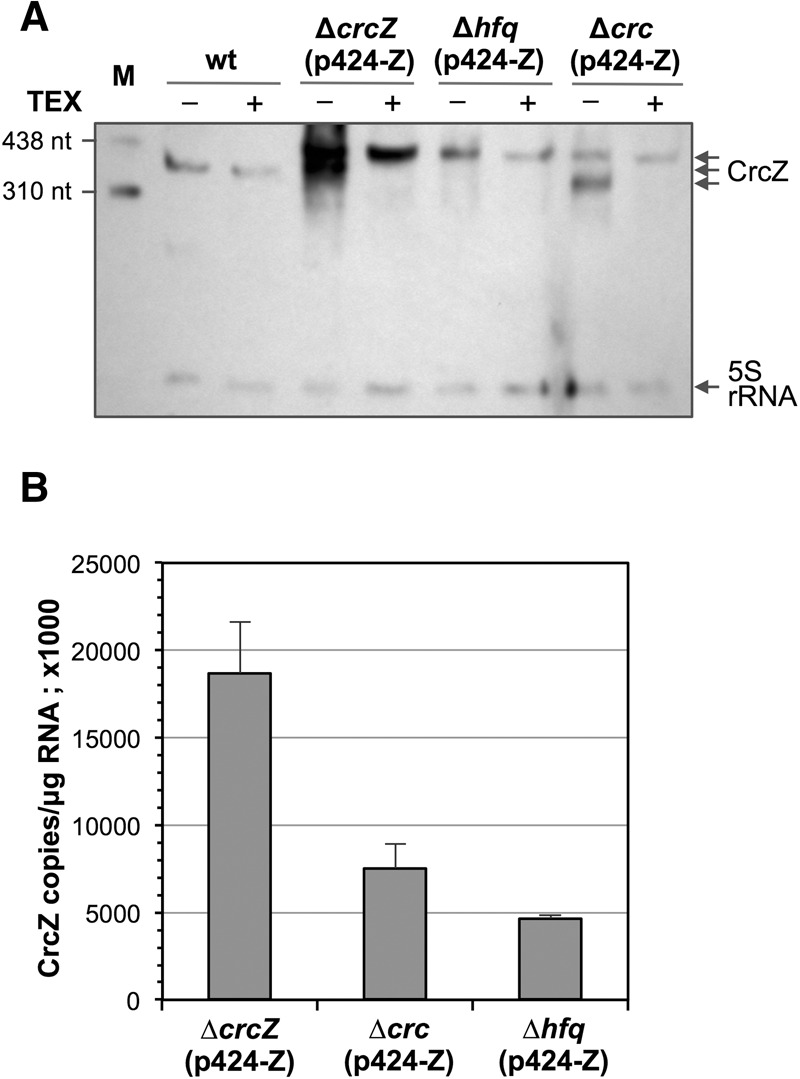
Overproduction of CrcZ in cells with wild-type, CrcZ-null, Hfq-null, or Crc-null genetic backgrounds. Plasmid p424-Z, which allows crcZ expression from the IPTG-inducible Ptrc promoter, was introduced into P. putida strains KT2440-Z (ΔcrcZ), KT2440Δhfq (Δhfq), and KTCRC (a Δcrc derivative of KT2440). Cells were cultured in LB medium with streptomycin and 1 mM IPTG. At mid-exponential phase (A600, 0.6), cells were collected and total RNA obtained. A wild-type KT2440 strain lacking plasmid p424-Z was cultured and processed in parallel. (A) RNA samples were untreated or TEX-treated, and CrcZ was analyzed by Northern blot as in Figure 4. Lane M shows an RNA size ladder. (B) The amount of CrcZ in RNA samples from strains bearing plasmid p424-Z, determined by real-time RT-PCR. Values show the mean ± SD for three independent assays.
To analyze the influence of Crc and Hfq on the stability of crcZ transcripts originated from plasmid p424-Z, P. putida strains KT2440-Z (CrcZ-null), KT2440Δhfq (Hfq-null), and KTCRC (Crc-null), all bearing plasmid p424-Z, were cultured in LB medium with IPTG, and transcription was blocked at mid-exponential phase by rifampicin addition. Samples were taken at different times, total RNA was purified from the cells, and crcZ transcript abundance was analyzed in each sample by Northern blot. The half-life of the primary crcZ transcript produced from the plasmid Ptrc promoter was ∼6 min in the strain containing both Crc and Hfq (strain ΔcrcZ), but decreased to ∼3.8 min in the strain lacking Hfq, and to ∼3 min when Crc was absent (Fig. 10A–C; summarized in Fig. 10D). The absence of Crc and Hfq thus reduces primary CrcZ stability by almost half. The pattern was similar for the processed crcZ transcript (higher electrophoretic mobility band in Northern blots); the absence of Crc reduced transcript half-life by almost half (from 6.8 to 3.8 min; Fig. 10E). It is worth noting that when Crc and Hfq proteins were both present, decay of the primary and the processed crcZ transcripts was delayed for ∼4 min, after which their levels began to diminish. In the absence of Crc or Hfq, RNA decay began immediately.
FIGURE 10.

Half-life of CrcZ produced from plasmid p424-Z in cells with CrcZ-null, Hfq-null, or Crc-null genetic backgrounds. (A−C) P. putida strains KT2440-Z (ΔcrcZ), KT2440Δhfq (Δhfq), and KTCRC (Δcrc) were cultured in LB medium with streptomycin and 1 mM IPTG. At a turbidity of 0.6 (A600), rifampicin was added to stop transcription. Aliquots were withdrawn at 0, 2, 4, 8, 16, and 32 min post-rifampicin addition, total RNA was isolated, resolved on a 6% polyacrylamide/7 M urea gel, and CrcZ and 5S rRNA detected by Northern blot with specific probes. Band intensities were quantified using a ChemiDoc XRS imager and Image Lab software (Bio-Rad). Values obtained for the larger (primary CrcZ transcript) or shorter bands (processed CrcZ transcript) detected with the CrcZ probe were normalized to those of the 5S rRNA of the corresponding sample. (D,E) Plots show the values for each time point expressed relative to that at time 0. Processed CrcZ was not detected in the Δhfq strain. Data shown as mean ± SD for three independent assays.
DISCUSSION
Many sRNAs originate from noncoding genes located within intergenic regions, between protein-coding genes (Miyakoshi et al. 2015). The CrcZ sRNA, present in all Pseudomonads for which the genome sequence is known, was thought to belong to this group, since it originates from a promoter (PcrcZ) located downstream from the cbrB gene. Here we show that crcZ can also be transcribed from the cbrB promoter, which generates a cbrB-crcZ transcript that is processed to render a sRNA very similar in size to that of the primary CrcZ produced from promoter PcrcZ. Processing occurred preferentially at a site 5–9 nt upstream of the 5′-end of the primary CrcZ. In a wild-type strain, these processed variants, collectively termed CrcZ*, account for a small percentage of the total CrcZ species. In the RpoN-null strain analyzed in this work, promoter PcrcZ is inactive and CrcZ* is the only species present, reaching levels similar to or higher than those of the primary CrcZ in the wild-type strain. The crcZ gene can thus be considered a 3′-untranslated region (3′-UTR) of cbrB that can generate a sRNA from PcrcZ or by cleavage of the cbrB mRNA 3′-UTR (see Fig. 11). Although these two sRNA variants have different 5′-ends, they share the transcriptional terminator at the crcZ 3′-end.
FIGURE 11.
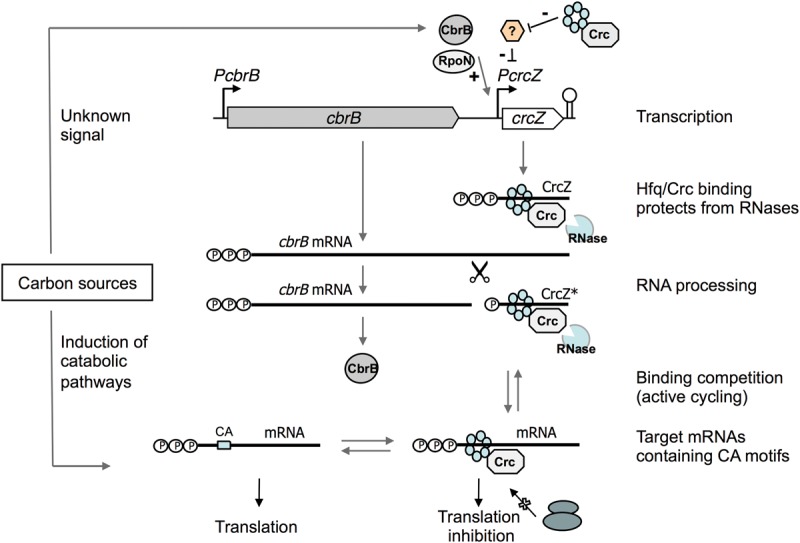
Model for the Hfq/Crc/CrcZ regulatory system. See Discussion for details.
We present evidence that an artificial CrcZ variant with an unrelated 62-nt extra tail at its 5′-end is also processed to render a sRNA similar in size to the primary CrcZ (Figs. 9, 10). This finding suggests that processing depends on a sequence or structure downstream from the cleavage site, and that the upstream sequences are less important. A similar observation was recently made for the E. coli GlmZ sRNA, in which a central stem–loop is decisive for cleavage by RNase E, whereas the precise sequence at the cleavage site was not important (Gopel et al. 2016). We did not detect processing of the CrcZ variant with 62 extra nucleotides at its 5′ end in the Hfq-null strain (Fig. 9), which implies that Hfq is involved in this processing or is needed to avoid transcript degradation by RNases. The Vibrio cholerae Hfq protein was similarly observed to participate in MicX transcript processing by RNaseE to generate a stable, more active sRNA (Davis and Waldor 2007).
In all Pseudomonas genomes analyzed to date, crcZ maps downstream from cbrB (Sonnleitner et al. 2009; Moreno et al. 2012; Filiatrault et al. 2013). Our finding that cbrB and crcZ are functionally linked and form a single transcriptional unit in P. putida suggests that cbrB-crcZ cotranscription, and possibly its processing to yield CrcZ*, might occur in other Pseudomonas species as well. The P. putida CrcY sRNA, which is very similar to CrcZ and functionally redundant, maps at a site distant from crcZ, downstream from the mvaB gene (Moreno et al. 2012). Like crcZ, crcY is transcribed from the CbrB-dependent PcrcY promoter, but transcripts that originate upstream of mvaB can run into crcY. This generates a transcript that can be processed to render sRNA similar in size to the primary CrcY generated from PcrcY (García-Mauriño et al. 2013). Therefore, CrcY can also be considered a 3′-UTR of mvaB. Pseudomonas syringae CrcZ and CrcX sRNAs are also proposed to arise from two promoters, one recognized by RpoN and the other by a different σ factor (Filiatrault et al. 2013). The coexistence of two forms of these sRNAs, a primary and a processed transcript of very similar sizes, could thus be common in Pseudomonas. Nesting a sRNA within a functionally related mRNA, with which it shares the intrinsic terminator, allows coupling a protein (a regulator) and a noncoding function (a sRNA) into a single expression unit that can be transferred horizontally to other bacteria (Miyakoshi et al. 2015).
There is evidence to support the idea that CrcZ* is a functional sRNA, able to antagonize the effect of Hfq and Crc. The growth of a ΔcrcZΔcrcY strain that lacks all forms of CrcZ and CrcY is strongly impaired when nonpreferred compounds such as citrate, benzoate, glucose, or succinate are provided as the sole carbon source in a minimal salts medium. This growth defect most likely derives from a hyperrepressing effect of the Hfq and Crc proteins on functions necessary to assimilate these compounds, an effect that cannot be controlled because CrcZ/CrcY and their processed variants are absent. Introduction of a plasmid that produces CrcZ*, but not CrcZ, into the ΔcrcZΔcrcY strain partially complemented the hyperrepressing phenotype, allowing growth on the mentioned nonpreferred compounds. This suggests that CrcZ* is functional and can antagonize the inhibitory effect of Hfq/Crc. Additional evidence that modifications at the 5′-end of CrcZ do not necessarily impair its ability to counteract Hfq/Crc activity derives from the observation that the individual overproduction of CrcZ or CrcY variants with 62 extra nt at the 5′-end, using an inducible heterologous promoter, reduces Hfq/Crc-dependent CCR in vivo (Moreno et al. 2012). This suggests that either the unprocessed 62-nt larger variants or their processed derivatives (see Fig. 9) are functional.
Although the amounts of CrcZ* provided in trans to the ΔcrcZΔcrcY strain could partially complement the hyperrepressing phenotype of this strain, they were not high enough to reduce the repressing effect of the Hfq/Crc proteins on benA expression in cells cultured in complete medium, a condition in which the Hfq/Crc-dependent CCR is very strong. In fact, our results show that under the conditions analyzed, CrcZ* is present at low and constant levels, produced from the weak and constitutive PcbrB promoter. On the contrary, the activity of promoter PcrcZ varies over an order of magnitude, being highest when cells use a poor carbon source such as oxaloacetate, and lowest when cells grow in a complete medium such as LB (Valentini et al. 2014). Even in LB medium promoter, PcrcZ shows some activity and the primary CrcZ is present, albeit its amounts are much lower than when PcrcZ is fully active (Moreno et al. 2012). The fact that processing of the cbrB-crcZ RNA generates a sRNA that is almost identical to CrcZ, and the observation that the processed sRNA is functional, suggest that it may play some role in helping CrcZ to antagonize Hfq/CrcZ activity. Since lack of all forms of CrcZ/CrcY is very detrimental to the cells, we propose that the processed CrcZ* sRNA might help in attaining basal levels of CrcZ/CrcZ* that are sufficient to protect the cell from an excessive Hfq/Crc-dependent repression. In addition, CrcZ* may also help to increase the total output of CrcZ, making the CrcZ-dependent response faster.
The considerable sequence similarity of CrcZ and CrcY suggests that CrcY* is also functional and could participate in controlling Hfq/Crc availability. In conditions that do not require CCR, the amounts of primary CrcZ and CrcY can increase rapidly by activation of promoters PcrcZ and PcrcY via the CbrB activator, thereby sequestering the Hfq/Crc proteins.
CrcZ and CrcY amounts are very low in Crc-null (García-Mauriño et al. 2013) and Hfq-null strains (this study). The use of transcriptional fusions to lacZ showed that Crc and Hfq have indirect influence on PcrcZ and PcrcY promoter activity, an effect that could derive from an as yet uncharacterized Hfq/Crc-regulated protein that represses transcription from these promoters (García-Mauriño et al. 2013). Our results nonetheless show that Crc and Hfq increase CrcZ stability, which adds an additional layer of control that might help to explain why inactivating the crc or hfq genes greatly reduces CrcZ and CrcY levels. The observation that Crc and Hfq increase CrcZ stability supports previous proposals that these proteins form a complex with CrcZ (Sonnleitner and Blasi 2014; Madhushani et al. 2015; Moreno et al. 2015). This complex might not only control Hfq/Crc availability, but could also protect CrcZ from degradation by RNases. Indeed, Hfq was observed to protect several RNAs from cleavage by RNaseE, consistent with the observation that Hfq can recognize targets very similar to those of RNase E (Møller et al. 2002; Folichon et al. 2003; Massé et al. 2003; Moll et al. 2003; for review, see Saramago et al. 2014). The half-life of CrcZ, CrcY, and their processed variants is central to determining the levels of these sRNAs in different growth conditions.
Combined with previous evidence, our results indicate that CrcZ abundance in P. putida is controlled by the complex interaction of several elements that ultimately affect transcription of crcZ, as well as the processing and stability of the crcZ transcript. Each component affects either the transcription or the stability of other components. The overall process would be similar for crcY. This complex autoregulation is summarized in Figure 11.
Since Hfq and Crc abundance in P. putida does not appear to vary greatly in distinct growth conditions (Moreno et al. 2015), one could predict that CrcZ/CrcY would compete with mRNAs bearing appropriate A-rich motives to bind available free Hfq/Crc molecules. Efficient transition from a situation in which Hfq and Crc proteins are bound to CrcZ/CrcY (no CCR control) to the opposite configuration, in which most Hfq and Crc proteins are bound to target mRNAs (strong CCR control), would be determined by the levels of CrcZ/CrcY and of target mRNAs. Hfq has high affinity for RNA and dissociates slowly, at least in vitro. To move rapidly among different RNAs, Hfq was proposed to follow an “active cycling” process, sliding from one RNA to another in a manner not limited by the slow dissociation rates, but rather driven by the concentration of free target RNAs (Wagner 2013). In cells growing in a complex medium such as LB, which is a mixture of preferred and nonpreferred carbon sources, PcrcZ and PcrcY promoter activity is low and CrcZ and CrcY levels are also comparatively low, such that Hfq/Crc can saturate target mRNAs, inhibiting their translation. The mRNAs that specify proteins needed for the induction, uptake, and assimilation of nonpreferred compounds frequently bear A-rich sites recognized by Hfq (Hernández-Arranz et al. 2013), and Hfq/Crc can therefore inhibit induction of these nonpreferred catabolic pathways. Hfq/Crc binding to these mRNAs would leave CrcZ/CrcY insufficiently protected and exposed to degradation by RNases. A decrease in the concentration of preferred compounds would lead to induction of promoters PcrcZ and PcrcY by the CbrB activator, increasing levels of CrcZ and CrcY, which also have A-rich sites. The equilibrium would switch toward a situation in which mRNAs containing A-rich sites lose Hfq/Crc, and can therefore be translated, while CrcZ/CrcY would bind to these proteins and thus be protected from RNases. The final configuration of the regulatory system would therefore depend on the relative levels of the target mRNAs and CrcZ/CrcY. The outcome of this complex and multilayered regulation is a rapid response that leads to the hierarchical regulation by available free Hfq/Crc proteins of several uptake and assimilation systems for different carbon sources, thereby organizing the best possible configuration of metabolism, which optimizes growth speed.
MATERIALS AND METHODS
Bacterial strains, culture media, and plasmids
E. coli and P. putida strains were cultured at 37°C and 30°C, respectively. Lysogeny broth (LB; 10 g/L tryptone; 5 g/L yeast extract, 10 g/L NaCl) was used as complete growth medium. M9 minimal salts medium (Sambrook and Russell 2001) was supplemented with trace elements (Bauchop and Eldsen 1960), and either 30 mM succinate, 30 mM citrate, 30 mM glucose, or 10 mM benzoate as the carbon source. When needed, antibiotics were added at the following concentrations: kanamycin (50 µg/mL), gentamicin (40 µg/mL), streptomycin (50 µg/mL, or 800 µg/mL when the host strain harbored a gentamicin-resistance determinant), ampicillin (100 µg/mL). Cell growth was followed by measuring turbidity at 600 nm.
The P. putida strains used were KT2440 (wild type), KT2442 (a spontaneous rifampicin-resistant derivative of strain KT2440 (Franklin et al. 1981), MPO401 (KT2442 cbrB::km; García-Mauriño et al. 2013), KT2442rpoN::Ωkm (Köhler et al. 1989), KT2440Δhfq (Arce-Rodriguez et al. 2015), KTCRC (KT2440 with a crc::tet allele; Hernández-Arranz et al. 2013), KT2440-Z (derives from KT2440 by replacing crcZ with an inactivated crcZ::tet allele; Moreno et al. 2012), KT2440-ZY (derived from KT2440 and lacks CrcZ and CrcY sRNAs; La Rosa et al. 2015), PBA1 (derived from KT2442; bears a PbenA::lacZ transcriptional fusion in the chromosome; Moreno et al. 2012), PBA1C (PBA1 with an inactivated crc::tet allele; Moreno et al. 2012), and PBAZY (PBA1 ΔcrcZΔcrcY; Moreno et al. 2012).
To search for promoters located upstream of promoter PcrcZ, a 1.7 kb DNA segment containing promoter PcbrB, the complete cbrB gene, and downstream sequences to nucleotide 118 downstream from the cbrB stop codon (excluding PcrcZ sequences and the CbrB binding site upstream of it), were PCR-amplified using oligonucleotides Fw1 and RV7 (Supplemental Table S1). This DNA fragment was cloned between the EcoRI and XbaI sites of plasmid pMP220 to generate a PcrcB-cbrB-lacZ transcriptional fusion; the plasmid obtained was termed pFW1. Similar plasmids were constructed in which this region was progressively deleted from the 5′-end, using oligonucleotides FW2 to FW7 as direct primers for the PCR reaction (Supplemental Table S1) and RV7 as the reverse primer; the constructs were termed pFW2, pFW3, pFW4, pFW5, pFW6, and pFW7.
To generate plasmid p421-Pwt, a DNA region that includes the cbrB and crcZ genes (including the PcbrB promoter) was PCR-amplified from KT2440 chromosomal DNA using the Pfu high-fidelity DNA polymerase (Promega) and oligonucleotides PcbrBEcoFW1 and CrcZ-XbaI-rv (Supplemental Table S1). The DNA fragment obtained was treated with EcoRI and XbaI and cloned between the EcoRI and XbaI restriction sites of plasmid pSEVA 421 (Martínez-García et al. 2015). Plasmid p421-Pmut is a variant of p421-Pwt in which the GG and GC nucleotides at the consensus −24 and −12 regions of promoter PcrcZ were modified to CC and TT, respectively. To construct it, the appropriate DNA segment was chemically synthesized by GeneArt (Thermo Fisher Scientific) and cloned between the EcoRI and XbaI restriction sites of plasmid pSEVA 421.
Plasmid pPcrcZ (Fonseca et al. 2013), which derives from plasmid pMP220 (Spaink et al. 1987), contains a PcrcZ-lacZ transcriptional fusion. Plasmid pPcrcZmut is equivalent to pPcrcZ except that the GG and GC nucleotides at the consensus −24 and −12 regions of promoter PcrcZ were modified to CC and TT, respectively. To obtain this plasmid, a DNA segment spanning from the end of cbrB to the start site of promoter PcrcZ was PCR-amplified from plasmid p421-Pmut with oligonucleotides PcrcZ-EcoRI and PcrcZ-rv-BamHI-2 (See Supplemental Table S1); the amplified fragment was treated with EcoRI and BamHI and cloned between the EcoRI and BamHI sites of the reporter plasmid pMP220, to generate a transcriptional fusion to the lacZ indicator gene.
Plasmid p424-Z contains crcZ under the influence of the Ptrc promoter, and allows overproduction of CrcZ sRNA using IPTG as inducer (Moreno et al. 2012). All plasmid constructs were sequenced and the plasmids transferred to P. putida by electroporation.
Total RNA purification from P. putida
Cells were grown at 30°C in aerated flasks containing LB medium. At mid-exponential phase (A600 = 0.6) or at the start of the stationary phase (A600 = 2.2), samples were collected, harvested by centrifugation, and frozen at −70°C. RNA was purified from cell pellets with the RNeasy RNA purification kit (Qiagen). Purified RNA was treated with RNase-free DNase I (Turbo RNA-free, Ambion). RNA integrity was analyzed by agarose gel electrophoresis. The absence of DNA was confirmed by real-time PCR using primers for rpoN or rpoD (Morales et al. 2006).
Real-time RT-PCR
Real-time RT-PCR assays were performed using total RNA preparations from three independent cultures (three biological replicas). RNA was reverse-transcribed into cDNA using the cDNA Archive kit (Applied Biosystems). Real-time PCR was performed essentially as described previously (Morales et al. 2006), except that the expression profile of each gene was analyzed by absolute quantitation using a standard curve. Standard curves were constructed by serial 10-fold dilutions from 107 to 103 copies of P. putida genomic DNA obtained using the G-NOME DNA purification kit (MP Biomedicals). The primers used are indicated in Supplemental Table S1.
Rapid amplification of cDNA ends (5′ RACE)
5′ RACE analysis was carried out using the Gene Racer Kit (Life Technologies). Briefly, total RNA obtained as described above was treated with terminator 5′-phosphate-dependent exonuclease (TEX). Reactions were terminated by extraction with phenol and chloroform, followed by ethanol precipitation. RNA samples were resuspended in water and treated with tobacco acid pyrophosphatase (TAP). The GeneRacer RNA oligo was ligated to the RNA 5′ end with T4 RNA ligase and samples were transformed to cDNA using random hexamers and SuperScript III reverse transcriptase (Life Technologies). To amplify the 5′ RACE product, a PCR reaction was performed using the GeneRacer 5′ primer and the indicated specific reverse primer (Supplemental Table S1). PCR products were then cloned into plasmid pCR4 and transformed into One Shot TOP10 E. coli using a TOPO PCR Cloning kit (Life Technologies). The junction (point of ligation) between the sequence corresponding to the RNA oligonucleotide and that of the cDNA 5′-end was determined by DNA sequencing in 14 independent transformants.
Analysis of CrcZ 5′-end heterogeneity by primer extension
The oligonucleotide use as primer for the extension reaction, end-labeled with [γ-32P]ATP and T4 polynucleotide kinase, was CrcZ-revPE (Supplemental Table S1). RNA was obtained as indicated above and, where indicated, treated with TEX enzyme, phenolized and precipitated with ethanol. Treated or untreated RNA (15 µg) were mixed with 4 pmol of the end-labeled primer and heated (80°C, 5 min). The oligonucleotide was allowed to anneal to the RNA by incubation (23°C, 5 min). Primer extension was performed with 200 U SuperScript III (Promega) at 55°C, as indicated by the supplier. The extended cDNA products were analyzed by electrophoresis on a denaturing 6% urea–polyacrylamide gel, in parallel with a DNA sequence ladder obtained by chemical sequencing (Maxam and Gilbert 1980) of a DNA fragment obtained by PCR. The gel was dried, exposed to a phosphorimager screen and visualized using the Personal Molecular Imager (Bio-Rad).
Assay for β-galactosidase
An overnight culture of the strain of interest was diluted to a final turbidity (A600) of 0.05 in fresh LB medium. Where indicated, 1 mM IPTG or 5 mM benzoate were added to induce transcription from promoters Ptrc or PbenA, respectively. Cells were allowed to grow at 30°C with vigorous aeration; aliquots were taken at various time points; and β-galactosidase activity was measured using as substrate o-nitrophenyl-β-d-galactoside (Miller 1972). Three independent assays were performed.
Northern blots
To generate an RNA probe against CrcZ, a 150-bp DNA segment spanning the central region of CrcZ (the least similar to CrcY) was PCR amplified using primers CrcZ-HindIII-fw and CrcZ EcoRI-rv (see Supplemental Table S1), which include restriction sites for endonucleases HindIII and EcoRI, respectively. The resulting DNA fragment was cloned between the HindIII and EcoRI sites of plasmid pSPT18 (Roche) to obtain pSPT18-Z. The plasmid was HindIII linearized and used as template for an in vitro transcription reaction in which the central region of crcZ was transcribed from the T7 promoter of the vector. Transcription was performed using the DIG RNA labelling kit (Roche) and T7 RNA polymerase. To generate a probe against the 5S ribosomal RNA, a DNA fragment corresponding to the gene specifying the 5S RNA was PCR amplified using primers 5S-HindIII-fw and 5S-EcoRI-rv (Supplemental Table S1), and the resulting DNA fragment was cloned between the HindIII and EcoRI sites of plasmid pSPT18 to generate plasmid pSPT18-5S. This plasmid was linearized with HindIII and used as template for an in vitro transcription reaction with T7 RNA polymerase. as indicated above.
For Northern blots, 5 µg total RNA purified from the indicated strain, and 60 ng of RNA Molecular Weight Marker III DIG-labelled (Roche), were resolved by electrophoresis on a 6% polyacrylamide gel containing 7 M urea and transferred to a Nylon Hybond N+ membrane (GE Healthcare Biosciences) with a semi-dry transfer unit (Trans-blot SD. Bio-Rad; 400 mA, 1 h). Membranes were UV-crosslinked and hybridized with a mixture of DIG-labeled probes against CrcZ and 5S (20 ng each). Bands that hybridized to the probes were detected using the DIG Luminescent Detection Kit (Roche), and signals were visualized by exposure to Agfa X-ray film. A digoxigenin-labeled RNA molecular weight marker (Roche) was used as a size marker.
Where indicated, the RNA was treated with TEX (terminator 5′-phosphate-dependent exonuclease, Epicentre) prior to Northern analyses. Briefly, 5 µg total RNA were incubated with 1 U of TEX (1 h, 30°C) and the reaction terminated by adding 1 µL of 100 mM EDTA (pH 8). Formamide buffer was added and the reaction was loaded into the 6% polyacrylamide 7 M urea gel.
Determination of CrcZ sRNA stability
P. putida strains KT2440-Z, KTCRC, and KT2440Δhfq containing plasmid p424-Z were cultured at 30°C in LB medium supplemented with streptomycin. To induce CrcZ synthesis, IPTG was added to a final concentration of 1 mM. When cultures reached a turbidity of 0.6 (A600), 0.3 mg/mL rifampicin was added; 10 mL aliquots were withdrawn at 0, 2, 4, 8, 16, and 32 min and mixed with two volumes of RNAprotect Reagent (Qiagen). Total RNA was isolated as above and 10 µg of the RNA obtained were resolved on a 6% polyacrylamide/7 M urea gel. The RNA bands were transferred to a nylon Hybond N+ membrane and detected by Northern blot as above. Band intensities were quantified using a ChemiDoc XRS imager and Image Lab software (Bio-Rad). The value obtained for CrcZ in each sample was normalized to that of the 5S rRNA of the same sample.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to V. de Lorenzo and I. Canosa for providing P. putida strains with inactivated hfq or cbrB genes, and to C. Mark for editorial assistance. S.H-A. and D.S-H. received predoctoral fellowships from the Spanish Ministry of Science and Competitiveness (MINECO). Work was supported by grants BFU2012-32797 (MINECO, Spain) and BIO2015-66203-P (MINECO/FEDER).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.058313.116.
REFERENCES
- Amador CI. 2011. “Characterization of the regulatory role of CbrB in Pseudomonas putida.” PhD thesis, Pablo de Olavide University, Seville, Spain. [Google Scholar]
- Arce-Rodriguez A, Calles B, Nikel PI, de Lorenzo V. 2015. The RNA chaperone Hfq enables the environmental stress tolerance super-phenotype of Pseudomonas putida. Environ Microbiol 10.1111/1462-2920.13052. [DOI] [PubMed] [Google Scholar]
- Bauchop T, Eldsen SR. 1960. The growth of microorganisms in relation to their energy supply. J Gen Microbiol 23: 457–469. [DOI] [PubMed] [Google Scholar]
- Bobrovskyy M, Vanderpool CK. 2013. Regulation of bacterial metabolism by small RNAs using diverse mechanisms. Annu Rev Genet 47: 209–232. [DOI] [PubMed] [Google Scholar]
- Davis BM, Waldor MK. 2007. RNase E-dependent processing stabilizes MicX, a Vibrio cholerae sRNA. Mol Microbiol 65: 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lay N, Schu DJ, Gottesman S. 2013. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J Biol Chem 288: 7996–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiatrault MJ, Stodghill PV, Wilson J, Butcher BG, Chen H, Myers CR, Cartinhour SW. 2013. CrcZ and CrcX regulate carbon source utilization in Pseudomonas syringae pathovar tomato strain DC3000. RNA Biol 10: 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folichon M, Arluison V, Pellegrini O, Huntzinger E, Regnier P, Hajnsdorf E. 2003. The poly(A) binding protein Hfq protects RNA from RNase E and exoribonucleolytic degradation. Nucleic Acids Res 31: 7302–7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca P, Moreno R, Rojo F. 2013. Pseudomonas putida growing at low temperature shows increased levels of CrcZ and CrcY sRNAs, leading to reduced Crc-dependent catabolite repression. Environ Microbiol 15: 24–35. [DOI] [PubMed] [Google Scholar]
- Franklin FC, Bagdasarian M, Bagdasarian MM, Timmis KN. 1981. Molecular and functional analysis of the TOL plasmid pWWO from Pseudomonas putida and cloning of genes for the entire regulated aromatic ring meta cleavage pathway. Proc Natl Acad Sci 78: 7458–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich KS, Vogel J. 2009. Activation of gene expression by small RNA. Curr Opin Microbiol 12: 674–682. [DOI] [PubMed] [Google Scholar]
- García-Mauriño SM, Pérez-Martínez I, Amador CI, Canosa I, Santero E. 2013. Transcriptional activation of the CrcZ and CrcY regulatory RNAs by the CbrB response regulator in Pseudomonas putida. Mol Microbiol 89: 189–205. [DOI] [PubMed] [Google Scholar]
- Gómez-Lozano M, Marvig RL, Molina-Santiago C, Tribelli PM, Ramos JL, Molin S. 2015. Diversity of small RNAs expressed in Pseudomonas species. Environ Microbiol Rep 7: 227–236. [DOI] [PubMed] [Google Scholar]
- Gopel Y, Khan MA, Gorke B. 2016. Domain swapping between homologous bacterial small RNAs dissects processing and Hfq binding determinants and uncovers an aptamer for conditional RNase E cleavage. Nucleic Acids Res 44: 824–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Arranz S, Moreno R, Rojo F. 2013. The translational repressor Crc controls the Pseudomonas putida benzoate and alkane catabolic pathways using a multi-tier regulation strategy. Environ Microbiol 15: 227–241. [DOI] [PubMed] [Google Scholar]
- Köhler T, Harayama S, Ramos JL, Timmis KN. 1989. Involvement of Pseudomonas putida RpoN σ factor in regulation of various metabolic functions. J Bacteriol 171: 4326–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa R, Nogales J, Rojo F. 2015. The Crc/CrcZ-CrcY global regulatory system helps the integration of gluconeogenic and glycolytic metabolism in Pseudomonas putida. Environ Microbiol 17: 3362–3378. [DOI] [PubMed] [Google Scholar]
- Madhushani A, Del Peso-Santos T, Moreno R, Rojo F, Shingler V. 2015. Transcriptional and translational control through the 5′-leader region of the dmpR master regulatory gene of phenol metabolism. Environ Microbiol 17: 119–133. [DOI] [PubMed] [Google Scholar]
- Mandin P, Guillier M. 2013. Expanding control in bacteria: interplay between small RNAs and transcriptional regulators to control gene expression. Curr Opin Microbiol 16: 125–132. [DOI] [PubMed] [Google Scholar]
- Martínez-García E, Aparicio T, Goñi-Moreno A, Fraile S, de Lorenzo V. 2015. SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res 43: D1183–D1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Escorcia FE, Gottesman S. 2003. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev 17: 2374–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxam AM, Gilbert W. 1980. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol 65: 499–560. [DOI] [PubMed] [Google Scholar]
- Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- Miyakoshi M, Chao Y, Vogel J. 2015. Regulatory small RNAs from the 3′ regions of bacterial mRNAs. Curr Opin Microbiol 24: 132–139. [DOI] [PubMed] [Google Scholar]
- Moll I, Afonyushkin T, Vytvytska O, Kaberdin VR, Blasi U. 2003. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA 9: 1308–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller T, Franch T, Hojrup P, Keene DR, Bachinger HP, Brennan RG, Valentin-Hansen P. 2002. Hfq: a bacterial Sm-like protein that mediates RNA–RNA interaction. Mol Cell 9: 23–30. [DOI] [PubMed] [Google Scholar]
- Morales G, Linares JF, Beloso A, Albar JP, Martínez JL, Rojo F. 2004. The Pseudomonas putida Crc global regulator controls the expression of genes from several chromosomal catabolic pathways for aromatic compounds. J Bacteriol 186: 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales G, Ugidos A, Rojo F. 2006. Inactivation of the Pseudomonas putida cytochrome o ubiquinol oxidase leads to a significant change in the transcriptome and to increased expression of the CIO and cbb3-1 terminal oxidases. Environ Microbiol 8: 1764–1774. [DOI] [PubMed] [Google Scholar]
- Moreno R, Rojo F. 2008. The target for the Pseudomonas putida Crc global regulator in the benzoate degradation pathway is the BenR transcriptional regulator. J Bacteriol 190: 1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno R, Rojo F. 2014. Features of pseudomonads growing at low temperatures: another facet of their versatility. Environ Microbiol Rep 6: 417–426. [DOI] [PubMed] [Google Scholar]
- Moreno R, Marzi S, Romby P, Rojo F. 2009. The Crc global regulator binds to an unpaired A-rich motif at the Pseudomonas putida alkS mRNA coding sequence and inhibits translation initiation. Nucleic Acids Res 37: 7678–7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno R, Fonseca P, Rojo F. 2012. Two small RNAs, CrcY and CrcZ, act in concert to sequester the Crc global regulator in Pseudomonas putida, modulating catabolite repression. Mol Microbiol 83: 24–40. [DOI] [PubMed] [Google Scholar]
- Moreno R, Hernández-Arranz S, La Rosa R, Yuste L, Madhushani A, Shingler V, Rojo F. 2015. The Crc and Hfq proteins of Pseudomonas putida cooperate in catabolite repression and formation of ribonucleic acid complexes with specific target motifs. Environ Microbiol 17: 105–118. [DOI] [PubMed] [Google Scholar]
- Nikel PI, Martínez-García E, de Lorenzo V. 2014. Biotechnological domestication of pseudomonads using synthetic biology. Nat Rev Microbiol 12: 368–379. [DOI] [PubMed] [Google Scholar]
- Poblete-Castro I, Becker J, Dohnt K, Dos Santos VM, Wittmann C. 2012. Industrial biotechnology of Pseudomonas putida and related species. Appl Microbiol Biotechnol 93: 2279–2290. [DOI] [PubMed] [Google Scholar]
- Romby P, Vandenesch F, Wagner EG. 2006. The role of RNAs in the regulation of virulence-gene expression. Curr Opin Microbiol 9: 229–236. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Saramago M, Bárria C, Dos Santos RF, Silva IJ, Pobre V, Domingues S, Andrade JM, Viegas SC, Arraiano CM. 2014. The role of RNases in the regulation of small RNAs. Curr Opin Microbiol 18: 105–115. [DOI] [PubMed] [Google Scholar]
- Sharma CM, Storz G. 2011. Interesting twists on small RNA themes in Pseudomonas aeruginosa. Mol Microbiol 80: 855–859. [DOI] [PubMed] [Google Scholar]
- Silby MW, Winstanley C, Godfrey SA, Levy SB, Jackson RW. 2011. Pseudomonas genomes: diverse and adaptable. FEMS Microbiol Rev 35: 652–680. [DOI] [PubMed] [Google Scholar]
- Sobrero P, Valverde C. 2012. The bacterial protein Hfq: much more than a mere RNA-binding factor. Crit Rev Microbiol 38: 276–299. [DOI] [PubMed] [Google Scholar]
- Sonnleitner E, Blasi U. 2014. Regulation of Hfq by the RNA CrcZ in Pseudomonas aeruginosa carbon catabolite repression. PLoS Genet 10: e1004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Haas D. 2011. Small RNAs as regulators of primary and secondary metabolism in Pseudomonas species. Appl Microbiol Biotechnol 91: 63–79. [DOI] [PubMed] [Google Scholar]
- Sonnleitner E, Schuster M, Sorger-Domenigg T, Greenberg EP, Bläsi U. 2006. Hfq-dependent alterations of the transcriptome profile and effects on quorum sensing in Pseudomonas aeruginosa. Mol Microbiol 59: 1542–1558. [DOI] [PubMed] [Google Scholar]
- Sonnleitner E, Abdou L, Haas D. 2009. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc Natl Acad Sci 106: 21866–21871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaink HP, Okker RJ, Wijffelman CA, Pees E, Lugtenberg BJ. 1987. Promoters in the nodulation region of the Rhizobium leguminosarum Sym plasmid pRL1JI. Plant Mol Biol 9: 27–39. [DOI] [PubMed] [Google Scholar]
- Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43: 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentini M, García-Mauriño SM, Pérez-Martínez I, Santero E, Canosa I, Lapouge K. 2014. Hierarchical management of carbon sources is regulated similarly by the CbrA/B systems in Pseudomonas aeruginosa and Pseudomonas putida. Microbiology 160: 2243–2252. [DOI] [PubMed] [Google Scholar]
- Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9: 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EG. 2013. Cycling of RNAs on Hfq. RNA Biol 10: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EG, Romby P. 2015. Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv Genet 90: 133–208. [DOI] [PubMed] [Google Scholar]
- Wassarman KM. 2002. Small RNAs in bacteria: diverse regulators of gene expression in response to environmental changes. Cell 109: 141–144. [DOI] [PubMed] [Google Scholar]
- Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136: 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Monchy S, Taghavi S, Zhu W, Ramos J, van der Lelie D. 2011. Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida. FEMS Microbiol Rev 35: 299–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.