

Abstract
CXCR4 is a stem/progenitor cell surface receptor specific for the cytokine stromal cell‐derived factor‐1 (SDF‐1α). There is evidence that bone marrow‐derived CXCR4‐expressing cells contribute to intimal hyperplasia (IH) by homing to the arterial subintima which is enriched with SDF‐1α. We have previously found that transforming growth factor‐β (TGFβ) and its signaling protein Smad3 are both upregulated following arterial injury and that TGFβ/Smad3 enhances the expression of CXCR4 in vascular smooth muscle cells (SMCs). It remains unknown, however, whether locally induced CXCR4 expression in SM22 expressing vascular SMCs plays a role in neointima formation. Here, we investigated whether elevated TGFβ/Smad3 signaling leads to the induction of CXCR4 expression locally in the injured arterial wall, thereby contributing to IH. We found prominent CXCR4 upregulation (mRNA, 60‐fold; protein, 4‐fold) in TGFβ‐treated, Smad3‐expressing SMCs. Chromatin immunoprecipitation assays revealed a specific association of the transcription factor Smad3 with the CXCR4 promoter. TGFβ/Smad3 treatment also markedly enhanced SDF‐1α‐induced ERK1/2 phosphorylation as well as SMC migration in a CXCR4‐dependent manner. Adenoviral expression of Smad3 in balloon‐injured rat carotid arteries increased local CXCR4 levels and enhanced IH, whereas SMC‐specific depletion of CXCR4 in the wire‐injured mouse femoral arterial wall produced a 60% reduction in IH. Our results provide the first evidence that upregulation of TGFβ/Smad3 in injured arteries induces local SMC CXCR4 expression and cell migration, and consequently IH. The Smad3/CXCR4 pathway may provide a potential target for therapeutic interventions to prevent restenosis. Stem Cells 2016;34:2744–2757
Keywords: CXCR4/SDF‐1α, TGFβ/Smad3, smooth muscle cell migration, smooth muscle cell specific CXCR4 knockout, intimal hyperplasia
Significance Statement.
CXCR4 is a stem/progenitor cell surface receptor specific for the cytokine stromal cell‐derived factor (SDF)‐1 alpha. While previous studies show that blood‐borne CXCR4‐expressing progenitor cells contribute to intimal hyperplasia by homing to the arterial subintima which is enriched with SDF‐1 alpha, it remains unclear whether CXCR4‐expressing cells from other sources also play a role. Here our studies using tissue‐specific CXCR4 knockout mice clarify that CXCR4 can be induced locally in the injured arterial wall and play a big part in the development of intimal hyperplasia by promoting smooth muscle cell migration to the SDF‐1 alpha rich subintima space.
Introduction
Over one million vascular reconstructions are performed in the United States each year. However, the long‐term success is plagued by the development of recurrent vascular disease either within the bypass or the treated artery. Intimal hyperplasia (IH) is a major component of this process which leads to vessel narrowing 1. A hallmark of IH is the proliferation of vascular smooth muscle cells (SMCs) and their migration to the subintima of the injured blood vessel wall. The underlying mechanism of IH is complex. One of the important and still unanswered questions is the origin of the cells that produce the neointimal lesion.
There is a large body of evidence revealing the importance of transforming growth factor‐β (TGFβ) in the development of IH. A number of studies have shown that exogenous TGFβ enhances, and blocking TGFβ inhibits IH 2. TGFβ plays an important role in a wide range of pathophysiological processes including cell proliferation, differentiation, and apoptosis 3. TGFβ signals via Smad‐dependent canonical and Smad‐independent noncanonical pathways. Canonically, TGFβ binds to its type II receptor leading to receptor phosphorylation and the subsequent activation of signaling proteins Smad2 and Smad3, which then form a heterotrimer with Smad4; this complex then translocates into the nucleus. Once inside the nucleus, Smad3 is able to regulate gene transcription by binding to consensus Smad‐binding elements (SBEs) in the promoter region of multiple target genes. We have demonstrated that levels of TGFβ and Smad3 are elevated after angioplasty both in animals as well as in humans 4, 5. TGFβ inhibits SMC proliferation and migration in vitro 6. However, in the context of elevated Smad3, TGFβ has the opposite effect 5, 7. Furthermore, our group has shown that enhancing Smad3 expression following rat carotid balloon injury aggravates IH, whereas inhibition of Smad2/3 suppresses the formation of neointimal lesion 2.
CXCR4 is a receptor for chemo‐attractant stromal cell‐derived factor‐1 (SDF‐1α). CXCR4 is expressed in stem/progenitor cells and lymphocytes but barely detectable in highly differentiated cells (such as SMCs). However, expression of CXCR4 is elevated under some pathological conditions. For example, during tumor metastasis CXCR4 plays an important role in directing cancer stem cell migration 8, 9, 10. CXCR4 has been shown to play a role in IH. It has been demonstrated that after vascular injury SDF‐1α is released from apoptotic cells from the arterial wall. Local SDF‐1α then induces chemotaxis, attracting CXCR4‐expressing progenitor cells to the site of injury 11. Binding of SDF‐1α to CXCR4 expressing progenitor cells initiates a series of changes including enhanced motility and proliferation as well as differentiation leading to a significant contribution of these cells to the growth of neointimal lesion.
The origin of neointimal cells has been a hotly debated issue 12. The current view is that medial SMCs migrate into the subintima and proliferate producing IH 2. A large body of literature supports that medial SMCs are the primary source of neointimal cells 2. However, there are also reports that myofibroblasts in the adventitia 13, or CXCR4‐expressing progenitor cells derived from bone marrow, migrate to the subintima and participate in the formation of this highly cellular neointima layer 11. The contribution of bone marrow‐derived cells to IH remains controversial. It has been reported that CXCR4‐expressing bone marrow‐derived cells are attracted to the subintima by SDF‐1α following balloon or wire‐induced arterial injury in mice 11. However, in studies employing rat carotid artery transplants following balloon injury, it appears that majority of cells in the neointima are derived from residential SMCs 14.
We have made the interesting observation that SMCs, when activated by TGFβ/Smad3 (which are both enhanced at the time of arterial injury), strongly express CXCR4. This finding raises the question as to whether SDF‐1α released at the time of injury might act as a chemoattractant of cells already within the arterial wall, that being CXCR4 expressing SMCs that have been activated by TGFβ/Smad3. This phenomenon could occur rather than or in addition to the effect of SDF‐1α on progenitor cells. To test this question, we fully evaluated both in vitro and in vivo, the ability of SMCs to express CXCR4. In addition, we created a new mouse line with CXCR4 selectively depleted in SM22‐expressing arterial wall SMCs. We found that IH was substantially reduced in wire‐injured femoral arteries where SMCs were lacking the CXCR4 receptor. In sum, we have identified a novel mechanism whereby SDF‐1α produces chemotaxis of local SM22 expressing vascular SMCs which have been induced to express CXCR4 in response to TGFβ/Smad3. These events in turn contribute to the development of injury‐induced restenosis.
Materials and Methods
Animals
Male Sprague‐Dawley rats were purchased from Charles River Laboratories (Wilmington, MA, http://www.criver.com/). In order to create a SMC‐specific CXCR4 knockout model, a mouse strain that expresses Cre (recombinase) under the control of SM22‐α promoter (SM22‐Cre) and another strain with loxP sites flanking the CXCR4 gene were acquired from Jackson Laboratories (Bar Harbor, ME, https://www.jax.org/). These two strains were crossed, and homozygous offspring without CXCR4 expression in the aorta were selected through RT‐PCR genotyping (Supporting Information Fig. S1). The experiments involving animal use were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Wisconsin‐Madison. Surgery was performed under isoflurane anesthesia (through inhaling, flow rate 2 ml/minute), and all efforts were made to minimize suffering. Animals were euthanized in a chamber gradually filled with CO2.
Reagents
Recombinant TGFβ1 and SDF‐1α were purchased from R&D Systems (Minneapolis, MN, https://www.rndsystems.com/). The MAPK inhibitors (PD98059 and U0126) were from Selleck Chemicals Company (Houston, TX, http://www.selleckchem.com/). Dulbecco's modified Eagle's medium (DMEM) and cell culture reagents were from Invitrogen (Carlsbad, CA, https://www.thermofisher.com/us/en/home.html). Other reagents, if not specified, were purchased from Sigma‐Aldrich (St. Louis, MO, https://www.sigmaaldrich.com/united-states.html).
SMC Culture and AdSmad3/TGFβ Treatment
Rat SMCs were isolated from the thoracoabdominal aorta of male Sprague‐Dawley rats. SMCs at passages 4‐5 were used for all experiments and maintained in DMEM supplemented with 10% fetal bovine solution (FBS) at 37°C with 5% CO2. Cell viability was >95% as assayed using the Trypan Blue exclusion method. Adenoviral vectors expressing Smad3 (AdSmad3) and green fluorescent protein (AdGFP control) were constructed as previously described 15. SMCs were infected for 4 hour with AdSmad3 (or AdGFP) (3×104 particles/cell) in DMEM containing 2% FBS, and recovered for 20 hour with 10% FBS, and then starved with 0.5% FBS for 24 hour followed by treatment with human recombinant TGFβ1 (5 ng/ml, R&D Systems, Minneapolis, MN) or equivalent amount of solvent (final 4 µM HCl and 1µg/ml BSA) for 6 or 24 hour.
Real‐Time PCR Analysis
RNA was isolated from cultured cells using a Trizol reagent (Invitrogen, Carlsbad, CA) following manufacturer's protocol. Potential contaminating genomic DNA was removed by using gDNA Eliminator columns provided in the kit. RNA (2 µg) was used for the first‐strand cDNA synthesis (Applied Biosystems, Carlsbad, CA, www.thermofisher.com/). Quantitative RT‐PCR was performed using the 7500 Fast Real‐Time PCR System (Applied Biosystems, Carlsbad, CA). Each cDNA template was amplified in triplicate using SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA) with gene specific primers.
Western Blotting Analysis
Cells were lysed in RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Nonidet P‐40 and 0.1% sodium dodecyl sulfate) containing Protease Inhibitor Cocktail I (Millipore, Billerica, MA, www.emdmillipore.com/). Protein concentration was determined using a Bio‐Rad DC Protein Assay kit (Hercules, CA). Thirty micrograms of proteins from each sample were separated by 10% SDS‐PAGE and transferred to nitrocellulose membranes. Immunoblotting was then performed to detect a protein specifically using the following antibodies: rabbit anti‐CXCR4 (Santa Cruz Biotechnology, Dallas, TX, http://www.scbt.com), rabbit anti‐phospho‐ERK, rabbit anti‐ERK (Cell Signaling Technology, Boston, MA), mouse antibodies for α‐smooth muscle actin (SMA) and β‐actin (Sigma, St. Louis, MO). After incubation with an appropriate primary antibody and then a horseradish peroxidase (HRP)‐conjugated secondary antibody, the specific protein band on the membrane was visualized by using enhanced chemiluminescence reagents (Pierce Biotechnology, Rockford, IL, www.thermfisher.com/). The signal was analyzed by using LAS 4000 mini (GE Healthcare Bio‐Sciences, Pittsburgh, PA).
Immunocytochemistry
Immunostaining was performed to detect in situ the expression of CXCR4 on SMCs, following our published method 16. Cells were fixed in 4% paraformaldehyde in PBS for 10 minutes, permeabilized with 0.1% triton x‐100 (in PBS), and then blocked with 5% BSA/5% donkey serum for 1 hour. A rabbit anti‐CXCR4 primary antibody (Abcam, Cambridge, MA, http://www.abcam.com/) was incubated with the cells overnight at 4°C, and then a secondary antibody was applied for 1 hour at room temperature followed by fluorescence microscopy.
Chromatin Immunoprecipitation
SMCs were crosslinked with 1% formaldehyde for 15 minute at room temperature, after which 125 mM glycine was added to quench formaldehyde. The cells were then lysed in 500 μl lysis buffer supplemented with a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland, http://www.roche.com/index.htm). Nuclei were pelleted at 3000 x g and 4°C for 5 minute and resuspended in 400 μl of nuclear lysis buffer. The samples were sonicated to yield DNA fragments between 200 and 700 bp, and lysates were cleared by centrifugation. The samples were then incubated with 2.5 μg of a rabbit anti‐Smad3 antibody (Abcam, Cambridge, MA) or control IgG (Upstate/Millipore, Billerica, MA) for 1 hour at 4°C. To reduce nonspecific association, 30 μg of sonicated salmon sperm DNA and 50 μg of BSA (Promega, Madison, WI, www.promega.com/) were included in each sample.
Immunoprecipitation was carried out using 50 μl of 50% (v/v) Protein A/G PLUS‐Agarose beads (Santa Cruz Biotechnology Inc., Dallas, TX)) at 4°C overnight. The immunoprecipitated complexes were washed sequentially with low‐salt buffer, high‐salt buffer, LiCl buffer, and TE buffer. The DNA‐protein complexes were eluted with 200 μl elution buffer (1.5% SDS in 50 mM NaHCO3), and then incubated at 65°C overnight to reverse formaldehyde crosslink. DNA was first extracted with phenol‐chloroform and then purified by ethanol precipitation and finally dissolved in 20 μl of 10 mM Tris‐HCl, pH 8.5 for quantitative RT‐PCR analysis of the CXCR4 promoter. The DNA sequence of SBE was from references 17, 18, 19.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) was performed using a Light Shift Chemiluminescent EMSA Kit (Pierce Biotechnology, Rockford, IL). A biotinylated DNA oligonucleotide containing the SBE in the CXCR4 promoter was synthesized in the University of Wisconsin Biotechnology Center (Madison, WI). Briefly, SMCs were infected with AdSmad3 and treated with TGFβ1 for 2 hour and then harvested for nuclei extraction. The extract (30 μg of nuclear protein) was incubated with 5 ng of biotin‐oligo at room temperature for 30 minute in 20 μl total volume of binding buffer containing 10 mM Tris‐HCl (pH 7.5), 50 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 4% glycerol (v/v), and 0.5 μg poly(deoxyinosinic‐deoxycytidylic acid). The reaction was stopped by adding gel loading buffer and subjected to nondenaturing 4% polyacrylamide gel electrophoresis in 0.5× TBE buffer (Tris‐borate‐EDTA). The gel was transferred to a nylon membrane provided in the kit and DNA was UV crosslinked to the nylon membrane. After incubating the membrane with streptavidin‐conjugated HRP, the biotinylated DNA bands were visualized using enhanced chemiluminescence reagents (Pierce Biotechnology, Rockford, IL).
SDF‐1α Binding Assay
SMCs infected with AdSmad3 and then treated with TGFβ1 (for 24 hour) were seeded in 96‐well plates at a density of 3000 cells/well. Biotinylated SDF‐1α was then added and incubated with the cells for 1 h at 4°C. The cells were washed three times with HBSS media, incubated with streptavidin‐conjugated HRP for 45 minute at 4°C, and then washed again three times with HBSS media. A colorimetric substrate, o‐phenylenediamine dihydrochloride in citrate buffer was added to assess streptavidin‐conjugated HRP bound with SDF‐1α/CXCR4 on the cell surface. Spectrophotometry was determined at 450 nm using a Flex Station 3 microplate reader (Molecular Devices, Sunnyvale, CA, www.moleculardevices.com/Microplates).
SMC Migration Assay
Migration assays were performed in Transwell plates (Corning Costar; Cambridge, MA) with a well diameter of 6.5 mm and filter pore size of 8 μm. SMCs infected with AdSmad3 (or AdGFP) and then treated with TGFβ1 (or solvent) for 24 hour were added to the upper chamber (1.5 × 104 cells), and then 600 μl of media with SDF‐1α (100 ng/ml) was added to the lower chamber. After 4 hour, the cells that migrated through the Transwell insert membrane were counted.
Rat Balloon Injury Model and In Vivo Gene Transfer
Male Sprague‐Dawley rats (∼350 g) underwent balloon injury of the left common carotid artery as described previously 14. Briefly, after induction of anesthesia with isofluorane, a 2‐French balloon catheter (Edwards Lifesciences, Irvine, CA, www.edwards.com/) was inserted through the left external carotid artery into the common carotid artery and insufflated with 2 atm of pressure three times. After injury, animals received intraluminal infusion of adenoviral vectors (2.5 × 109 plaque‐forming units) in 200 µl of PBS over 20 minute. The external carotid artery was then ligated, and blood flow was resumed. Rats were sacrificed at 3, 7, or 14 days after injury. Half the artery was processed for mRNA analysis and the other half was fixed in 4% paraformaldehyde overnight and then cryopreserved in the Optimal Cutting Temperature reagent.
Mouse Femoral Wire Injury Model
Mouse femoral artery wire injury was performed as described previously 20. Under anesthetization, a midline incision was made on the ventral left thigh to dissect the femoral artery. The distal and proximal ends of the femoral artery were looped with surgical suture for temporary control of blood flow. An arteriotomy was made on the femoral artery muscular branch, through which a 0.015″ guide wire (REF#C‐SF‐15‐15, Cook Medical, Bloomington, IN, https://www.cookmedical.com/products/di_tscf_webds/) was inserted and remained in the artery for 1 minute. After removal of the wire, the muscular branch was ligated and blood flow in the femoral artery was resumed. At 28 days after injury, femoral arteries were collected from anesthetized animals following perfusion fixation at a physiological pressure of 100 mmHg, the animals were then euthanized.
Morphometric Analysis of IH
Paraffin sections (5 μm thick) were excised at equally spaced intervals and stained with hematoxylin‐eosin (H&E) for morphometric analysis, as described in previous reports 21. Planimetric parameters as follows were measured on the sections and calculated using ImageJ: the area inside external elastic lamina (EEL area) or internal elastic lamina (IEL area), lumen area, intima area (= IEL area − lumen area), and media area (= EEL area – IEL area). Measurements were performed by a student blinded to the experimental conditions. IH was quantified as a ratio of intima area versus media area. Six to eight sections from each animal were used. The data from all sections were pooled to generate the mean for each animal. The means from all the animals in each treatment group were then averaged, and standard error of the mean (SEM) was calculated.
Immunohistochemistry
Immunostaining was performed on carotid or mouse femoral artery sections following our published method 21. Briefly, the sections were first incubated with each of the primary antibodies for 12 hour with dilution ratios as follows: rabbit anti‐CXCR4 (Abcam, Cambridge, MA), 1:200; rabbit anti‐SDF‐1α (Santa Cruz Biotechnology Inc., Dallas, TX), 1:200; mouse anti‐α‐SMA (Sigma‐Aldrich, St. Louis, MO), 1:1000. Normal IgG was used for background control. The detected proteins were then visualized by fluorescence microscopy after incubating the cells with species‐matched secondary antibodies conjugated with either Alexa Fluor 546 or Alexa Fluor 488 (Invitrogen, Carlsbad, CA).
For quantification, five immunostained sections from each animal were used. Positively stained cells were counted (based on fluorescence intensity) using ImageJ by a student blinded to experimental conditions. The numbers from all five sections were pooled to generate the mean for each animal. The means from all the animals in each treatment group were then averaged, and the SEM was calculated.
Statistical Analysis
All data are presented as mean ± SEM. One‐way or 2‐way ANOVA was performed, as appropriate, followed by Tukey's HSD post hoc test. p < .05 was considered statistically significant.
Results
TGFβ/Smad3 Stimulates CXCR4 Expression in Rat Aortic SMCs In Vitro
We have shown that TGFβ and its signaling protein Smad3 are upregulated in the arterial wall in response to injury and contribute to the development of IH 7, 21. In an effort to identify the molecular determinants that mediate TGFβ/Smad3‐stimulated IH, we performed gene arrays which revealed a dramatic increase in CXCR4 gene expression in SMCs in the presence of elevated TGFβ/Smad3 signaling 16. In order to further explore the influence of CXCR4 upregulation on SMC behavior and IH, we first confirmed that TGFβ/Smad3‐induced SMC CXCR4 expression by quantitative RT‐PCR. To mimic elevation of TGFβ and Smad3 in SMCs following vascular injury, cultured rat aortic medial SMCs were infected with adenovirus expressing Smad3 (AdSmad3) followed by stimulation with TGFβ. Cells infected with AdGFP served as a control. As shown in Figure 1A, compared to AdGFP control, TGFβ alone increased CXCR4 gene expression by 12‐fold, however, AdSmad3 infection increased CXCR4 expression by 18‐fold and TGFβ treatment (6 hour) following AdSmad3 infection further stimulated CXCR4 gene transcription by 60‐fold. To test whether TGFβ stimulated CXCR4 expression depends on Smad3, we incubated SMCs with Smad3 siRNA and then treated with TGFβ for 6 hour. The result showed that preincubation with Smad3 siRNA significantly decreased level of TGFβ‐induced CXCR4 mRNA elevation (Fig. 1B). We then determined the effect of TGFβ/Smad3 on CXCR4 protein levels by Western blotting at a later time point (24 hour TGFβ treatment). Consistent with our findings with gene expression, CXCR4 protein levels were also increased by treatment of either TGFβ or AdSmad3 alone, and were further elevated (4‐fold) by the combination of Smad3 and TGFβ (Fig. 1C). We then evaluated CXCR4 expression via immunocytochemistry which revealed intense fluorescence in SMCs stimulated by TGFβ/Smad3 (Fig. 1D). These data convincingly demonstrate that TGFβ/Smad3 signaling strongly enhances the expression of CXCR4 likely on the cell membrane of SMCs.
Figure 1.
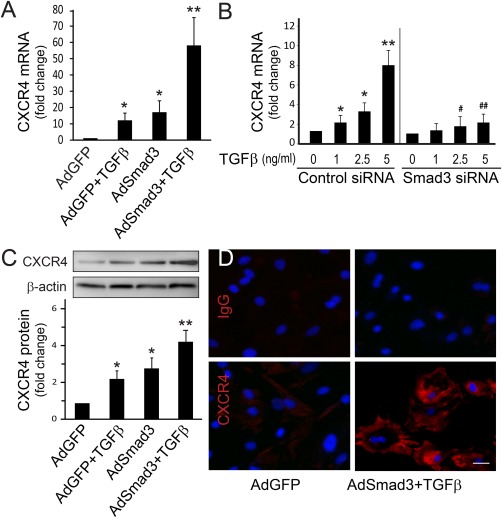
TGFβ/Smad3 stimulates CXCR4 expression in cultured rat aortic smooth muscle cells (SMCs). (A): Quantitative RT‐PCR. mRNA levels of CXCR4 were determined from SMCs infected with AdGFP (control) or AdSmad3 followed by treatment with solvent or TGFβ1 for 6 hour. Error bar represents SEM.*p < .05 compared with AdGFP control; **p < .05 compared with all other conditions; n = 3. (B): Quantitative RT‐PCR. mRNA levels of CXCR4 were determined with SMCs transfected with scrambled siRNA (control) or Smad3 siRNA followed by treatment with TGFβ1 for 6 hour. *p < .05 compared with solvent (0 ng/ml) in control siRNA group; **p < .01 compared with all other conditions in control siRNA group; #p < .05 compared with 2.5 ng/ml TGFβ1 treatment in control siRNA group; ##p < .05 compared with 5 ng/ml TGFβ1 treatment in control siRNA group; n = 3. (C): Western blotting. Protein levels of CXCR4 were assessed from SMCs infected with AdGFP or AdSmad3 followed by treatment with solvent or TGFβ1 (5 ng/ml) for 24 hour. *p < .05 compared with AdGFP control; **p < .05 compared with all other conditions; n = 3. (D): Immunocytochemistry. CXCR4 expression on SMCs was visualized (red) using SMCs treated with AdGFP or AdSmad3 followed by treatment with TGFβ1 (5 ng/ml) for 24 hour. Nuclei were stained blue by 4′,6‐diamidino‐2‐phenylindole (DAPI). Scale bar = 20 µm. Image magnification: 200×. Abbreviation: TGFβ, transforming growth factor‐β.
Smad3 Interacts with the CXCR4 Promoter in Rat Aortic SMCs
We next explored the molecular mechanism through which TGFβ/Smad3 stimulates expression of CXCR4. Since the CXCR4 promoter contains consensus SBE, we hypothesized that Smad3 may regulate CXCR4 expression by directly interacting with the CXCR4 promoter. Using chromatin immunoprecipitation (ChIP), we found a greater immunoprecipitation of the CXCR4 promoter from AdSmad3‐infected SMCs than from AdGFP control cells, which was further enhanced by stimulation with TGFβ (Fig. 2A). TGFβ alone also significantly increased immunoprecipitation of the CXCR4 promoter, likely through activation of endogenous Smad3. These results suggest that Smad3 can directly regulate CXCR4 through an interaction with its promoter. To further confirm the specific binding of Smad3 to the CXCR4 promoter, we incubated nuclear extracts from TGFβ/Smad3‐treated SMCs, with a biotinylated DNA oligo that contains the SBE present in the CXCR4 promoter. The mixture was resolved on a nondenaturing polyacrylamide gel and then subjected to transfer and detection of the biotinylated oligo using streptavidin‐conjugated HRP. Binding of Smad3 with the oligo was indicated by upshifting of the oligo band due to increased size of the Smad3/oligo complex (Fig. 2B). Our data show that the nuclear extracts from TGFβ/Smad3‐treated SMCs produced an upshift of the biotin‐oligo band, which was abolished by adding the same oligo but without the biotin label (which competes for binding with Smad3) to the mixture of the biotin‐oligo and nuclear extracts (Fig. 2C). A Smad3 antibody produced the oligo bands super shift while the biotinylated oligo precipitated Smad3 proteins (Fig. 2D and 2E). These results together with the ChIP assay support that there is a specific interaction between Smad3 and the CXCR4 promoter.
Figure 2.
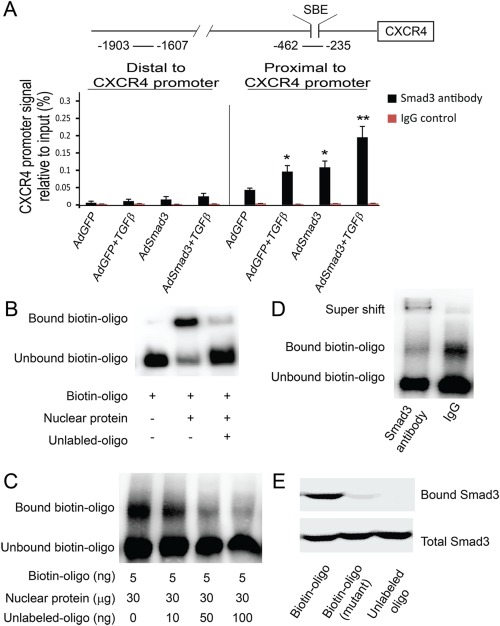
TGFβ/Smad3 treatment increases Smad3 binding to the CXCR4 promoter. (A): Schematic illustration of the rat CXCR4 promoter depicting the location of Smad binding element (SBE) and chromatin immunoprecipitation (ChIP) primer sets relative to the transcription start site. ChIP was performed using Smad3 antibody or normal IgG as described in the Methods section. Quantitative RT‐PCR was performed with precipitated DNA using primer sets flanking SBE amplifying the CXCR4 promoter proximal region. A primer set amplifying the distal region of CXCR4 promoter was used as a control. *p < .05, compared with AdGFP control; **p < .05, compared with AdGFP, AdGFP +TGFβ or AdSmad3; n = 3. (B): Electrophoretic mobility shift assay (EMSA) was performed as described in Methods. A synthetic biotinylated oligo containing the SBE from the CXCR4 promoter was used to react with nuclear protein extracts from AdSmad3‐infected and then TGFβ‐treated smooth muscle cells (SMCs). Shifted bands were detected with streptavidin‐conjugated horseradish peroxidase. The same DNA oligo without biotin (unlabeled oligo) was used to compete with the biotinylated oligo for binding with Smad3. (C): EMSA was performed with increasing amounts of unlabeled oligo to compete with the biotinylated oligo for binding with Smad3. (D): EMSA in the presence of a Smad3 antibody showed super shift. (E): A synthetic biotinylated oligo containing the SBE of the CXCR4 promoter was incubated with nuclear protein extracts from AdSmad3 infected and TGFβ1‐treated SMCs. Protein bound to biotinylated oligo was precipitated with avidin beads. Western blot was performed with a Smad3 antibody. A biotinylated oligo containing a mutated SBE or unlabeled SBE oligo were used as controls. Abbreviation: TGFβ, transforming growth factor‐β.
Stimulation of SMCs with TGFβ/Smad3 Promotes CXCR4‐Dependent SMC Migration Toward SDF‐1α
A well‐established cellular function of CXCR4 is its mediation of cell migration to SDF‐1α. We thus hypothesized that since TGFβ/Smad3 increase expression of CXCR4 this same stimulus through enhanced CXCR4 would promote SMC migration toward SDF‐1α. We first evaluated whether there was increased binding of SDF‐1α to SMCs treated with TGFβ/Smad3. SMCs were infected with AdGFP (control) or AdSmad3 followed by treatment with solvent or TGFβ1 for 24 hour, and then incubated with increasing concentrations of SDF‐1α conjugated with a biotin label. SDF‐1α binding to SMCs was detected via streptavidin‐conjugated HRP. As shown in Figure 3A, there was significantly more SDF‐1α bound to TGFβ/Smad3‐treated SMCs compared to the various controls at SDF‐1α concentrations >40 ng/ml.
Figure 3.
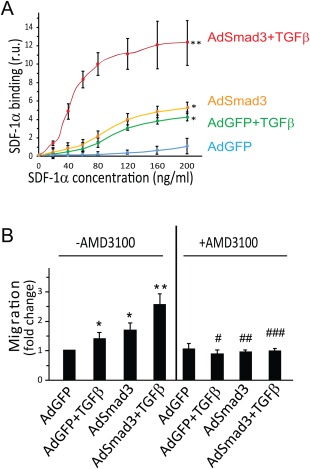
TGFβ/Smad3 treatment stimulates CXCR4‐dependent smooth muscle cell (SMC) migration toward SDF‐1α. (A): Binding of biotin‐SDF‐1α to CXCR4 expressed on the unpermeabilized SMC surface was assayed via streptavidin‐conjugated HRP, as described in Methods. Prior to the addition of biotin‐SDF‐1α, SMCs were infected with AdGFP (control) or AdSmad3 and then treated with solvent or TGFβ1 for 24 hour. Error bar represents SEM; *p < .05 compared with AdGFP control; **p < .05 compared with all other three conditions (n = 3). (B): Migration assay was performed as described in Methods. SMCs were infected with AdGFP (control) or AdSmad3 followed by treatment with solvent or TGFβ1 for 24 hour, and then transferred to a Boyden Transwell insert. AMD3100 (10 μM) or solvent were added to the upper chamber. Sixty minutes later, SDF‐1α was added to the lower chamber (100 ng/ml). After 4 hour, the cells that migrated through the Transwell membrane were counted. *p < .05 compared with AdGFP control; **p < .05 compared with all other conditions; #p < .05 compared with AdGFP+TGFβ treated group without AMD3100; ##p < .05 compared with AdSmad3 treated group without AMD3100; ###p < .05 compared with AdSmad3+TGFβ treated group without AMD3100; n = 3. Abbreviation: SDF‐1, stromal cell‐derived factor‐1; TGFβ, transforming growth factor‐β.
In order to assess the functional effect of TGFβ/Smad3‐stimulated CXCR4 production on SDF‐1α induced SMC migration, we performed a chemotaxis assay using a Transwell with treated SMCs in the upper chamber and SDF‐1α in the lower chamber. While treatment of SMCs with TGFβ or AdSmad3 alone slightly stimulated SMC migration in response to SDF‐1α, chemotaxis of SMCs stimulated with both TGFβ and Smad3 was enhanced by >2.5‐fold (Fig. 3B). The addition of a selective CXCR4 inhibitor (AMD3100, 10 µM) abolished SMC migration in response to SDF‐1α proving that the observed increase in chemotaxis was the consequence of CXCR4 expression. Finally, in separate assays, incubating SMCs with SDF‐1α did not alter CXCR4 mRNA levels (data not shown), ruling out the possibility that exposure to SDF‐1α during the chemotaxis assay induced SMC CXCR4 expression. Taken together, these results support the conclusion that TGFβ/Smad3 stimulates CXCR4 expression on SMCs which in turn promotes chemotaxis to SDF‐1α.
SDF‐1α Stimulates ERK Phosphorylation in TGFβ/Smad3‐Treated SMCs in a CXCR4‐Dependent Manner
In the forgoing studies, we have shown that TGFβ/Smad3 enhances the expression of CXCR4 in SMCs, which then leads to their migration in response to SDF‐1α. In order to determine the mechanism through which SDF‐1α binding to CXCR4 enhances SMC migration, we explored the ERK pathway. We and others have shown that activation of ERK can lead to SMC migration 22, 23 and it has previously been shown in cell types other than SMCs that SDF‐1α can enhance phosphorylation of ERK 24. We first determined the dose response of SDF‐1α‐induced ERK activation by Western blotting. SMCs were infected with AdGFP (control) or AdSmad3 followed by treatment with solvent or TGFβ1 (5 ng/ml) for 24 hour before SDF‐1α stimulation. In AdGFP‐treated control SMCs, SDF‐1α stimulation did not induce a detectable ERK MAPK activation (Fig. 4A). In AdGFP + TGFβ‐treated SMCs, stimulation with 100 ng/ml SDF‐1α led to a modest increase in ERK phosphorylation (Fig. 4B). Similarly, in AdSmad3‐treated SMCs, stimulation with 100 ng/ml SDF‐1α also increased ERK phosphorylation (Fig. 4 C). Then we tested in AdSmad3/TGFβ1 treated SMCs and found that the SDF‐1α induced ERK phosphorylation was markedly increased compared to cells treated with either AdGFP + TGFβ1 or AdSmad3 only (Fig. 4D). In the time course, study performing in AdSmad3‐infected and TGFβ1‐treated SMCs, we showed that SDF‐1α‐induced ERK activation became visible at 5 minute, peaked at 10 minute, and then declined but remained above the baseline for up to 60 minute (Fig. 4E). These results indicate that SDF‐1α‐stimulated ERK activation can be potentiated by TGFβ/Smad3 signaling. In order to verify that the enhancement of SDF‐1α‐stimulated ERK activation is the consequence of Smad3/TGFβ induction of CXCR4, we employed AMD3100 to inhibit CXCR4. AMD3100‐abolished ERK phosphorylation potentiated either by TGFβ, Smad3, or both (Fig. 4F). Next, we isolated SMCs from aorta of wild type or CXCR4 conditional knockout mice and treated the cells with solvent (4 mM HCl) or 5 ng/ml TGFβ1 for 24 hour. SDF‐1α (10 minute; 100 ng/ml) failed to induce ERK phosphorylation in the absence of CXCR4 (Fig. 4G), confirming that SDF‐1α‐induced ERK phosphorylation indeed through CXCR4. To prove the essential role of ERK phosphorylation in the SDF‐1α triggered SMC migration, we repeated the migration study in the presence or absence of the MEK inhibitor U0126 (5 µM) or PD98058 (10 µM). Both inhibitors abolished SMC migration toward SDF‐1α (Fig. 4H). These results, together with the foregoing, suggest a mechanism whereby TGFβ/Smad3 upregulates CXCR4 in SMCs, which in turn mediates the activation of the ERK pathway and the subsequent SMC migration toward SDF‐1α.
Figure 4.
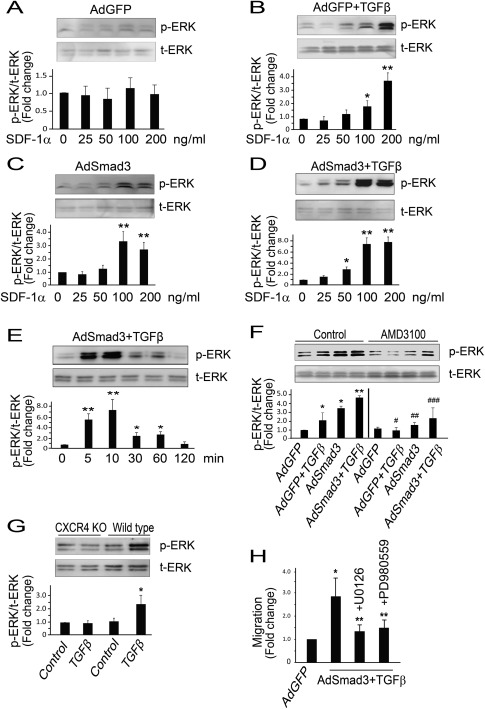
TGFβ/Smad3 treatment stimulates SDF‐1α‐induced, CXCR4‐dependent ERK phosphorylation in smooth muscle cells (SMCs). AdSmad3 infected and TGFβ1 (5 ng/ml) treated SMCs were stimulated with SDF‐1α and ERK phosphorylation was detected by Western blotting. (A‐D): Dose response of SDF1α induced (10 min) ERK phosphorylation in SMCs treated with AdGFP (A), AdGFP +TGFβ1 (B), AdSmad3 (C), and AdSmad3+TGFβ1 (D), respectively. *p < .05 compared with 0 ng/ml; **p < .01 compared with 0 ng/ml; n = 3. (E): AdSmad3 infected and TGFβ1 (5 ng/ml) treated SMCs were stimulated with SDF‐1α (100 ng/ml) for indicated time and ERK phosphorylation was detected by Western blotting. *p < .05 compared with 0 min; **p < .01 compared with 0 min; n = 3. (F): SDF‐1α‐induced (10 min, 100 ng/ml) ERK phosphorylation in SMCs without or with AMD3100 (10 µM). *p < .05 compared with AdGFP in control group; **p < .05 compared with all other conditions in control group; #p < .05 compared with AdGFP+TGFβ in control group; ##p < .05 compared with AdSmad3 in control group; ###p < .05 compared with AdSmad3+TGFβ in control group; n = 3. (G): SMCs were isolated from aorta of wild type or CXCR4 conditional knockout mice and treated with control (solvent, 4 mM HCl) or TGFβ1 (5 ng/ml) for 24 hour. SDF‐1α‐induced (10 min; 100 ng/ml) ERK phosphorylation was analyzed by Western blotting. *p < .05 compared with all other conditions, n = 3. (H): SMCs were infected with AdGFP (control) or AdSmad3 followed by treatment with TGFβ1 (5 ng/ml) for 24 hour, and then transferred to a Boyden Transwell insert. Cells were incubated with solvent or U0126 (5 µM) or PD989059 (10 µm) for 60 min, and then 100 ng/ml SDF‐1α was added to the lower chamber. The cells that migrated through the Transwell insert membrane after 4 hour were counted. *p < .05 compared with AdGFP; **p < .05 compared with AdSmad3+TGFβ without an inhibitor; n = 3. Abbreviations: SDF‐1, stromal cell‐derived factor‐1; TGFβ, transforming growth factor‐β.
Expression of CXCR4 Is Elevated in Balloon‐Injured Rat Carotid Arteries and Further Enhanced following Increased Expression of Smad3
We have previously shown that TGFβ and Smad3 are both upregulated following arterial injury 20. Having demonstrated the novel upregulation of CXCR4 by TGFβ/Smad3 in vascular SMCs, we next examined CXCR4 expression in vivo. We performed balloon angioplasty in male Sprague‐Dawley rats, followed by luminal infusion of AdSmad3 or AdGFP control. Animals were sacrificed at 3, 7, or 14 days and carotid arteries were collected and sectioned followed by immunostaining. Compared to the uninjured controls, levels of CXCR4 in the intima and media of the injured arteries were upregulated at all three time points. On day 14, the CXCR4 positive cells were primarily localized to the neointima. Moreover, CXCR4 expression was substantially increased in AdSmad3‐treated arteries compared to AdGFP control (Fig. 5A, 5B, 5C, 5F). Western blotting and immunostaining showed higher endogenous Smad3 levels after angioplasty, while AdSmad3 infusion further increased Smad3 level compared to AdGFP infected controls (Fig. 5D and 5E). As reported previously, the accumulation of SDF‐1α was evident 3 days after injury and sustained up to day 14. The SDF‐1α positivity was concentrated in the intima (Fig. 5G). Similarly, the expression of Ki67 and Smad3 also increased 3 days after injury and concentrated in the intimal area 7 and 14 days after angioplasty (Fig. 5H‐5K; H&E and phospho‐Smad3 IHC in Supporting Information Figure S4; Ki67 quantification in Supporting Information Figure S5).
Figure 5.
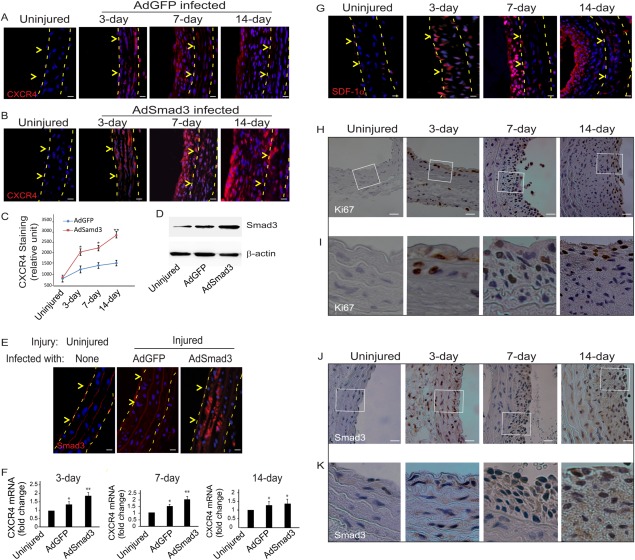
Enhanced Smad3 expression up‐regulates CXCR4 production in balloon‐injured rat carotid arteries. Balloon angioplasty was performed in rat carotid arteries followed by infusion of AdGFP or AdSmad3 (2.5 × 109 plaque‐forming units) for 20 minutes. Uninjured carotid arteries were used as control. Carotid arteries were retrieved at the indicated time points (3, 7, or 14 days) for preparation of cross sections. (A and B): Immunostaining was performed to detect CXCR4 (red) and DAPI was used to stain nuclei (blue). Dashed lines define the media layer; arrowheads mark internal elastic lamina (IEL). (C): Relative fluorescent intensity was measured using ImageJ software. Each bar is a mean ± SEM (n= 3‐5); *p < .05 compared with AdGFP control; **p < .05 compared with all other conditions. (D): Western blot was performed on proteins extracted from uninjured or injured with AdGFP or AdSmad3 infused carotid arteries 3 days after angioplasty. (E): Immunostaining of Smad3 on sections from uninjured, or injured with AdGFP or AdSmad3 infused carotid artery. Dashed lines define the media layer; arrowheads mark IEL. (F): RT‐PCR for CXCR4 levels after vascular injury. *p < .05 compared with uninjured; **p < .05 compared with uninjured or injured with AdGFP infused; n = 3. (G): Immunostaining of SDF‐1α on sections of carotid arteries on 3, 7, or 14 days after injury. Dashed lines define the media layer; arrowheads mark IEL. (H) and (J) are immunochemistry of Ki67 or Smad3 in injured sections of carotid arteries at the indicated time points (3, 7, or 14 days). (I) and (K) are magnified views of the boxed regions in H and J, respectively. Scale bar = 30 µm.
Selective Knockout of CXCR4 in Vascular SMCs Inhibits Intimal Hyperplasia in Wire‐Injured Mouse Femoral Arteries
Our hypothesis is that local expression of CXCR4 by vascular SMCs following arterial injury is enhanced by elevated TGFβ/Smad3. This in turn leads to SMC migration and contributes to the development of IH. To generate SMC‐specific CXCR4 knockout, we crossed a SM22‐driven Cre mouse line with a CXCR4‐floxed mouse strain to allow the CXCR4 gene to be deleted during genomic DNA recombination. PCR genotyping using samples taken from the aortic SMCs of the recombined SM22‐Cre/CXCR4‐Flox crossover strain confirmed the absence of CXCR4 (Supporting Information Figure S1). In contrast, CXCR4 was present in other organs including liver and tail, indicating that the CXCR4 knockout was specific to the SMCs. The wire‐injured femoral segments were retrieved 4 weeks after surgery. Representative H&E stained sections showed reduced neointima in CXCR4 conditional knockout mice compared to that in wild type. Immunostaining revealed abundant CXCR4 staining concentrated in the neointima of injured femoral artery in wild type mice (Fig. 6G and 6I). In contrast, the CXCR4 staining was essentially absent in the media and neointima of the conditional knockout mice (Fig. 6H and 6J). The SDF‐1α expression which is mainly localized to the luminal surface of the intima was not affected by the CXCR4 knockout (Fig. 6L and 6M). Similarly, SMA staining was comparable in both genotypes (Fig. 6N and 6O). Smad3 staining confirmed the elevation of this signaling protein in the intimal area and tunica media (Fig. 6Q and R; Ki67 and phospho‐Smad3 in Supporting Information Figure S6). However, morphometric analyses revealed drastic differences in intimal formation between the wild type and SMC‐specific CXCR4 knockout mice. At 4 weeks post injury, IH (measured as an I/M area ratio) was 60% less in knockout mice (0.29 ± 0.08) compared to their wild type littermates (0.74 ± 0.13). As the result, the lumen size increased by ∼80% in the knockout compared to the wild type (Fig. 6S). These in vivo findings demonstrate that injury‐induced local CXCR4 expression is an essential contributing factor to hyperplastic neointimal growth (Fig. 7).
Figure 6.
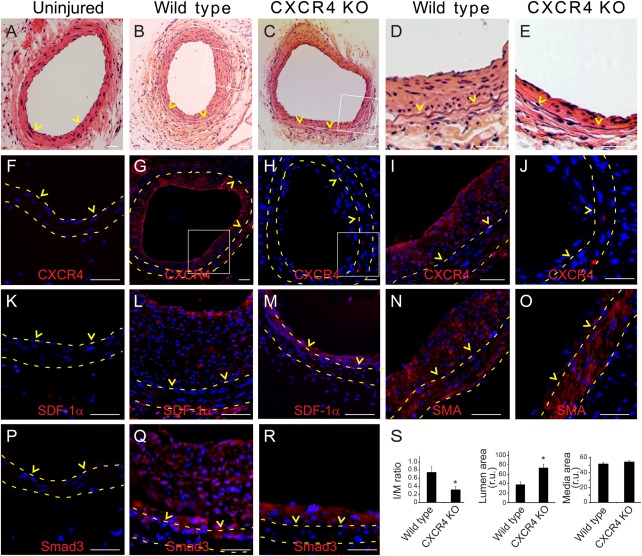
Selective knockout of CXCR4 in smooth muscle cells (SMCs) reduces intimal hyperplasia (IH) in wire‐injured mouse femoral arteries. SM22‐driven conditional CXCR4 knockout model was created and wire injury was performed in wild type and CXCR4 knockout mice, as described in Methods. (A–E): Representative H&E stained femoral arterial sections in uninjured (A), injured wild type (B) or injured knockout mice (C). (D) and (E) are enlarged views of the boxed areas in (B) and (C). (F–J): CXCR4 immunostaining in uninjured (F), injured wild type (G), or injured knockout mice (H). (I) and (J) are enlargement of the boxed regions from (G) and (H). (K‐M): SDF‐1α immunostaining in uninjured (K), injured wild type (L), or injured knockout mice (M). (N–O): SMA immunostaining in injured wild type (N) or injured knockout mice (O). (P–R): Smad3 immunostaining in uninjured (P), injured wild type (Q), or injured knockout mice (R). (S): IH (intima/media area ratio), lumen area, and media area were quantified as described in Methods. *p < .05 compared to wild type; n = 12‐13. Scale bar = 50 µm. Abbreviation: SDF‐1, stromal cell‐derived factor‐1.
Figure 7.
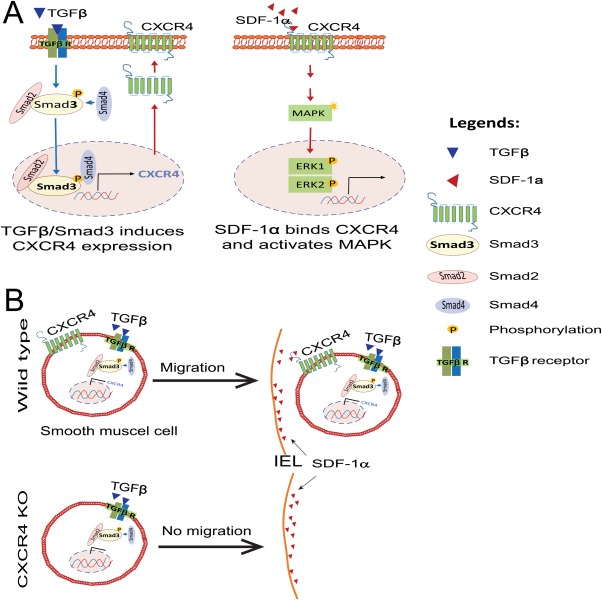
A schematic model showing the role of TGFβ/Smad3 regulated CXCR4 expression in SMC migration toward SDF‐1α. (A): TGFβ/Smad3 stimulates CXCR4 expression while SDF‐1α activates MAPK through CXCR4. (B): Elevated SDF‐1α in the subintima of injured artery attracts SMCs from wildtype mice but not from CXCR4 knockout mice. Abbreviations: SDF‐1, stromal cell‐derived factor‐1; SMCs, smooth muscle cells; TGFβ, transforming growth factor‐β.
Discussion
Our studies have produced several interesting findings. We have discovered that elevated TGFβ/Smad3 signaling markedly upregulates CXCR4 in vascular SMCs in vitro as well as in balloon‐injured rat carotid arteries. Moreover, enhanced CXCR4 expression in vitro results in the migration of SMCs toward SDF‐1α. Using a mouse line designed to deplete the CXCR4 gene in a SM22‐specific manner, we found that knocking out CXCR4 expression in vascular SMCs substantially reduced wire injury‐induced IH. These results suggest that local (residential) CXCR4‐expressing SMCs play an important role in the development of IH and suggest a novel mechanism of restenosis; i.e., in response to arterial injury, upregulated TGFβ/Smad3 activates the expression of CXCR4 in medial SMCs, which are then attracted to the subintima in response to high levels of SDF‐1α, thereby enhancing IH.
There exists considerable evidence that CXCR4‐expressing bone marrow‐derived cells, namely progenitor or mesenchymal stem cells, are attracted to the subintima in response to SDF‐1α, thus contributing to IH 11, 25, 26, 27. The findings of these studies have varied and are dependent upon the type of injury model used. M‐CSF mobilized CXCR4+ stem cells enhance neointimal formation 26 while bone marrow transplantation from CXCR4 knockout to wild type mice reduced the IH by ∼50% 11. These studies have demonstrated the importance of CXCR4+ cells in IH. Our study is the first to demonstrate that locally induced expression of CXCR4 in residential SMCs plays an important role in the development of IH. The most compelling evidence is from our wire injury experiments, where we employed a mouse strain deprived of CXCR4 in local SMCs within the arterial wall. We were able to demonstrate a pronounced diminution of IH in wire‐injured femoral arteries lacking CXCR4 in SM22‐expresssing cells. The fact that we do observe some positive staining for CXCR4 in the neointima of the knockout mice suggests that SDF‐1α acts as a chemoattractant of both internal CXCR4+ SMCs as well as external CXCR4+ bone marrow cells. Thus, it is plausible to propose that both residential and “foreign” CXCR4‐expressing cells contribute to the development of neointima, although we were unable to compare their pathogenic capacity in the current study.
In accordance with an important role of local inducible CXCR4 expression in the development of IH, elevated CXCR4 has also been documented in other vascular pathological conditions. CXCR4 expression has been found to be upregulated in atherosclerotic lesions 28, and also in the tunica media of rat carotid arteries in a type I diabetes model 29, although the identity of SMCs as the source of CXCR4 was not confirmed in either of these settings. The only previous observation of vascular SMCs expressing CXCR4 was in vitro studies showing high glucose stimulated CXCR4 expression with the resultant promotion of proliferation and migration of rat SMCs 29. Additionally, Schecter et al. 30 demonstrated that HIV gp120 activated the expression of tissue factor by SMCs through a mechanism that involved CXCR4, leading to thrombogencity of atherosclerotic plaques. In spite of these prior studies, an important role for CXCR4 expression in local SMCs has not been previously demonstrated.
Our hypothesis is that locally induced SMC CXCR4 leads to SDF‐1α‐mediated attraction of SMCs to the intima. This hypothesis is supported by the well‐established role of SDF‐1α/CXCR4 in chemotaxis. The most pertinent evidence might be that SDF‐1α attracts circulating, CXCR4‐expressing bone marrow‐derived progenitor or mesenchymal stem cells to the subintimal layer 11. These findings were confirmed by the reports that SDF‐1α neutralizing antibodies inhibit IH after femoral wire injury 31, 32. Moreover, the chemoattractant role of SDF‐1α has been extensively studied in development, cancer metastasis, as well as several other pathological processes. For example, the SDF‐1α/CXCR4 axis has been found to be important in the maintenance of hematopoietic stem cell pool in bone marrow 33, 34; this chemokine/receptor pair is also known to play an important role in directing cancer stem cell migration during tumor metastasis 8, 35. In the current study, while our in vitro data confirmed migration of CXCR4‐expressing SMCs toward SDF‐1α (Fig. 3B), we also observed abundant SDF‐1α in the intima of injured mouse arteries (Fig. 6L and 6M). Thus, it is reasonable to propose that following arterial injury, residential SMCs bearing a high content of induced CXCR4 are attracted to the subintima by SDF‐1α, thereby enhancing neointimal formation. In addition to CXCR4, CXCR7 is another potential receptor for SDF‐1α 36, 37. Since we did not observe significant effect of TGFβ/Smad3 treatment on CXCR7 expression (Supporting Information Fig. S3), we focused the current study on CXCR4.
Aside from stimulating chemotaxis, SDF‐1α/CXCR4 has been shown to influence cell proliferation, albeit its role varies with different cell types and experimental contexts. In some settings, CXCR4 induces proliferation; examples include a glioblastoma cell line 38, pancreatic cancer cell lines 39, and several types of progenitor and stem cells 40, 41. There is also a report suggesting that SDF‐1α can stimulate proliferation of vascular SMCs 29. In contrast, other investigators have found that CXCR4 negatively regulates proliferation (e.g., keratinocytes in IL‐23‐Mediated Psoriasiform Dermatitis) 42. Since neointimal SMCs are characterized by a higher proliferative rate, we asked whether the interaction of CXCR4‐expressing SMCs with SDF‐1α in the neointima would result in enhanced SMC proliferation. Despite the ability of SDF‐1α/CXCR4 to stimulate SMC chemotaxis, we did not demonstrate SDF‐1α‐enhanced proliferation of CXCR4‐expressing SMCs in vitro (Supporting Information Fig. S2). However, the lack of proliferation effects in vitro does not necessarily exclude the possibility that the SDF1/CXCR4 axis directly or indirectly contributes to the highly proliferative events taking place following vascular injury. Indeed, we noted a very similar temporal and spatial pattern of CXCR4+ and Ki67+ cells in the injured arteries. It is also possible that the high CXCR4 expression facilitates the phenotypic switch of SMCs, sensitizing them to the many mitogenic factors including PDGF‐BB and bFGF to which SMCs are exposed following injury.
Our study also presents a novel finding that the TGFβ/Smad3 axis is a potent stimulant of CXCR4, suggesting a new mechanism for the SDF‐1α/CXCR4 axis in the development of IH. Supporting evidence for the role of TGFβ/Smad3 in expression of CXCR4 can be found from studies in other disease systems. For example, rat hepatoma cells normally express low levels of CXCR4, but when treated with TGFβ, levels of CXCR4 are increased 43. In response to TGFβ1, rheumatoid synovial T cells also express high levels of CXCR4 and become responsive to SDF‐1α 44. In addition, Zhao and colleagues 45 showed that MCF‐7 breast cancer cells significantly increased CXCR4 expression after TGFβ1 treatment. Our group has previously shown that TGFβ and its signaling protein, Smad3, are upregulated at sites of arterial injury 20. In the presence of enhanced Smad3 expression, TGFβ appears to transform vascular SMCs in vitro from proapoptotic to proproliferative. We believe that the effect of TGFβ in promoting IH is related to coupregulation of both TGFβ and Smad3 following arterial injury, an observation we have made both in rats and in humans 5. In the current study, we have demonstrated that TGFβ/Smad3 stimulates CXCR4 expression in vascular SMCs, which ultimately leads to enhanced IH. While we believe that TGFβ stimulates SMC CXCR4 expression primarily through canonical Smad3‐dependent pathway, our work does not rule out a possible contribution of noncanonical pathways. It remains to be determined whether the inhibition of TGFβ signaling attenuates postinjury CXCR4 upregulation. Of note, other factors, such as HIF‐1α, have also been shown to contribute to the induction of CXCR4 expression in different cell types 46.
In search of a mechanism for the SDF‐1α‐stimulated migration of TGFβ/Smad3‐treated cells, we identified MAPK ERK as the intermediate. MAPK ERK is a known critical mediator of cell migration in multiple cell types. We have shown that TGFβ/Smad3 significantly enhances SDF‐1α‐induced ERK activation. Moreover, SMC migration in response to SDF‐1α is dependent upon both CXCR4 as well as MAPK ERK (Figs. 3 and 4). Binding of SDF‐1α to the CXCR4 receptor activates a myriad of downstream signaling pathways that comprise Akt, ERK, and mTORC1 to regulate chemotaxis of mature dendritic cells 47. The SDF‐1α/CXCR4 axis also regulates osteosarcoma cell migration through MEK1/2, ERK, and NF‐KB pathways 48. Regulation of SDF‐1α/CXCR4 for hematopoietic stem cell migration through PKC‐zeta, Pyk‐2 and ERK1/2 has also been documented 49. Our results indicated that MAPK ERK is critical for SDF‐1α‐induced SMC migration.
To explore the mechanism by which TGFβ/Smad3 stimulates CXCR4 expression in SMCs, we focused on the observation that the CXCR4 promoter has consensus SBEs. Previous computational analysis predicted that Smad3 might directly interact with the CXCR4 promoter 50. Our results from the ChIP assay reveal that Smad3 specifically binds to the CXCR4 promoter following TGFβ stimulation (Fig. 2). Our study is the first to report a link between TGFβ/Smad3 and CXCR4 in vascular SMCs. We also provide solid evidence supporting that Smad3 directly regulates CXCR4 expression by binding to SBEs in the promoter region of the CXCR4 gene.
In light of the fact that CXCR4 is a progenitor/stem cell marker, our observation of TGFβ/Smad3‐stimulated SMC CXCR4 expression is in accordance with recent reports suggesting that SMC de‐differentiation is a critical component of the response to vascular injury 51. In our recent work, we have shown that TGFβ in the presence of elevated Smad3 enhances SMC de‐differentiation by upregulating multiple stem or progenitor cell genes as well as by downregulating SMC markers or “differentiation” genes 16. Our new findings with CXCR4 reinforce this hypothesis in that we have shown in the presence of TGFβ/Smad3, that SMCs express CXCR4, a de‐differentiation marker. Thus, it is conceivable that following arterial injury, CXCR4 may together with other de‐differentiation genes transform SMCs into a phenotype that promotes IH.
Conclusion
We have identified a new pathway through which TGFβ stimulates the formation of IH. Following arterial injury, elevated levels of TGFβ and Smad3 induce expression of CXCR4 on medial vascular SMCs. These cells in turn are attracted to the neointima by the cytokine SDF‐1α (Fig. 7). Moreover, we have demonstrated the mechanism by which TGFβ/Smad3 controls the expression of CXCR4 as well as the mechanism used by CXCR4 to influence SMC migration. To confirm our hypothesis in vivo, we have created transgenic mice to selectively knockout CXCR4 in residential SMCs. Local depletion of CXCR4 in vascular SMCs can effectively mitigate IH in injured arteries. These data suggest a novel mechanism for restenosis. Thus, modulation of TGFβ/Smad3 function or local interruption of the CXCR4/SDF‐1α axis might lead to effective therapeutic strategies to prevent the development of the hyperplastic neointima that leads to restenosis.
Author Contributions
X.S., L.‐W.G., S.S., B.L., and K.C.K.: Designed experiments; X.S., S.S., T.T., B.W., M.Z., M.A.C., and Y.S.: Performed experiments; X.S., L.‐W.G., S.S., M.A.C., B.L., and K.C.K.: Analyzed data; X.S., L.‐W.G., B.W., S.R.F., B.L., and K.C.K.: Wrote manuscript; X.S., L.‐W.G., and S.S.: These authors contributed equally to this work.
Disclosures of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
Supporting information
Supplementary Information
Acknowledgments
This work was supported by a National Heart, Lung, and Blood Institute R01 grant (HL‐068673, to K.C.K), a T32 training grant (HL‐110853, to K.C.K.), an American Heart Association Grant‐In‐Aid award (14GRNT20380854, to K.C.K.), a Wisconsin Partnership Program award (ID2832, to L.‐W.G.) and an NIH grant (R01EY022678, to L.‐W.G.)
Contributor Information
Xudong Shi, Email: kent@surgery.wisc.edu.
Lian‐Wang Guo, Email: guo@surgery.wisc.edu.
K. Craig Kent, Email: shi@surgery.wisc.edu.
References
- 1. Mills B, Robb T, Larson DF. Intimal hyperplasia: slow but deadly. Perfusion 2012;27:520–528. [DOI] [PubMed] [Google Scholar]
- 2. Suwanabol PA, Kent KC, Liu B. TGF‐β and restenosis revisited: a smad link. J Surg Res 2011;167:287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akhurst RJ, Hata A. Targeting the TGFβ signaling pathway in disease. Nat Rev Drug Discov 2012;11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsai S, Butler J, Rafii S, et al. The role of progenitor cells in the development of intimal hyperplasia. J Vasc Surg 2009;49:502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Edlin RS, Tsai S, Yamanouchi D, et al. Characterization of primary and restenotic atherosclerotic plaque from the superficial femoral artery: potential role of Smad3 in regulation of SMC proliferation. J Vasc Surg 2009;49:1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF‐beta receptor in colon cancer cells with microsatellite instability. Science 1995;268:1336–1338. [DOI] [PubMed] [Google Scholar]
- 7. Kundi R, Hollenbeck ST, Yamanouchi D, et al. Arterial gene transfer of the TGF‐ b signalling protein Smad3 induces adaptive remodelling following angioplasty: a role for CTGF. Cardiovasc. Res 2009;326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang CY, Lee CY, Chen MY, et al. Stromal cell‐derived factor‐1/CXCR4 enhanced motility of human osteosarcoma cells involves MEK1/2, ERK and NF‐kappaB‐dependent pathways. J Cell Physiol 2009;221:204–212. [DOI] [PubMed] [Google Scholar]
- 9. Arya M, Ahmed H, Silhi N, et al. Clinical importance and therapeutic implications of the pivotal CXCL12‐CXCR4 (chemokine ligand‐receptor) interaction in cancer cell migration. Tumour Biol 2007;28:123–131. [DOI] [PubMed] [Google Scholar]
- 10. Mukherjee D, Zhao J. The role of chemokine receptor CXCR4 in breast cancer metastasis. Am J Cancer Res 2013;3:46–57. [PMC free article] [PubMed] [Google Scholar]
- 11. Zernecke A, Schober A, Bot I, et al. SDF‐1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res 2005;96:784–791. [DOI] [PubMed] [Google Scholar]
- 12. Nguyen AT, Gomez D, Bell RD, et al. Smooth muscle cell plasticity: fact or fiction? Circ Res 2013;112:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Si Y, Ren J, Wang P, et al. Protein kinase C‐delta mediates adventitial cell migration through regulation of monocyte chemoattractant protein‐1 expression in a rat angioplasty model. Arterioscler Thromb Vasc Biol 2012;32:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodriguez‐Menocal L, St‐Pierre M, Wei Y, et al. The origin of post‐injury neointimal cells in the rat balloon injury model. Cardiovasc Res 2009;81:46–53. [DOI] [PubMed] [Google Scholar]
- 15. Ryer EJ, Hom RP, Sakakibara K, et al. PKCdelta is necessary for Smad3 expression and transforming growth factor beta‐induced fibronectin synthesis in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2006;26:780–786. [DOI] [PubMed] [Google Scholar]
- 16. Shi X, DiRenzo D, Guo LW, et al. TGF‐β/Smad3 stimulates stem cell/developmental gene expression and vascular smooth muscle cell de‐differentiation. PLoS One 2014;9:e93995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Piek E, Ju WJ, Heyer J et al. Functional characterization of transforming growth factor β signaling in Smad2‐ and Smad3‐deficient fibroblasts. J Biol Chem 2001;276:19945–19953. [DOI] [PubMed] [Google Scholar]
- 18. Tang Y, Katuri V, Iqbal S et al. ELF a beta‐spectrin is a neuronal precursor cell marker in developing mammalian brain; structure and organization of the elf/beta‐G spectrin gene. Oncogene 2002;21:5255–5267. [DOI] [PubMed] [Google Scholar]
- 19. Wu PC, Lu JW, Yang JY et al. H3K9 histone methyltransferase, KMT1E/SETDB1, cooperates with the SMAD2/3 pathway to suppress lung cancer Metastasis. Cancer Res 2014;74:7333–7343. [DOI] [PubMed] [Google Scholar]
- 20. Sata M, Maejima Y, Adachi F, et al. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol 2000;32:2097–2104. [DOI] [PubMed] [Google Scholar]
- 21. Tsai S, Hollenbeck ST, Ryer EJ, et al. TGF‐ β through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am J Physiol Heart Circ Physiol 2009;297:H540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nelson PR, Yamamura S, Mureebe L et al. Smooth muscle cell migration and proliferation are mediated by distinct phases of activation of the intracellular messenger mitogen‐activated protein kinase. J Vas Surg 1998;27:117–25. [DOI] [PubMed] [Google Scholar]
- 23. Gerthoffer WT. 2007 Mechanisms of vascular smooth muscle cell Migration. Cic. Res 2007;100:607–621. [DOI] [PubMed] [Google Scholar]
- 24. Sun X, Wei L, Chen Q et al. CXCR4/SDF1 mediate hypoxia induced chondrosarcoma cell invasion through ERK signaling and increased MMP1 expression. Mol Cancer 2010;9:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sheng J, Cai WW, Fang NY, et al. Role of stromal‐derived factor‐1α/CXCR4 in neo‐intimal repair. Cardiovasc J Afr 2011;22:313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shiba Y, Takahashi M, Yoshioka T, et al. M‐CSF accelerates neointimal formation in the early phase after vascular injury in mice: the critical role of the SDF‐1‐CXCR4 system. Arterioscler Thromb Vasc Biol 2007;27:283–289. [DOI] [PubMed] [Google Scholar]
- 27. Li M, Yu J, Li Y, et al. CXCR4 positive bone mesenchymal stem cells migrate to human endothelial cell stimulated by ox‐LDL via SDF‐1alpha/CXCR4 signaling axis. Exp Mol Pathol 2010;88:250–255. [DOI] [PubMed] [Google Scholar]
- 28. Kodali R, Hajjou M, Berman AB, et al. Chemokines induce matrix metalloproteinase‐2 through activation of epidermal growth factor receptor in arterial smooth muscle cells. Cardiovasc Res 2006;69:706–715. [DOI] [PubMed] [Google Scholar]
- 29. Jie W, Wang X, Zhang Y, et al. SDF‐1α/CXCR4 axis is involved in glucose‐potentiated proliferation and chemotaxis in rat vascular smooth muscle cells. Int J Exp Pathol 2010;91:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schecter AD, Knarren S, Lietz M, et al. HIV envelope gp120 activates human arterial smooth muscle cells. Proc Natl Acad Sci USA 2001;98:10142–10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schober A, Knarren S, Lietz M, et al. Crucial role of stromal cell‐derived factor‐1alpha in neointima formation after vascular injury in apolipoprotein E‐deficient mice. Circulation 2003;108:2491–2497. [DOI] [PubMed] [Google Scholar]
- 32. Nemenoff RA, Horita H, Ostriker AC, et al. SDF‐1α induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury‐induced neointima formation. Arterioscler Thromb Vasc Biol 2011;31:1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fruehauf S, Seeger T, Maier P, et al. The CXCR4 antagonist AMD3100 releases a subset of G‐CSF‐primed peripheral blood progenitor cells with specific gene expression characteristics. Exp Hematol 2006;34:1052–1059. [DOI] [PubMed] [Google Scholar]
- 34. Smith‐Berdan S, Nguyen A, Hassanein D, et al. Robo4 cooperates with CXCR4 to specify hematopoietic stem cell localization to bone marrow niches. Cell Stem Cell 2011;8:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tseng D, Vasquez‐Medrano D, Brown JM. Targeting SDF‐1/CXCR4 to inhibit tumour vasculature for treatment of glioblastomas. Br J Cancer 2011;104:1805–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Balabanian K, Lagane B, Infantino S, et al. The chemokine SDF‐1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 2005;280:35760–35766. [DOI] [PubMed] [Google Scholar]
- 37. Burns JM, Summers BC, Wang Y, et al. A novel chemokine receptor for SDF‐1 and I‐TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 2006;203:2201–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Do Carmo A, Patricio I, Cruz MT, et al. CXCL12/CXCR4 promotes motility and proliferation of glioma cells. Cancer Biol Ther 2010; 9:56–65. [DOI] [PubMed] [Google Scholar]
- 39. Shen X, Artinyan A, Jackson D, et al. Chemokine receptor CXCR4 enhances proliferation in pancreatic cancer cells through AKT and ERK dependent pathways. Pancreas 2010;39:81–87. [DOI] [PubMed] [Google Scholar]
- 40. Kayali AG, Van Gunst K, Campbell IL, et al. The stromal cell‐derived factor‐1alpha/CXCR4 ligand‐receptor axis is critical for progenitor survival and migration in the pancreas. J Cell Biol 2003;163:859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kahn J, Byk T, Jansson‐Sjostrand L, et al. Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood 2004;103:2942–2949. [DOI] [PubMed] [Google Scholar]
- 42. Takekoshi T, Wu X, Mitsui H, et al. CXCR4 negatively regulates keratinocyte proliferation in IL‐23‐mediated psoriasiform dermatitis. J Invest Dermatol 2013;133:2530–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bertran E, Caja L, Navarro E, et al. Role of CXCR4/SDF‐1 alpha in the migratory phenotype of hepatoma cells that have undergone epithelial‐mesenchymal transition in response to the transforming growth factor‐beta. Cell Signal 2009;21:1595–1606. [DOI] [PubMed] [Google Scholar]
- 44. Buckley CD, Amft N, Bradfield PF, et al. Persistent induction of the chemokine receptor CXCR4 by TGF‐beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol 2000;165:3423–3429. [DOI] [PubMed] [Google Scholar]
- 45. Zhao X, Huang YY, Huang Y, et al. Transforming growth factor‐beta1 upregulates the expression of CXC chemokine receptor 4 (CXCR4) in human breast cancer MCF‐7 cells. Acta Pharmacol Sin 2010;31:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maltepe E, Krampitz GW, Okazaki KM, et al. Hypoxia‐inducible factor‐dependent histone deacetylase activity determines stem cell fate in the placenta. Development 2005;132:3393–3403. [DOI] [PubMed] [Google Scholar]
- 47. Delgado MC, Escribano C, Pablos JL, et al. Chemokine CXCL12 uses CXCR4 and a signaling core formed by bifunctional Akt, extracellular signal‐regulated kinase (ERK)1/2, and mammalian target of rapamycin complex 1 (mTORC1) proteins to control chemotaxis and survival simultaneously in mature dendriti. J Biol Chem 2011;286:37222–37236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang CY, Lee CY, Chen MY, et al. Stromal cell‐derived factor‐1/CXCR4 enhanced motility of human osteosarcoma cells involves MEK1/2, ERK and NF‐kappaB‐dependent pathways. J Cell Physiol 2009;221:204–212. [DOI] [PubMed] [Google Scholar]
- 49. Petit I, Goichberg P, Spiegel A, et al. Atypical PKC‐ζ regulates SDF‐1α‐ mediated migration and development of human CD34 + progenitor cells. J Clin Invest 2005;115:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Katoh M, Katoh M. Integrative genomic analyses of CXCR4: transcriptional regulation of CXCR4 based on TGFbeta, nodal, activin signaling and POU5F1, FOXA2, FOXC2, FOXH1, SOX17, and GFI1 transcription factors. Int J Oncol 2010;36:415–420. [DOI] [PubMed] [Google Scholar]
- 51. Tang Z, Wang A, Yuan F, et al. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun 2012;3:875. doi:10.1038/ncomms1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information