

Abstract
Fibrosis is a characteristic of Duchenne muscular dystrophy (DMD), yet the cellular and molecular mechanisms responsible for DMD fibrosis are poorly understood. Utilizing the Collagen1a1‐GFP transgene to identify cells producing Collagen‐I matrix in wild‐type mice exposed to toxic injury or those mutated at the dystrophin gene locus (mdx) as a model of DMD, we studied mechanisms of skeletal muscle injury/repair and fibrosis. PDGFRα is restricted to Sca1+, CD45− mesenchymal progenitors. Fate‐mapping experiments using inducible CreER/LoxP somatic recombination indicate that these progenitors expand in injury or DMD to become PDGFRα+, Col1a1‐GFP+ matrix‐forming fibroblasts, whereas muscle fibres do not become fibroblasts but are an important source of the PDGFRα ligand, PDGF‐AA. While in toxin injury/repair of muscle PDGFRα, signalling is transiently up‐regulated during the regenerative phase in the DMD model and in human DMD it is chronically overactivated. Conditional expression of the constitutively active PDGFRα D842V mutation in Collagen‐I+ fibroblasts, during injury/repair, hindered the repair phase and instead promoted fibrosis. In DMD, treatment of mdx mice with crenolanib, a highly selective PDGFRα/β tyrosine kinase inhibitor, reduced fibrosis, improved muscle strength, and was associated with decreased activity of Src, a downstream effector of PDGFRα signalling. These observations are consistent with a model in which PDGFRα activation of mesenchymal progenitors normally regulates repair of the injured muscle, but in DMD persistent and excessive activation of this pathway directly drives fibrosis and hinders repair. The PDGFRα pathway is a potential new target for treatment of progressive DMD. © 2016 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: PDGFRα signalling, fibrosis, collagen‐producing cells, Duchenne muscular dystrophy, skeletal muscle
Introduction
Duchenne muscular dystrophy (DMD) is a fatal disease primarily affecting skeletal and cardiac muscles. There are no approved treatments that halt or reverse the course of the disease. As the disease progresses, connective tissue accumulates in degenerating cardiac and skeletal muscles, a process commonly referred to as fibrosis 1. Fibrosis is the most prominent pathological feature, and a reliable determinant of disease progression, yet the cellular and molecular milieu that governs fibrosis in DMD remains largely uncharacterized 1, 2, 3. Skeletal muscle mesenchymal progenitors capable of differentiating into resident fibroblasts and adipocytes have been identified (also referred to as FAPs) 4, 5, 6. These mesenchymal progenitors are PDGFRα + cells that have been associated with the pathogenesis in mdx and DMD skeletal muscles 7, 8. Recently, these mesenchymal progenitors in the skeletal muscle and heart have been shown to express pro‐fibrotic genes in response to TGF‐β1 and PDGF‐AA in vitro, implicating them as potentially important cells in the fibrogenic process 7, 9.
Constitutive activation of PDGFRα has been reported to induce spontaneous systemic fibrosis in healthy mice, and pro‐fibrotic cells present in many organs express PDGFRα 7, 9, 10, 11, 12. In models of toxin injury to skeletal muscle, pro‐fibrotic cells also express PDGFRα 5, 7, 10. Recently, studies have highlighted a role for PDGFRα signalling in the pro‐fibrotic response of not only FAPs but also myogenic, haematopoietic, and endothelial cells 13. Imatinib, a broad spectrum tyrosine kinase inhibitor, has been reported to decrease fibrosis in mdx skeletal muscles by inhibiting c‐Abl and PDGFR signalling, suggesting that the PDGFR pathway may be important 6, 14, 15. Taken together, these reports suggest that pro‐fibrotic muscle cells can express PDGFRα and that this pathway has the potential to induce fibrosis. Despite this evidence, it remains unclear whether PDGFRα signalling directly promotes fibrosis and loss of function in DMD. In light of recent evidence indicating that multiple cell populations contribute to fibrosis 13, the proportion of PDGFRα‐expressing cells that produce fibrillar collagens and promote fibrosis in muscular dystrophy needs to be carefully examined.
Skeletal muscle fibrosis in DMD is characterized by the accumulation of type I and type III collagens 16. In cardiac muscle, fibroblasts are thought to be a heterogeneous population with various expression patterns and developmental origins, although very recent studies now suggest that the majority appear to derive from resident populations of mesenchymal cells 17, 18, 19, 20, 21. In skeletal muscles, populations including FAPs and endothelial, myogenic, and haematopoietic cells have been reported to express collagens 6, 13. However, the heterogeneity and the extent of collagen expression by various muscle populations have raised more questions which necessitate greater delineation. The identification of fibrotic cells is inherently hampered by the nature of collagen synthesis, which begins intracellularly, with the expression and production of pro‐collagens, and ends with the assembly of collagen fibrils in the extracellular space 22. Therefore, collagen‐producing cells are not easily distinguishable from other cell types, such as inflammatory cells, that also occupy damaged tissue and influence fibrosis in disease or injury 23.
In the present study, we used fate‐mapping techniques to study and dissect the roles of PDGFRα + cells and PDGFRα signalling in disease progression in mdx mice which harbour a mutated dystrophin gene, drawing direct comparison with human dystrophic muscles. We show that PDGFRα + cells are the primary source of fibrillar collagen matrix synthesis in disease. By comparing acute injury followed by repair in healthy skeletal muscle with chronic disease of mdx muscle, we dissected the differences and similarities between the regeneration process that results in restoration of function and the maladaptive repair process that characterizes muscle dystrophy.
Materials and methods
Expanded methods and details of mouse models, primers, and antibodies are listed in the supplementary material, Supplementary materials and methods and Tables S1–S5, and elsewhere 9, 24, 25, 26, 27, 28. Muscle injuries were caused by a single injection of a 10 nm solution of cardiotoxin (Sigma, St Louis, MO, USA) directly into the tibialis anterior (TA; 20 µl) or quadriceps (40 µl) 29. For inducible Cre activation, 100 µl of 20 mg/ml tamoxifen suspended in corn oil was injected intraperitoneally (i.p.) daily for five consecutive days and then allowed to resolve for a minimum of 5 days prior to cardiotoxin injury. Myography was conducted and specific isometric force was calculated as specified previously 29. Tissue processing, histology, and staining were in accordance with published procedures 9, 29, 30.
Cells for molecular analysis were isolated by FACS from single cell preparations prepared by digesting a collection of limb muscles (TA, gastrocnemius, and quadriceps) with collagenase and dispase (Worthington Biochemical Corp, Lakewood, NJ, USA) 26, 31. RNA was isolated from cells or tissue as described previously 29. Proteins were isolated from muscle tissues snap‐frozen with liquid nitrogen, ground into a fine powder, weighed, suspended in RIPA buffer containing protease and phosphatase inhibitors (Thermo/Pierce, Rockford, IL, USA), and then homogenized with a Tissue‐Tearor (BioSpec, Bartlesville, OK, USA). The cultured Col1a1‐GFP+ cells used for in vitro studies were generated as reported previously 9. Single muscle fibres were isolated and cultured as described elsewhere 32 from 4‐month‐old wild‐type (WT) and mdx extensor digitorum longus (EDL) muscles.
Measurement of the percentage area staining was performed using ImageJ software (NIH) 29, 33, 34. The minimum diameter of muscle fibres and densitometry were also measured by ImageJ 29. Graphs show the mean of replicates for each condition. Student's t‐test, or ANOVA when appropriate, was used for statistical analyses and computation of p values.
Results
Col1a1‐GFP+ cells are responsible for connective tissue accumulation in dystrophic muscles
To study the process of fibrosis in dystrophic skeletal muscles, we utilized mdx mice that harbour a reporter of collagen type I production, the Col1a1‐GFP transgene 9, 35. Col1a1‐GFP+ cells are abundant in regions of pathology in mdx diaphragms compared with diaphragms from wild‐type, healthy mice (Figure 1). Alterations in connective tissue and muscle architecture, highlighted by collagen type I staining and the presence of GFP+ cells, were prominent in diseased diaphragms and quadriceps of mdx mice (Figure 1A). This remodelling of the tissue architecture is characteristic of the cycles of degeneration and regeneration that occur with the progression of muscular dystrophy and presumably results in fibrosis as muscles lose their capacity to repair. Consistent with this, Col1a1‐GFP+ cells are closely associated with muscle fibres positive for sarcoplasmic fibronectin, an indicator of degenerating muscle 36, and α‐smooth muscle actin (αSMA), which is expressed by regenerating fibres 37 (Figure 1C, D). The cycles of damage and repair are associated with the accumulation of collagen type I and type III, the main components of connective tissue in mdx muscle fibrosis 38, 39, 40. Although expression of Col1a1‐GFP is indicative of likely pro‐collagen 1α1 production, it remained unclear if GFP+ cells were also the source of type III collagen. Therefore, we isolated Col1a1‐GFP‐positive and negative cells from mdx muscle digests by flow sorting to compare their expression of collagen mRNAs by RT‐qPCR. GFP+ cells isolated from mdx diaphragms expressed high levels of Col1a1 and Col3a1, whereas other muscle cells such as endothelial cells (CD31+, Sca1+, CD45−) and macrophages (CD45+, F4/80+) did not express these pro‐collagen mRNAs (Figure 1E).
Figure 1.

Col1a1‐GFP+ collagen‐producing cells accompany regeneration and degeneration in dystrophic skeletal muscles of mdx mice. (A, B) Immunofluorescence analysis of diaphragms and quadriceps from male wild‐type and mdx:Col1a1‐GFP mice (N = 3 mice per age group; 4, 14, and 20 months old) reveals that GFP+ cells and type I collagen progressively increase in dystrophic muscles. Staining for type I collagen highlights areas of muscle pathology where GFP+ cells accumulate. (C) Immunofluorescence from 4‐month‐old mdx diaphragms reveals that GFP+ cells are concentrated near regenerating α‐smooth muscle actin‐positive (αSMA+) muscle fibres (arrowhead) and degenerating fibres that stain positively for sarcomeric fibronectin (arrow). (D) Quantification reveals increased numbers of GFP+ cells in fields containing regenerating (αSMA+) or degeneration [fibronectin + (Fn+)] fibres versus regions within the same mdx diaphragm muscles negative for αSMA+ or Fn + fibres. Scale bars = 50 µm. NS = not significant; *p < 0.05; ***p < 0.0005 by Student's t‐test. (E) RT‐qPCR analysis of Col1a1 and Col3a1 expressed by FACS‐sorted cells from diaphragm muscles of 12‐month‐old mdx:Col1a1‐GFP male mice (N = 3). Relative to other populations implicated in collagen synthesis, endothelial cells (Sca1+, CD31+, CD45−) and macrophages (CD45+, F4/80+), Col1a1‐GFP+ cells expressed significantly higher levels of these pro‐collagen mRNAs. ***p < 0.0005 by single factor ANOVA. Error bars indicate ± SEM.
To understand the progression of fibrosis, we characterized collagen type III, which accumulates in dystrophic mdx:Col1a1‐GFP muscles 40. Diaphragms from mdx mice showed thickening of endomysial type III collagen by 4 months of age (Figure 2A). GFP+ cells were located in areas of collagen type III accumulation. In quadriceps muscles, which show less severe pathology (Figure 2B), accumulation of collagen type III was evident by 14 months of age (Figure 2B) 41. Collagen type III was also concentrated in the perimysium surrounding large vessels and associated with intense accumulation of Col1a1‐GFP+ cells (supplementary material, Figure S1A). In addition, a microfibrillar collagen, collagen type VI, which is up‐regulated in DMD 42, was also present in fibrotic regions occupied by Col1a1‐GFP+ cells (supplementary material, Figure S1B). The localization of both collagen type III and collagen type VI with GFP+ cells was also observed in aged hearts from the same mdx mice (supplementary material, Figure S2).
Figure 2.

Col1a1‐GFP cells accumulate in areas of fibrosis and expand with disease progression. (A) Immunodetection of type III collagen in mdx:Col1a1‐GFP muscles shows GFP+ cells accumulated in areas of pathological matrix deposition fibrosis that occurs with age and disease progression in mdx diaphragms. This accumulation is observed as early as 4 months in mdx versus wild‐type diaphragms. (B) In contrast, limb muscles are less affected in mdx mice. The build‐up of type III collagen and co‐localization with GFP+ cells were not obvious until 14 months in the quadriceps from the same mdx mice shown. (C) Quantification of Col1a1‐GFP+ cells and type III collagen in diaphragms and quadriceps reveals progressive increase with age. Scale bars = 50 µm.
Col1a1‐GFP matrix‐forming cells are PDGFRα+, Sca1+, CD45− in dystrophic muscles and derive from MSC progenitors
To characterize the markers of Col1a1‐GFP+ cells in skeletal muscle, we analysed single‐cell preparations of diaphragms from mdx mice by flow cytometry and compared our findings with immunohistological detection in tissue sections. GFP+ cells did not express the haematopoietic or endothelial markers CD45 or CD31, respectively (Figure 3A, B) 43, 44. Evaluation of the PDGFRα cellular compartments indicates that PDGFRα + cells expressing Col1a1‐GFP are also positive for Sca1 in both wild‐type and mdx diaphragms. In contrast, Col1a1‐GFP+ cells that are PDGFRα‐negative are negative for Sca1 (supplementary material, Figure S3). In mdx diaphragms, the majority of Col1a1‐GFP+ cells are PDGFRα+/Sca1+ (Figure 3A, B) and their proportion within the non‐haematopoietic population (CD45−) steadily increases with age, becoming the majority (>60%) at 20 months of age. This profiling suggests that a proportion of FAPs (PDGFRα+, Sca1+, CD45−) express Col1a1‐GFP in normal muscles, while the majority adopt a fibrogenic phenotype with the progression of dystrophy. In contrast, a large proportion of Col1a1‐GFP+ cells in wild‐type muscle show an absence of PDGFRα/Sca1, suggesting that these are pre‐committed cells in a state of inactivity.
Figure 3.
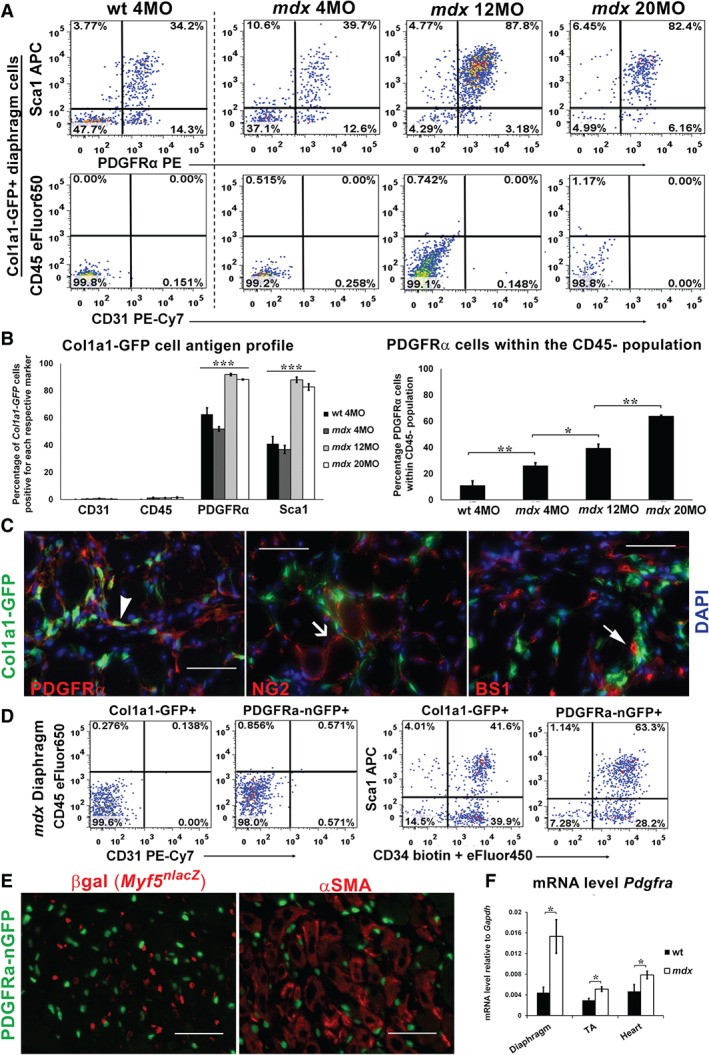
Col1a1‐GFP+ cells express PDGFRα but not endothelial or haematopoietic markers. (A, B) FACS analysis of the Col1a1‐GFP population in wild‐type and mdx diaphragms (N = 3 males per age group) reveals that GFP+ cells lack CD45 and CD31, but the majority express PDGFRα and Sca1 by 12 months of age in mdx mice. (C) Immunofluorescence analysis of mdx:Col1a1‐GFP diaphragms confirms that GFP+ cells co‐express PDGFRα + (arrowhead), but lack vascular markers NG2 (line arrow) and BS1 (filled arrow). (D) FACS analysis comparing single‐cell digests of diaphragms from Col1a1‐GFP and PDGFRa‐nGFP mice reveals a similar profile between GFP+ populations: negative for CD45 and CD31, but the majority co‐expressing Sca1 and CD34. (E) Images showing PDGFRa‐nGFP+ cells in mdx diaphragms concentrated in regions with regenerating fibres expressing αSMA+ as well as Myf5nlacZ‐positive myogenic cells in young mdx:PDGFRα‐nGFP:Myf5nlacZ/+ mice. Scale bars = 50 µm. (F) RT‐qPCR analysis for Pdgfra indicating increased expression in mdx versus wild‐type skeletal tibialis anterior (TA), diaphragm, and heart muscles. N = 3 per tissues were analysed from 12‐month‐old mdx and wild‐type mice. *p < 0.05. Bars show mean ± SEM.
Because DMD also directly causes cardiac fibrosis, we examined cardiac Col1a1‐GFP+ cells in the same way (supplementary material, Figure S4). In aged mdx heart muscle, more than 80% of Col1a1‐GFP+ cells co‐expressed PDGFRα but did not express CD31 or CD45. As previously observed, the majority of Col1a1‐GFP+, PDGFRα + cells were also Sca1+ in aged mdx diaphragm and heart muscles (Figure 3A and supplementary material, Figure S4) 9. By histological analysis, Col1a1‐GFP+ cells in mdx diaphragms co‐expressed PDGFRα+, but did not co‐localize with the vascular markers BS1 and NG2, which label endothelial cells and pericytes, respectively (Figure 3C) 45, 46. Therefore, the majority of Col1a1‐GFP+ cells in aged mdx skeletal muscles share close phenotypic similarity to collagen‐producing cells that have been characterized in the heart in DMD and in pressure overload cardiac failure 9, 19.
To validate the exclusive expression of PDGFRα by the matrix‐forming mesenchymal cells, we expressed nuclear GFP (nGFP) at the PDGFRα locus in mdx mice (PDGFRa‐nGFP). Similar to Col1a1‐GFP+ cells, PDGFRa‐nGFP+ cells co‐expressed CD34 and Sca1, but did not express CD45 and CD31 (Figure 3D). In addition, PDGFRa‐nGFP+ cells were also abundant around regenerating muscle fibres and myogenic cells (Figure 3E), as well as around degenerating, necrotic muscle fibres (supplementary material, Figure S5). Quantitative analysis of RNA established that PDGFRα mRNA abundance was significantly elevated in skeletal and cardiac muscles in mdx compared with wild‐type mice (Figure 3 F).
To study the response of the PDGFRα + mesenchymal progenitors to the onset and progression of dystrophy, we generated mdx mice carrying the PDGFRa‐Cre allele and mT/mG loxP reporter 47, 48. In mdx muscle, the presence of membrane‐EGFP+ cells at 6 weeks of age was similar to healthy muscle (supplementary material, Figure S6). By 8 weeks, however, there was a marked expansion of membrane‐EGFP+, PDGFRα fate‐mapped cells in diaphragmatic muscle. This timeframe of cell expansion is consistent with the commencement of muscular dystrophy fibrosis in mdx diaphragms 15, 40, 49. This patterning of Col1a1‐GFP+ and PDGFRa‐nGFP+ or PDGFRa‐Cre fate‐mapped populations highlights the tight cellular localization between collagen production and PDGFRα expression during the processes of skeletal muscle regeneration and degeneration.
PDGFRα signalling by Sca1+, CD45− cells is transiently activated in acute injury–repair of skeletal muscle
Toxin injury in non‐diseased muscle leads to a robust myogenic response that results in near‐complete regeneration within 14 days 50. Despite the severity of damage and alteration to the muscle architecture that occur with toxin‐induced injury, there is no persisting fibrosis, suggesting that collagen production is in balance with the need for muscle regeneration and restoration of damaged connective tissue 50, and that resorption of accumulated matrix during regeneration is coordinated and appropriate. With recurring damage in diseased mdx muscles, excessive deposition and thickening of the connective tissue occur, suggesting that fibrosis results from the unbalanced accumulation of collagen during chronic cycles of muscle regeneration and unresolved degeneration. Therefore, to gain insight into the process of connective tissue restoration, we examined the relationship between Col1a1‐GFP‐expressing cells and myogenic factor‐5‐expressing myogenic cells using the Myf5nlacZ allele following cardiotoxin (CTX) injury in wild‐type mice 51.
Following CTX injection, the myogenic response in wild‐type muscles peaks at day 3 post‐injury 31, 50. This was evident by the prominence of Myf5nlacZ/+ cells in CTX‐injured TA muscles compared with uninjured contralateral TA muscles (Figure 4A). Col1a1‐GFP+ cells were also abundant within regenerating areas of muscle and in close proximity to myogenic cells and newly regenerated muscle fibres expressing αSMA (supplementary material, Figure S7). This association indicates that Col1a1‐GFP+ cells are active in parallel with myogenesis. Such activation of Col1a1‐GFP+ cells and myogenic cells correlated with elevated levels of pro‐collagen mRNAs at day 3 and subsequent decline by 14 days post‐CTX injury (Figure 4A). Consistent with findings in mdx muscle, PDGFRa‐nGFP cells expanded in regenerating areas and expression levels of both Pdgfra and Pdgfa mRNAs showed the same pattern of increase during the peak of myogenesis at day 3, with subsequent decline at day 14, when regeneration is concluding (Figure 4B). These findings suggest that Col1a1‐GFP+ cells and PDGFRα + signalling in normal muscle repair result in restoration of the connective tissue surrounding regenerating myofibres, a process altered in muscular dystrophy.
Figure 4.

Col1a1‐GFP+ cells and Pdgfra expression increase with muscle injury to reconstitute the damaged connective tissue. (A) Images (left panels) of uninjured and cardiotoxin (CTX) injured TA muscles from a wt:Col1a1‐GFP:Myf5nlacZ/+ dual reporter mouse (1.5‐month‐old male) showing the response of GFP+ collagen‐producing cells alongside X‐gal + myogenic precursors to muscle injury. In uninjured muscle, Myf5nlaZ‐expressing cells (arrowhead) are separate from Col1a1‐GFP+ cells. In contrast, X‐gal + and GFP+ cells are abundant and in close proximity in damaged regions of muscle at the peak of myogenic cell response, 3 days following CTX injection. RT‐qPCR analysis (right panel) indicates the elevation of pro‐collagen mRNA levels in response to CTX injury and the decline as regeneration concludes 14 days following injury. (B) Images (left panels) of PDGFRa‐nGFP+ cells in the endomysium and perimysium near vessels (arrowhead) and collagen type I in normal muscle and response 3 days following cardiotoxin (CTX) injury (shown is TA muscle from a 1.5‐month‐old wt:PDGFRa‐nGFP male mouse). RT‐qPCR analysis (right panel) for Pdgfra and Pdgfa ligand in response to CTX injury and repair. Note levels are reduced close to uninjured levels as regeneration concludes, 14 days post injury. N = 3 TA muscles from 12‐month‐old males per time point. # p = 0.08; *p ≤ 0.05; **p < 0.005 by single factor ANOVA. Bars show mean ± SEM. (C, D) Representative photomicrographs and quantification of Picrosirius red and Fast Green‐stained quadriceps muscles from 3‐month‐old Cre‐flox mice. Animals received daily intraperitoneal injections of tamoxifen for five consecutive days. Fourteen days following the last dose of tamoxifen, muscles were injected with cardiotoxin (CTX) and collected 21 days post‐injury. Contralateral muscles were used as the uninjured controls. Note that tamoxifen‐induced, Cre‐mediated expression of the mutant (D842V), constitutively active form of PDGFRα at the endogenous locus (left column) resulted in fibrosis by day 21 following CTX injury, whereas expression of the constitutively active mutant (D536A) form of PDGFRβ resulted in much milder fibrosis (middle left) and incomplete regeneration following injury. In the absence of Cre recombination, neither PDGFR flox resulted in fibrosis (middle right and right). Scale bars = 50 µm.
Overactivation of PDGFRα signalling enhances fibrotic responses following muscle injury and characterizes human muscle dystrophy
PDGFR expression and activation are seen in mdx skeletal muscle 15, while expression and activation are also increased during muscle repair (Figure 4B). Therefore, in the context of skeletal muscle injury and repair, we tested whether constitutive activation of PDGFR signalling would result in excessive connective tissue deposition spontaneously or in response to injury. We utilized a Cre/loxP somatic cell recombination strategy to express the constitutively active mutant forms of the PDGFRs (PDGFRa ΔD842V 10 and PDGFRb ΔD536A 52) in PDGFRα‐expressing cells immediately prior to CTX‐mediated injury, using the PDGFRα‐CreER BAC transgene, which is activated by exposure to the oestrogen analogue tamoxifen 53. Histological analysis showed that in PDGFRa ΔD842V muscles, CTX injury led to excessive connective tissue reminiscent of mdx muscle pathology (Figure 4C, D). In contrast, injured PDGFRb ΔD536A mutant muscles showed slightly increased fibrosis. Uninjured muscles appeared similar between each model. Excessive PDGFRα activation during the process of muscle regeneration can therefore result in fibrosis. In turn, activation of PDGFRβ may also influence the response of collagen‐producing cells, but with less potency compared with PDGFRα. The elevated levels of fibrosis in mutant muscles injected with CTX correlated with smaller fibre size in injured areas (supplementary material, Figure S8), indicating that excessive PDGFRα signalling in the context of muscle injury can alter the normal processes of regeneration.
To confirm specificity of PDGFRa‐CreER and exclude the possibility of non‐specific Cre activation with CTX injury, we compared tamoxifen and vehicle‐treated PDGFRa‐CreER mice harbouring the mT/mG loxP flanked conditional reporter. In these studies, GFP+ cells, indicative of Cre‐mediated recombination, were observed only in animals pretreated with tamoxifen in both injured and uninjured muscles (supplementary material, Figure S9). In contrast, vehicle‐treated controls did not show detectable GFP+ cells in healthy conditions or following CTX injury (supplementary material, Figure S10).
Elevated numbers of PDGFRα + cells have been reported to co‐localize with fibrosis in human DMD muscles 8. Immunostaining of DMD muscles in our laboratory supports the presence of PDGFRα + cells in the regions of connective tissue thickening (Figure 5A). Collagen accumulation was pronounced in endomysium and perimysium surrounding vessels in DMD muscle (Figure 5B), sites where Col1a1‐GFP+ cells were also concentrated in mdx muscles (Figure 1). Furthermore, staining for the PDGF‐AA ligand revealed increased intensity in DMD and mdx muscle fibres (Figure 5C, D). Co‐cultures of myofibres with Col1a1‐GFP+ cells provided further evidence of PDGF production and paracrine signalling to mediate fibrogenesis. In this experiment, mdx myofibres showed significantly greater PDGF‐AA staining and co‐cultured Col1a1‐GFP+ cells produced a greater amount of collagen versus wild‐type fibre co‐cultures (supplementary material, Figure S11). These results highlight the similarities between human and mouse disease and suggest that activation of PDGFRα signalling in muscular dystrophy promotes connective tissue accumulation. Ominously, dystrophic muscle fibres may promote fibrosis in a paracrine fashion via production and secretion of the PDGF‐AA ligand.
Figure 5.
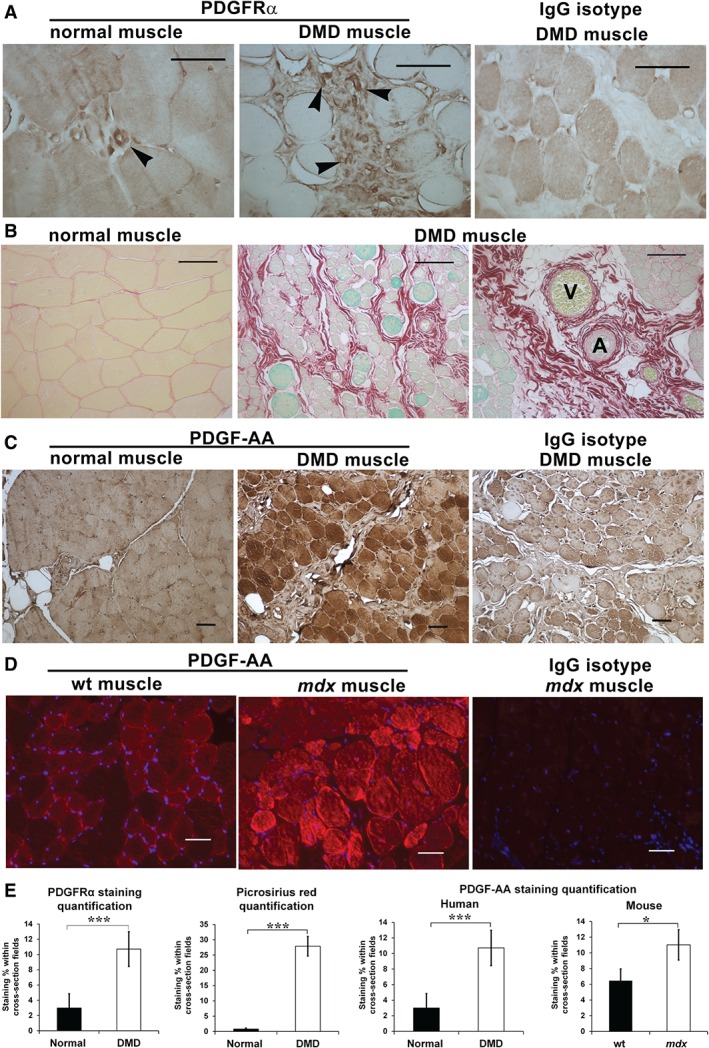
PDGFRα + cells and the PDGF‐AA ligand are up‐regulated in human DMD muscles. (A) PDGFRα + immunohistochemistry (brown) reveals increased numbers of PDGFRα + cells (arrowheads) in biopsies of DMD muscles compared with healthy quadriceps from males aged 8–10 years (N = 3). (B) Picrosirius red and Fast Green staining of the same muscles shows more collagen in the endomysium and perimysium, particularly near vessels, venules (V) and arterioles (A), where PDGFRα + cells are observed in human and mouse muscles. (C) PDGF‐AA immunohistochemistry (brown) on the same DMD and normal muscle tissue sections reveals a strong presence of this ligand in dystrophic muscles, expressed primarily in muscle fibres. (D) PDGF‐AA detection by immunofluorescence was also more intense in mdx than in wild‐type male mouse quadriceps at 18 and 24 months, respectively. Scale bars = 50 µm. (E) Quantification of staining indicates significant increases in PDGFRα and fibrosis in DMD muscle in addition to elevated PDGF‐AA staining in mdx and DMD muscles. *p < 0.05; ***p < 0.0005. Bars indicate mean ± SEM.
Inhibition of PDGFRα signalling ameliorates muscular dystrophy fibrosis and improves function
Crenolanib, a potent tyrosine kinase inhibitor selective for PDGFRα, is currently in phase II clinical trials for PDGFR‐associated cancers 54, 55, 56. Compared with imatinib, crenolanib has significantly greater avidity for the active form of PDGFRα, including mutant variants such as ΔD842V 54. Moreover, it has no activity against VEGFR2, Src or ABL, whose inhibition has been associated with cardiac toxicities 57. Therefore, we administered crenolanib to Col1a1‐GFP+ muscle fibroblasts initially in vitro, to evaluate whether PDGFRα inhibition might reduce the pro‐fibrotic activity of collagen‐producing cells. Col1a1‐GFP+ cells treated with 10 ng/ml PDGF‐AA had significantly increased relative expression of both Col1a1 and Col3a1 (supplementary material, Figure S12A). This response to PDGF‐AA was inhibited with the addition of 1 µm crenolanib. In contrast, exposure to the same concentration of PDGF‐BB did not result in the up‐regulation of pro‐collagen transcripts. Western blot analysis confirmed the inhibitory effect of crenolanib in reducing PDGFRα phosphorylation when cells were stimulated with PDGF‐AA (supplementary material, Figure S12B).
Next, we expanded our studies of PDGFRα inhibition to mdx mice in order to evaluate whether crenolanib can reduce the pro‐fibrotic response of collagen‐producing cells in vivo. Crenolanib or the vehicle was administered for 4 weeks in the drinking water ad libitum, beginning at 8 weeks of age, a timeframe when elevated numbers of PDGFRα‐expressing cells are evident (supplementary material, Figure S6), and just prior to the development of fibrosis in mdx diaphragms 15, 40. Based on published pharmacokinetic data 58, the concentration of crenolanib in the drinking water was set at 0.03 mg/ml, so that animals received an approximate daily dose of 5 mg/kg that would achieve inhibition of PDGFRα. Control mice were treated with an equal amount of the vehicle (DMSO) in their drinking water. Following 4 weeks of treatment, muscle strength was initially tested by ex vivo myography (Figure 6A), an assay commonly used to measure functional improvement in mdx muscles 59. Crenolanib‐treated mdx mice generated significantly greater isometric force in both EDL muscles and diaphragm strips.
Figure 6.

Inhibition of PDGFRα signalling and its downstream effector Src, but not c‐Abl, results in decreased fibrosis and improved muscle strength of mdx muscles. (A) Schema (left panel) depicting treatment of mdx male mice with crenolanib or vehicle (N = 6 per group). Myography (centre and right) of these treated mouse muscles records improved muscle strength in extensor digitorum longus (EDL) and diaphragm muscles following crenolanib. (B) Images and quantification of collagen deposits (fibrosis) detected by Picrosirius red staining following vehicle or crenolanib (N = 4 per group) treatment in mdx diaphragms. Scale bars = 50 µm. (C) RT‐qPCR for Col1a1 and Col3a1 mRNA showed a modest reduction in relative abundance. RT‐qPCR for mRNAs of PDGFs and PDGFRs showed no changes in relative abundance following crenolanib (N = 4 per group). (D) Western blots and normalized densitometry indicating a significant reduction in type III collagen and fibronectin in diaphragms from crenolanib‐treated mdx mice (N = 4 per group). (E) Western blots of treated diaphragms show that crenolanib reduced p‐PDGFRα and p‐Src, but not p‐Abl. *p ≤ 0.05 by Student's t‐test. Bars indicate mean ± SEM. (F) Summary and hypothesis: in contrast to acute PDGFRα activation in non‐diseased muscles that repair and lack fibrosis, chronic activation of PDGFRα signalling in muscular dystrophy potentiates connective tissue accumulation. To represent the pathogenesis in muscular dystrophy, the y‐axis depicts collagen accumulation versus time along the x‐axis.
To understand the factors and mechanisms that contributed to this functional gain, we examined changes in the muscles' connective tissue. Histopathology of diaphragm muscles showed reduced collagen deposition, assessed in serial cross‐sections as the percentage of Picrosirius red staining (Figure 6B). Whole tissue analysis indicated a significant reduction in the relative level of the Col3a1 transcript (Figure 6C). In contrast, the relative expression of Pdgf receptors and ligands did not change (Figure 6C), suggesting that the treatment does not result in the transcriptional ablation of the pathway. Accordingly, histological quantification of GFP and type III collagen immunofluorescence confirmed a reduction of collagen type III in crenolanib‐treated diaphragms, whereas the number of Col1a1‐GFP+ cells did not significantly change in comparison to vehicle‐treated diaphragms (supplementary material, Figure S13). Western blot analysis of diaphragm muscles confirmed a significant decline in collagen type III with crenolanib. Fibronectin, which also accumulates with fibrosis and can be present in necrotic fibres (Figure 1C), was also reduced (Figure 6D). An attenuation of necrotic areas was also noted with crenolanib (supplementary material, Figure S14). Such reductions in connective tissue and fibrotic components correlated with reduced phosphorylation of PDGFRα and its intracellular downstream effector, Src (Figure 6E). Indeed, phosphorylated Src levels are increased in mdx diaphragms in comparison to same‐age wild‐type diaphragms (supplementary material, Figure S15). In contrast, phosphorylation of c‐Abl, which is the main target of imatinib 15, did not change with crenolanib. Of note, a reduction in cardiac fibrosis and PDGFRα phosphorylation was also detected in the hearts of crenolanib‐treated mdx mice (supplementary material, Figure S16).
To discern whether a reduction in fibrosis permitted increased regeneration, we evaluated myogenic factors in crenolanib‐treated and vehicle‐treated mdx:Col1a1‐GFP muscles. As previously shown, collagen‐producing cells are involved in regeneration (Figure 4A, B). In these experiments, we again observed Col1a1‐GFP+ cells in close proximity to regenerating fibres expressing sarcoplasmic αSMA, which were greater in number and size with crenolanib (supplementary material, Figure S17). The Col1a1‐GFP+ cells were more abundant in regenerating areas of crenolanib‐treated diaphragm and EDL muscles (supplementary material, Figure S18A). Whole tissue levels of Acta2 transcripts in crenolanib‐treated diaphragm muscles were increased, in keeping with the reduction in fibrosis being more permissive for regeneration (supplementary material, Figure S18B). Crenolanib‐treated muscles showed significantly increased expression of the myogenic regulatory factor Pax7, a specific marker of satellite cells, but not of Myf5 or Myod, which are expressed by satellite cells and myoblasts 51, 60, 61 (supplementary material, Figure S18B). Such an increase in Pax7 transcripts suggests that the amelioration of fibrosis may result in greater satellite cell renewal during the course of treatment 60.
Discussion
Fibrosis in muscular dystrophy is widely recognized as a barrier to regeneration, yet remains understudied. Utilizing robust genetic models and molecular methods, in our present study we have identified an important role for PDGFRα signalling in collagen‐producing cells during damage to connective tissue. In the absence of disease, collagen‐producing cells restore the connective tissue in damaged muscles. This process is associated with transient elevations of mRNA levels for pro‐collagens and PDGFRα signalling components (the receptor and ligand), as Col1a1‐GFP+ cells respond to injury in a coordinated manner with myogenic precursors. In contrast, dystrophic muscles undergo continuous cycles of degeneration/regeneration, which maintains the response of Col1a1‐GFP+ cells and PDGFRα signalling. Increased PDGFRα phosphorylation 15 and PDGFRα expression by mesenchymal progenitors that have potential to express collagens 4, 5, 7, 8 suggest a role for this pathway in the pathology of muscular dystrophy.
To date, several populations of muscle cells, including FAPs and myogenic, haematopoietic, and endothelial cells, have been reported to express collagens and promote fibrosis 6, 13. Our results indicate that the majority of Col1a1‐GFP+ cells share a profile similar to FAPs (Sca1+, PDGFRα+, CD45−), suggesting that these populations are not mutually exclusive. In addition, a very small number (<3%) of Col1a1‐GFP+ cells expressed endothelial (CD31+, CD45−) or haematopoietic (CD45+) markers. Irrespective of cellular origin or classification, the pro‐fibrotic transition of muscle cells is frequently associated with PDGFRα expression. This tendency was also observed in our study, where the majority of Col1a1‐GFP+ cells were PDGFRα + in dystrophic but not normal muscles. However, whether PDGFRα‐expressing cells expanded or originated from various cell types that up‐regulate PDGFRα with disease or injury remains to be elucidated. Careful fate‐mapping of PDGFRα expression in conjunction with a reporter of collagen expression will be required to further delineate the origin and heterogeneity of pro‐fibrotic cells.
The expression of PDGFRα by collagen‐producing cells in vivo and the presence of the PDGF‐AA ligand in dystrophic muscles indicate a paracrine role for PDGF‐AA, which remains activated in response to chronic injury. PDGF‐AA is expressed by developing muscles but is absent in the somites of Myf5‐null mutants 62. Consequently, it is entirely possible that PDGF signalling in muscular dystrophy is a reactivation of the development programme that occurs in response to muscle repair. Therefore, we hypothesize that continuous activation of PDGFRα signalling in collagen‐producing cells and the resultant fibrosis are a consequence of the chronic injury and repair that occur in dystrophic muscles. In contrast, non‐diseased muscles down‐regulate PDGFRα signalling once the muscles' connective tissue has been restored, during the process of regeneration (modelled in Figure 6G). In addition to the aforementioned findings, the capability of PDGFRα signalling to promote fibrosis when constitutively activated during the repair process in non‐diseased muscles (Figure 6G) supports our hypothesis that excessive PDGFRα signalling promotes fibrosis and prevents normal repair. Consequently, inhibition of PDGFRα with crenolanib resulted in reduced fibrosis and functional improvement of mdx muscles (Figure 6A–F). Such results not only implicate PDGFRα signalling in the fibrotic response of collagen‐producing cells but also present a novel target for reducing connective tissue accumulation in patients with DMD.
Lemos et al 6 recently demonstrated another paracrine role mediated by macrophages, which in normal regenerating muscle secreted TNF to induce apoptosis of FAPs. In contrast, they showed that in dystrophic muscle, macrophages secreted TGF‐β, which prevented apoptosis but induced the fibrogenic programme of FAPs. Whether macrophages or muscle cells in addition to muscle fibres regulate PDGFRα signalling in FAPs and/or Col1a1‐GFP+ cells remains an important question worth pursuing.
Importantly, the intracellular signalling cascade by which PDGFRα stimulation leads to collagen production remains unknown. The reported reduction of mdx muscle fibrosis by imatinib has been attributed to PDGFR and c‐Abl tyrosine‐kinase inhibition 15. Our results indicate that selective inhibition of PDGFRα, independent of c‐Abl, can reduce fibrosis in mdx muscles and improve function (Figure 6). Herein, we demonstrated that Src, a direct downstream target of PDGFRα signalling, is increased in mdx diaphragms and that crenolanib treatment reduces phosphorylated Src. Recently, Pal et al 63 showed that oxidative stress results in persistent activation of Src in mdx muscles and impairs autophagy via mTOR. Crenolanib has 100‐fold higher selectivity for PDGFR than other tyrosine kinases including Src and c‐Abl 55. Thus, the observed reduction of phosphorylated Src following crenolanib treatment is mediated by direct inhibition of PDGFRα. Further studies are needed to define the signalling cascade by which PDGFRα‐Src mediates the pro‐fibrotic response of collagen‐producing cells in skeletal muscles. Although c‐Abl may also potentiate collagen transcription, c‐Abl inhibition with imatinib, and to a lesser extent with nilotanib, has been associated with cardiomyocyte toxicity and heart failure in cancer patients 57, 64. Such a side effect may compound the cardiomyopathy prevalent in the majority of DMD patients 65. Therefore, anti‐fibrotic therapies that selectively target PDGFRα‐Src signalling may prove efficacious and safer than broad‐spectrum tyrosine‐kinase inhibitors.
To conclude, we have further defined the role of PDGFRα +, Sca1+, CD45− mesenchymal progenitors in skeletal muscle, which become activated after injury to become pathological matrix‐forming cells. In settings of excessive PDGFRα signalling, these cells are a major population of matrix‐forming fibroblasts which promote muscle fibrosis and inhibit normal regeneration. Crenolanib, a specific PDGFRα/β inhibitor, reduces fibrosis in a mouse model of DMD and in turn improves muscle function.
Author contribution statement
The authors contributed in the following way: writing and editing: NI, JSD, and MR; study design: NI, MR, and JSD; experimentation: NI, AH, AM, and KJ; data analysis: NI, MR, JSD, AH, AP, and KJ.
SUPPLEMENTARY MATERIAL ONLINE .
Supplementary materials and methods
Supplementary figure legends
Figure S1. Col1a1‐GFP+ cells are expanded in areas of perimysial fibrosis of mdx quadriceps muscle, where type III and type VI collagens are deposited
Figure S2. GFP+ cells also expand in areas of type III and type VI collagen deposition in the myocardium of mdx:Col1a1‐GFP mice
Figure S3. Sca1 is concentrated in the PDGFRα + portion of the Col1a1‐GFP+ population in both wild‐type and mdx diaphragms
Figure S4. Col1a1‐GFP+ cells in the heart and diaphragm share a similar molecular profile
Figure S5. PDGFRα‐nGFP+ cells are concentrated in degenerating and regenerating regions of mdx muscles
Figure S6. Fate‐mapping reveals that PDGFRα‐expressing cells are prominent at the onset of dystrophy in mdx diaphragm muscle
Figure S7. Col1a1‐GFP+ cells are adjacent to both myogenic cells and regenerating fibres following acute cardiotoxin injury in healthy muscle
Figure S8. Histological analysis of mutant muscles shows reduced fibre size with excessive PDGF signalling following CTX injury
Figure S9. Tamoxifen‐treated PDGFRa‐CreER;mT/mG loxP mice show activation of the GFP reporter in both uninjured and injured muscle
Figure S10. Vehicle‐treated PDGFRa‐CreER;mT/mG loxP mice show no evidence of recombination in uninjured or injured muscle
Figure S11. Single fibre isolations and co‐culture with Col1a1‐GFP+ cells indicate that mdx fibres promote fibrosis by their production of PDGF‐AA
Figure S12. Crenolanib inhibits PDGF‐AA‐mediated expression of pro‐collagen mRNAs and phosphorylation of the PDGFRα in primary cultured Col1a1‐GFP+ cells
Figure S13. Crenolanib attenuates type III collagen accumulation in mdx diaphragms
Figure S14. Necrotic regions are smaller with crenolanib treatment
Figure S15. The phosphorylation of Src is increased in mdx diaphragms
Figure S16. Crenolanib reduces PDGFRα signalling and fibrosis in mdx hearts
Figure S17. The number and size of regenerating fibres are greater with crenolanib treatment
Figure S18. Crenolanib improves regeneration in mdx skeletal muscles
Table S1. Mouse strains used
Table S2. Antibodies and lectins used on tissue sections
Table S3. FACS antibodies and streptavidin conjugates
Table S4. RT‐qPCR primer sequences
Table S5. Antibodies used for western blotting
Supporting information
Supplementary materials and methods
Supplementary figure legends
Figure S1 Col1a1‐GFP+ cells are abundant in areas of perimysial fibrosis of mdx quadriceps muscle, where type III and VI collagens are deposited. Split panel immunofluorescence images of quadriceps and diaphragm muscles from male 20‐month‐old mdx:Col1a1‐GFP mice. (A) Type III collagen and αSMA. (B) Type VI collagen and αSMA. (C) IgG isotype control. Note the frequent Col1a1GFP+ cells in perimysial fibrosis, particularly surrounding αSMA‐positive medium‐sized vessels. Scale bars = 50 µm.
Figure S2 GFP+ cells are clustered in areas of type III and type VI collagen deposition in the myocardium of mdx:Col1a1‐GFP mice. (A) Split panel immunofluorescence images showing type III and type IV collagens, highlighting the proximity of Col1a1‐GFP+ cells with pathological matrix in the heart muscle of 14‐month‐old mdx:Col1a1‐GFP mice. (B) Representative panel images from mdx:Col1a1‐GFP mice highlight the accumulation of type III collagen with time and the gathering of GFP+ cells in these fibrotic areas. Scale bars = 50 µm.
Figure S3 Sca1 is concentrated in the PDGFRα + portion of the Col1a1‐GFP+ population in both wild‐type and mdx diaphragms. Col1a1‐GFP+ cells were gated as either PDGFRα‐positive or ‐negative to analyse marker expression within these two sub‐populations of GFP+ cells. As before, little or no CD45 or Sca1 was present. However, Sca1 was predominantly expressed in PDGFRα + but not PDGFRα − cells in both 4‐month‐old wild‐type (top two rows) and mdx diaphragms (bottom two rows).
Figure S4 Col1a1‐GFP+ cells in the heart and diaphragm share a similar molecular profile. Representative flow cytometry plots gated on Col1a1‐GFP+ cardiac mononuclear cells isolated from wild‐type (left) and mdx:Col1a1‐GFP (right) male mice (N = 3 per group), showing that Col1a1‐GFP+ cells are PDGFRα + and Sca1+, but remain CD45− or CD31 − .
Figure S5 PDGFRa‐nGFP+ cells are concentrated in degenerating and regenerating regions of mdx muscles. In mdx muscles, PDGFRa‐nGFP+ cells are concentrated near areas with degenerating fibres labelled by mouse IgG (top row, arrowhead) and regions of regeneration containing αSMA+ fibres and Myf5 nlacZ/+ expressing myogenic cells (bottom row, arrow). Shown are quadriceps from a 3‐month‐old male mdx:PDGFRa‐nGFP:Myf5 nlacZ/+ double reporter mouse. Scale bars = 50 µm.
Figure S6 Fate‐mapping reveals that PDGFRα‐expressing cells are prominent at the onset of dystrophy in mdx diaphragm muscle. (A) Schematic of Cre‐loxP generation. Mice carrying the PDGFRa‐Cre allele were crossed with mice carrying the mT/mG loxP allele to produce double transgenic progeny that harbour both alleles. In these double transgenic mice, cells that express the PDGFRa‐Cre transgene and their subsequent progeny are labelled with membrane‐localized EGFP. In contrast, Cre‐negative cells and their progeny will be labelled with membrane‐localized tdTomato. (B) Split panel images of diaphragm muscles from wt:PDGFRa‐Cre:mT/mG loxP male mice showing discrete GFP+ cells derived from cells which have expressed Pdgrfa in normal muscle. (C) In mdx:PDGFRa‐Cre:mT/mG loxP diaphragms at 6 weeks of age, just prior to the onset of dystrophy, there is a mild expansion of the PDGFRα lineage. By 8 weeks of age, there is a large expansion of the PDGFRα lineage coinciding with the onset of muscular dystrophy between 6 and 8 weeks of age. Note that the mT/mG loxP labels Cre‐expressing or ‐derived cells with membrane localized EGFP+, while Cre‐negative cells are labelled with membrane tdTomato. Scale bars = 50 µm.
Figure S7 Col1a1‐GFP+ cells are adjacent to both myogenic cells and regenerating fibres following acute cardiotoxin injury to healthy muscle. Split panel images in a Col1a1‐GFP;Myf5 LacZ/+ expressing TA muscle stained with X‐gal and labelled for αSMA, 3 days after cardiotoxin administration. Staining reveals Col1a1‐GFP+ cells adjacent to regenerating fibres labelled by sarcomeric αSMA (arrow) as well as Col1a1‐GFP+ cells adjacent to Myf5 LacZ/+ myogenic cells (arrowhead). Scale bars = 50 µm.
Figure S8 Histological analysis of mutant muscles shows reduced fibre size with excessive PDGF signalling following CTX injury. (A) H&E staining was used to measure the cross‐section fibre size in quadriceps muscles from the PDGFRa‐CreER and control mice depicted also in Figure 4C. Top row shows contralateral uninjured muscles; bottom row shows the injured muscles collected 21 days post‐CTX injection. Scale bars = 100 µm. (B) Size quantification was conducted by measuring the minimum diameter of individual fibres, a conservative metric for fibre size when using cross‐sections. In the absence of injury, fibre sizes were similar between mutant muscles (top graph). In contrast, injured muscles from both PDGFRa‐CreER:PDGFRa ∆D842V and PDGFRb ∆D536V had significantly smaller fibre diameters versus Cre‐negative controls (bottom graph). **p = < 0.005, *** p = < 0.0005 by Student's t‐test. NS = not significant. Error bars indicate ± SEM.
Figure S9 Tamoxifen‐treated PDGFRa‐CreER;mT/mG loxP mice show activation of the GFP reporter in both uninjured and injured muscle. Split panel images of TA muscle from wt:PDGFRa‐CreER:mT/mG loxP reporter mice versus Cre‐negative controls. Following i.p. injections of tamoxifen for five consecutive days, uninjured contralateral muscles (A) were compared with TA muscles injected with CTX and collected 14 days post‐injury (B). Note that following tamoxifen, there was specific appearance of GFP+ cells among tdTomato + cells localized to non‐myofibre areas. In addition, in the absence of Cre, we did not observe any recombination. Scale bars = 50 µm.
Figure S10 Vehicle‐treated PDGFRa‐CreER;mT/mG loxP mice show no evidence of recombination in uninjured or injured muscle. Split panel images of TA muscle from Cre‐negative and wt:PDGFRa‐CreER:mT/mG loxP reporter mice following i.p. injections of corn oil for five consecutive days. As portrayed in Figure S9, uninjured muscles (A) were compared with CTX‐injured TAs, 14 days after injury. (B) In the absence of tamoxifen, no GFP cells appeared within tdTomato + muscles. Scale bars = 50 µm.
Figure S11 Single fibre isolations and co‐culture with Col1a1‐GFP+ cells indicate that mdx fibres promote fibrosis by their production of PDGF‐AA. (A) Single fibres from wild‐type and mdx extensor digitorum longus (EDL) muscles were isolated and cultured in triplicate. Immediately following the fibre isolations, wild‐type ‐derived Col1a1‐GFP+ cells were added. Co‐cultures were incubated for 3 days and then fixed for staining against PDGF‐AA and collagens. Scale bars = 50 µm. (B) Picrosirius red staining was used to analyse collagen deposition by Col1a1‐GFP+ cells in co‐culture with wild‐type versus mdx fibres. Scale bars = 100 µm. (C) Quantification of the PDGF‐AA staining represented in panel A shows significantly more of this ligand in mdx fibre co‐cultures. (D) In turn, greater PDGF‐AA staining correlated with more collagen deposition in mdx fibre co‐cultures, quantified by Picrosirius red staining. ***p = < 0.0005 by Student's t‐test. Error bars indicate ± SEM.
Figure S12 Crenolanib inhibits PDGF‐AA‐mediated expression of pro‐collagen mRNAs and phosphorylation of PDGFRα in primary cultured Col1a1‐GFP+ cells. (A) Graphs show RT‐qPCR analysis for Col1a1 and Col3a1 in cultured, primary FACS‐sorted Col1a1‐GFP+ cells treated with DMEM with 5% FCS + Pen/Strep medium containing vehicle (DMSO), PDGF‐AA (10 ng/ml), PDGF‐BB (10 ng/ml), or PDGF‐AA + crenolanib (1 µm) for 4 days prior to isolation of RNA and protein extracts (N = 3 samples per group). *p < 0.05, ***p < 0.00005 by single factor ANOVA. Error bars indicate ± SEM. (B) Western blot and densitometry plot showing total PDGFRα and phosph‐PDGFRα in Col1a1‐GFP+ cells 30 min after stimulation with PDGF‐AA or PDGF‐AA + crenolanib (N = 1 per treatment).
Figure S13 Crenolanib attenuates type III collagen staining in mdx diaphragms. Histological analysis of diaphragms from mdx:Col1a1‐GFP mice (N = 4 per group) indicates that the area of type III collagen staining was reduced following 4 weeks of crenolanib treatment versus the vehicle. In contrast, the presence of GFP within these same diaphragms was not significantly different. **p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S14 Necrotic regions are smaller with crenolanib treatment. H&E staining highlights necrotic areas (arrows) in quadriceps from the vehicle‐ or crenolanib‐treated mdx mice depicted in Figure 6. Quantification of the necrotic versus total area within each field indicated that treatment with crenolanib mitigated the development of pathology in dystrophic muscles. Scale bars = 100 µm. *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S15 The phosphorylation of Src increases in mdx diaphragms. Total proteins from 12‐month‐old mdx and wild‐type diaphragms and hearts (N = 3 per group) were processed for western blot analysis. Quantification indicated that the phosphorylation of Src is significantly elevated in mdx diaphragms versus wild‐type (left panel). Although elevated, the phosphorylation of Src in hearts was not significantly different (right panel). *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S16 Crenolanib reduces PDGFRα signalling and fibrosis in mdx hearts. (A) Representative images of fibrosis in Picrosirius red‐stained hearts from mdx mice treated with the vehicle or crenolanib. Scale bars = 50 µm. (B) RT‐qPCR analysis of cardiac tissue (N = 4 per group) indicates a modest reduction in Col3a1 transcript levels, but no change for Col1a1. (C) Western blots (left) and normalized densitometry (right) showing that type III and type I collagen as well as fibronectin protein levels were significantly reduced with crenolanib treatment. (D) Western blots (left) and normalized densitometry (right) showing PDGFRα phosphorylation and phosphorylation of potential downstream effectors in crenolanib‐treated mdx mice. Note that although PDGFRα phosphorylation was reduced by systemic crenolanib, Src or Abl activation was not affected. *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S17 The number and size of regenerating fibres are greater with crenolanib treatment. Top graph: the number of muscle fibres positive for sarcoplasmic αSMA in regenerating regions of mdx diaphragms is significantly greater with crenolanib treatment. Bottom graph: correspondingly, in the same diaphragm muscles, the minimum diameter of αSMA+ fibres was also significantly greater with crenolanib. *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S18 Crenolanib improves regeneration in mdx skeletal muscles. (A) Split panel images of skeletal muscles (diaphragm and EDL) from mdx:Col1a1‐GFP mice treated with crenolanib or vehicle (from the same animal analysed in Figure 6). Note a greater number of αSMA+ regenerating fibres surrounded by Col1a1‐GFP+ cells in response to crenolanib treatment. Scale bars = 50 µm. (B) RT‐qPCR of RNA from diaphragm muscles showing enhanced Acta2 transcript with crenolanib treatment. (C) Graphs showing RT‐qPCR results for myogenic regulatory factors (Pax7, Myf5, Myod) indicate a trend towards increased expression of myogenic factor transcripts in response to crenolanib (N = 4 per group). *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Table S1. Mouse strains used
Table S2. Antibodies and lectins used on tissue sections
Table S3. FACS antibodies and streptavidin conjugates
Table S4. RT‐qPCR primer sequences
Table S5. Antibodies used for western blotting
Acknowledgements
We are grateful to Dr Luis Gonzalez‐Cuyar from the UW Neuropathology division for providing the human muscle sections. Funding that made this research possible was provided by the American Heart Association award 11BGIA7140028, the Muscular Dystrophy Association awards 255907 and 277543, the University of Washington (UW) Provost Bridge award to MR, NIH awards DK94768 and DK84077 to JSD, The UW Nathan Shock Center of Excellence in the Basic Biology of Aging Genetic Approaches to Aging Training award T32 AG000057, the Pancretan Association of America Venizelion Scholarship to NI, and the Anandamahidol Foundation Scholarship of Thailand to KJ. Support for the myography equipment was received from the Washington Research Foundation, the Duchenne Alliance, RaceMD, and Ryan's Quest.
Conflict of interest statement: The authors declare no competing interests. JSD is employed by Biogen and has stock in the company.
References
- 1. Klingler W, Jurkat‐Rott K, Lehmann‐Horn F, et al. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol 2012; 31 : 184–195. [PMC free article] [PubMed] [Google Scholar]
- 2. Kharraz Y, Guerra J, Pessina P, et al. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed Res Int 2014; 2014 : 965631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Desguerre I, Mayer M, Leturcq F, et al. Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J Neuropathol Exp Neurol 2009; 68: 762–773. [DOI] [PubMed] [Google Scholar]
- 4. Joe AW, Yi L, Natarajan A, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nature Cell Biol 2010; 12: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Uezumi A, Fukada S, Yamamoto N, et al. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nature Cell Biol 2010; 12: 143–152. [DOI] [PubMed] [Google Scholar]
- 6. Lemos DR, Babaeijandaghi F, Low M, et al. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF‐mediated apoptosis of fibro/adipogenic progenitors. Nature Med 2015; 21: 786–794. [DOI] [PubMed] [Google Scholar]
- 7. Uezumi A, Ito T, Morikawa D, et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci 2011; 124: 3654–3664. [DOI] [PubMed] [Google Scholar]
- 8. Uezumi A, Fukada S, Yamamoto N, et al. Identification and characterization of PDGFRα + mesenchymal progenitors in human skeletal muscle. Cell Death Dis 2014; 5: e1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ieronimakis N, Hays AL, Janebodin K, et al. Coronary adventitial cells are linked to perivascular cardiac fibrosis via TGFβ1 signaling in the mdx mouse model of Duchenne muscular dystrophy. J Mol Cell Cardiol 2013; 63: 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olson LE, Soriano P. Increased PDGFRα activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell 2009; 16: 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hayes BJ, Riehle KJ, Shimizu‐Albergine M, et al. Activation of platelet‐derived growth factor receptor alpha contributes to liver fibrosis. PLoS One 2014; 9: e92925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rock JR, Barkauskas CE, Cronce MJ, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A 2011; 108: E1475‐E1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pessina P, Kharraz Y, Jardi M, et al. Fibrogenic cell plasticity blunts tissue regeneration and aggravates muscular dystrophy. Stem Cell Rep 2015; 4: 1046–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito T, Ogawa R, Uezumi A, et al. Imatinib attenuates severe mouse dystrophy and inhibits proliferation and fibrosis‐marker expression in muscle mesenchymal progenitors. Neuromuscul Disord 2013; 23: 349–356. [DOI] [PubMed] [Google Scholar]
- 15. Huang P, Zhao XS, Fields M, et al. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J 2009; 23: 2539–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foidart M, Foidart JM, Engel WK. Collagen localization in normal and fibrotic human skeletal muscle. Arch Neurol 1981; 38: 152–157. [DOI] [PubMed] [Google Scholar]
- 17. Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res 2010; 107: 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial‐to‐mesenchymal transition contributes to cardiac fibrosis. Nature Med 2007; 13: 952–961. [DOI] [PubMed] [Google Scholar]
- 19. Moore‐Morris T, Guimaraes‐Camboa N, Banerjee I, et al. Resident fibroblast lineages mediate pressure overload‐induced cardiac fibrosis. J Clin Invest 2014; 124: 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Acharya A, Baek ST, Banfi S, et al. Efficient inducible Cre‐mediated recombination in Tcf21 cell lineages in the heart and kidney. Genesis 2011; 49: 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Acharya A, Baek ST, Huang G, et al. The bHLH transcription factor Tcf21 is required for lineage‐specific EMT of cardiac fibroblast progenitors. Development 2012; 139: 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Muiznieks LD, Keeley FW. Molecular assembly and mechanical properties of the extracellular matrix: a fibrous protein perspective. Biochim Biophys Acta 2013; 1832: 866–875. [DOI] [PubMed] [Google Scholar]
- 23. Mann CJ, Perdiguero E, Kharraz Y, et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle 2011; 1: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen X, Aravindakshan J, Yang Y, et al. Aberrant expression of PDGF ligands and receptors in the tumor prone ovary of follitropin receptor knockout (FORKO) mouse. Carcinogenesis 2006; 27: 903–915. [DOI] [PubMed] [Google Scholar]
- 25. Fukai N, Kenagy RD, Chen L, et al. Syndecan‐1: an inhibitor of arterial smooth muscle cell growth and intimal hyperplasia. Arterioscler Thromb Vasc Biol 2009; 29: 1356–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ieronimakis N, Balasundaram G, Reyes M. Direct isolation, culture and transplant of mouse skeletal muscle derived endothelial cells with angiogenic potential. PLoS One 2008; 3: e0001753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Au CG, Butler TL, Sherwood MC, et al. Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. Int J Exp Pathol 2011; 92: 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kenyon NJ, Ward RW, McGrew G, et al. TGF‐β1 causes airway fibrosis and increased collagen I and III mRNA in mice. Thorax 2003; 58: 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ieronimakis N, Pantoja M, Hays AL, et al. Increased sphingosine‐1‐phosphate improves muscle regeneration in acutely injured mdx mice. Skelet Muscle 2013; 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kiernan JA. Histological and Histochemical Methods: Theory and Practice (4th edn). Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2008. [Google Scholar]
- 31. Ieronimakis N, Balasundaram G, Rainey S, et al. Absence of CD34 on murine skeletal muscle satellite cells marks a reversible state of activation during acute injury. PLoS One 2010; 5: e10920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Danoviz ME, Yablonka‐Reuveni Z. Skeletal muscle satellite cells: background and methods for isolation and analysis in a primary culture system. Methods Mol Biol 2012; 798: 21–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hadi AM, Mouchaers KT, Schalij I, et al. Rapid quantification of myocardial fibrosis: a new macro‐based automated analysis. Cell Oncol (Dordr) 2011; 34: 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spurney CF, Sali A, Guerron AD, et al. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin‐deficient mdx mice. J Cardiovasc Pharmacol Ther 2011; 16: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin SL, Kisseleva T, Brenner DA, et al. Pericytes and perivascular fibroblasts are the primary source of collagen‐producing cells in obstructive fibrosis of the kidney. Am J Pathol 2008; 173: 1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gulati AK, Reddi AH, Zalewski AA. Distribution of fibronectin in normal and regenerating skeletal muscle. Anat Rec 1982; 204: 175–183. [DOI] [PubMed] [Google Scholar]
- 37. Springer ML, Ozawa CR, Blau HM. Transient production of alpha‐smooth muscle actin by skeletal myoblasts during differentiation in culture and following intramuscular implantation. Cell Motil Cytoskeleton 2002; 51: 177–186. [DOI] [PubMed] [Google Scholar]
- 38. Marshall PA, Williams PE, Goldspink G. Accumulation of collagen and altered fiber‐type ratios as indicators of abnormal muscle gene expression in the mdx dystrophic mouse. Muscle Nerve 1989; 12: 528–537. [DOI] [PubMed] [Google Scholar]
- 39. Goldspink G, Fernandes K, Williams PE, et al. Age‐related changes in collagen gene expression in the muscles of mdx dystrophic and normal mice. Neuromuscul Disord 1994; 4: 183–191. [DOI] [PubMed] [Google Scholar]
- 40. Zhou L, Porter JD, Cheng G, et al. Temporal and spatial mRNA expression patterns of TGF‐β1, 2, 3 and TβRI, II, III in skeletal muscles of mdx mice. Neuromuscul Disord 2006; 16: 32–38. [DOI] [PubMed] [Google Scholar]
- 41. Chamberlain JS, Metzger J, Reyes M, et al. Dystrophin‐deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J 2007; 21: 2195–2204. [DOI] [PubMed] [Google Scholar]
- 42. Sabatelli P, Gualandi F, Gara SK, et al. Expression of collagen VI α5 and α6 chains in human muscle and in Duchenne muscular dystrophy‐related muscle fibrosis. Matrix Biol 2012; 31: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Watt SM, Gschmeissner SE, Bates PA. PECAM‐1: its expression and function as a cell adhesion molecule on hemopoietic and endothelial cells. Leuk Lymphoma 1995; 17: 229–244. [DOI] [PubMed] [Google Scholar]
- 44. Charbonneau H, Tonks NK, Walsh KA, et al. The leukocyte common antigen (CD45): a putative receptor‐linked protein tyrosine phosphatase. Proc Natl Acad Sci U S A 1988; 85: 7182–7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yeh HI, Dupont E, Coppen S, et al. Gap junction localization and connexin expression in cytochemically identified endothelial cells of arterial tissue. J Histochem Cytochem 1997; 45: 539–550. [DOI] [PubMed] [Google Scholar]
- 46. Ozerdem U, Grako KA, Dahlin‐Huppe K, et al. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn 2001; 222: 218–227. [DOI] [PubMed] [Google Scholar]
- 47. Roesch K, Jadhav AP, Trimarchi JM, et al. The transcriptome of retinal Müller glial cells. J Comp Neurol 2008; 509: 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muzumdar MD, Tasic B, Miyamichi K, et al. A global double‐fluorescent Cre reporter mouse. Genesis 2007; 45: 593–605. [DOI] [PubMed] [Google Scholar]
- 49. DiMario JX, Uzman A, Strohman RC. Fiber regeneration is not persistent in dystrophic (MDX) mouse skeletal muscle. Dev Biol 1991; 148: 314–321. [DOI] [PubMed] [Google Scholar]
- 50. Yan Z, Choi S, Liu X, et al. Highly coordinated gene regulation in mouse skeletal muscle regeneration. J Biol Chem 2003; 278: 8826–8836. [DOI] [PubMed] [Google Scholar]
- 51. Beauchamp JR, Heslop L, Yu DS, et al. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J Cell Biol 2000; 151: 1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Olson LE, Soriano P. PDGFRβ signaling regulates mural cell plasticity and inhibits fat development. Dev Cell 2011; 20: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kang SH, Fukaya M, Yang JK , et al. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 2010; 68: 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heinrich MC, Griffith D, McKinley A, et al. Crenolanib inhibits the drug‐resistant PDGFRA D842V mutation associated with imatinib‐resistant gastrointestinal stromal tumors. Clin Cancer Res 2012; 18: 4375–4384. [DOI] [PubMed] [Google Scholar]
- 55. Lewis NL, Lewis LD, Eder JP, et al. Phase I study of the safety, tolerability, and pharmacokinetics of oral CP‐868,596, a highly specific platelet‐derived growth factor receptor tyrosine kinase inhibitor in patients with advanced cancers. J Clin Oncol 2009; 27: 5262–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dai J, Kong Y, Si L, et al. Large‐scale analysis of PDGFRA mutations in melanomas and evaluation of their sensitivity to tyrosine kinase inhibitors imatinib and crenolanib. Clin Cancer Res 2013; 19: 6935–6942. [DOI] [PubMed] [Google Scholar]
- 57. Kerkela R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nature Med 2006; 12: 908–916. [DOI] [PubMed] [Google Scholar]
- 58. Zimmerman EI, Turner DC, Buaboonnam J, et al. Crenolanib is active against models of drug‐resistant FLT3‐ITD‐positive acute myeloid leukemia. Blood 2013; 122: 3607–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Faulkner JA, Ng R, Davis CS, et al. Diaphragm muscle strip preparation for evaluation of gene therapies in mdx mice. Clin Exp Pharmacol Physiol 2008; 35: 725–729. [DOI] [PubMed] [Google Scholar]
- 60. Zammit PS, Relaix F, Nagata Y, et al. Pax7 and myogenic progression in skeletal muscle satellite cells. J Cell Sci 2006; 119: 1824–1832. [DOI] [PubMed] [Google Scholar]
- 61. Relaix F, Rocancourt D, Mansouri A, et al. A Pax3/Pax7‐dependent population of skeletal muscle progenitor cells. Nature 2005; 435: 948–953. [DOI] [PubMed] [Google Scholar]
- 62. Tallquist MD, Weismann KE, Hellstrom M, et al. Early myotome specification regulates PDGFA expression and axial skeleton development. Development 2000; 127: 5059–5070. [DOI] [PubMed] [Google Scholar]
- 63. Pal R, Palmieri M, Loehr JA, et al. Src‐dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nature Commun 2014; 5: 4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim TD, le Coutre P, Schwarz M, et al. Clinical cardiac safety profile of nilotinib. Haematologica 2012; 97: 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sultan A, Fayaz M. Prevalence of cardiomyopathy in Duchenne and Becker's muscular dystrophy. J Ayub Med Coll Abbottabad 2008; 20: 7–13. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Supplementary figure legends
Figure S1 Col1a1‐GFP+ cells are abundant in areas of perimysial fibrosis of mdx quadriceps muscle, where type III and VI collagens are deposited. Split panel immunofluorescence images of quadriceps and diaphragm muscles from male 20‐month‐old mdx:Col1a1‐GFP mice. (A) Type III collagen and αSMA. (B) Type VI collagen and αSMA. (C) IgG isotype control. Note the frequent Col1a1GFP+ cells in perimysial fibrosis, particularly surrounding αSMA‐positive medium‐sized vessels. Scale bars = 50 µm.
Figure S2 GFP+ cells are clustered in areas of type III and type VI collagen deposition in the myocardium of mdx:Col1a1‐GFP mice. (A) Split panel immunofluorescence images showing type III and type IV collagens, highlighting the proximity of Col1a1‐GFP+ cells with pathological matrix in the heart muscle of 14‐month‐old mdx:Col1a1‐GFP mice. (B) Representative panel images from mdx:Col1a1‐GFP mice highlight the accumulation of type III collagen with time and the gathering of GFP+ cells in these fibrotic areas. Scale bars = 50 µm.
Figure S3 Sca1 is concentrated in the PDGFRα + portion of the Col1a1‐GFP+ population in both wild‐type and mdx diaphragms. Col1a1‐GFP+ cells were gated as either PDGFRα‐positive or ‐negative to analyse marker expression within these two sub‐populations of GFP+ cells. As before, little or no CD45 or Sca1 was present. However, Sca1 was predominantly expressed in PDGFRα + but not PDGFRα − cells in both 4‐month‐old wild‐type (top two rows) and mdx diaphragms (bottom two rows).
Figure S4 Col1a1‐GFP+ cells in the heart and diaphragm share a similar molecular profile. Representative flow cytometry plots gated on Col1a1‐GFP+ cardiac mononuclear cells isolated from wild‐type (left) and mdx:Col1a1‐GFP (right) male mice (N = 3 per group), showing that Col1a1‐GFP+ cells are PDGFRα + and Sca1+, but remain CD45− or CD31 − .
Figure S5 PDGFRa‐nGFP+ cells are concentrated in degenerating and regenerating regions of mdx muscles. In mdx muscles, PDGFRa‐nGFP+ cells are concentrated near areas with degenerating fibres labelled by mouse IgG (top row, arrowhead) and regions of regeneration containing αSMA+ fibres and Myf5 nlacZ/+ expressing myogenic cells (bottom row, arrow). Shown are quadriceps from a 3‐month‐old male mdx:PDGFRa‐nGFP:Myf5 nlacZ/+ double reporter mouse. Scale bars = 50 µm.
Figure S6 Fate‐mapping reveals that PDGFRα‐expressing cells are prominent at the onset of dystrophy in mdx diaphragm muscle. (A) Schematic of Cre‐loxP generation. Mice carrying the PDGFRa‐Cre allele were crossed with mice carrying the mT/mG loxP allele to produce double transgenic progeny that harbour both alleles. In these double transgenic mice, cells that express the PDGFRa‐Cre transgene and their subsequent progeny are labelled with membrane‐localized EGFP. In contrast, Cre‐negative cells and their progeny will be labelled with membrane‐localized tdTomato. (B) Split panel images of diaphragm muscles from wt:PDGFRa‐Cre:mT/mG loxP male mice showing discrete GFP+ cells derived from cells which have expressed Pdgrfa in normal muscle. (C) In mdx:PDGFRa‐Cre:mT/mG loxP diaphragms at 6 weeks of age, just prior to the onset of dystrophy, there is a mild expansion of the PDGFRα lineage. By 8 weeks of age, there is a large expansion of the PDGFRα lineage coinciding with the onset of muscular dystrophy between 6 and 8 weeks of age. Note that the mT/mG loxP labels Cre‐expressing or ‐derived cells with membrane localized EGFP+, while Cre‐negative cells are labelled with membrane tdTomato. Scale bars = 50 µm.
Figure S7 Col1a1‐GFP+ cells are adjacent to both myogenic cells and regenerating fibres following acute cardiotoxin injury to healthy muscle. Split panel images in a Col1a1‐GFP;Myf5 LacZ/+ expressing TA muscle stained with X‐gal and labelled for αSMA, 3 days after cardiotoxin administration. Staining reveals Col1a1‐GFP+ cells adjacent to regenerating fibres labelled by sarcomeric αSMA (arrow) as well as Col1a1‐GFP+ cells adjacent to Myf5 LacZ/+ myogenic cells (arrowhead). Scale bars = 50 µm.
Figure S8 Histological analysis of mutant muscles shows reduced fibre size with excessive PDGF signalling following CTX injury. (A) H&E staining was used to measure the cross‐section fibre size in quadriceps muscles from the PDGFRa‐CreER and control mice depicted also in Figure 4C. Top row shows contralateral uninjured muscles; bottom row shows the injured muscles collected 21 days post‐CTX injection. Scale bars = 100 µm. (B) Size quantification was conducted by measuring the minimum diameter of individual fibres, a conservative metric for fibre size when using cross‐sections. In the absence of injury, fibre sizes were similar between mutant muscles (top graph). In contrast, injured muscles from both PDGFRa‐CreER:PDGFRa ∆D842V and PDGFRb ∆D536V had significantly smaller fibre diameters versus Cre‐negative controls (bottom graph). **p = < 0.005, *** p = < 0.0005 by Student's t‐test. NS = not significant. Error bars indicate ± SEM.
Figure S9 Tamoxifen‐treated PDGFRa‐CreER;mT/mG loxP mice show activation of the GFP reporter in both uninjured and injured muscle. Split panel images of TA muscle from wt:PDGFRa‐CreER:mT/mG loxP reporter mice versus Cre‐negative controls. Following i.p. injections of tamoxifen for five consecutive days, uninjured contralateral muscles (A) were compared with TA muscles injected with CTX and collected 14 days post‐injury (B). Note that following tamoxifen, there was specific appearance of GFP+ cells among tdTomato + cells localized to non‐myofibre areas. In addition, in the absence of Cre, we did not observe any recombination. Scale bars = 50 µm.
Figure S10 Vehicle‐treated PDGFRa‐CreER;mT/mG loxP mice show no evidence of recombination in uninjured or injured muscle. Split panel images of TA muscle from Cre‐negative and wt:PDGFRa‐CreER:mT/mG loxP reporter mice following i.p. injections of corn oil for five consecutive days. As portrayed in Figure S9, uninjured muscles (A) were compared with CTX‐injured TAs, 14 days after injury. (B) In the absence of tamoxifen, no GFP cells appeared within tdTomato + muscles. Scale bars = 50 µm.
Figure S11 Single fibre isolations and co‐culture with Col1a1‐GFP+ cells indicate that mdx fibres promote fibrosis by their production of PDGF‐AA. (A) Single fibres from wild‐type and mdx extensor digitorum longus (EDL) muscles were isolated and cultured in triplicate. Immediately following the fibre isolations, wild‐type ‐derived Col1a1‐GFP+ cells were added. Co‐cultures were incubated for 3 days and then fixed for staining against PDGF‐AA and collagens. Scale bars = 50 µm. (B) Picrosirius red staining was used to analyse collagen deposition by Col1a1‐GFP+ cells in co‐culture with wild‐type versus mdx fibres. Scale bars = 100 µm. (C) Quantification of the PDGF‐AA staining represented in panel A shows significantly more of this ligand in mdx fibre co‐cultures. (D) In turn, greater PDGF‐AA staining correlated with more collagen deposition in mdx fibre co‐cultures, quantified by Picrosirius red staining. ***p = < 0.0005 by Student's t‐test. Error bars indicate ± SEM.
Figure S12 Crenolanib inhibits PDGF‐AA‐mediated expression of pro‐collagen mRNAs and phosphorylation of PDGFRα in primary cultured Col1a1‐GFP+ cells. (A) Graphs show RT‐qPCR analysis for Col1a1 and Col3a1 in cultured, primary FACS‐sorted Col1a1‐GFP+ cells treated with DMEM with 5% FCS + Pen/Strep medium containing vehicle (DMSO), PDGF‐AA (10 ng/ml), PDGF‐BB (10 ng/ml), or PDGF‐AA + crenolanib (1 µm) for 4 days prior to isolation of RNA and protein extracts (N = 3 samples per group). *p < 0.05, ***p < 0.00005 by single factor ANOVA. Error bars indicate ± SEM. (B) Western blot and densitometry plot showing total PDGFRα and phosph‐PDGFRα in Col1a1‐GFP+ cells 30 min after stimulation with PDGF‐AA or PDGF‐AA + crenolanib (N = 1 per treatment).
Figure S13 Crenolanib attenuates type III collagen staining in mdx diaphragms. Histological analysis of diaphragms from mdx:Col1a1‐GFP mice (N = 4 per group) indicates that the area of type III collagen staining was reduced following 4 weeks of crenolanib treatment versus the vehicle. In contrast, the presence of GFP within these same diaphragms was not significantly different. **p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S14 Necrotic regions are smaller with crenolanib treatment. H&E staining highlights necrotic areas (arrows) in quadriceps from the vehicle‐ or crenolanib‐treated mdx mice depicted in Figure 6. Quantification of the necrotic versus total area within each field indicated that treatment with crenolanib mitigated the development of pathology in dystrophic muscles. Scale bars = 100 µm. *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S15 The phosphorylation of Src increases in mdx diaphragms. Total proteins from 12‐month‐old mdx and wild‐type diaphragms and hearts (N = 3 per group) were processed for western blot analysis. Quantification indicated that the phosphorylation of Src is significantly elevated in mdx diaphragms versus wild‐type (left panel). Although elevated, the phosphorylation of Src in hearts was not significantly different (right panel). *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S16 Crenolanib reduces PDGFRα signalling and fibrosis in mdx hearts. (A) Representative images of fibrosis in Picrosirius red‐stained hearts from mdx mice treated with the vehicle or crenolanib. Scale bars = 50 µm. (B) RT‐qPCR analysis of cardiac tissue (N = 4 per group) indicates a modest reduction in Col3a1 transcript levels, but no change for Col1a1. (C) Western blots (left) and normalized densitometry (right) showing that type III and type I collagen as well as fibronectin protein levels were significantly reduced with crenolanib treatment. (D) Western blots (left) and normalized densitometry (right) showing PDGFRα phosphorylation and phosphorylation of potential downstream effectors in crenolanib‐treated mdx mice. Note that although PDGFRα phosphorylation was reduced by systemic crenolanib, Src or Abl activation was not affected. *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S17 The number and size of regenerating fibres are greater with crenolanib treatment. Top graph: the number of muscle fibres positive for sarcoplasmic αSMA in regenerating regions of mdx diaphragms is significantly greater with crenolanib treatment. Bottom graph: correspondingly, in the same diaphragm muscles, the minimum diameter of αSMA+ fibres was also significantly greater with crenolanib. *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Figure S18 Crenolanib improves regeneration in mdx skeletal muscles. (A) Split panel images of skeletal muscles (diaphragm and EDL) from mdx:Col1a1‐GFP mice treated with crenolanib or vehicle (from the same animal analysed in Figure 6). Note a greater number of αSMA+ regenerating fibres surrounded by Col1a1‐GFP+ cells in response to crenolanib treatment. Scale bars = 50 µm. (B) RT‐qPCR of RNA from diaphragm muscles showing enhanced Acta2 transcript with crenolanib treatment. (C) Graphs showing RT‐qPCR results for myogenic regulatory factors (Pax7, Myf5, Myod) indicate a trend towards increased expression of myogenic factor transcripts in response to crenolanib (N = 4 per group). *p = < 0.05 by Student's t‐test. Error bars indicate ± SEM.
Table S1. Mouse strains used
Table S2. Antibodies and lectins used on tissue sections
Table S3. FACS antibodies and streptavidin conjugates
Table S4. RT‐qPCR primer sequences
Table S5. Antibodies used for western blotting