

Abstract
Recent discoveries of new classes of self‐cleaving ribozymes in diverse organisms have triggered renewed interest in the chemistry and biology of ribozymes. Functional analysis and engineering of ribozymes often involve performing biochemical assays on multiple ribozyme mutants. However, because each ribozyme mutant must be individually prepared and assayed, the number and variety of mutants that can be studied are severely limited. All of the single and double mutants of a twister ribozyme (a total of 10 296 mutants) were generated and assayed for their self‐cleaving activity by exploiting deep sequencing to count the numbers of cleaved and uncleaved sequences for every mutant. Interestingly, we found that the ribozyme is highly robust against mutations such that 71 % and 30 % of all single and double mutants, respectively, retain detectable activity under the assay conditions. It was also observed that the structural elements that comprise the ribozyme exhibit distinct sensitivity to mutations.
Keywords: deep sequencing, high-throughput assays, ribozymes, RNA
Bioinformatic and biochemical investigations have revealed the wide presence of self‐cleaving ribozymes in the genomes of diverse organisms.1 Although some ribozymes are known to be involved in viral RNA processing, the biological functions of most ribozymes remain largely unknown.2 However, that has not hindered the efforts to elucidate their structures and catalytic mechanisms, as well as to engineer ribozymes for biotechnological applications.3 Such efforts often require individual preparation of ribozyme mutants and functional assays using techniques such as electrophoresis or chromatography. Since these conventional methods are intrinsically low‐throughput, the number and variety of the mutants studied are highly limited or biased based on the researcher's interests. To address these challenges, we recently reported a high‐throughput strategy to assay ribozyme mutants that are transcribed in vitro as a mixture. By exploiting deep sequencing to both sequence and count the number of cleaved and uncleaved reads for every mutant, we successfully assayed circa 1 500 locally randomized ribozyme variants.4
We propose that an unbiased and comprehensive functional analysis of ribozyme mutants can complement the existing structural and biochemical information of ribozymes and provide novel insights. In this work, we extended our sequencing‐based ribozyme assay to exhaustively assay all of the single and double mutants of a 48‐nucleotide section of the 54‐nucleotide Oryza sativa Osa‐1‐4 ribozyme sequence belonging to the twister ribozyme class recently discovered by the Breaker group.5 Crystal structures of Osa‐1‐46 and other variants7 adapting a characteristic double‐pseudoknot structure have been described recently (Figure 1 A) and their catalytic mechanisms have been scrutinized.8 Ten positions are reported to be more than 97 % conserved among the twister ribozyme variants.5 By exhaustive mutagenesis, we sought to elucidate which bases in the ribozyme are essential for ribozyme activity, as well as to understand the global tolerance of the ribozyme to mutations.
Figure 1.
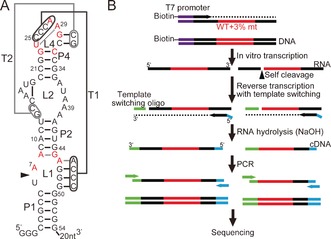
A) Structure of the Osa‐1‐4 ribozyme based on the crystal structure described by Liu et al.6 The nucleotides are numbered according to Ref. 6. The arrowhead indicates the cleavage site. The bases shown in red are more than 97 % conserved among twister ribozymes.5 B) llustration of the library preparation procedure.
To generate all single and double mutants of Osa‐1‐4 positions downstream of the cleavage site (all 48 nucleotides between A7 and G54), a DNA template was prepared in which the positions targeted for mutagenesis were synthesized by coupling 97 % of the wild‐type bases doped with 1 % each of the remaining three bases. The doped ribozyme‐coding template was transcribed in vitro into a mixture of ribozyme mutants for 2 h at 37 °C during which some ribozymes self‐cleaved co‐transcriptionally. The ribozymes were subsequently reverse‐transcribed into cDNAs along with a template‐switching oligonucleotide (TSO)9 to attach partial adapter sequences for deep sequencing. Finally, polymerase chain reaction (PCR) was performed to prepare the sequencing sample (Figure 1 B).
As each template is sequenced, the identity of the mutant is recorded along with its cleavage status based on the presence or absence of the sequence upstream of the cleavage site. By counting the numbers of cleaved and uncleaved templates (N clv, N unclv, respectively) for each ribozyme mutant, its activity can be expressed as the fraction cleaved (FC), which is calculated from Eq. (1) as follows:
| (1) |
FC values were further normalized by the corresponding activity of the wild‐type sequence (FCwt=0.821) to obtain the relative activity (RA), as shown in Eq. (2):
| (2) |
Deep sequencing of the sample yielded a total of 45 396 995 effective reads. Of these, 13.6 % (6 193 111 reads) were the wild‐type, while 144 single mutants were read on average 93 541 times each, and 10 152 double mutants were read on average 1365 times each. A summary of the sequencing results is given in the Supporting Information (Table S1).
We selected 13 ribozyme variants including the wild‐type to validate the sequencing results against the conventional assay based on gel electrophoresis. The FC values based on the sequencing and gel assays display a good linear correlation (Supporting Information, Figure S1). Some systematic differences between the absolute FC values obtained by the two assays can be attributed to the systematic biases introduced during the sequencing library preparation. For example, the efficiency of template switching during reverse transcription can be different for the cleaved and the uncleaved RNAs. However, normalization of the FC values by that of the wild‐type (that is, RA) results in a useful semiquantitative picture of the global mutational profile of the twister ribozyme sequence.
One interesting observation is that a significant fraction of the mutants retain at least some ribozyme activity (Table 1). For example, 42 % and 11 % of single and double mutants, respectively, exhibit RA >0.80. Seventy‐one‐percent of the single mutants and 30 % of the double mutants show RA >0.20. This suggests that despite the compact and complex folding of the ribozyme, it is highly robust against mutations, which may have resulted in an evolutionary advantage.
Table 1.
Fractions of active ribozyme mutants.[a]
| Relative activity | Single mutants (144[b]) | Double mutants (10 152[b]) |
|---|---|---|
| >0.80 | 60 (42 %) | 1067 (11 %) |
| >0.60 | 75 (52 %) | 1627 (16 %) |
| >0.40 | 91 (63 %) | 2145 (21 %) |
| >0.20 | 102 (71 %) | 3027 (30 %) |
[a] The ribozyme reactions were performed co‐transcriptionally in the transcription buffer (40 mm Tris‐HCl pH 8.0, 6 mm MgCl2, 2 mm spermidine, 10 mm dithiothreitol) for 2 h at 37 °C. See Experimental Details in the Supporting Information for details. [b] Total number of mutants.
Only four positions (G13, G30, A46, and G48) were found for which substitution with any other base resulted in RA <0.20 (Figure 2 A). All of these nucleotides are involved in one of the two pseudoknots T1 and T2. A closer examination of single mutants in T1 and T2 highlights the sensitivity of the ribozyme to mutations in these regions. While most mutations in T1 and T2 are highly disruptive, some C‐to‐U mutations that transform a G–C Watson–Crick base pair to a G–U wobble base pair (C14U, C25U, C26U, and C31U) are tolerated (Figure 2 A).
Figure 2.
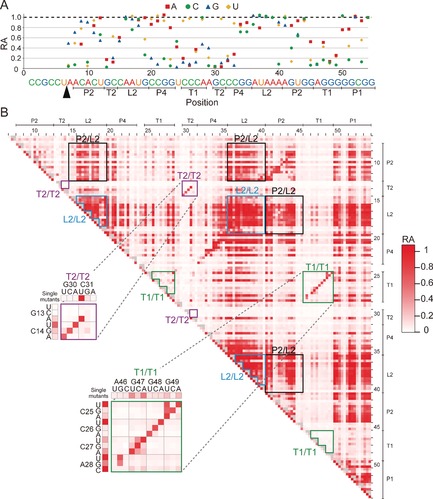
Visualizations of the activity of the ribozyme mutants. A) Relative activity (RA) of 144 single mutants of the Osa‐1‐4 ribozyme. B) Two‐dimensional RA matrix of all single and double mutants of the Osa‐1‐4 ribozyme. Single mutants are represented on the diagonal side. Several characteristic clusters of double mutants are shown in the boxes and are discussed in the main text. The full RA matrix with numerical RA data and mutations is provided in the Supporting Information.
A contrasting picture emerges for mutations in the canonical helices P1, P2, and P4. In these regions, mutations that result in a disruption of a Watson–Crick base pair are often tolerated, with 33 out of 69 single mutants in these helices exhibiting RA >0.80 (Figure 2 A). In particular, robustness of the P1 mutants is consistent with the recent finding that the P1 stem, though phylogenetically conserved, is not essential for catalytic activity.8b
Perhaps more intuitively, mutations in the central loop region L2 are less critical for the ribozyme function. Nucleotides in L2 are not only phylogenetically less conserved among the twister ribozymes, but some subclasses of the twister ribozymes accommodate additional P3 and P5 helices within this region.5 Therefore, it is reasonable that most mutants in L2 exhibit a relatively high RA. One intriguing exception is A40C (RA=0.09). We speculate that A40C can extend the P2 helix by one base pair (G14–C40), which in turn disrupts the formation of T2.
Overall, the complete RA profile of the single mutations reveals a surprising robustness of the twister ribozyme even in well‐conserved nucleotides. One caveat is that RA does not correlate linearly with the intrinsic activity (that is, k cat) of the ribozyme because the speed of the wild‐type ribozyme reported under similar conditions (k cat=1–10 min−1, corresponding to a half‐life of 1 min or less)6, 7b is much faster than the timescale of our experiment (120 min). Therefore, a moderate reduction in RA actually corresponds to a more drastic decrease in the rate constant. In fact, many mutations in the conserved bases do result in significant reductions in RA (Figure 2 A). Therefore, while some of these bases may not be essential for activity, their conservation may afford an evolutionary advantage.
The comprehensive RA data of the 10 152 double mutants provide further insights into the sequence–function relationship. The global matrix of the activity of the double mutants (Figure 2 B and the Supporting Information) displays distinct clusters of mutants where double mutations are well‐tolerated. Not surprisingly, double mutations within L2 are well‐tolerated as indicated by the clusters of active mutants corresponding to this region (L2/L2 in Figure 2 B). Combinations of single mutants in the mutationally robust elements (P1, P2, P4, and L2) also tend to be relatively active. For example, double mutants with one mutation in P2 and another mutation in L2 are represented as clusters of mutants with a relatively high activity (P2/L2 in Figure 2 B).
Double mutants within the pseudoknots T1 or T2 display different characteristics. In T2, the Watson–Crick base pair C14–G30 can be replaced with any of the three other Watson–Crick base pairs and retain RA >0.60, while all the other single and double mutations at this position abolish ribozyme activity. A similar observation can be made for T1, in which most base pairs can be replaced by another base pair and still retain activity while bases incapable of pairing diminish activity. These regions appear as a diagonal stretch of active double mutants on the mutational matrix (Figure 2 B).
We recently demonstrated our high‐throughput assay strategy to measure the activity of approximately 1500 ribozyme variants in which a short (4 or 5 bases) sequence of contiguous nucleotides were randomized.4 In our present work, we successfully harnessed the power of massively parallel sequencing to map the complete sequence–function relationship of a ribozyme and all of its 10 296 single and double mutants. We expect that a complete coverage of triple mutants may be possible for small ribozymes under optimized conditions, and double mutants of a ribozyme as large as 379 bases can also be assayed by the existing technology (see Experimental Details in the Supporting Information).
Recently, in vitro selection of ribozyme mutants and subsequent sequencing of the selected variants have been used to infer the fitness landscapes of various ribozymes.10 However, these techniques do not provide direct and quantitative information about the activities of the ribozyme mutants, and the results are based on the sequences that survive an arbitrary and potentially biased selection pressure imposed by the researcher. Our strategy does not involve selection and thus complements these selection‐based methods by providing an unbiased and semiquantitative activity profile for all single and double mutants of a ribozyme of interest regardless of their activity. More precise determination of the rate constant (k cat) should also be possible by modifying the library preparation procedure to assay ribozyme activity at multiple time points. As briefly reviewed above, however, our current semiquantitative (but more biologically relevant) co‐transcriptional cleavage activity data still yield rich sequence–function information that is difficult to obtain by conventional methodologies.
In summary, we obtained a comprehensive mutational landscape of the Osa‐1‐4 twister ribozyme by measuring the activity of all 144 single and 10 152 double mutants by deep sequencing. We discovered that the ribozyme is surprisingly robust to mutations despite its compactness and complex structure, and observed that individual structural elements exhibit different sensitivity to mutations. We believe that our strategy can provide new comprehensive and quantitative insights into the sequence–function relationships of ribozymes.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Hiroki Goto and OIST DNA Sequencing Section for sequencing support and helpful discussions. The research was funded by Okinawa Institute of Science and Technology Graduate University.
S. Kobori, Y. Yokobayashi, Angew. Chem. Int. Ed. 2016, 55, 10354.
References
- 1.
- 1a. Hammann C., Lupták A., Perreault J., de la Peña M., RNA 2012, 18, 871–885; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Webb C.-H. T., Riccitelli N. J., Ruminski D. J., Lupták A., Science 2009, 326, 953; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Weinberg Z., Kim P. B., Chen T. H., Li S., Harris K. A., Lünse C. E., Breaker R. R., Nat. Chem. Biol. 2015, 11, 606–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jimenez R. M., Polanco J. A., Lupták A., Trends Biochem. Sci. 2015, 40, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frommer J., Appel B., Müller S., Curr. Opin. Biotechnol. 2015, 31, 35–41. [DOI] [PubMed] [Google Scholar]
- 4. Kobori S., Nomura Y., Miu A., Yokobayashi Y., Nucleic Acids Res. 2015, 43, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roth A., Weinberg Z., Chen A. G., Kim P. B., Ames T. D., Breaker R. R., Nat. Chem. Biol. 2014, 10, 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu Y., Wilson T. J., McPhee S. A., Lilley D. M., Nat. Chem. Biol. 2014, 10, 739–744. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Eiler D., Wang J., Steitz T. A., Proc. Natl. Acad. Sci. USA 2014, 111, 13028–13033; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Ren A., Košutić M., Rajashankar K. R., Frener M., Santner T., Westhof E., Micura R., Patel D. J., Nat. Commun. 2014, 5, 5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Gaines C. S., York D. M., J. Am. Chem. Soc. 2016, 138, 3058–3065; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Košutić M., Neuner S., Ren A., Flür S., Wunderlich C., Mairhofer E., Vušurović N., Seikowski J., Breuker K., Höbartner C., Patel D. J., Kreutz C., Micura R., Angew. Chem. Int. Ed. 2015, 54, 15128–15133; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15343–15348; [Google Scholar]
- 8c. Wilson T. J., Liu Y., Domnick C., Kath-Schorr S., Lilley D. M., J. Am. Chem. Soc. 2016, 138, 6151–6162; [DOI] [PubMed] [Google Scholar]
- 8d. Zhang S., Ganguly A., Goyal P., Bingaman J. L., Bevilacqua P. C., Hammes-Schiffer S., J. Am. Chem. Soc. 2015, 137, 784–798; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Ucisik M. N., Bevilacqua P. C., Hammes-Schiffer S., Biochemistry 2016, 55, 3834–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Zhu Y. Y., Machleder E. M., Chenchik A., Li R., Siebert P. D., Biotechniques 2001, 30, 892–897; [DOI] [PubMed] [Google Scholar]
- 9b. Picelli S., Faridani O. R., Björklund Å. K., Winberg G., Sagasser S., Sandberg R., Nat. Protoc. 2014, 9, 171–181. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Ameta S., Winz M. L., Previti C., Jäschke A., Nucleic Acids Res. 2014, 42, 1303–1310; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Curtis E. A., Bartel D. P., RNA 2013, 19, 1116–1128; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Pitt J. N., Ferré-D′Amaré A. R., Science 2010, 330, 376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary