

Abstract
A novel DNA‐based hybrid catalyst comprised of salmon testes DNA and an iron(III) complex of a cationic meso‐tetrakis(N‐alkylpyridyl)porphyrin was developed. When the N‐methyl substituents were placed at the ortho position with respect to the porphyrin ring, high reactivity in catalytic carbene‐transfer reactions was observed under mild conditions, as demonstrated in the catalytic enantioselective cyclopropanation of styrene derivatives with ethyl diazoacetate (EDA) as the carbene precursor. A remarkable feature of this catalytic system is the large DNA‐induced rate acceleration observed in this reaction and the related dimerization of EDA. It is proposed that high effective molarity of all components of the reaction in or near the DNA is one of the key contributors to this unique reactivity. This study demonstrates that the concept of DNA‐based asymmetric catalysis can be expanded into the realm of organometallic chemistry.
Keywords: carbenes, cyclopropanation, DNA, hybrid catalysts, porphyrins
Inspired by enzymes, second‐coordination‐sphere interactions are increasingly being recognized as key elements for the design of highly active and selective catalysts.1 The concept of DNA‐based catalysis, in which a transition‐metal complex is embedded in a DNA scaffold that provides the chiral second coordination sphere, is a powerful illustration of this principle.2 DNA‐based catalysis has been applied successfully in a variety of copper(II)‐catalyzed (that is, Lewis acid catalyzed) reactions with high catalytic activity and enantioselectivity.3 However, expansion of the reaction scope beyond Lewis acid catalysis has so far proven to be challenging. Jäschke and co‐workers reported a catalytic allylic amination with a DNA‐based catalyst comprising a covalently attached iridium complex.4 Although catalysis was demonstrated, the enantioselectivity of the reaction was low. Recently, enantioselective sulfoxidation with a DNA G‐quadruplex based copper catalyst was described.5 Previously, we reported the first example of a DNA‐based copper‐catalyzed intramolecular cyclopropanation reaction.6 Although this transformation demonstrated the feasibility of DNA‐based organometallic catalysis, it also illustrated the challenges that are involved, since the reactions required strictly anaerobic conditions, the yields of products were low, and the substrate scope was limited. We now introduce a novel DNA/cationic‐iron‐porphyrin‐based hybrid catalyst that enables highly DNA accelerated catalysis of carbene‐transfer reactions, such as enantioselective cyclopropanation, under mild conditions and an ambient atmosphere.
[Fe(TPP)Cl] (TPP=5,10,15,20‐tetraphenyl‐21H,23H‐porphine) and related iron porphyrins have been employed successfully in carbene‐transfer reactions, even in aqueous media.7 Elegant recent studies with engineered P4508 or myoglobin artificial metalloenzymes9 and in combination with living cells10 have demonstrated that these reactions are biocompatible: excellent catalysis of carbene‐transfer reactions, such as enantioselective cyclopropanation, has been reported, albeit mostly under strictly anaerobic conditions.
For this study, a variety of iron(III) complexes of cationic porhyrins, that is, meso‐tetrakis(N‐alkylpyridyl)porphyrins differing in the position of the N‐alkyl substituent and the length of the alkyl chain, were selected as catalysts (Scheme 1 b). This class of iron(III) porphyrins has been studied extensively, for example, as mimics of the enzyme Superoxide dismutase.11 They are known DNA binders, and it has been reported that their interaction with DNA, as well as their physical properties, are very much dependent on the position of the N‐alkyl moiety and the length of the alkyl chain.11, 12
Scheme 1.
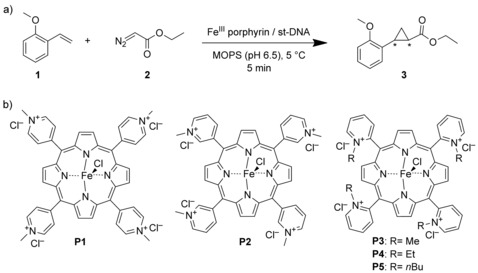
a) Enantioselective cyclopropanation reaction catalyzed by a DNA‐based iron porphyrin. b) Iron(III) complexes of meso‐tetrakis(N‐alkylpyridyl)porphyrins used in this study.
The cyclopropanation of o‐methoxystyrene (1) with ethyldiazoacetate (EDA, 2) as the carbene precursor was selected as the benchmark reaction (Scheme 1 a). The DNA‐based catalyst was self‐assembled by combining the iron(III) porphyrin (75 μm, 1.5 mol %) with salmon testes DNA (st‐DNA, 6 mm in base pairs) in 20 mm 3‐(N‐morpholino)propanesulfonic acid (MOPS) buffer at pH 6.5, which was found to be the optimal pH value with regard to both activity and selectivity (see Table S1 in the Supporting Information). The ortho‐substituted porphyrins P3–P5 11 were used as mixtures of atropoisomers.
The catalytic reactions were performed at 5 °C under an ambient atmosphere with a 10‐fold excess of 2, and the reaction mixture was mixed by continuous inversion. First, the three isomeric iron porphyrins P1–P3 with N‐methyl groups were evaluated as catalysts. In the absence of DNA, the reaction was sluggish and resulted in low yields of the cyclopropanation product 3 (Table 1, entries 1, 3 and 5), even after prolonged reaction times. In the presence of st‐DNA (6 mm in base pairs), low activity was again observed in the case of the para and meta isomers P1 and P2 (Table 1, entries 2 and 4). Surprisingly, with the ortho isomer P3, immediate and vigorous evolution of N2 was observed, which ceased after approximately 5 min. It was found that at this time all EDA had been consumed. The trans isomer of the cyclopropanation product 3 (trans/cis≥12:1)13 was obtained in 14 % yield, which amounts to 9 turnovers of the Fe catalyst, with a promising ee value of 42 % (Table 1, entry 6). A large fraction of the EDA was converted into side products, the most important detectable side product being diethyl maleate, which results from the dimerization of EDA.
Table 1.
Enantioselective cyclopropanation of 1 with EDA under the catalysis of iron porphyrins in the presence and absence of st‐DNA.[a]
| Entry | Porphyrin | st‐DNA [mm] | Yield [%][b] | TTN[c] | ee [%][b] |
|---|---|---|---|---|---|
| 1 | P1 | – | ≤3 | – | – |
| 2 | P1 | 6 | ≤3 | – | – |
| 3 | P2 | – | ≤3 | – | – |
| 4 | P2 | 6 | ≤3 | – | – |
| 5 | P3 | – | ≤3 | – | – |
| 6 | P3 | 6 | 14±2 | 9 | 42 |
| 7[d] | P3 | 6 | 16±4 | 22 | 40 |
| 8[e] | P3 | – | ≤3 | – | – |
| 9 | P3 | 3 | 22±1 | 14 | 33 |
| 10 | P3 | 1.5 | 25±0 | 17 | 24 |
| 11 | P4 | – | 33±3 | 22 | – |
| 12 | P4 | 6 | 15±0.5 | 10 | 23 |
| 13 | P5 | – | 26±0 | 17 | – |
| 14 | P5 | 6 | 11±1 | 7 | 5 |
[a] The experiments were carried out with 1 (5 mm), 2 (50 mm), st‐DNA (6 mm in base pairs), and the FeIII porphyrin (75 μm) in 20 mm MOPS buffer (pH 6.5) containing acetonitrile (3 % v/v) for 5 min at 5 °C, unless otherwise specified. Yields and ee values are based on the areas of HPLC and GC peaks as compared to those of 2‐methyl anisole as an internal standard. All data were averaged over two or more experiments. Errors reported are standard deviations. [b] The yield and ee value (reproducibility: ±3 % ee) of the trans isomer are given. The diastereomeric ratio of the product ranged from 88:12 to 95:5. [c] TTN=total turnover number. [d] Metalloporphyrin concentration: 37.5 μm. [e] Metalloporphyrin concentration: 7 μm.
Interestingly, the Fe porphyrins P4 and P5, which contain an N‐ethyl and an N‐butyl moiety, respectively, showed high activity, even in the absence of DNA (Table 1, entries 11–14). With these porphyrins, the yield of 3 was found to be lower in the presence of DNA, whereas the ee value of the product 3 was lower than that observed with the corresponding N‐methyl derivative P3. Since the highest enantioselectivity was observed with P3, in combination with the intriguing acceleration effect of DNA on the catalysis, we decided to focus our further studies on this complex.
We varied the concentration of P3 with the concentration of st‐DNA fixed at 6 mm in base pairs. When the concentration of the metalloporphyrin was halved, the total number of turnovers increased, whereas the enantioselectivity remained the same (Table 1, entry 7). Therefore, 37.5 μm was used as the optimal concentration for our catalysis. Since a solution of DNA with a concentration of 6 mm in base pairs is very viscous, the DNA concentration was also varied. Lowering of the concentration of st‐DNA at a fixed concentration of porphyrin resulted in an increase in yield, which is tentatively ascribed to increased diffusion of the substrates owing to the lower viscosity of the solution. This increase in yield was accompanied, however, by a decrease in enantioselectivity (Table 1, entries 9 and 10).
The scope of the reaction with respect to the styrene substrate was investigated by using p‐methoxystyrene (4), p‐chlorostyrene (5), and styrene (6) in the reaction with 2 as the diazo compound (Scheme 2, Table 2). In all cases, a significant amount of the cyclopropanation product was only obtained in the presence of DNA (entries 1–6). The trend observed is consistent with an electrophilic carbene as the active species, that is, styrenes 1 and 4 containing electron‐donating groups were converted into the corresponding cyclopropanation products (3 and 10) in higher yield than those substrates containing electron‐withdrawing groups. The somewhat lower activity towards 1 (Table 1, entry 7) as compared to 4 (Table 2, entry 2) is attributed to steric hindrance by the ortho methoxy group. The ee values observed ranged from 40 % ee in the case of 1 (Table 1, entry 7) to 53 % ee in the case of styrene (Table 2, entry 6).
Scheme 2.
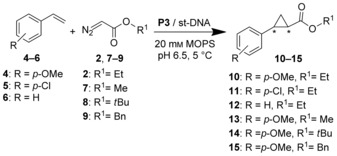
Investigation of the scope of the enantioselective cyclopropanation under DNA‐based iron porphyrin catalysis. Bn=benzyl.
Table 2.
Investigation of the scope of st‐DNA/P3‐catalyzed enantioselective cyclopropanation reactions with 2.[a]
| Entry | Styrene | Product | DNA [mm] | Yield [%][b] | TTN | ee [%][b] |
|---|---|---|---|---|---|---|
| 1 | 4 | 10 | – | 3±3 | 4 | – |
| 2 | 4 | 10 | 6 | 32±6 | 43 | 50 |
| 3 | 5 | 11 | – | ≤3 | – | – |
| 4 | 5 | 11 | 6 | 4±1 | 5 | 41 |
| 5 | 6 | 12 | – | ≤3 | – | – |
| 6 | 6 | 12 | 6 | 12 | 16 | 53 |
[a] The experiments were carried out with the styrene substrate (5 mm), 2 (50 mm), st‐DNA (6 mm in base pairs), and P3 (37.5 μm) in 20 mm MOPS buffer (pH 6.5) containing acetonitrile (3 % v/v) for 5 min at 5 °C. Yields and ee values were determined by HPLC and GC with 2‐methyl anisole as an internal standard. All data were averaged over two or more experiments. Errors reported are standard deviations. [b] The yield and ee value (reproducibility: ±3 % ee) of the trans isomer are given.
The scope of the reaction with respect to the diazo compound was investigated in the reaction of 4 with alkyl diazoacetates 2, 7, 8, and 9 (Scheme 2). It was found that bulkier groups R1 resulted in lower or even no yield of the cyclopropanation product (see Table S2). Conversely, with methyl diazoacetate, the corresponding product was obtained in 44 % yield (TTN: 59) with 50 % ee (see Table S2). Notably, in this case activity was observed in the absence of DNA (TTN: 31; see Table S2), albeit significantly lower than in the presence of DNA.
The observed DNA acceleration of catalysis was further studied by determining the kinetics of the reaction of 4 with EDA in presence and absence of DNA. However, the analysis is complicated by the fact that the cyclopropane is not the only product formed in the reaction: a significant amount of side products was formed. The most important detectable side product was diethyl maleate, resulting from the reaction of the carbene with another molecule of 2.14 Therefore, the formation of diethyl maleate over time was monitored by GC both in the presence and absence of st‐DNA and/or 4 (see Figure S1 in the Supporting Information). After a reaction time of 5 min, diethyl maleate was obtained in approximately 30 % yield (TTN: ca. 200) in the presence of st‐DNA versus 1 % yield in the absence of st‐DNA. In the absence of 4, the same trend was observed, but the yields were slightly lower.
The formation of N2 was measured during 5 min by using a gas burette (see Figure S2). For experimental reasons, it was necessary to switch from continuous inversion to orbital mixing, which is a less efficient method of mixing and, as a consequence, gave rise to a decrease in the yield of 10. However, the trends in reactivity in the presence and absence of DNA and the enantioselectivity remained the same (see Table S3).15 In the absence of DNA, hardly any evolution of N2 was observed (Figure 1). In contrast, in the presence of st‐DNA, after a short lag phase, a significant amount of released N2 (3.9 mL) was measured. The lag phase is tentatively ascribed to slow release of N2 from the solution owing to its viscosity.
Figure 1.
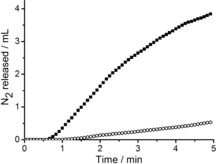
Evolution of N2 over time in the P3‐catalyzed decomposition of EDA in the presence (solid squares) and absence (open circles) of st‐DNA.
Arguably one of the intriguing aspects of this reaction is the observed DNA‐induced rate acceleration in catalysis by P3. At present it is difficult to quantify the effect of DNA on the reaction rate owing to the presence of several competing side reactions that are each affected differently by the presence of DNA. However, on the basis of the data presented above, the acceleration by DNA can be estimated to be at least between one and two orders of magnitude. Although it is not possible at present to explain this phenomenon in full molecular detail, a few important observations can be made:
1) The reaction does take place in or close to the DNA, as is evidenced by the enantioselectivity observed in the formation of the cyclopropanation product.
2) It is unlikely DNA accelerates the catalysis by providing an activating axial ligand to the iron ion, as has been observed for P450‐catalyzed cyclopropanation reactions8b and in the [FeIII(TPP)Cl]‐catalyzed dimerization of 2, in which case a significantly increased reaction rate was observed in the presence of pyridine.16 If anything, the presence of the ortho N‐methyl groups is known to reduce the interaction of metalloporphyrins with DNA owing to steric hindrance.11b, 17 Moreover, in control experiments in the absence of st‐DNA, but in the presence of a mixture of the four DNA nucleobases (adenine, thymine, guanine, and cytosine), benzyl amine and pyridine in all cases showed no release of N2, and the results of the catalysis were comparable to those observed in absence of st‐DNA.
3) The DNA acceleration effect was observed in the case of the ortho‐substituted N‐methylpyridinium porphyrin P3, but not with meta and para isomers P1 and P2, which gave rise to sluggish reactions both in the presence and absence of DNA. This result suggests that the different DNA‐binding mode of the iron porphyrins is important in catalysis.12 P3 predominantly interacts with DNA through groove binding in AT‐rich regions.12b Groove binding of the catalyst would make the catalytic site readily accessible for substrates, since it is more at the periphery of the DNA structure.
4) Conversely, iron porphyrin derivatives with larger alkyl substituents at the pyridyl nitrogen atom, such as P4 and P5, already promote very fast reactions in the absence of DNA, thus suggesting that a more hydrophobic environment around the metal site is beneficial for catalysis.
On the basis of these combined observations, we propose that the binding of P3 to DNA gives rise to the formation of hydrophobic pockets along the DNA in which the reaction partners, that is, the EDA and the styrene derivative, are concentrated. The resulting high effective molarity of all reaction components then is a key contributor to the observed rate acceleration of catalysis. This hypothesis is in agreement with the micellar rate enhancement observed in iron‐metalloporphyrin‐catalyzed cyclopropanation10, 17 and mechanistically similar epoxidation reactions.18 Also, the significant rate acceleration observed in cyclopropation reactions catalyzed by artificial metalloenzymes that present a large hydrophobic cavity8, 9, 19 and cobalt porphyrins encapsulated in hydrophobic metallocages supports this hypothesis.20 Finally, effective molarity has been proposed to be important in DNA‐accelerated Lewis acid catalyzed reactions as well,21 thus suggesting that this feature is a general hallmark of the concept of DNA‐based catalysis.
In conclusion, we have described the use of a novel DNA/cationic iron porphyrin hybrid catalyst for carbene‐transfer reactions in water under mild conditions and an ambient atmosphere. This system was applied to the catalytic enantioselective cyclopropanation of styrene derivatives, and the corresponding cyclopropanation products were obtained with promising ee values. However, arguably the most intriguing observation in this study is the large DNA‐induced rate acceleration in the case of P3. It is proposed that high effective molarity of the reaction partners in hydrophobic pockets provided by the DNA/P3 hybrid as a result of the unique DNA‐binding mode of P3 is a key contributor to the observed rate acceleration. The results presented herein convincingly demonstrate that the concept of DNA‐based asymmetric catalysis can be expanded beyond Lewis acid catalyzed reactions. In view of the broad catalytic scope of metalloporphyrins, it is envisioned that the novel design presented herein is general and will allow further expansion of the concept of DNA‐based catalysis into the realm of organometallic chemistry.
Experimental Section
Representative procedure: Salmon testes DNA (6.0 mg mL−1; 9.0 mm in base pairs) was dissolved in a 20 mm solution of MOPS buffer (pH 6.5) 2 days before use. The following final concentrations were used in catalysis: 5 mm of the substrate 4‐methoxystyrene (4), 50 mm of ethyl diazoacetate (2), 37.5 μm of the iron porphyrin, and 6 mm of DNA (in base pairs). A 1.125 mm solution of the iron porphyrin (250 μL) was added to MOPS buffer (pH 6.5, 2.0 mL) in a 15 mL Greiner tube. Then, 5 mL of the st‐DNA solution was added, and the mixture was mixed by continuous inversion at 5 °C. After incubation for 30 min, 50 μL of a 750 mm solution of 4‐methoxystyrene (4) in acetonitrile and 200 μL of a 1.875 m solution of ethyl diazoacetate (2) in acetonitrile were added to start the catalytic reaction. After 5 min, the product was extracted with diethyl ether (3×5 mL). The organic layer was washed with brine (1×5 mL). After drying (Na2SO4) and evaporation of the solvent, the crude product was analyzed by HPLC and GC with 2‐methylanisole as an internal standard.
Dedicated to Professor Ben L. Feringa on the occasion of his 65th birthday
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support from NRSC‐Catalysis, the European Research Council (ERC starting grant 280010), the Ramón Areces Foundation (postdoctoral grant to A.R.‐M.), and the Netherlands Ministry of Education, Culture, and Science (Gravitation program no. 024.001.035) is gratefully acknowledged.
A. Rioz-Martínez, J. Oelerich, N. Ségaud, G. Roelfes, Angew. Chem. Int. Ed. 2016, 55, 14136.
References
- 1. Rosati F., Roelfes G., ChemCatChem 2010, 2, 916–927. [Google Scholar]
- 2.
- 2a. Roelfes G., Feringa B. L., Angew. Chem. Int. Ed. 2005, 44, 3230–3232; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3294–3296; [Google Scholar]
- 2b. Boersma A. J., Megens R. P., Feringa B. L., Roelfes G., Chem. Soc. Rev. 2010, 39, 2083–2092; [DOI] [PubMed] [Google Scholar]
- 2c. Park S., Sugiyama H., Angew. Chem. Int. Ed. 2010, 49, 3870–3878; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3960–3969; [Google Scholar]
- 2d. Rioz-Martínez A., Roelfes G., Curr. Opin. Chem. Biol. 2015, 25, 80–87. [DOI] [PubMed] [Google Scholar]
- 3.For selected recent examples, see:
- 3a. Amirbekyan K., Duchemin N., Benedetti E., Joseph R., Colon A., Markarian S. A., Bethge L., Vonhoff S., Klussmann S., Cossy J., Vasseur J.-J., Arseniyadis S., Smietana M., ACS Catal. 2016, 6, 3096–3105; [Google Scholar]
- 3b. Park S., Okamura I., Sakashita S., Yum J. H., Acharya C., Gao L., Sugiyama H., ACS Catal. 2015, 5, 4708–4712; [Google Scholar]
- 3c. Dey S., Jäschke A., Angew. Chem. Int. Ed. 2015, 54, 11279–11282; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11432–11436; [Google Scholar]
- 3d. Li Y., Cheng M., Hao J., Wang C., Jia G., Li C., Chem. Sci. 2015, 6, 5578–5585; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Wang J., Benedetti E., Bethge L., Vonhoff S., Klussmann S., Vasseur J.-J., Cossy J., Smietana M., Arseniyadis S., Angew. Chem. Int. Ed. 2013, 52, 11546–11549; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11760–11763; [Google Scholar]
- 3f. Li Y., Wang C., Jia G., Lu S., Li C., Tetrahedron 2013, 69, 6585–6590; [Google Scholar]
- 3g. Park S., Ikehata K., Watabe R., Hidaka Y., Rajendran A., Sugiyama H., Chem. Commun. 2012, 48, 10398–10400; [DOI] [PubMed] [Google Scholar]
- 3h. Megens R. P., Roelfes G., Chem. Commun. 2012, 48, 6366–6368; [Google Scholar]
- 3i. Boersma A. J., Coquière D., Geerdink D., Rosati F., Feringa B. L., Roelfes G., Nat. Chem. 2010, 2, 991–995. [DOI] [PubMed] [Google Scholar]
- 4. Fournier P., Fiammengo R., Jäschke A., Angew. Chem. Int. Ed. 2009, 48, 4426–4429; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4490–4493. [Google Scholar]
- 5. Cheng M., Li J., Zhou J., Jia G., Lu S.-M., Yang Y., Li C., Chem. Commun. 2016, 52, 9644–9647. [DOI] [PubMed] [Google Scholar]
- 6. Oelerich J., Roelfes G., Chem. Sci. 2013, 4, 2013–2017. [Google Scholar]
- 7.
- 7a. Morandi B., Dolva A., Carreira E. M., Org. Lett. 2012, 14, 2162–2163; [DOI] [PubMed] [Google Scholar]
- 7b. Nicolas I., Le Maux P., Simonneaux G., Tetrahedron Lett. 2008, 49, 5793–5795. [Google Scholar]
- 8.
- 8a. Coelho P. S., Brustad E. M., Kannan A., Arnold F. H., Science 2013, 339, 307–310; [DOI] [PubMed] [Google Scholar]
- 8b. Coelho P. S., Wang Z. J., Ener M. E., Baril S. A., Kannan A., Arnold F. H., Brustad E. M., Nat. Chem. 2014, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Bordeaux M., Tyagi V., Fasan R., Angew. Chem. Int. Ed. 2015, 54, 1744–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1764–1768; [Google Scholar]
- 9b. Tyagi V., Fasan R., Angew. Chem. Int. Ed. 2016, 55, 2512–2516; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2558–2562. [Google Scholar]
- 10.
- 10a. Wallace S., Balskus E. P., Angew. Chem. Int. Ed. 2016, 55, 6023–6027; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6127–6131; [Google Scholar]
- 10b. Wallace S., Balskus E. P., Angew. Chem. Int. Ed. 2015, 54, 7106–7109; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7212–7215. [Google Scholar]
- 11.
- 11a. Tovmasyan A., Weitner T., Sheng H., LU M., Rajic Z., Warner D. S., Spasojevic I., Roucas J. S., Benov L., Batinic-Haberle I., Inorg. Chem. 2013, 52, 5677–5691; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Batinić-Haberle I., Spasojević I., Hambright P., Benov L., Crumbliss A. L., Fridovich I., Inorg. Chem. 1999, 38, 4011–4022. [Google Scholar]
- 12.
- 12a. Yun B. H., Jeon S. H., Cho T.-S., Yi S. Y., Sehlstedt U., Kim S. K., Biophys. Chem. 1998, 70, 1–10; [DOI] [PubMed] [Google Scholar]
- 12b. Pasternack R. F., Gibbs E. J., Villafranca J. J., Biochemistry 1983, 22, 2406–2416; [DOI] [PubMed] [Google Scholar]
- 12c. Carvlin M. J., Fiel R. J., Nucleic Acids Res. 1983, 11, 6121–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The trans/cis ratio in all experiments was >9:1; in many cases, the yield and ee value of the minor cis isomer could not be reliably quantified, and therefore it was left out of consideration.
- 14.Only minor quantities of diethyl fumarate were observed.
- 15.In contrast, magnetic stirring did cause a significant change in the results of catalysis (see Table S3).
- 16. Baumann L. K., Mbuvi H. M., Du G., Woo L. K., Organometallics 2007, 26, 3995–4002. [Google Scholar]
- 17. Batinić-Haberle I., Benov L., Spasojević I., Fridovich I., J. Biol. Chem. 1998, 273, 24521–24528. [DOI] [PubMed] [Google Scholar]
- 18. Omagari T., Suzuki A., Akita M., Yoshizawa M., J. Am. Chem. Soc. 2016, 138, 499–502. [DOI] [PubMed] [Google Scholar]
- 19. Srivastava P., Yang H., Ellis-Guardiola K., Lewis J. C., Nat. Commun. 2015, 6, 8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Otte M., Kuijpers P. F., Troeppner O., Ivanović-Burmazović I., Reek J. N. H., de Bruin B., Chem. Eur. J. 2014, 20, 4880–4884. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Draksharapu A., Boersma A. J., Browne W. R., Roelfes G., Dalton Trans. 2015, 44, 3656–3663; [DOI] [PubMed] [Google Scholar]
- 21b. Boersma A. J., Feringa B. L., Roelfes G., Angew. Chem. Int. Ed. 2009, 48, 3346–3348; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3396–3398; [Google Scholar]
- 21c. Boersma A. J., Klijn J. E., Feringa B. L., Roelfes G., J. Am. Chem. Soc. 2008, 130, 11783–11790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary