

Abstract
Supramolecular split‐enzyme complementation restores enzymatic activity and allows for on–off switching. Split‐luciferase fragment pairs were provided with an N‐terminal FGG sequence and screened for complementation through host‐guest binding to cucurbit[8]uril (Q8). Split‐luciferase heterocomplex formation was induced in a Q8 concentration dependent manner, resulting in a 20‐fold upregulation of luciferase activity. Supramolecular split‐luciferase complementation was fully reversible, as revealed by using two types of Q8 inhibitors. Competition studies with the weak‐binding FGG peptide revealed a 300‐fold enhanced stability for the formation of the ternary heterocomplex compared to binding of two of the same fragments to Q8. Stochiometric binding by the potent inhibitor memantine could be used for repeated cycling of luciferase activation and deactivation in conjunction with Q8, providing a versatile module for in vitro supramolecular signaling networks.
Keywords: cooperativity, cucurbit[8]uril, split-luciferase, supramolecular chemical biology, switching
The field of supramolecular chemistry has long been inspired by biological systems.1, 2 Supramolecular systems have become increasingly complex, creating new opportunities for interfacing with biology.3, 4, 5 As evidence of this, supramolecular architectures have been generated that function as platforms for the dimerization, assembly, or functional modulation of proteins, providing orthogonal control and reversible switching.6, 7, 8, 9, 10 The generation of orthogonal synthetic systems mimicking cellular components, via in vitro synthetic biological networks, constitutes a landmark objective.11, 12 By combining proteins with synthetic supramolecular systems, we stand to benefit from the unique structural and functional features of both, and broaden the functionality of signaling systems. Notwithstanding the level of sophistication and control in natural systems,13, 14 impressive first examples of supramolecular systems on the way to these goals have been reported.15, 16, 17
Cucurbit[8]uril (Q8) is a cyclic glycoluril‐derived supramolecular host system capable of binding simultaneously to two N‐terminal phenylalanine residues.18 Q8 has demonstrated significant potential as a scaffold for the formation of supramolecular protein complexes,19, 20 as well as to modulate the function of biomaterials.21, 22, 23 Here Q8 is used to reconstitute a split‐protein system, split‐luciferase,24 via selective stabilization of the native split‐protein heterocomplex, enabling reversible signal generation, a large dynamic range, and a generic approach to control protein activity in an in vitro setting (Scheme 1).
Scheme 1.
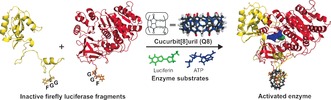
Overview of the designed supramolecular split‐luciferase complementation system. Two inactive split‐firefly luciferase fragments are complemented in a controlled manner through Q8‐binding, forming a ternary complex.
Luciferases and their split variants have mainly been used in cellular systems by fusing the split‐luciferase elements to proteins of interest.25, 26, 27, 28 The specific advantages of split‐luciferases, namely a large dynamic range and a real‐time (direct) signal of complex formation, make them very attractive read‐out signals for in vitro signaling networks. The limited number of studies on purified split‐luciferase fragments and their potential limited stability has thus far hampered their broad application to in vitro systems and limited the molecular insights in split‐luciferase complementation.23, 24, 25, 26 Therefore, we first set out to discover appropriate split‐luciferase fragments that can be bacterially expressed and purified without large stabilizing fusion proteins. Reported examples of split‐firefly luciferase pairs (Fluc) were used as a starting point to explore three N‐ and C‐terminal fragments Fluc(1–416)/(398–550),29 Fluc(1–437)/(438–550),30 and Fluc(1–475)/(265–550) (Table S1).31 First, these fragment pairs were linked via a flexible (GGS)12 amino acid linker. Only the Fluc(1–416)/(398–550) and Fluc(1–437)/(438–550) combinations were obtained in sufficient expression yields and enzymatic activity (Supporting Information, Figures S1, S2). The N‐ and C‐terminal fragments (NFluc and CFluc) of these constructs were subsequently expressed individually, featuring an N‐terminal FGG sequence motif, amenable to Q8‐binding (Supporting Information, Table S1). The NFluc437, CFluc438, and CFluc398 split fragments expressed well (Supporting Information, Figure S3).
For selection of the optimal split‐luciferase pair, Q8‐mediated complementation was screened for by mixing the split‐luciferase fragments with Q8 and recording light emission on addition of luciferase assay reagent (Promega) (Scheme 1). While each combination showed some background enzyme activity (Supporting Information, Figure S4), the NFluc437‐CFluc438 combination did not respond to Q8 addition, which is potentially related to the impossibility to bridge the two N‐termini of these split fragments. In contrast, the combination of NFluc437 with CFluc398 resulted in a 9‐fold increase in enzymatic activity upon addition of Q8 (Supporting Information, Figure S4). The CFluc398 fragment, alone or in presence of Q8, did not show activity, while only minor background activity was observed for the NFluc437 fragment (Supporting Information, Figure S5), independent of Q8 and in line with previous reports on N‐terminal luciferase domains.29, 32, 33
The selected NFluc437 and CFluc398 split‐luciferase fragments were expressed on a milligram scale (Supporting Information, Figure S6) and their intrinsic affinity was determined by monitoring the luciferase activity of 0.5 μm NFluc437 as a function of CFluc398 concentration (Figure 1). This revealed a weak background affinity between the two luciferase fragments. Fitting the light intensity at t=50 minutes using a one‐site specific binding model yields a dissociation constant of K d NC=112±12 μm. This low affinity makes the split‐luciferase pair ideally suited for Q8‐mediated complementation, as strong binding would result in Q8‐independent complementation, whereas an absence of intrinsic affinity would not support functional active site complementation.
Figure 1.
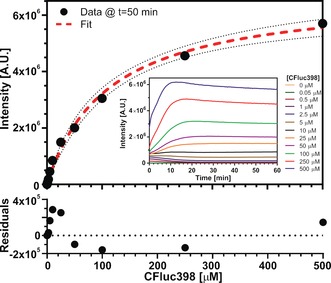
Titration of CFluc398 to NFluc437. The intensity at t=50 minutes at the different CFluc398 concentrations minus the intensity in the absence of CFluc398 was fitted using a one‐site‐specific binding model (dotted red line). The dotted black line represents the 95 % confidence band and the bottom graph shows the residuals of the fit. The NFluc437 was present at 0.5 μm. Inset: full time‐traces at different concentrations of CFluc398. Luciferase assay reagent (LAR) (Promega) in 20 mm sodium phosphate, 150 sodium chloride, 1 mm EDTA, pH 7, 30 °C.
The enzymatic and spectral characteristics of the NFluc437‐CFluc398 pair were compared with those of full length luciferase (see the Supporting Information, Figure S7 for protein characterization). The split‐luciferase, expectedly, showed lower enzymatic activity compared to the full‐length protein. The full‐length luciferase showed the typical burst kinetics on the seconds time scale, whereas the steady‐state kinetics of the split‐luciferase was only achieved after several minutes. While luciferase burst kinetics are not well understood,38 the observed difference could be explained by a combination of slower accumulation of inhibitory products due to a lower turnover number39 and differences in protein folding and complementation kinetics during the substrate oxidation process.40 The emission spectrum of both systems was identical in shape and emission maximum at 555 nm (Supporting Information, Figure S8). This shows that the enzyme becomes functional after complementation and that the amino acid surrounding of the substrate is conserved in the split‐luciferase construct.34, 35, 36, 37
Next we investigated Q8‐mediated split‐luciferase complementation by measuring the luciferase activity of an equimolar NFluc437–CFluc398 mixture as a function of Q8 concentration (Figure 2 A). The maximum enhancement of the luciferase activity (20‐fold) was attained on addition of 2.5 μm Q8 to 0.5 μm of each of the split‐luciferase fragments. A further increase in Q8 concentration resulted in down‐regulation of the luciferase activity, with concomitant changes in the kinetic profile (Supporting Information, Figure S9). At such high concentrations, the Q8 is expected to bind to other aromatic amino acids of the split fragments in addition to the two N‐terminal phenylalanine residues, thereby influencing the split‐luciferase conformation and activity.41, 42, 43, 44 In the absence of one of the FGG motifs, no responsiveness to Q8 was observed (Supporting Information, Figure S10).
Figure 2.
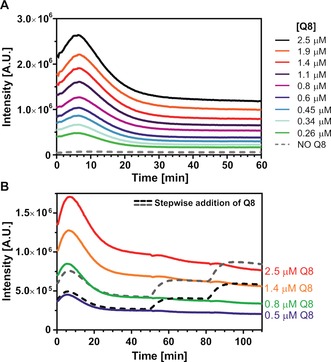
Q8‐dependence of split‐luciferase activity. A) Pre‐incubation with increasing Q8 concentrations results in a higher enzymatic activity. B) The enzymatic activity of the split‐luciferase can be controlled step‐wise by Q8. The colored lines depict the activity of different amounts of premixed Q8 with 0.5 μm of each split‐luciferase fragment. Addition of extra Q8 at steady state kinetics results in an increase of enzyme activity to the levels reached upon direct addition of the final overall concentration. Conditions: see Figure 1 caption.
The supramolecular nature of the split‐protein complementation enables the step‐wise upregulation of split‐luciferase enzymatic activity (Figure 2 B). The data reveal that the steady state kinetics correlate with the total amount of Q8, independent of the order of addition. This precise regulation of the amount of active enzyme, independent of the initial Q8 concentration, bodes well for usage as a switching platform.
The reversibility of the supramolecular split‐luciferase complementation was explored using two Q8 inhibitors; the weak Q8‐binding tripeptide FGG (forming Q8‐(FGG)2 with Kter=1.5×1011 m −2)18 and the strong Q8‐binding small molecule memantine (forming Q8‐memantine with K a=4.3×1011 m −1).45 Different concentrations of inhibitors were added when luciferase activity of the Q8⋅split‐luciferase complex had reached steady‐state level, resulting in a concentration dependent decrease in enzymatic activity (Figure 3). The enzymatic activity after partial disassembly remained constant for at least one hour, showing the high stability of the system. Complete inhibition was observed upon addition of an equimolar concentration of memantine (0.5 μm), consistent with the high affinity of memantine for Q8. For the FGG tripeptide, a 500 μm concentration was required to fully revert the split‐luciferase activity. This reveals strong cooperativity in the supramolecular protein complex formation.
Figure 3.
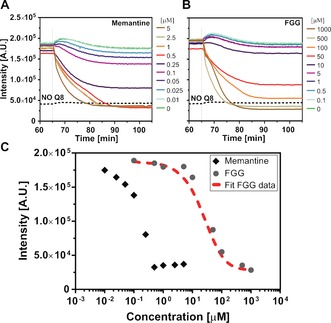
Q8‐mediated split‐luciferase complexes disassembled using Q8 inhibitors. A),B) At steady state conditions the inhibitors A) memantine and B) FGG were added at different concentrations. The split‐luciferase fragments and Q8 were present at 0.5 μm each. The temporary activity increase at t=65 min. results from a temperature recalibration upon inhibitor addition. C) The steady‐state light intensities at t=100 min are plotted against the inhibitor concentration. Using a binding model, the thermodynamic parameters that describe ternary homo‐ and heterocomplex formation were determined from the titration data using FGG as an inhibitor (red dotted line). Conditions: see Figure 1 caption.
The need of a large excess of the short FGG peptide to completely disassemble the Q8 assembled split‐luciferase complexes indicates that the Q8‐induced assemblies are additionally stabilized by interactions between the two split‐luciferase fragments.19 To obtain quantitative information on the strength of these interactions, we developed a thermodynamic model that describes the formation of the various Q8‐mediated complexes for a mixture of NFluc and CFluc in the presence of FGG competitor. The model contained two cooperativity parameters σ1 and σ2 to distinguish between split‐protein homo‐complex (NFluc‐NFluc and CFluc‐CFluc) and heterocomplex (NFluc‐CFluc) formation (Supporting Information). A non‐linear least‐squares fit of the FGG titration data (Figure 3 C) yielded a 300‐fold higher cooperativity parameters σ 2 for formation of the ternary heterocomplex compared to a complex consisting of two of the same fragments bound to Q8. A ternary equilibrium constant for heterocomplex formation could be derived (K d ter_het=9.1×10−12 m 2) which was much higher than that for homo‐complex formation (K d ter_hom=5.5×10−9 m 2). There is thus a strong cooperativity between the Q8‐mediated binding to the split‐protein N‐termini and the intrinsic affinity of the two luciferase fragments. This results in efficient formation of the active supramolecular split‐heterocomplex over split‐homocomplex.
The tight binding of memantine to Q8 is ideal for reversible switching of the split‐luciferase activity. The alternated addition of Q8 and memantine allows formation and disassembly of the split‐protein heterodimer (Figure 4). Addition of memantine to Q8‐activated split‐luciferase lowered the enzymatic activity back to background activity. The effect of inhibition was independent of the order of addition (Figure 4, compare green and purple lines). Subsequent addition of fresh Q8 in two‐fold excess allowed the recomplementation of the split‐luciferase complex and concomitant full enzymatic activation. The very strong memantine‐Q8 interaction thus enables repeated cycling of split‐luciferase assembly, with only small excesses of either of the two components.
Figure 4.
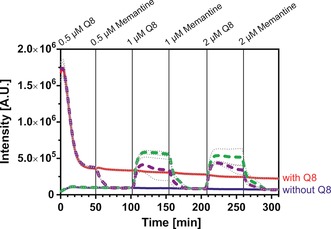
Assembly and disassembly of the supramolecular split‐luciferase complex over several cycles upon sequential addition of the inhibitor memantine and Q8. Red and blue lines represent the system in the constant presence and absence of Q8. The dotted lines represent the assembly and disassembly processes. The purple dotted line started from a complex were Q8 was present at t=0. For the green dotted line Q8 was added at t=100 min. The split fragments were present at 0.5 μm each. The average of three measurements was depicted; the dotted gray line represents the standard deviation. Conditions: see Figure 1 caption.
This work represents the first example of synthetic supramolecular control over a split‐protein system. The intrinsic weak residual affinity between the two fragments of the split‐luciferase pair is ideal for templated control of protein complementation. The affinity is weak enough to ensure low background activity, yet strong enough to ensure efficient heterocomplementation in the presence of the Q8 template. The binding of Q8 to the N‐termini of the split‐luciferase fragments results in an efficient formation of the reconstituted protein with functional enzymatic activity at sub‐micromolar concentrations. The supramolecular nature of the system allows for exact control over the level of enzyme activation and the tight Q8‐binder memantine allows for repeated enzyme activation and deactivation. This Q8‐mediated split‐luciferase system could potentially be used to monitor dynamic supramolecular signaling pathways. The general concept of stabilizing weak protein heterodimerization at low concentrations using a short genetically encoded tag and a small host molecule could potentially find use in a wide range of applications, from enzyme catalysis to signal transduction control.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funded by the Netherlands Organization for Scientific Research (NWO) via Complexity program 645.000.006 and Gravity program 024.001.035.
R. P. G. Bosmans, J. M. Briels, L.-G. Milroy, T. F. A. de Greef, M. Merkx, L. Brunsveld, Angew. Chem. Int. Ed. 2016, 55, 8899.
References
- 1. Bromfield S. M., Wilde E., Smith D. K., Chem. Soc. Rev. 2013, 42, 9184–9195. [DOI] [PubMed] [Google Scholar]
- 2. Klärner F.-G., Schrader T., Acc. Chem. Res. 2013, 46, 967–978. [DOI] [PubMed] [Google Scholar]
- 3. Caruso F., Hyeon T., Rotello V. M., Chem. Soc. Rev. 2012, 41, 2537–2538. [DOI] [PubMed] [Google Scholar]
- 4. Uhlenheuer D. A., Petkau K., Brunsveld L., Chem. Soc. Rev. 2010, 39, 2817–2826. [DOI] [PubMed] [Google Scholar]
- 5. Oohora K., Hayashi T., Curr. Opin. Chem. Biol. 2014, 18, 154–161. [DOI] [PubMed] [Google Scholar]
- 6. Zhang L., Wu Y., Brunsveld L., Angew. Chem. Int. Ed. 2007, 46, 1798–1802; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1830–1834. [Google Scholar]
- 7. Bier D., Rose R., Bravo-Rodriguez K., Bartel M., Ramirez-Anguita J. M., Dutt S., Wilch C., Klärner F.-G., Sanchez-Garcia E., Schrader T., Ottmann C., Nat. Chem. 2013, 5, 234–239. [DOI] [PubMed] [Google Scholar]
- 8. Petkau-Milroy K., Uhlenheuer D. A., Spiering A. J. H., Vekemans J. A. J. M., Brunsveld L., Chem. Sci. 2013, 4, 2886–2891. [Google Scholar]
- 9. Sankaran S., Kiren M. C., Jonkheijm P., ACS Nano 2015, 9, 3579–3586. [DOI] [PubMed] [Google Scholar]
- 10. Omichi M., Asano A., Tsukuda S., Takano K., Sugimoto M., Saeki A., Sakamaki D., Onoda A., Hayashi T., Seki S., Nat. Commun. 2014, 5, 3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hockenberry A. J., Jewett M. C., Curr. Opin. Chem. Biol. 2012, 16, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Roekel H. W. H., Rosier B. J. H. M., Meijer L. H. H., Hilbers P. A. J., Markvoort A. J., Huck W. T. S., de Greef T. F. A., Chem. Soc. Rev. 2015, 44, 7465–7483. [DOI] [PubMed] [Google Scholar]
- 13. Camacho-Soto K., Castillo-Montoya J., Tye B., Ghosh I., J. Am. Chem. Soc. 2014, 136, 3995–4002. [DOI] [PubMed] [Google Scholar]
- 14. Stein V., Alexandrov K., Proc. Natl. Acad. Sci. USA 2014, 111, 15934–15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peters R. J. R. W., Marguet M., Marais S., Fraaije M. W., van Hest J. C. M., Lecommandoux S., Angew. Chem. Int. Ed. 2014, 53, 146–150; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 150–154. [Google Scholar]
- 16. Betz K., Malyshev D. A., Lavergne T., Welte W., Diederichs K., Romesberg F. E., Marx A., J. Am. Chem. Soc. 2013, 135, 18637–18643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lygina A. S., Meyenberg K., Jahn R., Diederichsen U., Angew. Chem. Int. Ed. 2011, 50, 8597–8601; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8756–8760. [Google Scholar]
- 18. Heitmann L. M., Taylor A. B., Hart P. J., Urbach A. R., J. Am. Chem. Soc. 2006, 128, 12574–12581. [DOI] [PubMed] [Google Scholar]
- 19. Dang D. T., Nguyen H. D., Merkx M., Brunsveld L., Angew. Chem. Int. Ed. 2013, 52, 2915–2919; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2987–2991. [Google Scholar]
- 20. Nguyen H. D., Dang D. T., van Dongen J. L. J., Brunsveld L., Angew. Chem. Int. Ed. 2010, 49, 895–898; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 907–910. [Google Scholar]
- 21. Smith L. C., Leach D. G., Blaylock B. E., Ali O. A., Urbach A. R., J. Am. Chem. Soc. 2015, 137, 3663–3669. [DOI] [PubMed] [Google Scholar]
- 22. Rowland M. J., Atgie M., Hoogland D., Scherman O. A., Biomacromolecules 2015, 16, 2436–2443. [DOI] [PubMed] [Google Scholar]
- 23. Si C., Li J., Luo Q., Hou C., Pan T., Li H., Liu J., Chem. Commun. 2016, 52, 2924–2927. [DOI] [PubMed] [Google Scholar]
- 24. Shekhawat S. S., Ghosh I., Curr. Opin. Chem. Biol. 2011, 15, 789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Binkowski B. F., Butler B. L., Stecha P. F., Eggers C. T., Otto P., Zimmerman K., Vidugiris G., Wood M. G., Encell L. P., Fan F., Wood K. V., ACS Chem. Biol. 2011, 6, 1193–1197. [DOI] [PubMed] [Google Scholar]
- 26. Takakura H., Hattori M., Takeuchi M., Ozawa T., ACS Chem. Biol. 2012, 7, 901–910. [DOI] [PubMed] [Google Scholar]
- 27. Macdonald-Obermann J. L., Piwnica-Worms D., Pike L. J., Proc. Natl. Acad. Sci. USA 2012, 109, 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thorne N., Inglese J., Auld D. S., Chem. Biol. 2010, 17, 646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luker K. E., Smith M. C. P., Luker G. D., Gammon S. T., Piwnica-Worms H., Piwnica-Worms D., Proc. Natl. Acad. Sci. USA 2004, 101, 12288–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paulmurugan R., Umezawa Y., Gambhir S. S., Proc. Natl. Acad. Sci. USA 2002, 99, 15608–15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Paulmurugan R., Gambhir S. S., Anal. Chem. 2005, 77, 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paulmurugan R., Gambhir S. S., Anal. Chem. 2007, 79, 2346–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohmuro-Matsuyama Y., Chung C.-I., Ueda H., BMC Biotechnol. 2013, 13, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paley M. A., Prescher J. A., MedChemComm 2014, 5, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Branchini B. R., Magyar R. A., Murtiashaw M. H., Anderson S. M., Helgerson L. C., Zimmer M., Biochemistry 1999, 38, 13223–13230. [DOI] [PubMed] [Google Scholar]
- 36. Branchini B. R., Southworth T. L., Khattak N. F., Michelini E., Roda A., Anal. Biochem. 2005, 345, 140–148. [DOI] [PubMed] [Google Scholar]
- 37. Branchini B. R., Ablamsky D. M., Rosenman J. M., Uzasci L., Southworth T. L., Zimmer M., Biochemistry 2007, 46, 13847–13855. [DOI] [PubMed] [Google Scholar]
- 38. Marques S. M., Esteves da Silva J. C. G., IUBMB Life 2009, 61, 6–17. [DOI] [PubMed] [Google Scholar]
- 39. da Silva L. P., Esteves da Silva J. C. G., Photochem. Photobiol. Sci. 2011, 10, 1039. [DOI] [PubMed] [Google Scholar]
- 40. Zako T., Ayabe K., Aburatani T., Kamiya N., Kitayama A., Ueda H., Nagamune T., Biochim. Biophys. Acta Proteins Proteomics 2003, 1649, 183–189. [DOI] [PubMed] [Google Scholar]
- 41. Yang H., An Q., Zhu W., Li W., Jiang Y., Cui J., Zhang X., Li G., Chem. Commun. 2012, 48, 10633–10635. [DOI] [PubMed] [Google Scholar]
- 42. Sonzini S., Ryan S. T. J., Scherman O. A., Chem. Commun. 2013, 49, 8779–8781. [DOI] [PubMed] [Google Scholar]
- 43. Smith L. C., Leach D. G., Blaylock B. E., Ali O. A., Urbach A. R., J. Am. Chem. Soc. 2015, 137, 3663–3669. [DOI] [PubMed] [Google Scholar]
- 44. Bosmans R. P. G., Hendriksen W. E., Verheijden M., Eelkema R., Jonkheijm P., van Esch J. H., Brunsveld L., Chem. Eur. J. 2015, 21, 18466–18473. [DOI] [PubMed] [Google Scholar]
- 45. Liu S., Ruspic C., Mukhopadhyay P., Chakrabarti S., Zavalij P. Y., Isaacs L., J. Am. Chem. Soc. 2005, 127, 15959–15967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary