

Summary
Chromosomal inversions can provide windows onto the cytogenetic, molecular, evolutionary and demographic histories of a species. Here we investigate a paracentric 1.17‐Mb inversion on chromosome 4 of Arabidopsis thaliana with nucleotide precision of its borders. The inversion is created by Vandal transposon activity, splitting an F‐box and relocating a pericentric heterochromatin segment in juxtaposition with euchromatin without affecting the epigenetic landscape. Examination of the RegMap panel and the 1001 Arabidopsis genomes revealed more than 170 inversion accessions in Europe and North America. The SNP patterns revealed historical recombinations from which we infer diverse haplotype patterns, ancient introgression events and phylogenetic relationships. We find a robust association between the inversion and fecundity under drought. We also find linkage disequilibrium between the inverted region and the early flowering Col‐FRIGIDA allele. Finally, SNP analysis elucidates the origin of the inversion to South‐Eastern Europe approximately 5000 years ago and the FRI‐Col allele to North‐West Europe, and reveals the spreading of a single haplotype to North America during the 17th to 19th century. The ‘American haplotype’ was identified from several European localities, potentially due to return migration.
Keywords: chromosome rearrangement, Arabidopsis thaliana, transposon, phylogenetic relationship, introgression, haplotype pattern
Significance Statement
Structural rearrangements redefine chromosomes, shape genome diversity and can have profound effects on selection, adaptation and spread. Here we elucidate the history of a paracentric inversion in Arabidopsis thaliana, including its origin a few thousand years ago, its maintenance under certain environmental conditions and its migration patterns, from Europe to North America and back.
Introduction
Eukaryotic chromosomes evolve through a combination of processes, associated with changes in number, morphology and organization. Most dramatic events follow nuclear restitution or unequal chromosome segregation leading to loss or gain of complete genomes or chromosomes. Likewise, breakage and ligation of chromosomal segments can lead to rearrangements of the chromosome structure, such as inversions, translocations, centric split and fusion, duplications and deletions. Comparative cytogenetic studies have revealed chromosomal rearrangements in several species, in particular fruit flies and mosquitos (Coluzzi et al., 2002; Hoffmann et al., 2004; Bhutkar et al., 2008; Kirkpatrick, 2010), but also in mammals including humans (Stefansson et al., 2005; Bhatt et al., 2009; Knebel et al., 2011) and plants (Laufs et al., 1999; Linc et al., 1999; Jiang et al., 2007; Lowry and Willis, 2010; Lukaszewski et al., 2011; Szinay et al., 2012). Recent developments in high‐throughput genome technologies have provided us with a wealth of information about SNP and structural diversity both between and within species (Feuk et al., 2006; Faraut, 2008; Rowan et al., 2015). Chromosome painting studies have allowed for the detection and visualization of many translocations and inversions, for example, in the Brassicaceae family (Lysak et al., 2003, 2006, 2010), the Solanaceae family (Iovene et al., 2008; Tang et al., 2008; Anderson et al., 2010; Lou et al., 2010; Wu and Tanksley, 2010; Szinay et al., 2012) and the grass family (Hasterok, 2006; Febrer et al., 2010; Betekhtin et al., 2014). Furthermore, de novo genome assemblies of related species enabled multiple genome alignment studies giving rise to unprecedented comparative genomics with detailed information of inversions, translocations and other structural variants (Kurtz et al., 2004; Pop et al., 2004; Ohtsubo et al., 2008; Cheung et al., 2009; Darling et al., 2010; Zapata et al., 2016).
In natural populations inversions seem to be the most prevalent large‐scale structural chromosome variants, and are found in subspecies, accessions and related wild genotypes (Madan, 1995). However, when a chromosomal inversion arises as a low‐frequency novel mutation, it mostly exists in the heterozygous state where single crossover events inside the inversion loop create unviable gametes or progenies with aberrant chromosomes. Given that an inversion may be deleterious when rare, several hypotheses explain its high frequency in populations despite the initial detrimental effect. It may increase by genetic drift, especially in a self‐fertilizing species where heterozygotes are rare. It may increase by the hitchhiking with a linked adaptive allele. The inversion event may also create advantageous effects by creating a new open reading frame, disrupting an existing gene, or changing the expression profile or epigenetic marks of genes near the breakpoints (for example, position‐effect variegation; Muller, 1930). Finally, the inversion could capture adaptive alleles of multiple genes and prevent maladaptive recombination, creating a ‘super‐gene’ affecting multiple adaptive traits (Dobzhansky, 1970; Kirkpatrick and Barton, 2006). These factors are not mutually exclusive, and an inversion may be driven to high frequency by the combination of several factors. Indeed, the advantageous effects of chromosomal inversions have been reported. For example, the contribution of an inversion to local adaptation followed by ecological reproductive isolation between perennial and annual ecotypes of Mimulus guttatus has been demonstrated for the yellow monkey‐flower (Lowry and Willis, 2010). Furthermore, a ±50‐Mb‐sized inversion has been described in wild subspecies of Zea mays (parviglumis and mexicana), but not in domesticated maize. This inversion polymorphism shows evidence of adaptive evolution, as it demonstrates a strong altitudinal cline with multiple environmental and phenotypic traits (Fang et al., 2012).
Previous work identified a heterochromatic knob and associated centromere rearrangements on the short‐arm of chromosome 4 of the model plant Arabidopsis thaliana (Fransz et al., 2000; The Cold Spring Harbor Laboratory, 2000). Specifically, the knob was identified in the Columbia accession that was used for reference genome sequencing (The Arabidopsis Genome Initiative, 2000). It was speculated that the knob resulted from a paracentric inversion event; however, this has not yet been definitively demonstrated. Also, it is unknown how many accessions of Arabidopsis carry the putative inversion polymorphism. The widespread distribution of the inversion would have important implications for genetic mapping and/or genome‐wide association studies (GWAS) of Arabidopsis, as a large, essentially non‐recombining, part of the genome could bear extensive long‐range linkage disequilibrium (LD), reducing the resolution of genetic mapping. Early studies using Arabidopsis did in fact identify extensive LD on the top of the short‐arm of chromosome 4 associated with loss of function mutations to the flowering time gene FRIGIDA (Nordborg et al., 2002). There are multiple independent knockout mutations of FRI, including a partial gene deletion in the Columbia allele (FRI‐Col) that is known to exist in multiple accessions (Shindo et al., 2005). GWAS using diverse sets of Arabidopsis accessions have found associations between early‐flowering and FRI‐mutations, but also associations with genetic markers several Mb away near the centromere and the putative inversion (Li et al., 2010). With recent developments in cytogenetics and the (near‐) completion of the Arabidopsis 1001 genomes project, it is now possible to examine the mechanistic, evolutionary and demographic implications of an inversion in Arabidopsis.
Here we present the molecular mechanism of how the inversion on chromosome 4 of A. thaliana has arisen through activity of a Vandal transposable element. We describe genetic and epigenetic effects of this inversion on flanking chromosomal regions, and show ancient recombination events that define accession‐specific haplotypes pointing at historical introgressions between ancestors of the current accessions. We report the worldwide distribution of the inversion haplotype to more than 170 European and North American accessions. Using genome‐wide association mapping we propose that the presence of the inverted regions may have provided increased fitness under certain drought stress conditions. In addition, selection for early‐flowering, via linked mutations to the flowering time gene FRIGIDA, may have contributed to the maintenance and/or spread of the inversion. The inversion most likely originated in South‐East Europe and spread to other populations through repeated introgression events. The unique recombination sites enabled us to track migration both to and returning from North America.
Results and discussion
An inversion event spanning 1.17 Mb leads to the formation of the heterochromatic knob hk4S
The heterochromatic knob (hk4S) had earlier been mapped to the short‐arm of chromosome 4 in the accessions Col‐0 and Ws‐2, but absent from Ler and C24 (Fransz et al., 1998). The knob was supposed to be the result of a chromosomal rearrangement (Fransz et al., 2000; The Cold Spring Harbor Laboratory, Washington University Genome Sequencing Center, and PE Biosystems Arabidopsis Sequencing Consortium*, 2000), possibly an inversion; if so, it would explain the lack of recombination in the genetic maps of Col × Ler (Schmidt et al., 1995; Drouaud et al., 2006; Giraut et al., 2011). We confirmed the lower recombination frequency in crosses between Col and Ws‐2 versus the knobless accession Ler. We observed considerably lower recombination frequencies between the classical markers ga1, close to hk4S, and bp in the crosses Ler × Col and Ler × Ws‐2 compared with the Ler (wt) × Ler triple mutant (ga1, bp and tt11) cross (Table S1). Meiosis in the latter segregating population is likely not affected by any structural rearrangements (Koornneef et al., 1983). A more detailed amplified fragment length polymorphism (AFLP) analysis using crosses between Col and Ler revealed a 1.13 Mb region at 340 kb from ga1 in which no recombination was found (Figure S1).
To assess the nature and the exact position of the rearrangement in chromosome 4, we applied BAC‐fluorescent in situ hybridization (FISH) painting on pachytene chromosomes (Figure 1a and b) of knob accession Ws‐2 and knobless accessions (C24, Ler, Zh, No‐0), and compared the results with the reference sequence map of the knob accession Col‐0. BAC contig F9H3‐T5L23, which maps to the distal border of the knob hk4S in Col, gave a bright signal near the knob in Ws‐2, whereas in C24 the T5L23 signal is located in pericentromere heterochromatin, indicating that T5L23 is involved in the rearrangement. Similar results were obtained with the BAC contig T27D20‐T19B17 proximal from the knob. Any combination of BACs from the hk4S region or the proximal euchromatin showed an inversed position comparing knob accessions with knobless accessions (Figure S1). The data confirm with unrefutable evidence that a paracentric inversion event has occurred that moved the distal part of the pericentromere heterochromatin into interstitial euchromatin, thus generating the heterochromatic knob hk4S. Microscopic analysis of microsporocytes at pachytene stage in the F1 from Ler × Col revealed an opposite orientation of BAC pool signals in the chromosome arm 4S, supporting a paracentric inversion (Figure S1). We did not observe clear inversion loops and only few anaphase I bridges. Similarly, Lamb et al. (2007a) and Schneider et al. (2016), who studied inversions in centromere regions of maize, found no evidence for loop formation and did not find recombination events within the inversion breakpoints. The chromosomal rearrangement event having one of the breakpoints within a heterochromatic region is a good explanation for the formation of interstitial heterochromatic knobs, instead of other arguments, such as: (i) spontaneous or transposon driven transfer of distal satellite repeats (Zhong et al., 1998; Szinay, 2010); (ii) DNA methylation and heterochromatinization (Soppe et al., 2002; Golyshev et al., 2008; Zhang et al., 2008; Kejnovsky et al., 2009); and (iii) (retro)transposon accumulation (Ananiev et al., 1998; Lamb et al., 2007b).
Figure 1.
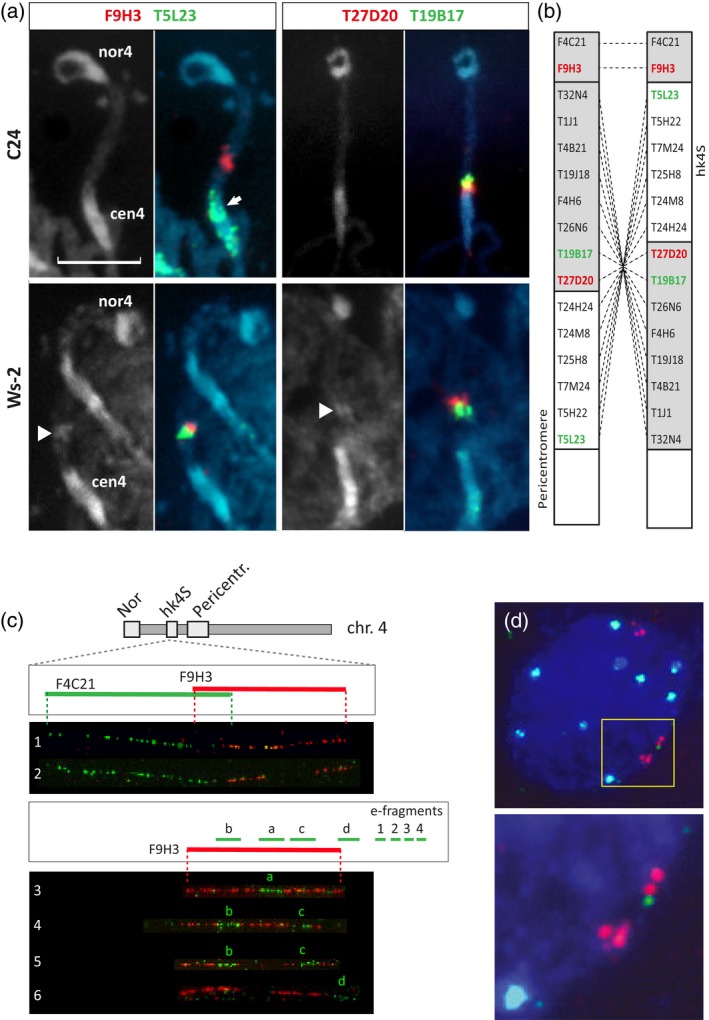
Cytogenetic characterization of the short‐arm of chromosome 4.
(a) DAPI‐stained images of the short‐arm of chromosome 4 in C24 (top row) and Ws‐2 (bottom row). Note the presence of the knob hk4S in the accession Ws‐2 (arrowheads). The fluorescent in situ hybridization (FISH) signals show hybridization with probes from BACs F9H3, T27D20, T19B17 and the repeat‐rich BAC T5L23 to C24 (top row) and Ws‐2 (bottom row). The arrow points at the green repeat‐rich T5L23 signals in the pericentromere of C24.
(b) Relative map positions of the BAC DNA clones that are used for the FISH. The right diagram shows the map for Col‐0 and the left diagram represents a non‐inversion accession. Gray rectangles are euchromatin, white rectangles are heterochromatin.
(c) FISH to DNA fibers from Col‐0 (1, 3, 4) and C24 (2, 5, 6) using BACs F9H3 (red), F4C21 (green in 1,2) and 10 kb DNA fragments (green in 3–6). The a, b, c and d signals in the DNA fibers 3–6 correspond to the small DNA fragments (a–d) as shown in the top drawing.
(d) Interphase FISH to Col‐0 using F9H3 (red) as reference probe and fragment e3 (green) mapped proximal from F9H3 (see schematic drawing in c). The top image shows the whole nucleus with one green signal adjacent to reference probe F9H3 (red) shown in the yellow frame. The bottom image is a magnification of the part in the yellow frame. All pachytene and interphase nuclei preparations are counterstained with DAPI (blue). cen4, centromere of chromosome 4; nor4, nucleolar organizer of chromosome 4. Scale bar: 3 μm.
Because hk4S forms the distal breakpoint of the inversion, we applied fiber‐FISH using F9H3 as the reference BAC to fine map the boundary of the inversion. Knobless accessions showed a discontinuity in the F9H3 signal of approximately 32 kb, indicating a rearrangement in this region between knob and knobless accessions (Figure 1c; Table S2). Sub‐fragments of F9H3 flanking the 32 kb region, however, revealed no differences between knob and knobless accession, indicating that the inversion breakpoint is proximal of the F9H3 region. Further examination of the flanking T5L23 region by fiber‐FISH was, however, hampered by the presence of repetitive elements in the knob (The Cold Spring Harbor Laboratory, Washington University Genome Sequencing Center, and PE Biosystems Arabidopsis Sequencing Consortium*, 2000). Hence, we hybridized small fragments of T5L23 (e1–e4; Table S3) to interphase nuclei and quantified the frequency of their juxtaposition to the reference BAC F9H3 (Figure 1d; Table 1). The interphase‐FISH data of the fragments e1, e2, e3 and e4 showed almost 100% juxtaposition to F9H3 in Col‐0. In contrast, in the knobless accessions Ler and C24, only fragments e1 and e2 are adjacent to F9H3 in more than 90% of the cases. Signals of fragment e3 flank the F9H3 signal in 44% frequency of the cases, while the most proximal fragment e4 shows only few signals adjacent to F9H3. These results indicate that the breakpoint of the paracentric inversion maps to the e3 fragment. Hence, the distal breakpoint of the inversion maps at position 1614 ± 2 kb. Similarly, we estimate the proximal border of the inversion at 2800 ± 50 kb. These positions match the AFLPs (Figure S1) that flank the cold spot of recombination.
Table 1.
Percentage of FISH signals from e‐fragments associated with BAC F9H3
| PCR fragment | e1 | e2 | e3 | e4 |
|---|---|---|---|---|
| Col (n = 50) | 97% | 100% | 100% | 100% |
| C24 (n = 50) | 94% | 90% | 48% | 4% |
| Ler (n = 50) | 94% | 90% | 44% | 8% |
The inversion is generated by Vandal transposon activity
A BLAST search (www.arabidopsis.org/Blast/cereon.jsp) revealed a match between fragment e3 and Ler fragment ATL8C23344. Strikingly, this 2.8 kb Ler sequence corresponds to both distal and proximal breakpoints of the inversion. More precisely, two major fragments of ATL8C23344 (857 bp and 1176 bp, respectively) show 98% identity to the distal (At4 g03635, score = 1507 bits) and proximal (At4 g05500, score = 2078 bits) breakpoint regions of the inversion in Col‐0 (Figure S2). The Ler fragment thus provided us the precise map positions of the distal and proximal breakpoints at 1 612 609 bp and 2 782 618 bp, respectively. The de novo assembly of the Ler‐0 sequence confirmed the two breakpoint positions (Zapata et al., 2016). The entire inversion spans 1.17 Mb and contains 145 protein‐coding genes.
Strikingly, comparison of the Ler sequence and the reconstructed Col‐0 sequence showed no differences at the proximal and distal breakpoints of the inversion, indicating that the transposition generated a clean split in the F‐box gene (Figures S3 and S4). The high level identity between the two accessions further suggests that the sequence around the breakpoints has not changed since the inversion event. Moreover, a 945 bp fragment of this region, covering the distal breakpoint, is 95% identical with the putative protein‐coding gene [ARALYDRAFT_911822 a DNA fragment (XM_002874807.1)] of the related species A. lyrata (Figure S4), indicating that the sequence spanning the distal breakpoint has not significantly changed since A. thaliana and A. lyrata diverged.
Interestingly, several sequences that flank the distal side of the two breakpoints are annotated as F‐box protein‐coding genes of the RNI‐like superfamily (https://www.arabidopsis.org), while the sequences at the proximal flank of both breakpoints are all Vandal5 transposon elements. Both the F‐box protein‐coding genes and the Vandal elements are present at the breakpoint positions in the Ler genome (Zapata et al., 2016), from which we can deduce that these elements are also in the knobless ancestor. The presence of Vandal5, a Mutator‐like (Mule) transposon, at the two breakpoints suggests that the inversion was created by the activity of the transposon. Based on position and the opposite orientation of the F‐box protein‐coding genes at the distal and proximal breakpoints, we infer that the 5′‐ end of the Vandal5 transposon inserted into the 3rd exon of the original F‐box protein‐coding gene, while the other side of the transposon remained attached to the original donor site. Rejoining of the two free ends resulted in an inversion (Figures 2 and S5). A similar transposition event, in which donor and recipient sites remain associated, has been described for a Tam3 element in Antirrhinum majus (Robbins et al., 1989). The Vandal insertion has generated a stop codon, giving rise to a gene that is similar to the F‐box coding gene annotated as At4 g05480. Whether the inversion is caused by Vandal5 transposition activity or whether it is due to a spontaneous double‐strand break at a repeat sequence is not clear. The latter option seems to be rather infrequent as shown in a molecular and computational study of 29 inversions in Drosophila, of which only two inversions were linked to repeat sequences (Ranz et al., 2007). The absence of sequence duplication at the transposon target insertion site is unusual, but not unique (Fu et al., 2013).
Figure 2.
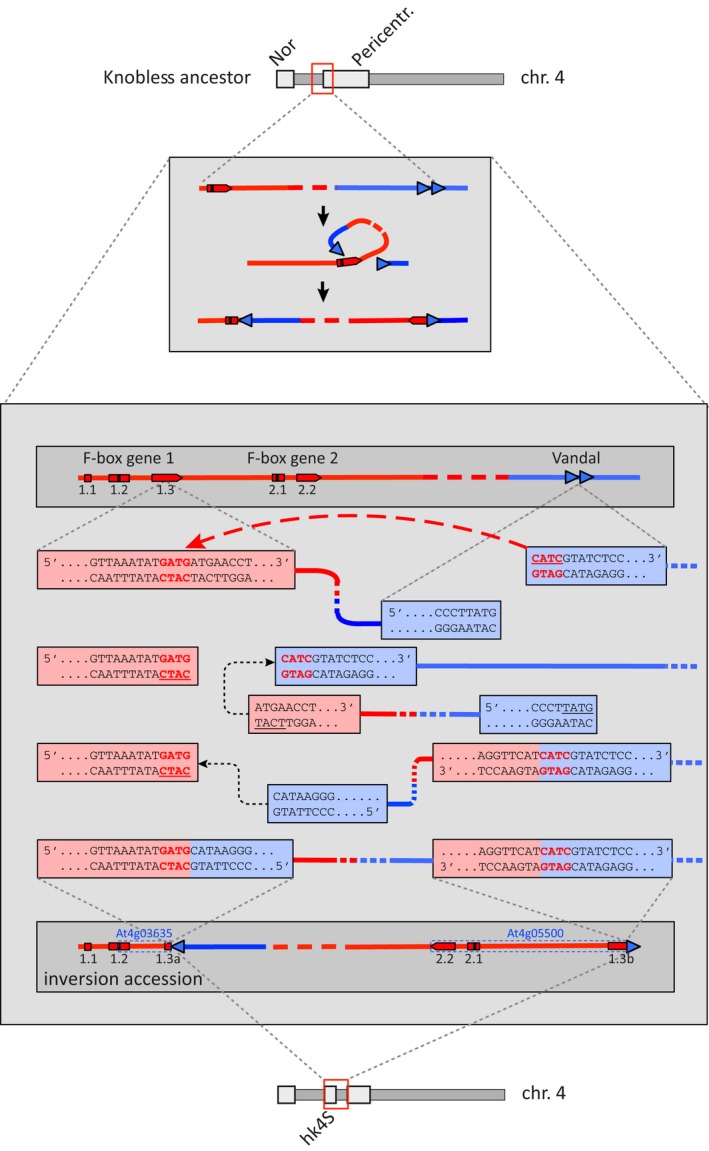
Reconstruction of the inversion event.
The rearrangement event is supposed the action of two Vandal5 transposon elements and one F‐box protein‐coding gene. The 5′‐CATC end of one Vandal5 transposon element inserted into an F‐box protein‐coding gene next to a 5′‐CTAC fragment of exon 3, and recombines with the 3′‐TACT end of the F‐box fragment. The other end of the Vandal5 transposon remains attached to its origin. Meanwhile, the free 3′‐GTA end of the flanking Vandal5 element recombines with the free 5′‐CATC end of the remaining F‐box fragment. The two resulting F‐box gene fragments, annotated as At4 g03635 and At4 g05500, reside at the distal and proximal end of the inversion, respectively.
Novel heterochromatin‐euchromatin transitions are formed at the borders of the inversion
The formation of the heterochromatic knob hk4S by the paracentric inversion implies that two new euchromatin‐heterochromatin boundaries have been formed in Col‐0 and Ws‐2: the distal breakpoint at the distal side of the knob and the proximal breakpoint, which is the new boundary of the pericentric heterochromatin. The original pericentric boundary before the inversion has become the proximal side of the knob. To find out if the new boundaries at the breakpoints have changed flanking euchromatin characteristics, we examined the expression and chromatin profiling data generated (http://chromatin.cshl.edu/cgi-bin/gbrowse/epivariation; Lippman et al., 2004; Vaughn et al., 2007; Tanurdžić et al., 2008). The heterochromatin markers 5‐methylcytosine and H3K9me2 revealed a sharp transition at the distal border of the knob at 1612 kb (Figure S6), and coincide exactly with the distal breakpoint of the inversion. At the sequence level the border is marked by a sharp contrast in repeat sequences. The same accounts for the proximal breakpoint located at the border of pericentromere heterochromatin. Hence, we conclude that the paracentric inversion breakpoints coincide with new, distinct heterochromatin‐euchromatin transitions. The concurrency of these transitions with the inversion breakpoints indicates that the inversion did not change epigenetic marks in the newly generated euchromatin‐heterochromatin borders. Moreover, no major differences in the epigenetic profiles of methylated DNA and methylated histone H3K9 around the inversion breakpoints have been found between Col‐0 and Ler, suggesting that there has been no shift in heterochromatin‐euchromatin transition along the chromosome (Figure S6). In addition, gene expression profiles revealed no major differences in the expression patterns between Col‐0 and Ler (Figure 3), suggesting that the close proximity of heterochromatin after the inversion did not affect the transcriptional activity of genes in knob‐flanking euchromatin of Col‐0 under the experimental conditions. Apparently, silencing by heterochromatin spreading into flanking chromatin, for example, position effect variegation as found in Drosophila (Elgin and Reuter, 2013) or Schizosaccharomyces pombe (Gottschling et al., 1990; Allshire et al., 1994; Allshire and Ekwall, 2015), did not occur at the boundary of this inversion. Neither has the presence of nearby transposon elements (TEs) a silencing effect on the genes nearby the inversion breakpoint. We also examined whether the presence of knob heterochromatin has resulted in changes in the compaction of flanking chromatin. A comparison between Col‐0 and Ws‐2 versus C24 and Ler using BAC‐FISH showed no significant differences in the condensation patterns of knob‐flanking regions. Hence, there seems to be no major differences between knob‐containing and knobless accessions with respect to chromatin compaction of knob‐flanking euchromatin in leaf nuclei, further supporting the suggestion that the heterochromatin boundary of the knob is stable and that epigenetic marks in the knob do not affect the structure of flanking euchromatin.
Figure 3.
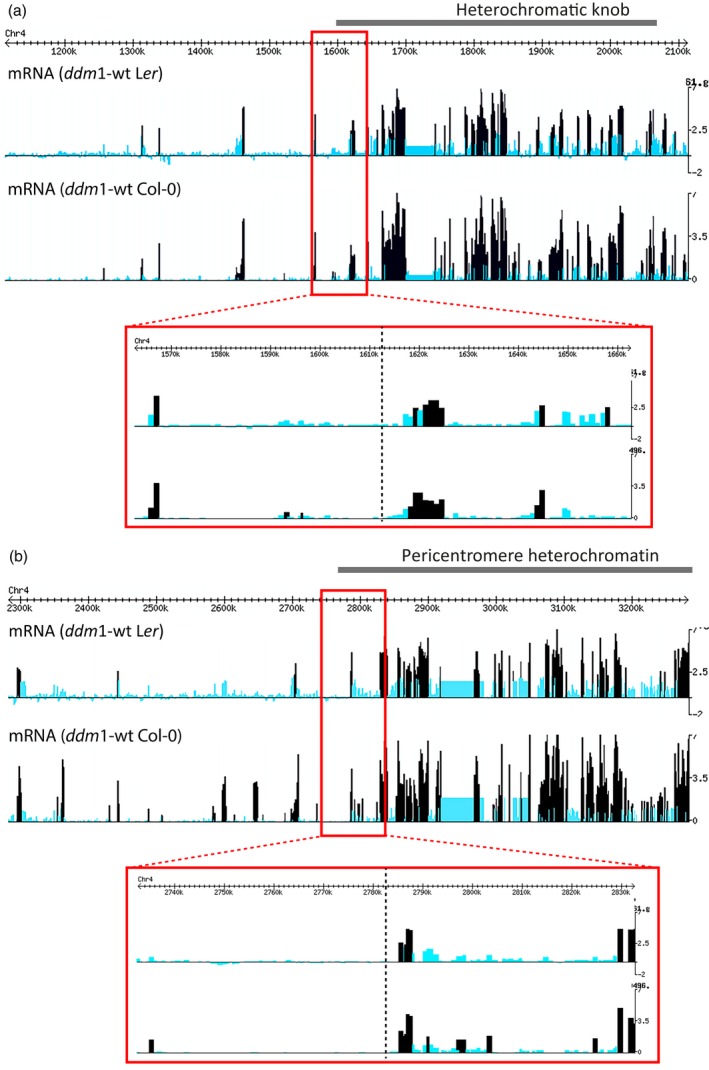
Gene expression profiles (Log Ration ddm1/wt) at the boundaries of the inversion.
Transcription profiles (light blue) at the distal (a) and proximal (b) breakpoints of the inversion in Col‐0 and Ler are similar. The transcription profile of the DNA methylation mutant (dark blue), ddm1, is also similar in the Col‐0 and the Ler background. Significance was assessed using both a family wise error rate (FWER) and a false discovery rate (FDR). A step‐down multiple comparisons procedure was used to control the FWER below a = 0.01, while a step‐up procedure was used to control the FDR below a = 0.01.
The inversion has spread into European and American accessions
Because recombination in the inverted region in plants heterozygous for the inversion is limited to gene conversion (and single crossover events in this region are not viable), we have an excellent opportunity to investigate sequence divergence in a well‐documented, genetically fixed 1.17 Mb ‘allele’. We first examined the presence of 233 Col+/ Ler− AFLP markers in the Ws‐2, C24 and Cvi‐0 (Peters et al., 2001), and found that Col‐0 and Ws‐2 have high sequence similarity in the inverted region compared with the knobless accessions. We further investigated the sequence divergence between knob and knobless accessions using 2653 SNPs (~0.23% of the total inversion sequence) that are a subset of the 250k SNPs included on the Arabidopsis SNPChip. The RegMap project used the SNPChip on a worldwide collection of 1307 accessions that included Col‐0 and Ws‐2 (Horton et al., 2012). We found that these two inversion accessions differed at only 55 (2%) of the subset of Chip SNPs in the inverted region. This high similarity is not the result of high sequence identity genome‐wide, however. For example, by examining similarity in an arbitrarily chosen 1.883 Mb region between positions 13.214 Mb and 15.097 Mb on chromosome 4, we found that this pair differed at 392 of 2954 (13%) Chip SNPs. Furthermore, we identified an additional 172 accessions (from 67 distinct locations) that putatively contain the inversion due to their high sequence similarity to Col‐0 and Ws‐2 at the 250k SNPs in the inverted region (Figure S7; Tables S4 and S5).
Using the extensive sequence data in the 1001 Genomes Project (http://1001genomes.org/) (1001 Genome Consortium, 2016), we accessed the genomic inversion sequence across all accessions. Clustering distance based on sequences across the 1.2 Mb inverted region was used to construct maximum likelihood (ML) trees by a modified approach with introgression browser (iBrowser; Aflitos et al., 2015), where a total of 72 906 polymorphic positions was identified (from a total of 12 898 225). The ML tree clearly shows a monophyletic clade of 132 inversion accessions (marked in red) from 41 individual locations and low levels of polymorphism between inversion accessions, strongly supporting a single origin of the inversion (Figure 4a). Similar results were obtained in an analysis of the 2653 SNPs from the 1307 accession panel (Figure S7). The inversion accessions identified overlap with most of the worldwide distribution of Arabidopsis (Figure 4d), but are particularly concentrated in Central and North‐West Europe and in North America. Furthermore, the North American inversion accessions form a monophyletic sub‐clade with very few polymorphic sites. This strongly suggests a single recent migration of the inversion haplotype from Europe to North America. The majority of American accessions analyzed carry the inversion. The origin of the inversion and spread are discussed in greater detail below.
Figure 4.
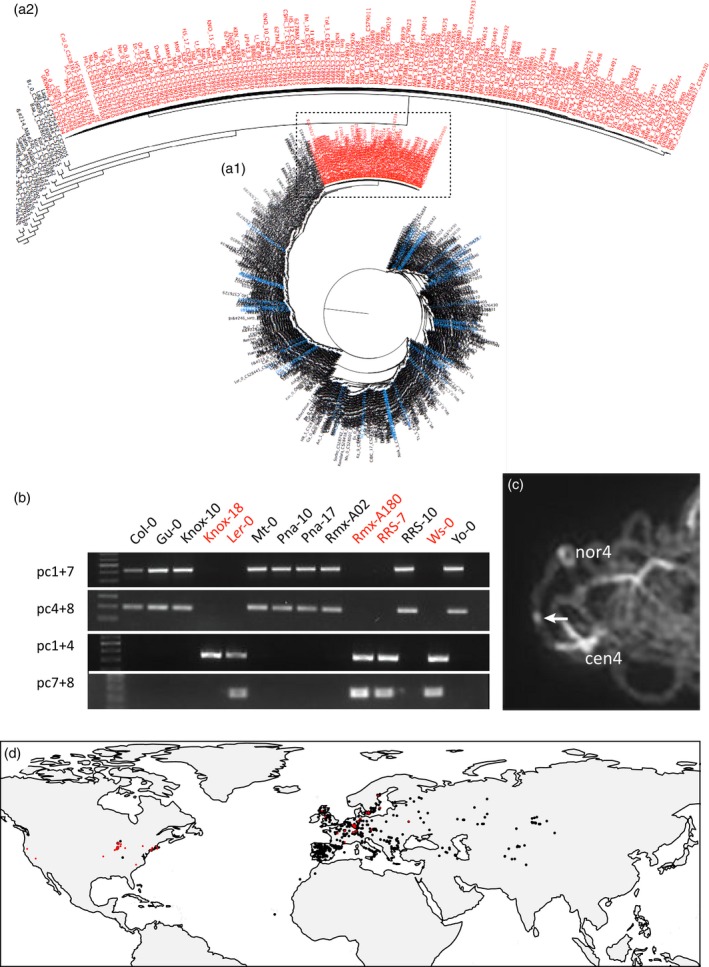
Identification of inversion accessions.
(a) Phylogenetic tree of all accessions from the 1001 genome database based on the 1.17 Mb inversion sequence. The inversion accessions (red names) form a monophyletic clade separated from all others. (a1) shows 1130 accessions, (a2) is a magnification of the clade with 132 inversion accessions.
(b) Identification of inversion accessions by PCR analysis of the inversion breakpoints using primers that flank the distal (pc1+7) and proximal (pc4+8) side of the inversion. The absence of the inversion breakpoints (accession name in red) gives rise to PCR fragments in case the primers pc1+4 or pc7+8 are used. The absence of a pc7+8 band in Knox‐18 is probably due to an aberrant nucleotide in this non‐inversion accession.
(c) DAPI staining of pachytene bivalent of accession Gu‐0 showing the heterochromatin knob hk4S (arrow) on chromosome 4 indicating the presence of the inversion.
(d) Global distribution of inversion (red) and non‐inversion accessions (black) from the 1001 Genomes Project. The inversion haplotype of 110 American inversion accessions is similar to UKSWO6‐333 (1), DIR‐9 (2) and NOZ‐6 (3) located in the UK and France.
To confirm that candidate inversion accessions indeed have the same paracentric inversion found in Col‐0 and Ws‐2, we tested the presence of the breakpoints in a subset of 30 European and North American accessions using PCR analysis with primers that flank the proximal and distal breakpoints. All tested ‘putative inversion’ accessions showed PCR fragments similar to Col‐0/Ws‐2, indicating the presence of breakpoint sequences (Figures 4b and S8). We further tested 20 Arabidopsis accessions, nine of which with putative inversion, for the presence of the inverted region by microscopic analysis of pachytene chromosomes. All nine ‘putative inversion’ accessions indeed contained the heterochromatic knob hk4S of chromosome 4 (Figures 4c and S9).
Historical meiotic recombination sites reveal inversion introgression events
Although we cannot exclude the rare occurrence of double crossovers in plants heterozygous for the inversion, we expect that the sequence polymorphism within the inverted region among the inversion accessions would be limited to new mutations that have accumulated subsequent to the inversion event or derived from gene conversion events with non‐inversion haplotypes. In contrast, outside the inversion, clear transitions from nearly non‐polymorphic regions to highly polymorphic regions can be identified (Figure 5a). These transitions represent historical recombination events, and thus mark the introgression boundaries and transfers into new genomic backgrounds.
Figure 5.

Haplotype analysis based on all SNPs in chromosome 4.
(a) Heatmap of Arabidopsis chromosome 4 representing genetic distances between different accessions based on SNPs from the 1001 Arabidopsis consortium (http://1001genomes.org/). Sequences with low SNP density (compared with the reference Col‐0) are shown as white blocks. The inversion accessions are ranked according to the left introgression boundary. The accessions in the intermittent black rectangle form the North American group. Due to space limitations, 55 accessions with high similarity to 627RMX_1MN4 and 22 accessions with high similarity to Gre‐0 are not shown in the North American group. The inverted region and the pericentromere are indicated by blue and red rectangles, respectively.
(b) SNP density profile in chromosome arm 4S of TAAL7 with Ler‐0 (blue) or Col‐0 (red) as reference. Note the switch in identity in this German accession from a ‘Col‐0’ haplotype to a ‘Ler’ haplotype and back to ‘Col‐0’. The black bar indicates the position of the inversion.
Most left and right introgression borders are positioned at approximately 1350 kb and 5000 kb, respectively, of the reference sequence. The latter corresponds to the boundary of the pericentromere in the long arm (Figure S10). This is not surprising, as the proximal breakpoint of the inversion is directly flanked by the pericentric heterochromatin that represses meiotic recombination. Some inversion haplotypes share the exact same recombination positions, suggesting that they are derived from the same historical recombination event and thus share the same ancestral donor accession for this region (Figure 5a; Table S6).
Based on the genomic positions of the left introgression sites, we can group several accessions (Table S6). The Col group (n = 8) has the Col‐0 haplotype for the entire short‐arm 4S. Four accessions in this group show high similarity to Col‐0 for all chromosomes and thus represent potential contaminants of the reference accession from various collections (such as described by Anastasio et al., 2011). The 1344 group (n = 10) contains accessions of which the left recombination site is at position 1344 kb, whereas the right introgression border shows more site variation (potentially signaling that the 1344 kb recombination site is an older shared event). The 1344 group consists predominantly of mid‐European accessions, in particular those that were found near Frankfurt. The members of the Northern American group (n = 110) have the same left and right recombination sites at 1354 kb and 5166 kb, respectively, and show little haplotype variation along the short‐arm. It is most likely that this introgression haplotype is derived from a single knob accession that was introduced into America. Indeed, it was previously reported that a large fraction of North American populations comprise a set of near‐isogenic lines that would have evolved in isolation and near‐clonally after colonization (Hagmann et al., 2015; Exposito‐Alonso et al., 2016). The Swedish‐German group (n = 4) has their left introgression site at 1588 kb, which is close to the inversion. These accessions show more SNPs in the inverted region, compared with the reference genome, than other inversion accessions. This suggests that the inversion haplotype of this group may have split off from the major haplotype at an early phase after the inversion event.
The Swedish accession Taal‐07 shows a remarkable distribution of SNPs, with regions highly similar to Col‐0 then alternating with regions highly similar to Ler ‐0. The right border of the introgression maps in the long arm at position 14 821 kb (Figure 5b; Table S6). From there up to the left introgression site at 1391 kb Taal‐07 has a ‘Col’‐like haplotype. From 1391 to 738 kb it has a ‘Ler’‐like haplotype, and from 738 kb towards the telomere it is like ‘Col’ again. Apparently, Taal‐07 has introgressed ‘Col’‐like and ‘Ler’‐like haplotypes. The ‘Ler’‐like haplotype shows less similarity with Ler‐0 than the ‘Col’ haplotype does with Col‐0, which suggests that the introgression of the ‘Ler’‐like haplotype is an older event or that Taal‐07 has introgressed a more ancient ‘Ler’‐like haplotype. The double ‘Col’ haplotype (one around the inversion and a distal one towards the telomere) is also shown in other inversion accessions, although less pronounced. Interestingly, the distal region with ‘Col’ similarity, which seems to be more common in inversion accessions (Figure 5a: white strip in, for example, US accessions), encompasses the area flanking the well‐studied FRIGIDA gene.
LD between Col‐FRIGIDA allele and inversion, and associations with fecundity
To examine the allelic status of the FRIGIDA locus relative to the allelic status of the inversion, we constructed three ML trees using iBrowser (Aflitos et al., 2015) in subsets of SNPs along the upper arm of chromosome 4 (Figure 6a). Accessions that carry the inversion are marked in red, as also shown in Figure 4a, and form a monophyletic clade for the analysis based on SNPs within the inversion (Figure 6a). The ML tree constructed from the 2615 SNPs derived in the 50 kb window around the FRIGIDA locus extracted using iBrowser showed that most inversion accessions have a FRI‐Col early flowering allele and thus form a monophyletic clade. All genotypes carrying the FRI‐Col allele (but not the inversion) are marked in blue. When we analyze the ML trees that are in the in‐between recombinant zone, the association between FRI‐Col and the inversion is clearly less pronounced.
Figure 6.
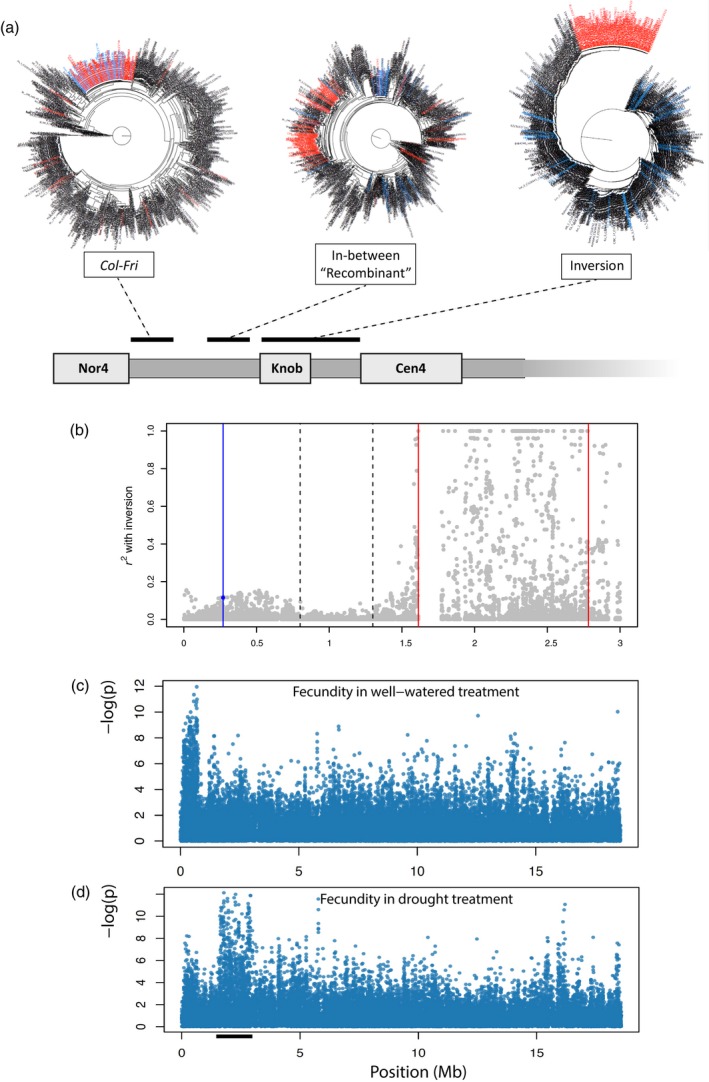
Phylogeny, linkage disequilibrium (LD) and genome‐wide association studies (GWAS) for fitness under abiotic conditions in chromosome 4.
(a) Phylogenetic trees of regions around FRIGIDA (FRI), within the inversion, and the region between them. Red: accessions with inversion. Blue: accessions with the Col allele of FRI but not the inversion.
(b) LD (r 2) between the inversion (as a binary status) and SNPs in the short‐arm of chromosome 4. Red lines: inversion breakpoints. Dashed lines: the recombinant region between inversion and FRI. Blue line: the FRI gene. The blue dot near FRI denotes LD between inversion and the FRI‐Col, a binary status.
(c) GWAS for chromosome 4 of plant fecundity in well‐watered environment.
(d) GWAS for chromosome 4 of plant fecundity in drought treatment.
The above results show LD between the inverted region and FRI. However, there apparently is more recombination, and thus a lack of LD, between the two regions. Given that many inversion accessions are from North America, this pattern could simply be caused by the founder events of early Arabidopsis immigrants to North America. To test this, we estimated the magnitude of LD (r2) for all SNPs up to 3 Mb position on chromosome 4 with the binary inversion status (with inversion or not) in all Eurasian accessions. The LD decreases sharply outside of the inversion, consistent with the clear breakpoints defining the left recombination border discussed above (Table S6) and suggesting ample historical recombination since the origin of this inversion. If selection were acting only on the inversion (or only around FRI‐Col), one would expect LD to decrease further away from the inversion (or FRI‐Col region). However, we find that the region around FRI is in moderate LD with the inversion (Figure 6b). This suggests joint selection for the two genomic regions with a region of relaxed (or neutral) selection in between. However, it is unknown what phenotypic effects the inversion might have.
We identified the inversion locus in a new, separate study of genome‐wide associations with phenotypes under experimental terminal drought (phenotypes published in Kenney et al., 2014). The inversion emerged in this work due to strong association between fecundity under drought (but not well‐watered conditions) and SNPs across the length of the inversion. We tested associations between ~200k SNPs (Horton et al., 2012) and total fruit length (a proxy for fecundity; Kenney et al., 2014) for 167 accessions (25 with inversion). Twenty‐one of the top 50 strongest SNP associations with fecundity under drought were all found in a 1.5 Mb region surrounding the inversion, from ~1.5 to ~3 Mb on chromosome 4 (Figure 6c). A follow‐up analysis showed that the inversion itself was significantly associated with higher fecundity in the drought treatment (general linear model, inversion accession total fruit length 66 mm or 33% greater than non‐inversion, SE = 22, P = 0.00258) even when accounting for FRI functionality (Lovell et al., 2013; inversion effect 47 mm, SE = 21, P = 0.0294) or when accounting for a random effect of kinship (Kang et al., 2008; EMMA, P = 0.01814854). High fecundity in the Kenney et al. (2014) drought experiment was strongly associated with faster flowering time, and thus we asked whether flowering time (possibly due to causal FRI variants) was causing inversion associations with fecundity. We found that the inversion was associated with greater fecundity even after incorporating the effect of flowering time on fecundity (general linear model, inversion effect 35 mm, SE = 15, P = 0.025). To further explore the association of the inversion with fecundity, we tested associations with fecundity data from five field common garden experiments across Europe from Spain to Finland (Fournier‐Level et al., 2011; Hancock et al., 2011). We found that in one garden in Lille, France, the inversion (11/164 accessions had inversion) was again associated with higher fecundity (inversion accession total fruit length 1240 mm or 72% greater than non‐inversion, SE = 465, P = 0.00842), even after accounting for the influence of imputed FRI functionality (Lovell et al., 2013; inversion effect 1194 mm, SE = 459, P = 0.0101), but not when accounting for a random effect of kinship (EMMA, P = 0.905724). The inversion was not associated with fecundity in any of the other four common gardens (general linear models, all P > 0.05). While these associations with higher fecundity in certain environments indicate that the inversion might be under positive selection locally, the effects of natural allelic variation at this specific locus have not been experimentally demonstrated. We tested whether the distribution of the inversion could be explained by climatic variation. The inversion was not significantly associated with any of ~100 climate variables or with the first 10 principal component axes of climatic variation among accessions (lowest P = 0.14; Lasky et al., 2012). Note the distribution of the inversion is consistent with this lack of climate association; it is found in diverse climates from Scandinavia to North Africa (see map below, red indicates inversion). This, however, does not mean the inversion has no effect. The lack of climatic association may simply reflect it being a young mutation and does not have enough time to reach migration‐selection balance in the species‐wide scale.
Origin, distribution and migration patterns of the inversion haplotype
To further explore the distribution, history and spread of the inversion, we analyzed population variation in sequence data of the short‐arm of chromosome 4 of Arabidopsis. Given the observed LD between inversion and FRI‐Col and their effect on fitness and phenology, we aimed to identify their locations of origin and investigate their evolutionary history. Assuming the inversion event only happened once, the location where the event happened would have local non‐inverted haplotypes genetically closest to the inversion. We therefore calculated the genetic distance of all Eurasian non‐inversion to inversion and extrapolated the distance across the map. Although currently the inversion is most abundant in central Germany (Figure 4d), local non‐inversion haplotypes in southern Italy are genetically more similar to the inversion than those in central Germany (Figure 7a), suggesting its southern origin between 2000 and 5000 years ago (estimated from genetic distances among Eurasian inversions). To investigate whether spatial uneven sampling of accessions affects this individual‐based result, we divided the Eurasia map into 5° latitude‐by‐longitude grids (Figure S11) and estimated the mean genetic distance of all non‐inversion accessions in each grid to the inversion. The grid‐based method showed consistent result: non‐inversions from southern Italy have the lowest genetic distance to the inversion (Figure S11). With the same method, the origin of FRI‐Col of FRI is estimated to be in northwestern Europe (Figure 7c), and the majority of American Arabidopsis accessions is also estimated to originate from there (using the recombining region between FRI and inversion compared with this region from American inversion accessions; Figure 7b). This supports the hypothesis of a Northwest European origin of North American A. thaliana, consistent with genetic structure from genome‐wide data (1001 Genomes Consortium, 2016).
Figure 7.
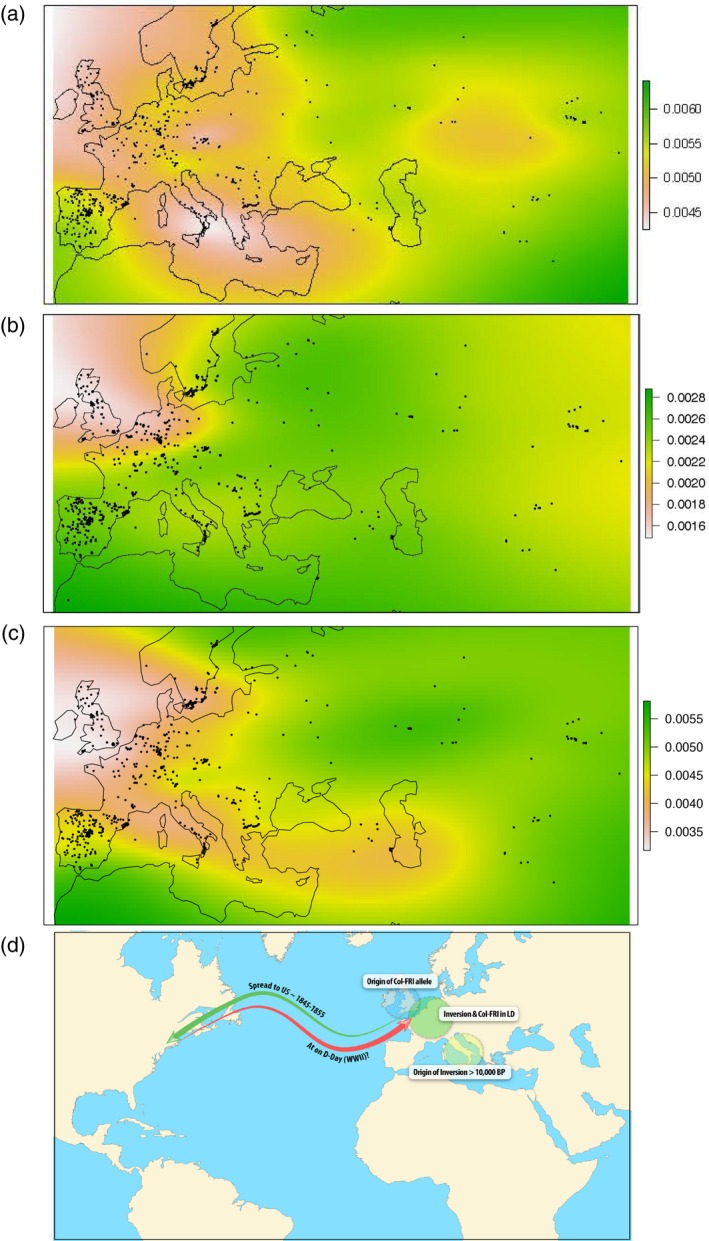
Evolution and demography of FRI‐Col and the inversion.
(a) Likely geographic origin of the inversion. Shown is the extrapolation of local non‐inversion accessions’ genetic distance to the inversion, within the inverted region. (a)–(c) Regions with the lowest genetic distance (white) are the likely origin.
(b) Likely geographic origin of the majority of North American haplotypes in chromosome 4. Shown is the extrapolation of local accessions’ genetic distance to Knox‐10 (an accession representing the majority of North American haplotypes) in the recombining region between FRIGIDA and inversion.
(c) Likely geographic origin of FRI‐Col. Shown is the extrapolation of local accessions’ genetic distance to Columbia‐0 in the FRIGIDA gene.
(d) The ancestor of the inversion originated from southern Europe about 5000 years ago and was later in LD with FRI‐Col, which originated near northwestern Europe. The majority of the North American haplotypes were descended from the migration event about 200–300 years ago (green arow). At least three ‘American’ accessions were detected in Europe: DIR‐9 (Brest, France), NOZ‐6 (Marseilles, France) and UKSW06‐333 (Cornwall, UK). These locations correspond to sites of American activities at the end of WWII, which may point to a possible time of return (red arrow).
Taken together, the results suggest that while the inversion and FRI‐Col originated from different parts of Europe, they encountered each other in Central Germany; and while in moderate LD with FRI‐Col (Figure 6b), the inversion reached a higher frequency there. Alternatively, the FRI‐Col allele could have arisen in an inversion accession. In approximately 1847 (estimated at approximately 169 years ago from genetic distances among North American inversions), accessions with both the inversion and FRI‐Col haplotypes migrated to North America. Using an alternative mutation rate estimate from Exposito‐Alonso et al. (2016), the introduction is approximately 17th century, consistent with their time estimate. The introduction of A. thaliana, commonly found in agricultural settings, into Northern America is potentially associated with European immigration. Further, among accessions carrying the North American colonizer haplotype, three accessions, considered as non‐contaminants (data in Figure S12), are identified in Europe [two of them coinciding with locations of intense American activity during WWII; Cornwall (UK) and Bretagne (France)].
While the results suggest that inversion originated from southern Europe, it remains unclear why its current frequency is relatively high in central Germany but not in southern Europe. Given the potential adaptive effects of inversion and FRI‐Col in certain environments, it is possible that, when in linkage, their combined effects gave the inversion enough advantage to increase in frequency. With the lack of FRI‐Col in southern Europe, local inversions might go extinct because their fitness effect alone may not be large enough to overcome genetic drift and the deleterious effect when occasionally in heterozygotes. Alternatively, this could be due to the changing climatic conditions since the end of the last Ice Age: southern inversions and non‐inversions might both migrate from southern Europe into Germany, and later the migrant non‐inversions recombined with Germany non‐inversions (which is generically distant from the inversion), leaving those non‐inversions staying in the south currently closest to the inversions. Finally, we cannot rule out the possibility that the inversion originated in Germany, after which its non‐inversion closest relatives in Germany all went extinct, leaving southern Italian non‐inversions the closest.
To summarize, our data are suggestive of the following model for the origin and spread of the inversion (Figure 7d): the inversion arose in southern Italy approximately 5000 years ago, later moving into Central‐Europe; the FRI‐Col allele originated in NW Europe and recombined with an inversion line in Central‐Europe (or possibly occurred in an inversion genotype); the inversion + FRI‐Col haplotype spread to several European populations with recombination occurring between the two regions (with the Swedish‐German group representing a relatively old separation); a particular inversion + FRI‐Col haplotype from NW‐Europe migrated to North America during the 17–19th century, an era of massive human migration both from Europe and across North America; and finally the return migration to three European localities during the 20th century. The model will need additional genotyping data from targeted locations and/or historical herbarium collections to verify. The potential causal gene(s) associated with fitness under drought within the inversion, epistastic interactions with FRI‐Col and their G × E interactions can be tested with population genetic modeling, empirical work and functional genetics. We have only begun to peak through this inversion window, it is clear there is still much to be learned.
Experimental procedures
Plant material
Plants of the A. thaliana accessions, crosses and mutants were grown under standard conditions in the greenhouse, unless otherwise noted. The accessions are: Ak‐1, Ba‐1, C24, Ca‐0, Col‐0, Cu‐0, Cvi, En‐2, Gifu‐2, Gre‐0, Gu‐0, Ha‐0, Kas‐2, Knox‐10, Knox‐18, Kro‐0, Ler‐0, Mh‐0, Mt‐0, No‐0, Nw‐0, Ob‐0, Pna‐10, Pna‐17, Ragl‐1, Rmx‐A02, Rmx‐A180, RRS‐7, RRS‐10, Si‐0, Tol‐0, Tul‐0, Ws‐0, Ws‐2, Yo‐0. Seeds from all accessions were obtained from the Nothingham Stock Centre (NASC). The F1 cross Col × Ler was used for cytogenetic analysis. The triple mutant (ga1, bp, tt11) in a Ler background (Koornneef et al., 1983) was used for the genetic analysis. The mutant ddm1 (in Col‐0 and Ler‐0 background) was used for ChIP and RNA analysis.
Cytogenetic analysis
Young flower buds were fixed in freshly prepared ethanol/acetic acid (3:1) for chromosome spreads of interphase nuclei and pachytene complements following the protocol by Ross et al. (1996). Extended DNA fibers were prepared from isolated leaf nuclei following the method of Fransz et al. (1996).
The BAC DNA clones used in this study were constructed from the Col genome. The following clones were used: F4C21, F9H3, F4H6 (IGF library; Mozo et al., 1998), and T5L23, T5H22, T7M24, T25H8, T24M8, T24H24, T27D20, T19B17, T26N6, T4B21, T1J1, T32N4 (TAMU library; Choi et al., 1995) and C6L9. All DNA clones were labeled with either biotin‐dUTP or digoxigenin‐dUTP, using a standard Nick translation kit (Roche).
Fluorescent in situ hybridization to pachytene chromosomes and to interphase nuclei was carried out using the methods described by Fransz et al. (1998), with minor modifications. Chromosome preparations were heated at 60°C for 30 min, incubated with RNase (10 μg mL−1 in 2 × SSC) and rinsed in 2 × SSC for 2 × 5 min and in PBS [10 mm sodium phosphate (pH 7.0), 143 mm NaCl] for 2 × 5 min. The slides were than fixed in 1% (v/v) paraformaldehyde in PBS for 10 min, rinsed in 1 × PBS for 2 × 5 min, dehydrated through an ethanol series and air dried. To each preparation, 20 μL of denaturation buffer [70% formamide, 2 × SSC, 50 mm sodium phosphate (pH 7.0)] was added and denatured on the heat block at 80°C for 2 min. After removing the coverslips, the slides were washed in ice‐cold ethanol series (70%, 90% and 100%, each for 2 min) and air‐dried. The hybridization mix contained 50 ng labeled probe in 50% formamide, 2 × SSC, 50 mm sodium phosphate (pH 7.0) and 10% dextran sulfate. Slides were examined under a Zeiss Axioplan 2 imaging photomicroscope equipped with epifluorescence illumination, and small band pass filters for DAPI, FITC, Cy3 and Cy3.5/Texas Red fluorescence. Selected images were captured using Photometrics or Spot CCD camera. The images were further processed for multicolor imaging with Adobe Photoshop software. DNA Fiber FISH was carried out following the protocol by Fransz et al. (1996).
PCR primers and amplification conditions
For the amplification of the fragments ‘a, b, c, d, e’, we designed primers from the Col‐0 genome DNA information (Table S3). PCR amplification was carried out on a GeneAmp PCR System 2700 (Applied Biosystem, Foster City, CA, USA) in 50 μL, in the presence of MgCl2, 200 μm of each dNTP, 200 pmol of each primer and 1 unit of Taq DNA polymerase. PCR conditions were 95°C for 5 min; 95°C, 1 min, 60°C, 1 min and 72°C, 2 min (35 cycles); 72°C for 5 min. For the amplification of the fragments at the inversion breakpoints (Figure S8), the PCR was carried out in a Bio‐Rad iCycler Thermal Cycler. The PCR reaction was set at 1 min at 95°C, followed by 35 cycles with 30 sec of DNA denaturation at 94°C, annealing at 58°C for 30 sec, and extension at 72°C for 1 min, and final extension at 72°C for 2 min.
Recombination analysis of the inverted region
Two approaches were followed to determine the recombination frequency around and within the paracentric inverted region that causes the heterochromatic knob (hk4S) on the short‐arm of chromosome 4 of Arabidopsis. For the first approach, a triple mutant (ga1, bp, tt11) in Ler background was crossed with wild‐types of three genetic backgrounds, Ler, Ws‐2 and Col (Koornneef et al., 1983). Ws‐2 and Col accessions contain the heterochromatic knob, whereas Ler is a knobless accession. By selfing the resulting F1 plants, three corresponding F2 populations were obtained. Phenotyping of the F2 individuals will reveal the recombination frequencies between the three loci in each of the three F2 populations. The area between ga1 (gibberellin mutant 1, At4 g02780) and bp (Brevipedicellus, At4 g08150) includes the paracentric inversion, and tt11 (transparent testa 11) lies 37.5 cM south of bp (Figure 4a). For the second more precise approach, we made another two BC1 crosses, [(Col × Ler) × Col] and [(Ler × Col) × Col], with 112 and 120 individuals, respectively. Recombination events between six AFLP markers covering the inverted region were determined (Figure S1).
AFLP analysis
The genomic DNA needed for AFLP analysis was prepared by collecting leaves of young seedlings in 1.2 mL collection microtubes (Qiagen, Hilden, Germany) that were placed in 96‐well racks. Each tube containing one tungsten carbide bead (3 mm, Qiagen) was kept in liquid nitrogen until further processing. The frozen leaves were grinded in the mixer mill MM300 (Retsch GmbH, Haan, Germany) for 30 sec at 30 Hz. Immediately after grinding, 500 μL preheated (60°C) extraction buffer (100 mm Tris‐HCl, pH 7.5; 700 mm NaCl; 10 mm EDTA; 1% w/v CTAB) was added, and the samples were incubated at 60°C for 10 min. To each tube, 250 μL chloroform/isoamylalcohol (24:1) was added and mixed for 10 min. The watery phase was separated from the chloroform phase by centrifugation (4 min at 6000 rpm in a Sigma 4‐15C centrifuge), and 200 μL of the watery phase was transferred to new collection tubes containing 200 μL isopropanol. After mixing, the samples were centrifuged for 10 min at 6000 rpm to spin down the DNA. The pellet was washed in 70% ethanol, vacuum dried and dissolved in 200 μL water or TE buffer (10 mm Tris‐HCl, pH 8.0; 0.1 mm EDTA).
Amplified fragment length polymorphism analysis was performed according to Vos et al. (1995) using the restriction enzymes SacI and MseI (Peters et al., 2001). The primer combinations used were SM18, SM38, SM76, SM202, SM207 and SM239, and can be found in table VI of Peters et al. (2001). The selective amplification step was performed with IRDye 700‐ or 800‐labeled SacI+2 primers, and the products were resolved in 25‐cm gels (0.25 mm spacer thickness) containing 20 mL 6.5% gel matrix (KBplus, LI‐COR, Lincoln, USA). Before loading, an equal amount of loading buffer (95% formamide; 20 mm EDTA, pH 8.0; 1 mg mL−1 Bromophenol Blue) was added to the AFLP reaction, which was subsequently denaturated for 3 min at 90°C and transferred to ice. The samples were run on a two‐dye, model 4200 LI‐COR automated DNA sequencer. Run parameters were as follows: 1500 V, 40 mA, 40 W, 45°C, scan speed 3‐moderate. A detailed protocol for AFLP analysis on a LI‐COR sequencer was described by Myburg et al. (2001).
Profiling of RNA, H3K9me3 and 5 m‐cytosine
RNA, DNA methylation and H3K9me2 profiles were determined using the 1 kb microarray representing 18.6 Mb of chromosome 4 (Vaughn et al., 2007).
For RNA profiling, total RNA was extracted from 9‐day‐old seedlings (WT and ddm1). Polyadenylated RNA was then purified using either MicroPoly(A)purist (Ambion) or the Dynabeads kit [oligo(dT)25, Dynal]. cDNA samples were generated with reverse transcriptase and labeled with Cy5 or Cy3 (Amersham) following standard procedures. All details of the RNA analysis can be found in Lippman et al. (2004; Supplementary Online Material).
For H3K9me2 profiling, leaves from 14‐day‐old Col‐0 plants were fixed in 1% formaldehyde. Chromatin was purified and target DNA was isolated by chromatin immunoprecipitation using anti‐dimethyl‐H3K9 (Upstate/Milipore). Extensive details of the ChIP procedure can be found in Gendrel et al. (2005) and Lippman et al. (2005).
For DNA methylation profiling, genomic DNA was isolated from 9‐day‐old seedlings (Col‐0 and Ler). DNA samples were either treated or non‐treated with the DNA methylation‐dependent restriction enzyme McrBC, which allows the detection of densely located 5 m‐cytosines in several sequence contexts. After removal of the methylated DNA fragments from the ‘treated’ sample via size fractionation, both the ‘treated’ and ‘untreated’ DNA samples were differentially labeled (Cy3 and Cy5), mixed and hybridized to the microarray. An extensive description of the methylated DNA analysis can be found in Lippman et al. (2004, 2005).
Methodology of SNP analysis
250K SNP chip data from RegMap accessions
RegMap genotype data were downloaded from the Bergelson lab website (http://bergelson.uchicago.edu/wp-content/uploads/2015/04/call_method_75.tar.gz). Genotype calls specific to the inversion region or a ‘control’ region were extracted from the full dataset. The proportion of SNPs in each region that differed between each pair of accessions was computed, and arranged in a dissimilarity matrix. These matrices were used as input to generate unweighted pair group method with arithmetic mean trees via the hierarchical clustering function in R [hclust((as.dist(matrix)),method=“average”)]. Custom Perl scripts were written to describe the patterns of SNP polymorphism within the inversion and non‐inversion classes of accessions.
One‐thousand, one‐hundred and thirty‐five VCF files available at the Arabidopsis 1001 Genomes Project (http://1001genomes.org/) were analyzed using iBrowser (Aflitos et al., 2015; v. d228c22) with 50 kbp window size. Genomic clustering using concatenated homozygous SNPs was created using FastTree2 (v2.17) for regions in chromosome 4 named: upstream (0 bp to 1.6 Mbp), short‐arm (0 bp to 3 Mbp), inversion (1.6–3 Mbp), centromere (3–5.5 Mbp), downstream (> 3 Mbp) and long arm (> 5.5 Mbp). Trees were plotted using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Geographical origin of inversion and FRI‐Col
Within the inverted region, we calculated pairwise genetic distances among all 1135 accessions, published by the Arabidopsis 1001 genomes project (1001 Genomes Consortium, 2016). We used sites with less than 10% missing data, and an accession was inferred to have the inversion if its genetic distance to the reference genome (Col‐0) is low. As shown in Figure S13, the genetic distance to Col showed a clear bimodal pattern, allowing us to unambiguously assign which accession has the inversion and which has not. To confirm the inversion status, we investigated the original Illumina paired‐end reads and identified read pairs supporting either karyotype. The Illumina‐based and genetic‐distance‐based results are consistent, except for two accessions: UKSE06‐252 and UKNW06‐233. The former has four pairs supporting inversion and 16 supporting non‐inversion, while the latter has 27 and 28 pairs, respectively. These two accessions are not inversion heterozygotes as their SNP heterozygosity is low within the inversion, and therefore they are likely to have further rearrangements or duplications near the breakpoints. Given it is difficult to unambiguously assign their inversion status, we removed these two accessions for further analyses.
Assuming that the inversion event happened only once, in the geographical region where this event happened, the local non‐inverted accessions should have closest genetic distance to the inverted accessions than those elsewhere in the species range do. We therefore interpolated non‐inverted accessions’ genetic distances to inverted ones across the Eurasia map, using the thin plate spline method as implemented in function ‘Tps’ of the library ‘fields’ in R (https://www.r-project.org/). To check whether accessions with excess amounts of missing data affected the results, we performed the same analysis only with accessions with more than 400 000 sites in this inverted region (excluding 38 accessions).
To confirm whether this result was robust to uneven sampling of accessions across the map, we further performed a grid‐based analysis. The whole Eurasia map was divided into 5° latitude by 5° longitude grids and, within a grid, the mean genetic distance of all non‐inverted accessions to inversion was used to infer the origin of this inversion. This simple method controlled for spatial uneven sampling (e.g. too many accessions from southern Sweden) and avoided the complexity of thin plate spline while allowing us to investigate the difference among geographical grids (Figure S11).
We used the same method to investigate the possible geographical origin of the Col‐like allele of FRI, and also the origin of American accessions from the recombining region between FRI and inversion. To estimate the age inversion, we calculated pairwise genetic distances among the inversion haplotypes, divided the distances by two, and scaled them with the estimated mutation rate of 7 × 10−9 per base pair per generation, assuming one generation per year (Ossowski et al., 2010). The lower bound of the age of inversion is estimated by mean pairwise distances (assuming a star phylogeny), and the upper bound is estimated by the maximum of all pairwise distances among Eurasian inversions. For American inversions, we used the mean pairwise distances among all American inversions to estimate the age of the most common haplotype. We did not use the maximum pairwise distance because the value can be easily affected by multiple haplotypes migrating from Europe.
LD between FRI and inversion
We used bi‐allelic SNPs from beginning of chromosome 4 up to 3 Mb to investigate the pattern of LD within this region. Only accessions from Eurasia were used, and sites with more than 10% missing data were excluded. Accessions were first coded according to their binary inversion/non‐inversion status, and the LDs of all SNPs to the inversion were estimated in r2.
Phenotype associations
In a previous study (Kenney et al., 2014), fecundity (i.e. total estimated fruit length) was measured in well‐watered and dry treatments in a greenhouse on 167 accessions matched with accessions among the 1307 RegMap panel (Horton et al., 2012). Some accessions originated from the same collection site but not the same individual collected plant in the two studies (Horton et al., 2012; Kenney et al., 2014). Arabidopsis exhibits isolation by distance and individuals from the same site tend to be closely related (Platt et al., 2010) and so, to improve overlap between panels, we used SNP data from RegMap for accessions in Kenney et al. (2014) that originated from the same site (even if they were not from the same collected plant), resulting in 167 accessions. We tested fecundity associations with SNPs having minor allele frequency of at least 0.1. We used the mixed‐model association approach of Kang et al. (2008), which controls for genome‐wide identity in state among accessions, putatively representing population structure. After having identified the inversion as the likely source of the many SNPs across large distances with strong drought fecundity associations, we tested fecundity associations with imputed inversion status based on 174 inversion accessions of the RegMap panel. We tested for the effect of FRI functionality using genotyping described in Lovell et al. (2013). Tests of association between inversion allele and fecundity were done either with general linear models or with a mixed‐effects model accounting for kinship (Kang et al., 2008).
Supporting information
Figure S1. Genetic and physical map positions of markers in chromosome 4.
Figure S2. Dot plot comparison of Ler and Col‐0 fragments at the inversion breakpoints.
Figure S3. Specification of DNA sequences at the breakpoints of the inversion.
Figure S4. Sequence alignment of A. lyrata (XM_002874807.1), Ler (ATL8C23344) and the reconstructed Col‐0 region (At4 g03635 + At4 g05500).
Figure S5. Alignment of the F‐box genes in A. lyrata, Ler and the reconstructed Col‐0.
Figure S6. Chromatin profile at the boundary of the inversion.
Figure S7. Genetic relationships of RegMap accessions.
Figure S8. Identification of inversion accessions by PCR analysis of the inversion breakpoints.
Figure S9. DAPI‐stained pachytene complements.
Figure S10. Scatter plot of left against right introgression sites based on the major haplotype shared by the majority of inversion accessions.
Figure S11. Arabidopsis accessions in 5° latitude‐by‐longitude grids.
Figure S12. Phylogenetic tree of all accessions from the 1001 genome database based on the long arm of chromosome 4.
Figure S13. The genetic distance of 1135 Arabidopsis accessions to Col‐0 in the inverted region, showing clear bimodal pattern.
Table S1. Recombination frequencies (±SD) between ga1, bp and tt11.
Table S2. Estimated molecular lengths in kb based on fiber‐FISH analysis (n = 20–30 per accession).
Table S3. List of primers for the fragments a–e.
Table S4. List of 174 accessions inferred to have the inversion.
Table S5. Patterns of 250K chip SNP variation in the inversion region and in a ‘Control’ region, 13.214–15.097 Mb on chromosome 4.
Table S6. List of 53 representative European and North American inversion accessions ranked according to their left and right introgression site.
Acknowledgements
The authors greatly acknowledge A.V. Gendrel and Z. Lippman for their help in chromatin profile analysis, T. Peterson, S. Peters and E. Wijnker for stimulating discussions, D. Weigel and M. Exposito‐Alonso for helpful comments to the manuscript, and RijkZwaan Breeding, Fijnaart, the Netherlands, for financial support of X.J. G.L. was supported by the Marie Curie Individual Fellowship (OTKA K 108555). C.R.L. was supported by the EMBO Long‐Term Fellowship. T.E.J. was supported by the National Science Foundation (grant DEB‐0618347). Contribution no. 16‐302‐J from the Kansas Agricultural Experiment Station (C.T.).
Contributor Information
Paul Fransz, Email: p.f.fransz@uva.nl.
Michael E. Schranz, Email: eric.schranz@wur.nl
References
- Aflitos, S.A. , Sanchez‐Perez, G. , de Ridder, D. , Fransz, P. , Schranz, M.E. , de Jong, H. and Peters, S.A. (2015) Introgression browser: high‐throughput whole‐genome SNP visualization. Plant J. 82, 174–182. [DOI] [PubMed] [Google Scholar]
- Allshire, R.C. and Ekwall, K. (2015) Epigenetic regulation of chromatin states in Schizosaccharomyces pombe . Cold Spring Harb. Perspect. Biol. 7, a018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire, R.C. , Javerzat, J.P. , Redhead, N.J. and Cranston, G. (1994) Position effect variegation at fission yeast centromeres. Cell, 76, 157–169. [DOI] [PubMed] [Google Scholar]
- Ananiev, E.V. , Phillips, R.L. and Rines, H.W. (1998) Complex structure of knob DNA on maize chromosome 9. Retrotransposon invasion into heterochromatin. Genetics, 149, 2025–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasio, A.E. , Platt, A. , Horton, M. , Grotewold, E. , Scholl, R. , Borevitz, J.O. , Nordborg, M. and Bergelson, J. (2011) Source verification of mis‐identified Arabidopsis thaliana accessions. Plant J. 67, 554–566. [DOI] [PubMed] [Google Scholar]
- Anderson, L.K. , Covey, P.A. , Larsen, L.R. , Bedinger, P. and Stack, S.M. (2010) Structural differences in chromosomes distinguish species in the tomato clade. Cytogenet. Genome Res. 129, 24–34. [DOI] [PubMed] [Google Scholar]
- Betekhtin, A. , Jenkins, G. and Hasterok, R. (2014) Reconstructing the evolution of Brachypodium genomes using comparative chromosome painting. PLoS ONE, 9, e115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt, S. , Moradkhani, K. , Mrasek, K. , et al. (2009) Breakpoint mapping and complete analysis of meiotic segregation patterns in three men heterozygous for paracentric inversions. Eur. J. Hum. Genet. 17, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutkar, A. , Schaeffer, S.W. , Russo, S.M. , Xu, M. , Smith, T.F. and Gelbart, W.M. (2008) Chromosomal rearrangement inferred from comparisons of 12 Drosophila genomes. Genetics, 179, 1657–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, F. , Trick, M. , Drou, N. , et al. (2009) Comparative analysis between homoeologous genome segments of Brassica napus and its progenitor species reveals extensive sequence‐level divergence. Plant Cell, 21, 1912–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. , Creelman, R.A. , Mullet, J.E. and Wing, R.A. (1995) Construction and characterization of a bacterial artificial chromosome library of Arabidopsis thaliana . Plant Mol. Biol. Rep. 13, 124–128. [Google Scholar]
- Coluzzi, M. , Sabatini, A. , della Torre, A. , Di Deco, M.A. and Petrarca, V. (2002) A polytene chromosome analysis of the Anopheles gambiae species complex. Science, 298, 1415–1418. [DOI] [PubMed] [Google Scholar]
- Darling, A.E. , Mau, B. and Perna, N.T. (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE, 5, e11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky, T. (1970) Genetics of the Evolutionary Process. New York: Columbia University Press. [Google Scholar]
- Drouaud, J. , Camilleri, C. , Bourguignon, P.‐Y. , et al. (2006) Variation in crossing‐over rates across chromosome 4 of Arabidopsis thaliana reveals the presence of meiotic recombination “hot spots”. Genome Res. 16, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgin, S.C. and Reuter, G. (2013) Position‐effect variegation, heterochromatin formation, and gene silencing in Drosophila . Cold Spring Harb. Perspect. Biol. 5, a017780–a017780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exposito‐Alonso, M. , Becker, C. and Schuenemann, V.J. et al. (2016) The rate and effect of de novo mutations in natural populations of Arabidopsis thaliana . bioRxiv. doi: http://dx.doi.org/10.1101/050203. [Google Scholar]
- Fang, Z. , Pyhajarvi, T. , Weber, A.L. , Dawe, R.K. , Glaubitz, J.C. , Sánchez González, Jde J. , Ross‐Ibarra, C. , Doebley, J. , Morrell, P.L. and Ross‐Ibarra, J. (2012) Megabase‐scale inversion polymorphism in the wild ancestor of maize. Genetics, 191, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraut, T. (2008) Addressing chromosome evolution in the whole‐genome sequence era. Chromosome Res. 16, 5–16. [DOI] [PubMed] [Google Scholar]
- Febrer, M. , Goicoechea, J.L. , Wright, J. , et al. (2010) An integrated physical, genetic and cytogenetic map of Brachypodium distachyon, a model system for grass research. PLoS ONE, 5, e13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk, L. , Carson, A.R. and Scherer, S.W. (2006) Structural variation in the human genome. Nat. Rev. Genet. 7, 85–97. [DOI] [PubMed] [Google Scholar]
- Fournier‐Level, A. , Korte, A. , Cooper, M.D. , Nordborg, M. , Schmitt, J. and Wilczek, A.M. (2011) A map of local adaptation in Arabidopsis thaliana . Science, 334, 86–89. [DOI] [PubMed] [Google Scholar]
- Fransz, P.F. , Alonso‐Blanco, C. , Liharska, T.B. , Peeters, A.J. , Zabel, P. and de Jong, J.H. (1996) High‐resolution physical mapping in Arabidopsis thaliana and tomato by fluorescence in situ hybridization to extended DNA fibres. Plant J. 9, 421–430. [DOI] [PubMed] [Google Scholar]
- Fransz, P. , Armstrong, S. , Alonso‐Blanco, C. , Fischer, T.C. , Torres‐Ruiz, R.A. and Jones, G. (1998) Cytogenetics for the model system Arabidopsis thaliana . Plant J. 13, 867–876. [DOI] [PubMed] [Google Scholar]
- Fransz, P.F. , Armstrong, S. , de Jong, J.H. , Parnell, L.D. , van Drunen, C. , Dean, C. , Zabel, P. , Bisseling, T. and Jones, G.H. (2000) Integrated cytogenetic map of chromosome arm 4S of A. thaliana: structural organization of heterochromatic knob and centromere region. Cell, 100, 367–376. [DOI] [PubMed] [Google Scholar]
- Fu, Y. , Kawabe, A. , Etcheverry, M. , Ito, T. , Toyoda, A. , Fujiyama, A. , Colot, V. , Tarutani, Y. and Kakutani, T. (2013) Mobilization of a plant transposon by expression of the transposon‐encoded anti‐silencing factor. EMBO J. 32, 2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendrel, A.V. , Lippman, Z. , Martienssen, R. and Colot, V. (2005) Profiling histone modification patterns in plants using genomic tiling microarrays. Nat. Meth. 2, 213–218. [DOI] [PubMed] [Google Scholar]
- 1001 Genomes Consortium (2016) 1135 sequenced natural inbred lines reveal the global pattern of polymorphism in Arabidopsis thaliana . Cell, 166, 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraut, L. , Falque, M. , Drouaud, J. , Pereira, L. , Martin, O.C. and Mezard, C. (2011) Genome‐wide crossover distribution in Arabidopsis thaliana meiosis reveals sex‐specific patterns along chromosomes. PLoS Genet. 7, e1002354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golyshev, S.A. , Vichreva, P.N. , Sheval, E.V. , Kiryanov, G.I. and Polyakov, V.Y. (2008) Role of DNA methylation and histone modifications in structural maintenance of heterochromatin domains (chromocenters). Cell Tiss. Biol. 2, 590–600. [PubMed] [Google Scholar]
- Gottschling, D.E. , Aparicio, O.M. , Billington, B.L. and Zakian, V.A. (1990) Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell, 16, 751–762. [DOI] [PubMed] [Google Scholar]
- Hagmann, J. , Becker, C. , Müller, J. , et al. (2015) Century‐scale methylome stability in a recently diverged Arabidopsis thaliana lineage. PLoS Genet. 11, e1004920–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, A.M. , Brachi, B. , Faure, N. , Horton, M.W. , Jarymowycz, L.B. , Sperone, F.G. , Toomajian, C. , Roux, F. and Bergelson, J. (2011) Adaptation to climate across the Arabidopsis thaliana genome. Science, 334, 83–86. [DOI] [PubMed] [Google Scholar]
- Hasterok, R. (2006) Alignment of the genomes of Brachypodium distachyon and temperate cereals and grasses using bacterial artificial chromosome landing with fluorescence in situ hybridization. Genetics, 173, 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, A. , Sgro, C. and Weeks, A. (2004) Chromosomal inversion polymorphisms and adaptation. Trends Ecol. Evol. 19, 482–488. [DOI] [PubMed] [Google Scholar]
- Horton, M.W. , Hancock, A.M. , Huang, Y.S. , et al. (2012) Genome‐wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet. 44, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovene, M. , Wielgus, S.M. , Simon, P.W. , Buell, C.R. and Jiang, J. (2008) Chromatin structure and physical mapping of chromosome 6 of potato and comparative analyses with tomato. Genetics, 180, 1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, L. , Zhang, W. , Xia, Z. , Jiang, G. , Qian, Q. , Li, A. , Cheng, Z. , Zhu, L. , Mao, L. and Zhai, W. (2007) A paracentric inversion suppresses genetic recombination at the FON3 locus with breakpoints corresponding to sequence gaps on rice chromosome 11L. Mol. Genet. Genom. 277, 263–272. [DOI] [PubMed] [Google Scholar]
- Kang, H.M. , Zaitlen, N.A. , Wade, C.M. , Kirby, A. , Heckerman, D. , Daly, M.J. and Eskin, E. (2008) Efficient control of population structure in model organism association mapping. Genetics, 178, 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kejnovsky, E. , Hobza, R. , Cermak, T. , Kubat, Z. and Vyskot, B. (2009) The role of repetitive DNA in structure and evolution of sex chromosomes in plants. Heredity, 102, 533–541. [DOI] [PubMed] [Google Scholar]
- Kenney, A.M. , McKay, J.K. , Richards, J.H. and Juenger, T.E. (2014) Direct and indirect selection on flowering time, water‐use efficiency (WUE, delta (13)C), and WUE plasticity to drought in Arabidopsis thaliana . Ecol. Evol. 4, 4505–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, M. (2010) How and why chromosome inversions evolve. PLoS Biol. 8, e1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, M. and Barton, N. (2006) Chromosome inversions, local adaptation and speciation. Genetics, 173, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knebel, S. , Pasantes, J.J. , Thi, D.A.D. , Schaller, F. and Schempp, W. (2011) Heterogeneity of pericentric inversions of the human Y chromosome. Cytogenet. Gen. Res. 132, 219–226. [DOI] [PubMed] [Google Scholar]
- Koornneef, M. , van Eden, J. , Hanhart, C.J. , Stam, P. , Braaksma, F.J. and Feenstra, W.J. (1983) Linkage map of Arabidopsis thaliana . J. Heredity, 74, 265–272. [Google Scholar]
- Kurtz, S. , Phillippy, A. , Delcher, A.L. , Smoot, M. , Shumway, M. , Antonescu, C. and Salzberg, S.L. (2004) Versatile and open software for comparing large genomes. Gen. Biol. 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, J.C. , Meyer, J.M. and Birchler, J.A. (2007a) A hemicentric inversion in the maize line knobless Tama flint created two sites of centromeric elements and moved the kinetochore‐forming region. Chromosoma, 116, 237–247. [DOI] [PubMed] [Google Scholar]
- Lamb, J.C. , Meyer, J.M. , Corcoran, B. , Kato, A. , Han, F. and Birchler, J.A. (2007b) Distinct chromosomal distributions of highly repetitive sequences in maize. Chrom. Res. 15, 33–49. [DOI] [PubMed] [Google Scholar]
- Lasky, J.R. , Des Marais, D.L. , McKay, J.K. , Richards, J.H. , Juenger, T.E. and Keitt, T.H. (2012) Characterizing genomic variation of Arabidopsis thaliana: the roles of geography and climate. Mol. Ecol. 21, 5512–5529. [DOI] [PubMed] [Google Scholar]
- Laufs, P. , Autran, D. and Traas, J. (1999) A chromosomal paracentric inversion associated with T‐DNA integration in Arabidopsis . Plant J. 18, 131–139. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Huang, Y. and Bergelson, J. (2010) Association mapping of local climate‐sensitive quantitative trait loci in Arabidopsis thaliana . Proc. Natl Acad. Sci. USA, 107, 21199–21204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linc, G. , Friebe, B.R. , Kynast, R.G. , Molnár‐Láng, M. , Koszegi, B. , Sutka, J. and Gill, B.S. (1999) Molecular cytogenetic analysis of Aegilops cylindrica Host. Genome, 42, 497–503. [PubMed] [Google Scholar]
- Lippman, Z. , Gendrel, A.V. , Black, M. , et al. (2004) Role of transposable elements in heterochromatin and epigenetic control. Nature, 430, 471–476. [DOI] [PubMed] [Google Scholar]
- Lippman, Z. , Gendrel, A.V. , Colot, V. and Martienssen, R. (2005) Profiling DNA methylation patterns using genomic tiling microarrays. Nat. Meth. 2, 219–224. [DOI] [PubMed] [Google Scholar]
- Lou, Q. , Iovene, M. , Spooner, D.M. , Buell, C.R. and Jiang, J. (2010) Evolution of chromosome 6 of Solanum species revealed by comparative fluorescence in situ hybridization mapping. Chromosoma, 119, 435–442. [DOI] [PubMed] [Google Scholar]
- Lovell, J.T. , Juenger, T.E. , Michaels, S.D. , Lasky, J.R. , Platt, A. , Richards, J.H. , Yu, X. , Easlon, H.M. , Sen, S. and McKay, J.K. (2013) Pleiotropy of FRIGIDA enhances the potential for multivariate adaptation. Proc. Royal Soc. London B: Biol. Sci. 280, 20131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, D.B. and Willis, J.H. (2010) A widespread chromosomal inversion polymorphism contributes to a major life‐history transition, local adaptation, and reproductive isolation. PLoS Biol. 8, e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewski, A.J. , Kopecky, D. and Linc, G. (2011) Inversions of chromosome arms 4AL and 2BS in wheat invert the patterns of chiasma distribution. Chromosoma, 121, 201–208. [DOI] [PubMed] [Google Scholar]
- Lysak, M.A. , Pecinka, A. and Schubert, I. (2003) Recent progress in chromosome painting of Arabidopsis and related species. Chrom. Res. 11, 195–204. [DOI] [PubMed] [Google Scholar]
- Lysak, M.A. , Berr, A. , Pecinka, A. , Schmidt, R. , McBreen, K. and Schubert, I. (2006) Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc. Natl Acad. Sci. USA, 103, 5224–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysak, M.A. , Mandakova, T. and Lacombe, E. (2010) Reciprocal and multi‐species chromosome BAC painting in crucifers (Brassicaceae). Cytogenet. Gen. Res. 129, 184–189. [DOI] [PubMed] [Google Scholar]
- Madan, K. (1995) Paracentric inversions: a review. Hum. Genet. 96, 503–515. [DOI] [PubMed] [Google Scholar]
- Mozo, T. , Fischer, S. , Meier‐Ewert, S. , Lehrach, H. and Altmann, T. (1998) Use of the IGF BAC library for physical mapping of the Arabidopsis thaliana genome. Plant J. 16, 377–384. [DOI] [PubMed] [Google Scholar]
- Muller, H.J. (1930) Types of visible variations induced by X‐rays in Drosophila . J. Genet. 22, 299–334. [Google Scholar]
- Myburg, A.A. , Remington, D.L. , O'Malley, D.M. , Sederoff, R.R. and Whetten, R.W. (2001) High‐throughput AFLP analysis using infrared dye‐labeled primers and an automated DNA sequencer. Biotechnology, 30, 348–352, 354, 356, 357. [DOI] [PubMed] [Google Scholar]
- Nordborg, M. , Borevitz, J.O. , Bergelson, J. , et al. (2002) The extent of linkage disequilibrium in Arabidopsis thaliana . Nat. Genet. 30, 190–193. [DOI] [PubMed] [Google Scholar]
- Ohtsubo, Y. , Ikeda‐Ohtsubo, W. , Nagata, Y. and Tsuda, M. (2008) GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinform. 9, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski, S. , Schneeberger, K. , Lucas‐Lledo, J.I. , Warthmann, N. , Clark, R.M. , Shaw, R.G. , Weigel, D. and Lynch, M. (2010) The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana . Science, 327, 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J.L. , Constandt, H. , Neyt, P. , Cnops, G. , Zethof, J. , Zabeau, M. and Gerats, T. (2001) A physical amplified fragment‐length polymorphism map of Arabidopsis . Plant Physiol. 127, 1579–1589. [PMC free article] [PubMed] [Google Scholar]
- Platt, A. , Horton, M. , Huang, Y.S. , et al. (2010) The scale of population structure in Arabidopsis thaliana . PLoS Genet. 6, e1000843–e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop, M. , Phillippy, A. , Delcher, A.L. and Salzberg, S.L. (2004) Comparative genome assembly. Brief. Bioinform. 5, 237–248. [DOI] [PubMed] [Google Scholar]
- Ranz, J.M. , Maurin, D. , Chan, Y.S. , von Grotthuss, M. , Hillier, L.W. , Roote, J. , Ashburner, M. and Bergman, C.M. (2007) Principles of genome evolution in the Drosophila melanogaster species group. PLoS Biol. 5, e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, T.P. , Carpenter, R. and Coen, E.S. (1989) A chromosome rearrangement suggests that donor and recipient sites are associated during Tam3 transposition in Antirrhinum majus . EMBO J. 8, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, K.J. , Fransz, P. and Jones, G.H. (1996) A light microscopic atlas of meiosis in Arabidopsis thaliana . Chrom. Res. 4, 507–516. [DOI] [PubMed] [Google Scholar]
- Rowan, B.A. , Patel, V. and Weigel, D. (2015) Rapid and inexpensive whole‐genome genotyping‐by‐sequencing for crossover localization and fine‐scale genetic mapping. G3: Genes Genomes Genetics, 5, 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, R. , West, J. , Love, K. , Lenehan, Z. , Lister, C. , Thompson, H. , Bouchez, D. and Dean, C. (1995) Physical map and organization of Arabidopsis thaliana chromosome 4. Science, 270, 480–483. [DOI] [PubMed] [Google Scholar]
- Schneider, K.L. , Xie, Z. , Wolfgruber, T.K. and Presting, G.G. (2016) Inbreeding drives maize centromere evolution. Proc. Natl Acad. Sci. 113, E987–E996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo, C. , Aranzana, M.J. , Lister, C. , Baxter, C. , Nicholls, C. , Nordborg, M. and Dean, C. (2005) Role of FRIGIDA and FLOWERING LOCUS C in determining variation in flowering time of Arabidopsis . Plant Physiol. 138, 1163–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppe, W. , Jasencakova, Z. , Houben, A. , Kakutani, T. , Meister, A. , Huang, M.S. , Jacobsen, S.E. , Schubert, I. and Fransz, P.F. (2002) DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis . EMBO J. 21, 6549–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson, H. , Helgason, A. , Thorleifsson, G. , et al. (2005) A common inversion under selection in Europeans. Nat. Genet. 37, 129–137. [DOI] [PubMed] [Google Scholar]
- Szinay, D. (2010) Dynamics and characterisation of the two major repeat families in tomato (Solanum lycopersicum) In The Development of FISH Tools for Genetic, Phylogenetic and Breeding Studies in Tomato (Solanum lycopersicum). PhD thesis, Wageningen University. ISBN 978‐90‐8585‐635‐1. [Google Scholar]
- Szinay, D. , Wijnker, E. , van den Berg, R. , Visser, R.G.F. , de Jong, H. and Bai, Y. (2012) Chromosome evolution in Solanum traced by cross‐species BAC‐FISH. New Phytol. 195, 688–698. [DOI] [PubMed] [Google Scholar]
- Tang, X. , Szinay, D. , Lang, C. , et al. (2008) Cross‐species bacterial artificial chromosome‐fluorescence in situ hybridization painting of the tomato and potato chromosome 6 reveals undescribed chromosomal rearrangements. Genetics, 180, 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanurdžić, M. , Vaughn, M.W. , Jiang, H. , Lee, T.J. , Slotkin, R.K. , Sosinski, B. , Thompson, W.F. , Doerge, R.W. and Martienssen, R.A. (2008) Epigenomic consequences of immortalized plant cell suspension culture. PLoS Biol. 6, 2880–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature, 408, 796–815. [DOI] [PubMed] [Google Scholar]
- The Cold Spring Harbor Laboratory, Washington University Genome Sequencing Center, and PE Biosystems Arabidopsis Sequencing Consortium (2000) The complete sequence of a heterochromatic island from a higher eukaryote. Cell, 100, 377–386. [PubMed] [Google Scholar]
- Vaughn, M.W. , Tanurdzic, M. , Lippman, Z. , et al. (2007) Epigenetic natural variation in Arabidopsis thaliana . PLoS Biol. 5, e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos, P. , Hogers, R. , Bleeker, M. , et al. (1995) AFLP: a new technique for DNA fingerprinting. Nucl. Acids Res. 23, 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. and Tanksley, S.D. (2010) Chromosomal evolution in the plant family Solanaceae . BMC Genom. 11, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata, L. , Ding, J. and Willing, E.M. et al. (2016) Chromosome‐level assembly of Arabidopsis thaliana Ler reveals the extent of translocation and inversion polymorphisms. Proc. Natl Acad. Sci. 113, E4052–E4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Liu, Z. , Liu, C. , Yang, Z. , Deng, K. , Peng, J. , Zhou, J. , Li, G. , Tang, Z. and Ren, Z. (2008) Analysis of DNA methylation variation in wheat genetic background after alien chromatin introduction based on methylation‐sensitive amplification polymorphism. Chin. Sci. Bull. 53, 58–69. [Google Scholar]
- Zhong, X.B. , Fransz, P.F. , Wennekes‐Eden, J. , Ramanna, M.S. , van Kammen, A. , Zabel, P. and Hans de Jong, J. (1998) FISH studies reveal the molecular and chromosomal organization of individual telomere domains in tomato. Plant J. 13, 507–517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genetic and physical map positions of markers in chromosome 4.
Figure S2. Dot plot comparison of Ler and Col‐0 fragments at the inversion breakpoints.
Figure S3. Specification of DNA sequences at the breakpoints of the inversion.
Figure S4. Sequence alignment of A. lyrata (XM_002874807.1), Ler (ATL8C23344) and the reconstructed Col‐0 region (At4 g03635 + At4 g05500).
Figure S5. Alignment of the F‐box genes in A. lyrata, Ler and the reconstructed Col‐0.
Figure S6. Chromatin profile at the boundary of the inversion.
Figure S7. Genetic relationships of RegMap accessions.
Figure S8. Identification of inversion accessions by PCR analysis of the inversion breakpoints.
Figure S9. DAPI‐stained pachytene complements.
Figure S10. Scatter plot of left against right introgression sites based on the major haplotype shared by the majority of inversion accessions.
Figure S11. Arabidopsis accessions in 5° latitude‐by‐longitude grids.
Figure S12. Phylogenetic tree of all accessions from the 1001 genome database based on the long arm of chromosome 4.
Figure S13. The genetic distance of 1135 Arabidopsis accessions to Col‐0 in the inverted region, showing clear bimodal pattern.
Table S1. Recombination frequencies (±SD) between ga1, bp and tt11.
Table S2. Estimated molecular lengths in kb based on fiber‐FISH analysis (n = 20–30 per accession).
Table S3. List of primers for the fragments a–e.
Table S4. List of 174 accessions inferred to have the inversion.
Table S5. Patterns of 250K chip SNP variation in the inversion region and in a ‘Control’ region, 13.214–15.097 Mb on chromosome 4.
Table S6. List of 53 representative European and North American inversion accessions ranked according to their left and right introgression site.