

Abstract
Removal of the 5′ cap on mRNA by the decapping enzyme Dcp2 is a critical step in 5′-to-3′ mRNA decay. Understanding the structural basis of Dcp2 activity has been a significant challenge because Dcp2 is dynamic, with weak affinity for cap substrate. Here we present a 2.6-Å-resolution crystal structure of a heterotrimer of fission yeast Dcp2, its essential activator Dcp1, and the human NMD cofactor PNRC2, in complex with a tight-binding cap analog. Cap binding is accompanied by a conformational change of Dcp2 to form a composite nucleotide binding site using conserved residues on the catalytic and regulatory domains. Kinetic analysis of PNRC2 reveals a conserved short linear motif enhances both substrate affinity and the catalytic step of decapping. These findings explain why Dcp2 requires a conformational change for efficient catalysis and reveals that coactivators can promote RNA binding and the catalytic step of decapping, possibly through different conformational states.
INTRODUCTION
All eukaryotic mRNA transcripts carry a 7-methyl-guanosine cap at the 5′ end that is critical for splicing, export, translation, and mRNA stability.1,2 Removal of the 5′ cap inhibits translation initiation and generally commits an mRNA transcript to rapid degradation via 5′-to-3′ mRNA decay pathways.3 Cap cleavage is carried out by the conserved decapping enzyme Dcp2, a Nudix hydrolase family member that catalyzes the hydrolysis of the m7GpppN cap structure to release m7GDP and 5′ monophosphate RNA.4–6 Decapped, monophosphate RNA transcripts are then targeted and fully degraded by conserved 5′-to-3′ exonucleases.7 Decapping plays an important role in a variety of cellular pathways including bulk 5′-to-3′ mRNA decay,4,5 decay of lncRNA,8 miRNA-mediated decay,9,10 nonsense-mediated decay,11–13 Staufen-mediated decay,14,15 transcription termination,16 and decay of transcripts containing non-optimal codons.17 Recent studies in mammalian cells link Dcp2 function to diseases related to RNA quality control such as spinal muscular atrophy,18 and to the innate immunity pathway and interferon reponse.19 Dcp2 activity is regulated by a network of protein-protein interactions that include the essential activator Dcp1,20 and general and pathway-specific coactivators such as Edc1–4,21–26 PNRC1&2,27 Pat1/Lsm1–7,28–30 and Dhh1.31 Together with RNA and coactivator proteins, the Dcp1–Dcp2 complex forms the catalytic core of decapping ribonucleoprotein complexes that play a critical role in regulating cellular mRNA stability and gene expression.
Dcp2 is a bilobed enzyme consisting of a conserved N-terminal regulatory domain that binds the activator Dcp1, a catalytic Nudix domain that binds RNA and carries out cap hydrolysis chemistry, and a disordered C-terminal extension of variable length that contains protein-protein interaction motifs.20,32–35 The regulatory and catalytic domains of Dcp2 are connected by a short, flexible linker, which allows Dcp2 to adopt multiple conformations in solution.36 Although the Nudix domain can bind RNA and carry out cap hydrolysis in isolation, both Dcp1 and the regulatory domain of Dcp2 are essential for decapping in yeast.3 Moreover, Dcp1 and the regulatory domain of Dcp2 enhance catalytic activity by 10-fold and 100-fold respectively, in vitro.37 Biochemical and NMR data suggest that the regulatory domain harbors a binding site for the 5′ cap and that cap recognition occurs via a composite active site composed of residues on both the Nudix and regulatory domains of Dcp2.37 However, very weak affinity of Dcp2 for cap (~10mM),37 coupled with the enzyme’s conformational dynamics,36 have made characterization of the substrate-bound, catalytically-active state of the enzyme challenging, and a molecular-level understanding of Dcp2 catalysis has been elusive.
Dcp1 acts a platform for binding decapping coactivators that contain short linear interaction motifs found in proteins conserved from yeast to humans.38 These include the enhancer of decapping proteins (Edc1 and Edc2), human PNRC2 and likely PNRC1.21,22,27 These proteins contain a proline-rich motif that binds a hydrophobic cleft on Dcp1 and residues N-terminal to this motif to enhance RNA binding and substrate catalysis by the Dcp1–Dcp2 complex. Recently, Valkov et al showed that a similar activator in fission yeast, Sp Edc1 (also known as Dbs1), can promote a large conformational change in the Dcp1–Dcp2 complex that brings the RNA binding helix of the Dcp2 Nudix domain in proximity to a positively charged surface of Dcp1, likely creating a channel that binds RNA.39 Based on this new conformation of Dcp1–Dcp2, it was proposed that activation of the decapping complex occurs primarily by promoting a conformation of the enzyme that enhances affinity for RNA. However, this structure, and all previous structures of Dcp2 or Dcp1–Dcp2, do not have cap substrate bound to the enzyme, and so it remains unclear how the decapping complex recognizes and hydrolyzes the 5′ cap of RNA.
We recently described the discovery and characterization of a tight-binding two-headed cap analog that binds to Dcp2 with 20-fold tighter affinity than native cap or any previously tested cap analogs.40,41 We sought to use this tight-binding cap analog, in combination with Edc or PNRC coactivators, as tools to trap the substrate-bound, catalytically-active conformation of the Dcp1–Dcp2 decapping complex. Here we show that binding of cap analog to a PNRC2–Dcp1–Dcp2 complex induces a heretofore unknown conformation of Dcp2 that reveals a composite substrate binding site composed of residues on the regulatory and Nudix domains. A conformational change brings together conserved cap-binding residues that are either buried or far apart in all other structures of Dcp2 or Dcp1–Dcp2. Furthermore, in this cap-bound structure a positively charged tunnel extends from the RNA binding helix on the Nudix domain to the composite cap-binding site, suggesting a new path for RNA binding. Finally, we show that a conserved, short-linear motif in PNRC and Edc1,2 coactivator proteins activates decapping at the level of substrate binding and the catalytic step. Our cap-bound structure of Dcp1–Dcp2 provides the first molecular-level picture of cap recognition by Dcp2, explains a wealth of prior biochemical and biophysical data, and allows us to begin to interpret the different conformational states of Dcp2 within the broader context of the enzyme’s catalytic cycle.
RESULTS
Cap binding requires a unique conformational change in Dcp2
Fission yeast Dcp1–Dcp2 has been crystallized in prior studies and we found that coactivator human PNRC2 could robustly stimulate decapping of this complex in vitro (Supplementary Fig. 1a,b). Accordingly, we set out to determine structures of Sp Dcp1–Dcp2 in complex with coactivator Hs PNRC2 and in the presence of a tight-binding ‘two-headed’ cap analog, in order to trap and structurally characterize a substrate-bound, activated conformation of the decapping enzyme. For crystallization studies, we found that human PNRC2(91-121) was sufficient to activate fission yeast Dcp1–Dcp2, and that single chain constructs of Hs PNRC2(91-121) covalently tethered to the N-terminus of Sp Dcp2(1-243), in complex with Sp Dcp1 (hereafter denoted Dcp1–scPNRC2Dcp2), were both catalytically active and amenable to crystallization (Supplementary Fig. 1c,d). To explore the role of PNRC2 in activation of decapping, we first determined a 3.0 Å crystal structure of the Dcp1–scPNRC2Dcp2 complex in the absence of cap analog (Fig. 1a, Table 1, Supplementary Fig. 2a). Although the crystal form of this complex is different from other known structures of Dcp1–Dcp2, the conformation of the apo Dcp1–Dcp2 heterodimer is very similar to the previously published “closed”, ATP-bound conformation of the enzyme,20 and is nearly identical to a more recently published structure of Dcp1–Dcp2 (Supplementary Fig. 2b,c).39 As we have reported previously, this conformation of the enzyme is incompatible with substrate binding and catalysis.37 Our structure also shows residues 100–115 of Hs PNRC2 bound to the aromatic cleft of Sp Dcp1 in a similar conformation as Hs PNRC2 binds to Hs Dcp1.27 Though residue 100 of PNRC2 points towards the Nudix domain, density for residues 91–99 of PNRC2 was unobservable, presumably due to disorder. This disordered region of PNRC2 contains a conserved YAGxxF motif shared with coactivators Edc1&2 that is critical for Dcp2 activation in yeast.22
Figure 1.
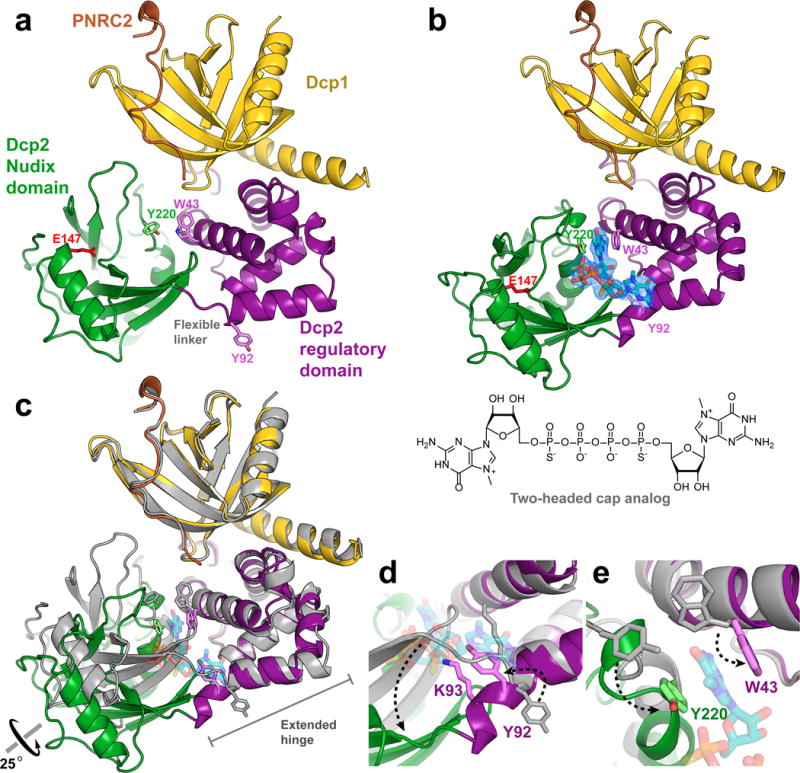
Cap binding requires a conformational change in Dcp2. (a) Crystal structure of apo Dcp1–scPNRC2Dcp2. (b) Crystal structure of two-headed cap analog-bound Dcp1–scPNRC2Dcp2. Fo-Fc omit map is shown for the cap analog at 2σ, cap-binding aromatic residues Y220, W43 and Y92 are shown as sticks, along with the general base E147 in red. (c) Structural alignment of apo (gray) and cap-analog bound (colored) conformations of Dcp1–Dcp2 shows that cap binding results in a 25° rotation of the Nudix domain and restructuring of the extended hinge. PNRC2 peptide is not shown. (d) Close-up of structural alignment showing restructuring of the extended hinge region. Extension of the C-terminal helix of the Dcp2 regulatory domain flips Y92 and repositions K93 for cap binding. (e) Close-up of structural alignment showing conformational and rotameric changes of the aromatic cap binding residues Y220 and W43 required for cap recognition.
Table 1.
Crystallographic data collection and refinement statistics.
| Dcp1–scPNRC2Dcp2 (PDB 5KQ1) |
Dcp1–scPNRC2Dcp2 with two-headed cap bound (PDB 5KQ4) | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 45.62, 118.73, 93.17 | 43.07, 120.75, 91.85 |
| α, β, γ (°) | 90.00, 102.77, 90.00 | 90.00, 97.90, 90.00 |
| Resolution (Å) | 50.0 – 3.00 (3.05 – 3.00)a |
50.0 – 2.55 (2.61 – 2.55) |
| Rmerge | 0.106 (0.745) | 0.115 (0.940) |
| I/σI | 24.2 (1.90) | 25.6 (1.74) |
| CC1/2 | 94.3 (79.6) | 87.9 (62.6) |
| Completeness (%) | 99.0 (91.9) | 100.0 (99.9) |
| Redundancy | 3.7 (3.4) | 4.0 (3.9) |
| Refinement | ||
| Resolution (Å) | 44.9 – 3.00 | 45.5 – 2.56 |
| No. reflections | 19,125 | 29,862 |
| Rwork/Rfree | 0.2101/0.2474 | 0.2065/0.2510 |
| No. atoms | ||
| Protein | 6274 | 6347 |
| Ligand | – | 114b |
| Water | – | 27 |
| B-factors | ||
| Protein | 128 | 83 |
| Ligand | – | 117b |
| Water | – | 64 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.003 | 0.003 |
| Bond angles (°) | 0.616 | 0.655 |
Each data set was collected from a single crystal.
Values in parentheses are for highest-resolution shell.
Two molecules of two-headed cap analog.
To determine how cap interacts with the decapping complex, we soaked a tight-binding two-headed cap analog into the aforementioned Dcp1–scPNRC2Dcp2 crystals and solved a 2.6 Å substrate-analog-bound structure of the heterotrimer. (Fig. 1b, Table 1, Supplementary Fig. 3). The two-headed cap analog consists of two m7G nucleotides linked by a tetraphosphate bridge, which binds 20-fold more tightly than native cap and is readily hydrolyzed by Dcp2 as the cap analog alone, or when incorporated at the 5′ end of RNA.40 Binding of this cap analog induces a significant conformational change in Dcp2 within the crystal, consisting primarily of a 25° rotation of the Nudix domain away from Dcp1, relative to the apo structure (Fig. 1c, Supplementary Video 1). Reorientation of the Nudix domain for cap binding is accommodated by a restructuring of the C-terminal helix of the Dcp2 regulatory domain and the flexible linker that connects the regulatory and Nudix domains, which together form an extended hinge region. A twisting motion in the extended hinge elongates the regulatory domain C-terminal helix by a full turn and reorients the conserved residue Y92 (Fig. 1d). In this position Y92 points toward the interior of the protein, rather than toward solvent, where it binds one of the m7G nucleotides of two-headed cap analog. Extension of the C-terminal helix on the regulatory domain also shifts conserved residue K93 from a flexible linker into a helical secondary structure, where it is positioned for substrate binding. Finally, rotation of the Nudix domain brings conserved residues Y220 and W43 into alignment, and a rotameric change of W43 allows these two aromatic residues to bind the second m7G nucleotide of the two-headed cap analog (Fig. 1e).
Although we have not yet trapped the transition state of decapping, as the catalytic base E147 is too far from substrate to carry out chemistry, our cap analog-bound structure of Dcp2 reveals a new conformation of the decapping enzyme that for the first time defines the structural basis of cap binding and recognition. Critically, the conserved aromatic residues (W43, Y220, Y92) that form the basis for cap binding are either occluded or separated by large distances in all other Dcp1–Dcp2 structures, including the recently published “activated” Sp Edc1–Dcp1–Dcp2 structure (Supplementary Fig. 4).39 The new Dcp2 conformation in our cap analog-bound Dcp1–Dcp2 complex suggests that previously identified conformations of Dcp2 are incapable of binding and hydrolyzing cap substrate and must undergo large conformational changes in order to recognize the 5′ mRNA cap.
Cap is recognized by a composite substrate binding site
The conformational change observed in Dcp2 upon cap analog binding results in a repositioning of residues at the Dcp2 interdomain interface to form a composite substrate binding site in which conserved, catalytically important residues on both the Nudix and regulatory domains of Dcp2 recognize cap (Fig. 2a; Supplementary Fig. 5). One m7G nucleotide of the two-headed cap analog is sandwiched by the conserved aromatic residues W43 and Y220, with D47 making hydrogen bonds with the 7-methyl-guanine ring that mimic Watson-Crick base pairing (Fig. 2b). This nucleotide binding mode, very similar to m7G cap recognition motifs in eIF4E and CBP20,42,43 explains why W43 and D47 are strongly perturbed by m7GDP in NMR titration experiments, why W43 is required for nucleotide-dependent compaction of Dcp2 in SAXS experiments,37 and why these residues on the regulatory domain are critical for cap binding and catalysis (Supplementary Table 1). Mutation of W43 impairs the catalytic step of decapping by 20-fold, while mutation of D47 reduces the catalytic step by 80-fold, nearly the same as deletion of the entire regulatory domain in vitro.37 Both W43 and D47 are critical for activation by Dcp1 and coactivator Edc1, and their mutation results in large decapping defects in vivo.32,36 Y220 and K127, which both have modest (~2-fold) effects on decapping,33 make hydrogen bond contacts with the phosphate backbone of cap. Similarly, E39 appears to make a hydrogen bond contact with the 2′ OH of the ribose ring, and its mutation has a ~2-fold effect on decapping.32
Figure 2.
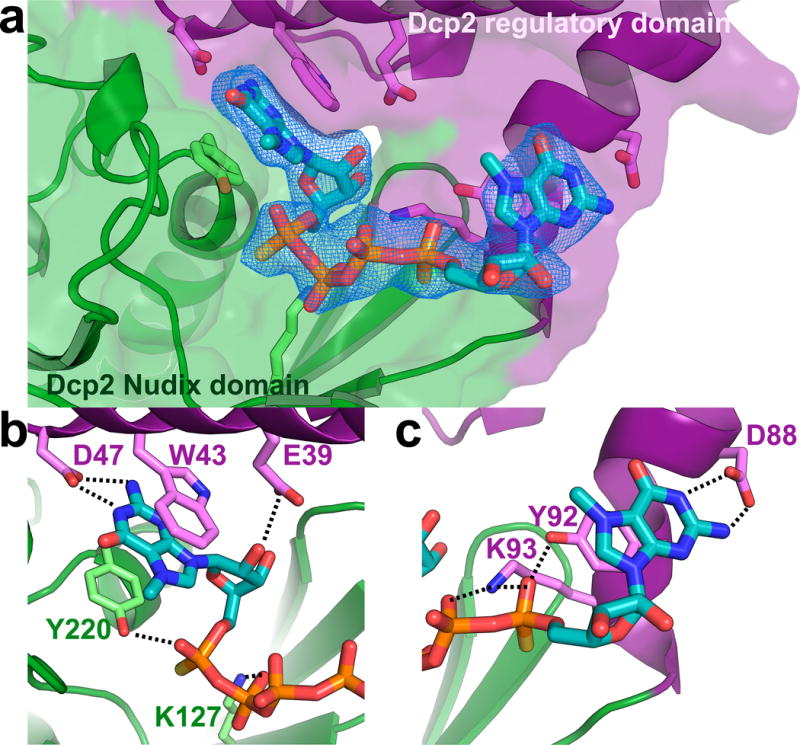
Cap is recognized by a composite nucleotide binding site on Dcp2. (a) Dcp2 interdomain interface showing two-headed cap analog bound by residues on both the regulatory (purple) and Nudix (green) domains. Fo-Fc omit map is shown for cap analog at 2σ. (b) Close-up view showing interactions between m7G nucleotide of two-headed cap analog and residues on both domains of Dcp2. The aromatic m7G is sandwiched between conserved residues W43 and Y220, and D47 makes hydrogen bonds to the 7-methyl-guanine base. Y220, K127 and E39 make additional hydrogen bond contacts with the phosphate chain and ribose ring. (c) Close-up view showing interactions between the other m7G nucleotide of two-headed cap analog and the extended hinge that links Dcp2 regulatory and Nudix domains. The m7G ring stacks against Y92, while Y92 and K93 make hydrogen bond contacts with the phosphate chain. D88 makes hydrogen bonding interactions with the 7-methyl-guanine base. Distances for these interactions are shown in Supplementary Fig. 5.
The second m7G nucleotide of cap analog is bound by a cluster of residues located in the extended hinge region between the regulatory and catalytic domains of Dcp2 (Fig. 2c). The conserved residue Y92 stacks against the m7G base and hydrogen bonds with the phosphate chain of cap analog. Mutation of Y92 results in a ~5-fold defect in decapping in vivo.32 Interestingly, Y92 resonances are not perturbed by addition of m7GDP or two-headed cap analog in NMR titration experiments with the isolated Dcp2 regulatory domain,37,40 suggesting conformational changes in the hinge region and its coupling with Nudix domain motions, are critical for cap recognition. Upon cap binding, conserved residue K93 moves from a flexible linker to a structured helix and likely makes multiple hydrogen bond contacts with the phosphate chain of cap; its mutation results in modest decapping defects both in vitro and in vivo.32,33 Finally, D88 makes hydrogen bond contacts with the 7-methyl-guanine base of cap analog that mimic Watson-Crick base pairing. In summary, cap is bound by a composite nucleotide binding site consisting primarily of conserved residues on both the regulatory and Nudix domains, and in the extended hinge region between the two domains of Dcp2. The existence of a composite cap binding site, as predicted by solution NMR and kinetic analyses, explains the nearly 100-fold enhancement of catalysis contributed by the regulatory domain of Dcp2 in vitro, and the catalytic importance of many conserved residues at the Dcp2 interdomain interface.37
Cap analog binding reveals possible RNA binding tunnel
The rotation of the Dcp2 Nudix domain that composes the composite cap binding site also creates a positively-charged tunnel that leads from the dorsal helix of the Nudix domain, through a cavity between the regulatory and catalytic domains of Dcp2, directly to the cap binding site (Fig. 3). The dorsal helix harbors the positively charged Box B motif, whose conserved lysine or arginine residues are critical for RNA binding.20,35 The flexible, lysine-rich loop on the dorsal surface of the catalytic domain (K208-K216) has also been shown to be important for RNA binding,33 and several lysine residues in this loop (K208 and K209) sit just below the entrance to the positively charged tunnel. Strikingly, the cap-bound conformation of Dcp2 shifts K216 from a flexible, solvent-exposed loop, to a more structured helical turn that points the sidechain of K216 directly into the positively charged tunnel (Fig. 3a, inset). This raises the intriguing possibility that the RNA body may bind the dorsal surface of the Nudix domain and then thread through a composite tunnel between the regulatory and Nudix domains to access the cap binding site.
Figure 3.
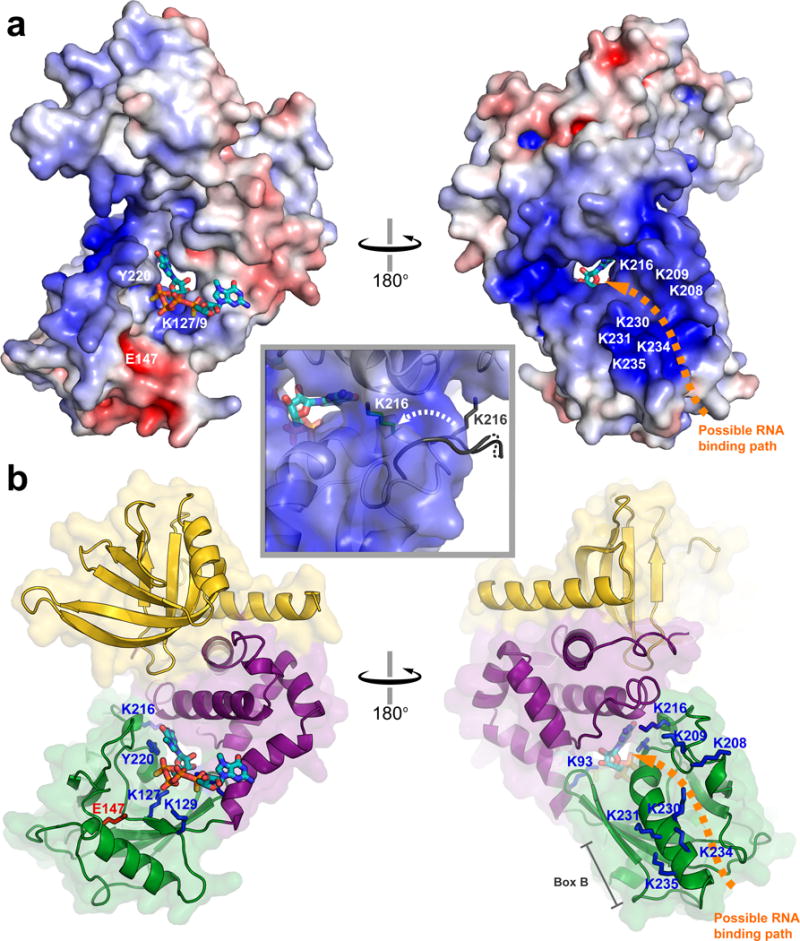
The cap-bound conformation of Dcp2 harbors a positively-charged tunnel that may bind RNA. (a) Electrostatic surface potential of the cap-bound conformation of Dcp1–Dcp2 (−7 to +7 kT/e; PNRC2 is not shown). The location of RNA binding residues are labeled in white, and an orange arrow shows a possible path for RNA binding through the tunnel to the cap binding site. The inset shows a close-up view of the positively charged tunnel, and the movement of K216 from a solvent-exposed flexible loop in the apo structure (gray) to a more structured conformation that points into the putative RNA binding tunnel in the cap-bound structure (green stick). (b) Cartoon views of the cap-bound Dcp1–Dcp2 structure oriented as in (a), Dcp1 is yellow, Dcp2 regulatory domain is purple, Dcp2 Nudix domain is green, residues important for RNA binding are highlighted as blue sticks, and the catalytic base E147 is shown in red.
If RNA does thread through the positively-charged tunnel, this suggests that the nucleotide of two-headed cap analog bound by W43/Y220/D47 represents the first transcribed nucleotide of RNA, and the nucleotide bound by Y92 is the m7G of cap. Several lines of evidence support this hypothesis. First, W43 and D47 bind both m7GDP and GDP in NMR titrations with the regulatory domain.37 Second, Y220 binds the adenine base of ATP in a previous structure of Dcp1–Dcp2.20 Third, several other Nudix enzymes, including Ap4A and MTH1, use a conserved aromatic at the Y220 position to bind substrate nucleotides.44,45 These observations together with our structure, suggest that the W43/D47/Y220 composite nucleotide binding site may bind the first transcribed nucleotide of mRNA and that the RNA body may thread through the positively charged tunnel to the RNA binding residues located on the dorsal surface of the Dcp2 Nudix domain. However, we cannot rule out the possibility that RNA takes an external route around the side of the Nudix domain and that the m7G of cap is bound by W43/Y220/D47 near the center of the complex (Supplementary Figure 6). Distinguishing these RNA binding modes will require an RNA-bound structure of Dcp2.
A conserved activation motif enhances substrate binding and catalysis
Coactivators such as PNRC2, Edc1 and Edc2 share two conserved, short linear motifs: a C-terminal proline-rich Dcp1 binding motif, and a YAGxxF Dcp2 activating motif located 8–10 amino acids N-terminal to the Dcp1 binding motif that in Sc Edc1 is required for activation of Dcp2 (Fig. 4a).22 Sequence alignment reveals that the fission yeast protein Sp Edc1 (or Dbs1) also contains the tandem Dcp2-activating/Dcp1-binding sequences in a predicted disordered region of the protein, and we found that this protein robustly activates decapping (Supplementary Fig. 7a). Valkov et al recently showed that Sp Edc1 peptides containing both motifs can also promote a new conformation of the Dcp1–Dcp2 decapping complex, in which the YAGxxF motif extends a beta-sheet on the Nudix domain of Dcp2 to stabilize a conformation of the enzyme that may enhance RNA binding.39
Figure 4.
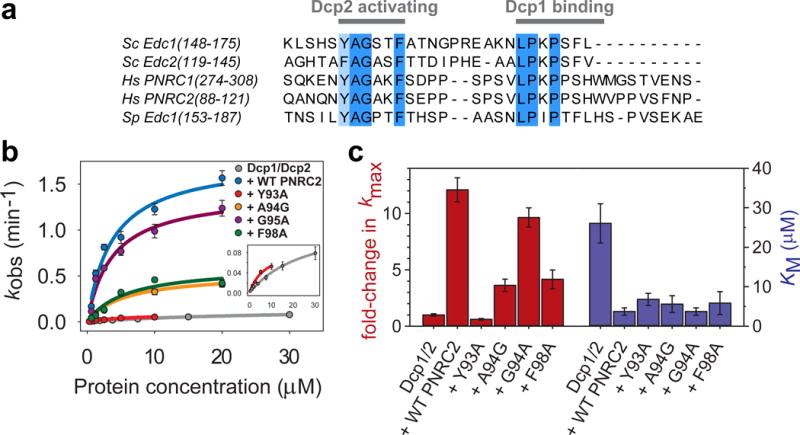
A conserved short-linear motif enhances the catalytic step of decapping. (a) Sequence alignment of decapping coactivators that share the tandem Dcp2-activating motif and Dcp1-binding motif. (b) Fits of kobs versus protein concentration under single-turnover conditions to determine kmax and KM for Dcp1–Dcp2 and WT or mutant complexes of Hs PNRC2(1-121) with Sp Dcp1–Dcp2(1-243). The single point mutation Y93A in the PNRC2 Dcp2-activating motif completely abolishes its ability to activate decapping chemistry. Errors are the standard error of individual fits to determine kobs. (c) Bar graph showing effects of PNRC2 and point mutants in the Dcp2-activating motif on fold-activation of Dcp1–Dcp2 kmax (red), or the KM (blue) determined for each protein complex. Errors are the standard error of individual fits to determine kmax and KM. Primary kinetics data used to construct the plots in (b) and bar graphs in (c) can be found in Supplementary Dataset 1.
In our Dcp1–scPNRC2Dcp2 structures, the N-terminus of the PNRC2 peptide that contains the YAGxxF activation motif was disordered and could not be modeled. Because our structure had no apparent density for the Dcp2 activation motif, we turned to kinetic analysis to understand how this motif might be stimulating Dcp2. We co-expressed and purified complexes of Hs PNRC2(1-121) – Sp Dcp1–Dcp2 in which each of the four conserved residues in the Dcp2 activating motif were individually mutated to alanine (or glycine in the case of A94), and determined KM and kmax for decapping of a 355mer RNA substrate in vitro under single-turnover conditions (Fig. 4b,c; Supplementary Fig. 7b,c; Supplementary Dataset 1).46 Addition of WT Hs PNRC2(1-121) to Sp Dcp1–Dcp2 robustly activates decapping and increases kmax by 12-fold, and decreases KM by 7-fold, suggesting that PNRC2 enhances both the catalytic step and substrate binding. A single point mutation in the activation motif, Y93A, completely abolishes any activation of kmax by PNRC2, while leaving KM unchanged. Point mutants A94G and F98A had an intermediate effect on PNRC2-mediated activation, providing only a 4-fold stimulation of kmax compared to Dcp1–Dcp2 alone. The G95A mutation had no effect on kmax. For all of the single point mutations in the Dcp2 activating motif, the KM remained unchanged and PNRC2 retained its ability to lower the KM of Dcp1–Dcp2. This is consistent with previous studies of coactivator Sc Edc1, in which it was found that residues N-terminal to the YAGxxF motif were primarily responsible for changes in KM, while peptides containing both the Dcp2 activating and Dcp1 binding motifs were sufficient to fully stimulate kmax.22 Our kinetic analysis suggests that coactivators containing the conserved Dcp2 activating and Dcp1 binding motifs stimulate decapping by enhancing both the catalytic step (increasing kmax) and substrate affinity (decreasing KM), and that mutations in the conserved activation motif affect only PNRC2-mediated activation of the catalytic step.
DISCUSSION
Over the last decade, the decapping enzyme Dcp2 has been crystallized in a number of very different conformations and extensively characterized biochemically. However, a major obstacle to understanding how different conformational states affect catalysis has been its weak affinity for 5′ cap substrate and the consequent lack of a substrate-bound Dcp2 structure. We crystallized a complex of Hs PNRC2 and Sp Dcp1–Dcp2 (Dcp1–scPNRC2Dcp2) bound to a synthetic two-headed cap analog that for the first time shows how Dcp2 engages cap substrate (Fig. 1). Our structure reveals a new conformation of the enzyme that reorganizes conserved, catalytically-critical residues at the interface of the regulatory and Nudix domains of Dcp2 to form a composite nucleotide binding site (Fig. 2). This is consistent with previous studies showing that both the regulatory and Nudix domains of Dcp2 bind the 5′ cap and that cap recognition occurs during the catalytic step of decapping.37 Our cap-bound structure rationalizes why the regulatory domain contributes 100-fold to kmax in vitro, and why conserved residues D47 and W43 are critical for cap binding and contribute 80-fold and 20-fold to kmax, respectively. The role of W43 in cap recognition is also consistent with its importance for nucleotide-dependent compaction of the Dcp1–Dcp2 complex observed in SAXS experiments.37 Furthermore, we identify additional conserved residues that make contacts with cap such as Y92, K93, Y220, and E39, which explains why mutation of these residues results in in vitro and/or in vivo decapping defects.32,33,37 Cap recognition is accompanied by a conformational change of Dcp2 that exposes conserved cap-binding residues that are occluded in other structures of Dcp1–Dcp2.
A recent crystal structure of Sp Dcp1–Dcp2 in complex with coactivator Sp Edc1 by Valkov et al trapped Dcp2 in a very different conformation in which the RNA binding helix on Dcp2, which harbors the “Box B” motif critical for substrate binding, is positioned near a positively-charged surface of Dcp1.39 Coactivator Sp Edc1 appears to promote this conformation by bridging an extended surface of the Nudix and regulatory domains of Dcp2. Based on this structure and data showing that Edc1 and the positively charged surface of Dcp1 enhance RNA binding, the authors concluded that this conformation represents the active conformation of the decapping enzyme and that activation by Edc1-like coactivators and Dcp1 is primarily accomplished by improved binding of RNA substrate. However, our cap-bound conformation of Dcp1–Dcp2 suggests that the Edc1-bound conformation solved by Valkov et al is incompatible with 5′ RNA cap recognition and therefore incompatible with catalysis. Residues that compose the composite nucleotide binding site in our structure are either buried, or separated by large distances in the Edc1–Dcp1–Dcp2 structure (Supplementary Fig. 4c). For example, in the Edc1-bound structure D47 and W43, which are critical for cap binding and catalysis, are buried in the protein-protein interface between the regulatory domain and Edc1, and are thus unavailable for cap binding. Likewise, W43 and Y220, residues on the regulatory and Nudix domains of Dcp2 that come together in our structure to bind one nucleotide of cap, are separated by nearly 20 Å in the Edc1-bound conformation.
The apparent conflict between the cap-bound conformation of Dcp2 identified in this study, and the Edc1-bound conformation of Dcp2 identified by Valkov et al, may be reconciled by our kinetic analysis of the conserved Dcp2-activating YAGxxF motif found in PNRC2 and Edc1 coactivators (Fig. 4). Consistent with previous kinetic data on Sc Edc1, we show that Hs PNRC2 activates decapping catalysis by both enhancing substrate binding (~10-fold decrease in KM) and accelerating the catalytic step (~12-fold increase in kmax). This raises the possibility that the different conformational states of Dcp2 may reflect different steps in the enzyme’s catalytic cycle, and that Edc1-like coactivators may promote these different steps (Fig. 5). Indeed, it was previously shown that conformational dynamics are crucial for Dcp2 catalysis and activation of catalysis by Sc Edc1, and that these dynamics depend critically upon the “gatekeeper” residue W43,36 which plays very different roles in the cap-bound versus Edc1-bound structures. The Edc1-bound conformation characterized by Valkov et al may represent an early step in the catalytic cycle in which Edc1 binding promotes a conformation of the decapping complex primed for RNA binding, and possibly scanning along the RNA body, which would account for Edc1’s ability to enhance RNA binding. In a subsequent step, recognition of the RNA 5′ cap target may result in a large conformational change of Dcp2 to compose the composite nucleotide binding site identified in our cap analog-bound structure. Finally, a further conformational change of Dcp2 will be required to access the transition state to decapping. Edc1-like coactivators will also act during this step, possibly by contacting the Nudix domain or cap itself, because the primary role of the conserved YAGxxF Dcp2-activating motif is to promote the catalytic step, not substrate binding.
Figure 5.
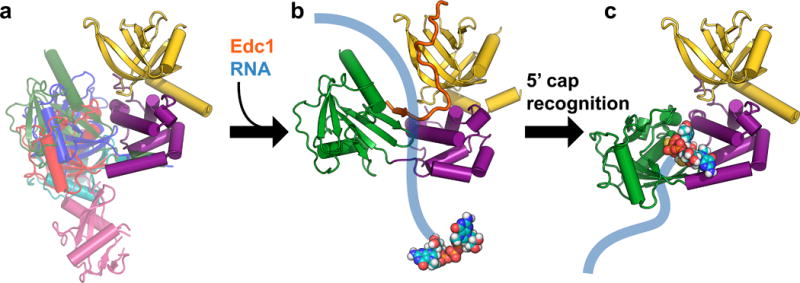
Proposed model for initial steps in the Dcp2 catalytic cycle. (a) The Dcp1–Dcp2 complex exists in an ensemble of conformations in the absence of substrate or coactivators. Dcp1 is colored yellow, the regulatory domain of Dcp2 is colored purple, and different known conformations of the Dcp2 Nudix domain are overlaid in different colors: magenta is the “open” conformation of PDB 2QKM,20 cyan is PDB 2A6T,32 red is PDB 5KQ1 (apo structure from this study; similar to “closed” conformation of PDB 2QKM20), blue is the cap analog-bound conformation from PDB 5KQ4 (this study), and green is the Edc1-bound conformation of PDB 5J3T.39 (b) Coactivators such as Edc1 can bind to the Dcp1–Dcp2 complex and may initially promote a conformation of the apo enzyme that is primed for binding, and possibly subsequent scanning along, RNA. The structure shown in this step is PDB 5J3T,39 with Sp Edc1 coactivator colored orange, and schematic RNA in blue with the 5′ cap shown as spheres. (c) Recognition of the 5′ cap structure on RNA is accompanied by a large conformational change in which the Nudix domain rotates 90° from the Edc1-bound structure shown in (b) to expose previously occluded, conserved cap binding residues to form a composite binding site for cap. The structure shown in this step is PDB 5KQ4, the cap analog-bound conformation from this study. A further conformational change will be required to access the transition state for decapping.
This model for RNA recognition by Dcp1–Dcp2 has precedence in other nucleic acid binding proteins, such as transcription factors, restriction enzymes or DNA repair enzymes, which use scanning or facilitated diffusion to quickly search long stretches of DNA for their target sequences.47,48 Many of these proteins, for example the lac repressor, use one conformation to carry out nonspecific DNA binding during scanning and switch to a different conformation upon specific target recognition.49 While further experiments will be required to test these possibilities for the decapping complex, regulation of mRNA decapping may be controlled by protein-protein interactions that manipulate the conformational transitions of Dcp2 to promote or inhibit different steps in the catalytic cycle. The cap-analog-bound structure of Dcp2 presented here defines the structural basis of cap recognition and lays the groundwork for understanding how different conformational states of Dcp2 might be leveraged by the cell to control decapping and mRNA stability.
ONLINE METHODS
Protein expression and purification
Single chain his-TRX-tev-HsPNRC2(91-121)-GGGGS-SpDcp2(1-243) constructs used for crystallization experiments were obtained as E. coli codon-optimized DNA sequences from Integrated DNA Technologies (his is hexahistadine affinity tag, TRX is thioredoxin solubility tag, tev is Tobacco Etch Virus protease cleavage site). The single chain scPNRC2Dcp2 fragment was cloned into MCS1 of a Novagen pETduet expression vector containing GB1-tev- SpDcp1(1-127) in MCS2. The Dcp1–scPNRC2Dcp2 construct was expressed in E. coli BL21-star DE3 cells (Invitrogen) grown in LB media with an induction time of 18 hours at 20 °C. Cells were harvested at 5000g, lysed by sonication, and clarified at 16,000g in lysis buffer (50 mM sodium phosphate pH 7.5, 300 mM sodium chloride, 10 mM imidazole, 5% glycerol, 10 mM 2-mercaptoethanol, Roche EDTA-free protease inhibitor cocktail). The protein complex was purified by Ni-NTA affinity chromatography and solubility/affinity tags were cleaved by treatment with TEV protease overnight at room temperature. Dcp1–scPNRC2Dcp2 complex was separated from cleaved tags by size exclusion chromatography on a GE Superdex 200 16/60 gel filtration column in crystallization buffer (10 mM HEPES pH 7, 150 mM NaCl, 1 mM DTT). The purified complex was concentrated to 14 mg/mL and flash frozen in liquid nitrogen for crystallization experiments.
Complexes of Hs PNRC2(1-121) – Sp Dcp1(1-127)–Dcp2(1-243) for kinetic experiments were prepared by separately expressing Sp Dcp2(1-243) on a Novagen pACYC vector, and his-TRX-tev-PNRC2(1-121) – his-GB1-tev-Dcp1(1-127) on a Novagen pETduet vector in E. coli BL21-star DE3 cells. Cells were grown in LB media with a 5 hour, 30 °C induction. Clarified lysates (prepared as above) for Dcp2 and his-TRX-PNRC2–his-GB1-Dcp1 were mixed in a 1:1 ratio with NiNTA resin and nutated at 4 °C for 3 hours to form the ternary complex while binding to Ni resin. After Ni-NTA purification, the complex was cleaved with TEV protease overnight, then separated from cleaved tags by size exclusion chromatography on a GE Superdex 200 16/60 gel filtration column in storage buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 5% glycerol, 5 mM DTT). The complex was concentrated, glycerol added to a final concentration of 20%, and flash frozen in liquid nitrogen; final concentration was 200 μM. Point mutants of the Hs PNRC2(1-121) – Sp Dcp1–Dcp2 complex were made by whole plasmid PCR with mutagenic divergent primers, and purified as above.
Crystallization of protein complexes
Purified single chain Dcp1–scPNRC2Dcp2 at 6 mg/mL was combined in a 1:1 ratio with well solution in a hanging drop vapor diffusion experiment at 20 °C. The well solution contained 150 mM trilithium citrate, 13% PEG 3350, and 0.1% mellitic acid. ~100 μm × 50 μm plates grew within 1–2 days and were flash frozen in liquid nitrogen using a cryoprotectant consisting of well solution with 20% glycerol.
To obtain the cap analog-bound Dcp1–scPNRC2Dcp2 structure, the single chain complex at 10 mg/mL was combined in a 1:1 ratio with well solution in a hanging drop vapor diffusion experiment at 20 °C. The well solution contained 150 mM trilithium citrate, 13% PEG 3350, and 0.05% mellitic acid. ~100 μm × 50 μm plates grew within 1–2 days, and 1uL of 10 mM cap analog dissolved in well solution was added to the crystallization drop, to give a final concentration of ~3.5 mM cap analog. The crystals were soaked overnight, and flash frozen in liquid nitrogen using a cryoprotectant consisting of well solution with 20% glycerol and ~3 mM cap analog.
Crystallographic data collection, structure determination, and refinement
All data were collected at Lawrence Berkeley National Lab, Beamline 8.3.1 at the Advanced Light Source on an ADSC Quantum 315r CCD detector at 100 K and a wavelength of 1.115869 Å. Data were indexed, integrated and scaled using HKL2000.50 The structures were solved in space group P21 by molecular replacement using Phaser in the CCP4 suite.51 We used chains A and B of PDB 2QKM as a molecular replacement model,20 first placing Dcp1–Dcp2(1-94), then Dcp2(95-243). After a preliminary round of refinement including rigid-body refinement in PHENIX,52 the structure was manually adjusted in COOT53 and iteratively refined in PHENIX. Final refinements used Non-Crystallographic Symmetry (NCS), Translation-Libration-Screw-rotation (TLS), stereochemical, and Atomic Displacement Parameter (ADP) restraints. For the final model of Dcp1–scPNRC2Dcp2, all residues were in the favored (92.7%), allowed (6.8%), or generously allowed (0.4%), regions of the Ramachandran plot generated by ProCheck,54 with none disallowed. For the final model of Dcp1–scPNRC2Dcp2 with two-headed cap analog bound, a single residue was in the disallowed region of the Ramachandran plot generated by ProCheck (0.1%, D91 chain D), with the remaining residues falling within the favored (92%), allowed (7.7%), or generously allowed (0.1%) regions.
All structural figures were prepared using PyMol version 1.7. Electrostatic surfaces were calculated using PDB2PQR & APBS webservers,55,56 using the AMBER94 forcefield to calculate charges. Electrostatic surfaces were visualized in PyMol using APBS tools 2.1.
Accession codes
Coordinates and structure factors were deposited in the Protein Data Bank with accession codes PDB 5KQ1 (Dcp1–scPNRC2Dcp2 complex), and PDB 5KQ4 (Dcp1–scPNRC2Dcp2 complex with bound two-headed cap analog).
Kinetic decapping assays
Single turnover in vitro decapping assays were carried out as previously described.46 Kinetics of PNRC2–Dcp1–Dcp2 complexes were too fast to follow by manual pipetting under our normal buffer conditions, so a modified decapping buffer was used: 50 mM HEPES pH 7, 100 mM KCl, 10% glycerol, 5 mM MgCl2, with RNase inhibitor and 0.1 mg/mL acetylated BSA. A32P-labeled, capped 355-mer RNA substrate derived from MFA2 was used for all decapping assays. Reactions were initiated by combining 30 μL 3× protein solution with 60 μL 1.5× RNA solution at 4 °C; final protein concentrations ranged from 0.6 μM to 30 μM, and final RNA concentration was <100 pM. Time points were quenched by the addition of excess EDTA, TLC was used to separate RNA from product m7GDP, and the decapped fraction was quantified using a GE Typhoon scanner and ImageQuant software as previously described.46 Plots of fraction m7GDP product versus time were fit to a first order exponential to obtain kobs; in the case of some PNRC2 point mutants, and Dcp1–Dcp2 alone, when decapping kinetics were too slow at lower concentrations to obtain reliable exponential fits kobs was obtained from a linear fit of initial rates (less than 25% decapped), by dividing the slope by the average endpoint of 0.85. Errors on kobs are shown as the standard error from individual fits used to obtain kobs. To obtain kmax and KM, kobs was plotted versus protein concentration and fit to the model: kobs = kmax[E]/KM + [E].
Supplementary Material
Acknowledgments
The authors would like to thank Drs. Xi Liu, Jennifer Binning, and Chris Waddling at UCSF for valuable help and advice on crystallography experiments, and Drs. James Holton and George Meigs at Lawrence Berkeley National Lab, Advanced Light Source beamline 8.3.1 for help with X-ray data collection. We also thank Dr. Joanna Kowalska for helpful discussions on the design and synthesis of the two-headed cap analog. This work was supported by the US National Institutes of Health (R01 GM078360 to J.D.G. and NRSA fellowship F32 GM105313 to J.S.M.) and the National Science Centre, Poland (Grant No. UMO-2012/05/E/ST5/03893 to J.J. and Fellowship No. UMO-2014/12/T/NZ1/00528 to M.Z.). The Advanced Light Source is supported by the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
CONTRIBUTIONS
J.S.M. designed and purified all protein constructs, carried out crystallization experiments, collected and refined crystallographic data, carried out decapping kinetics experiments, wrote the manuscript, and prepared the figures. M.Z. and J.J. designed and synthesized the two-headed cap analog. J.D.G. supervised the project and experimental design and guided manuscript preparation and editing. All authors read and commented on the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Moore MJ. From Birth to Death: The Complex Lives of Eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 2.Topisirovic I, Svitkin YV, Sonenberg N, Shatkin AJ. Cap and cap-binding proteins in the control of gene expression. Wiley Interdiscip Rev RNA. 2011;2:277–298. doi: 10.1002/wrna.52. [DOI] [PubMed] [Google Scholar]
- 3.Parker R. RNA Degradation in Saccharomyces cerevisae. Genetics. 2012;191:671–702. doi: 10.1534/genetics.111.137265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arribas-Layton M, Wu D, Lykke-Andersen J, Song H. Structural and functional control of the eukaryotic mRNA decapping machinery. Biochim Biophys Acta BBA - Gene Regul Mech. 2013;1829:580–589. doi: 10.1016/j.bbagrm.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Kiledjian M. Regulation of mRNA decapping. Wiley Interdiscip Rev - RNA. 2010;1:253–265. doi: 10.1002/wrna.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mildvan AS, et al. Structures and mechanisms of Nudix hydrolases. Arch Biochem Biophys. 2005;433:129–143. doi: 10.1016/j.abb.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 7.Nagarajan VK, Jones CI, Newbury SF, Green PJ. XRN 5′ → 3′ exoribonucleases: Structure, mechanisms and functions. Biochim Biophys Acta BBA - Gene Regul Mech. 2013;1829:590–603. doi: 10.1016/j.bbagrm.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geisler S, Lojek L, Khalil AM, Baker KE, Coller J. Decapping of long non-coding RNAs regulates inducible genes. Mol Cell. 2012;45:279–291. doi: 10.1016/j.molcel.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 11.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Popp MW-L, Maquat LE. Organizing Principles of Mammalian Nonsense-Mediated mRNA Decay. Annu Rev Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho H, Kim KM, Kim YK. Human Proline-Rich Nuclear Receptor Coregulatory Protein 2 Mediates an Interaction between mRNA Surveillance Machinery and Decapping Complex. Mol Cell. 2009;33:75–86. doi: 10.1016/j.molcel.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 14.Cho H, et al. Staufen1-Mediated mRNA Decay Functions in Adipogenesis. Mol Cell. 2012;46:495–506. doi: 10.1016/j.molcel.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Park E, Maquat LE. Staufen-mediated mRNA decay. Wiley Interdiscip Rev RNA. 2013;4:423–435. doi: 10.1002/wrna.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brannan K, et al. mRNA Decapping Factors and the Exonuclease Xrn2 Function in Widespread Premature Termination of RNA Polymerase II Transcription. Mol Cell. 2012;46:311–324. doi: 10.1016/j.molcel.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Presnyak V, et al. Codon Optimality Is a Major Determinant of mRNA Stability. Cell. 2015;160:1111–1124. doi: 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shukla S, Parker R. Quality control of assembly-defective U1 snRNAs by decapping and 5′-to-3′ exonucleolytic digestion. Proc Natl Acad Sci. 2014;111:E3277–E3286. doi: 10.1073/pnas.1412614111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Dai J, Song M, Fitzgerald-Bocarsly P, Kiledjian M. Dcp2 Decapping Protein Modulates mRNA Stability of the Critical Interferon Regulatory Factor (IRF) IRF-7. Mol Cell Biol. 2012;32:1164–1172. doi: 10.1128/MCB.06328-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.She M, et al. Structural Basis of Dcp2 Recognition and Activation by Dcp1. Mol Cell. 2008;29:337–349. doi: 10.1016/j.molcel.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunckley T, Tucker M, Parker R. Two related proteins, Edc1p and Edc2p, stimulate mRNA decapping in Saccharomyces cerevisiae. Genetics. 2001;157:27–37. doi: 10.1093/genetics/157.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borja MS, Piotukh K, Freund C, Gross JD. Dcp1 links coactivators of mRNA decapping to Dcp2 by proline recognition. RNA. 2011;17:278–290. doi: 10.1261/rna.2382011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fromm SA, et al. The structural basis of Edc3- and Scd6-mediated activation of the Dcp1:Dcp2 mRNA decapping complex. EMBO J. 2012;31:279–290. doi: 10.1038/emboj.2011.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fromm SA, et al. In Vitro Reconstitution of a Cellular Phase-Transition Process that Involves the mRNA Decapping Machinery. Angew Chem Int Ed. 2014;53:7354–7359. doi: 10.1002/anie.201402885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harigaya Y, Jones BN, Muhlrad D, Gross JD, Parker R. Identification and Analysis of the Interaction between Edc3 and Dcp2 in Saccharomyces cerevisiae. Mol Cell Biol. 2010;30:1446–1456. doi: 10.1128/MCB.01305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang CT, Bercovich N, Loh B, Jonas S, Izaurralde E. The activation of the ecapping enzyme DCP2 by DCP1 occurs on the EDC4 scaffold and involves a conserved loop in DCP1. Nucleic Acids Res. 2014;42:5217–5233. doi: 10.1093/nar/gku129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai T, et al. Structural Basis of the PNRC2-Mediated Link between mRNA Surveillance and Decapping. Structure. 2012;20:2025–2037. doi: 10.1016/j.str.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Chowdhury A, Mukhopadhyay J, Tharun S. The decapping activator Lsm1p-7p–Pat1p complex has the intrinsic ability to distinguish between oligoadenylated and polyadenylated RNAs. RNA. 2007;13:998–1016. doi: 10.1261/rna.502507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chowdhury A, Tharun S. Activation of decapping involves binding of the mRNA and facilitation of the post-binding steps by the Lsm1–7–Pat1 complex. RNA. 2009;15:1837–1848. doi: 10.1261/rna.1650109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coller J, Parker R. General Translational Repression by Activators of mRNA Decapping. Cell. 2005;122:875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sweet T, Kovalak C, Coller J. The DEAD-Box Protein Dhh1 Promotes Decapping by Slowing Ribosome Movement. PLoS Biol. 2012;10:e1001342. doi: 10.1371/journal.pbio.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.She M, et al. Crystal structure and functional analysis of Dcp2p from Schizosaccharomyces pombe. Nat Struct Mol Biol. 2006;13:63–70. doi: 10.1038/nsmb1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deshmukh MV, et al. mRNA Decapping Is Promoted by an RNA-Binding Channel in Dcp2. Mol Cell. 2008;29:324–336. doi: 10.1016/j.molcel.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 34.He F, Jacobson A. Control of mRNA decapping by positive and negative regulatory elements in the Dcp2 C-terminal domain. RNA. 2015 doi: 10.1261/rna.052449.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piccirillo C, Khanna R, Kiledjian M. Functional characterization of the mammalian mRNA decapping enzyme hDcp2. RNA. 2003;9:1138–1147. doi: 10.1261/rna.5690503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Floor SN, Borja MS, Gross JD. Interdomain dynamics and coactivation of the mRNA decapping enzyme Dcp2 are mediated by a gatekeeper tryptophan. Proc Natl Acad Sci. 2012;109:2872–2877. doi: 10.1073/pnas.1113620109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Floor SN, Jones BN, Hernandez GA, Gross JD. A split active site couples cap recognition by Dcp2 to activation. Nat Struct Mol Biol. 2010;17:1096–1101. doi: 10.1038/nsmb.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonas S, Izaurralde E. The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev. 2013;27:2628–2641. doi: 10.1101/gad.227843.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valkov E, et al. Structure of the Dcp2-Dcp1 mRNA-decapping complex in the activated conformation. Nat Struct Mol Biol. 2016;23:574–579. doi: 10.1038/nsmb.3232. [DOI] [PubMed] [Google Scholar]
- 40.Ziemniak M, et al. Two-headed tetraphosphate cap analogs are inhibitors of the Dcp1/2 RNA decapping complex. RNA. 2016;22:518–529. doi: 10.1261/rna.055152.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ziemniak M, et al. Phosphate-modified analogues of m(7)GTP and m(7)Gppppm(7)G-Synthesis and biochemical properties. Bioorg Med Chem. 2015;23:5369–5381. doi: 10.1016/j.bmc.2015.07.052. [DOI] [PubMed] [Google Scholar]
- 42.Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK. Cocrystal Structure of the Messenger RNA 5′ Cap-Binding Protein (eIF4E) Bound to 7-methyl-GDP. Cell. 1997;89:951–961. doi: 10.1016/s0092-8674(00)80280-9. [DOI] [PubMed] [Google Scholar]
- 43.Mazza C, Segref A, Mattaj I, Cusack S. Large-scale induced fit recognition of an m7GpppG cap analogue by the human nuclear cap-binding complex. EMBO J. 2002;21:5548–5557. doi: 10.1093/emboj/cdf538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bailey S, et al. The Crystal Structure of Diadenosine Tetraphosphate Hydrolase from Caenorhabditis elegans in Free and Binary Complex Forms. Structure. 2002;10:589–600. doi: 10.1016/s0969-2126(02)00746-3. [DOI] [PubMed] [Google Scholar]
- 45.Svensson LM, et al. Crystal structure of human MTH1 and the 8-oxo-dGMP product complex. FEBS Lett. 2011;585:2617–2621. doi: 10.1016/j.febslet.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 46.Jones BN, Quang-Dang DU, Oku Y, Gross JD. In: Methods in Enzymology. Maquat Lynne E, Kiledjian Megerditch., editors. Vol. 448. Academic Press; 2008. pp. 23–40. [DOI] [PubMed] [Google Scholar]
- 47.Smith NC, Matthews JM. Mechanisms of DNA-binding specificity and functional gene regulation by transcription factors. Curr Opin Struct Biol. 2016;38:68–74. doi: 10.1016/j.sbi.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 48.von Hippel PH. From ‘Simple’ DNA-Protein Interactions to the Macromolecular Machines of Gene Expression. Annu Rev Biophys Biomol Struct. 2007;36:79–105. doi: 10.1146/annurev.biophys.34.040204.144521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalodimos CG, et al. Structure and Flexibility Adaptation in Nonspecific and Specific Protein-DNA Complexes. Science. 2004;305:386–389. doi: 10.1126/science.1097064. [DOI] [PubMed] [Google Scholar]
- 50.Otwinowski Z, Minor W. Methods in Enzymology. In: Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography. Vol. 276. New York: Academic Press; 1997. pp. 307–326. Part A. [Google Scholar]
- 51.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot Acta Crystallogr. D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 55.Dolinsky TJ, et al. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.