

SUMMARY
Common sequence variants in cis-regulatory elements (CREs) are suspected etiological causes of complex disorders. We previously identified an intronic enhancer variant in the RET gene disrupting SOX10 binding and increasing Hirschsprung disease (HSCR) risk 4-fold. We now show that two other functionally independent CRE variants, one binding Gata2 and the other binding Rarb, also reduce Ret expression and increase risk 2- and 1.7-fold. By studying human and mouse fetal gut tissues and cell lines, we demonstrate that reduced RET expression propagates throughout its gene regulatory network, exerting effects on both its positive and negative feedback components. We also provide evidence that the presence of a combination of CRE variants synergistically reduces RET expression and its effects throughout the GRN. These studies show how the effects of functionally independent non-coding variants in a coordinated gene regulatory network amplify their individually small effects, providing a model for complex disorders.
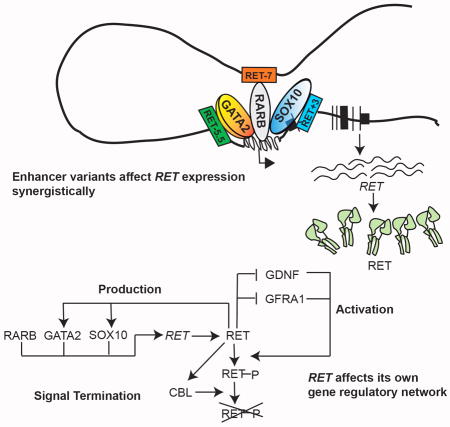
Graphical abstract
INTRODUCTION
Regulatory control of gene expression is the fundamental basis for cell differentiation during organismal development. Gene expression control is modular, using a current set of biochemical signals to alter the expression of a specific set of target genes affecting subsequent cellular functions (Davidson, 2006). These modules are not arbitrary but have adapted during evolution to have specific motifs that encode classes of gene expression control (Alon, 2007). Thus, the function of a gene should not be considered in isolation but in the context of its gene regulatory network (GRN), i.e., the set of genes and their products that interact to control specific cell functions. GRNs are coordinated through cis-regulatory elements (CRE), chiefly enhancers, which integrate signals from numerous transcription factors (TFs), lineage markers and signaling proteins within a cell-type to modulate target gene expression (Alon, 2007; Davidson, 2006). Therefore, perturbations of these GRNs are highly relevant to human disease. This can be particularly relevant for multifactorial disorders that arise mostly from non-coding variants within cell-type specific CREs (Bauer et al., 2013; Emison et al., 2010; Emison et al., 2005; Maurano et al., 2012; Trynka et al., 2013), unlike Mendelian disorders in which disease-associated variants are largely coding (Antonarakis et al., 2010).
Mendelian and multifactorial diseases also differ in the magnitude of the effect of disease-associated variants: the latter have small-to-modest risks making their detection difficult (Manolio et al., 2009), and, leading to suspicion that their biological roles are unimportant (Goldstein, 2009). However, three recent advances strongly argue against this view: (1) the genomic segment relevant to transcriptional control of a gene is highly organized into a topologically associated domain (TAD) within which CREs interact with greater frequency than with elements outside (Dixon et al., 2012; Rao et al., 2014); (2) epigenomic data suggests that each TAD contains multiple enhancers (Bernstein et al., 2010; Consortium, 2012); and (3) multiple enhancer use for a specific gene is dynamic and interactive during development (Andrey et al., 2013; Davidson, 2006). This suggests the hypothesis that combinations of sequence variants within multiple enhancers of a gene, depending on temporal use and transcriptional effect, not individual variants, are the primary yet variable risk factors in a complex disease. The first evidence of multiple CREs controlling mammalian gene expression arose from studies of beta hemoglobin (Grosveld et al., 1987), shown subsequently to be deleted in some thalassemias (Driscoll et al., 1989). More recently, mutations in an evolutionary conserved limb enhancer of sonic hedgehog was shown to cause preaxial polydactyly in humans and other species (Lettice et al., 2003). Here, we extend these observations to multifactorial Hirschsprung disease (HSCR) by demonstrating that the causal defect arises from the synergistic effects of polymorphisms in multiple RET enhancers that reduce RET gene expression and affects its entire GRN through feedback mechanisms.
HSCR or congenital intestinal aganglionosis is a severe but common (~15/100,000 live births) developmental disorder of the enteric nervous system (ENS) in which the gut fails to get innervated and loses motility: the resulting aganglionosis is always caudal to rostral, partial but contiguous (Chakravarti A, 2001). Genetic studies have identified rare, high-penetrance coding variants in 14 genes (RET, GDNF, NRTN, SOX10, EDNRB, EDN3, ECE1, ZFHX1B, TCF4, PHOX2B, KBP, L1CAM, SEMA3C, SEMA3D) that together explain ~10% of cases, usually syndromic and severe forms (Alves et al., 2013; Chakravarti A, 2001; Jiang et al., 2015). All known HSCR genes are expressed in enteric neuroblasts or its supporting mesenchymal cells, are involved in the early stages of fate determination of enteric neural crest cells (ENCCs) and affect their subsequent survival, proliferation, migration and differentiation into neurons (Heanue and Pachnis, 2007). Of these, the most frequent coding mutations occur in RET, encoding a receptor tyrosine kinase (Emison et al., 2010; Emison et al., 2005). Surprisingly, significantly greater risk to the more common, isolated, non-syndromic HSCR arises from three polymorphic, low-penetrance non-coding variants at RET (rs2435357, rs2506030) and SEMA3C/D (rs11766001), in patients of European ancestry (Kapoor et al., 2015). Of these, we have shown that the RET intronic variant rs2435357 has high (~24%) allele frequency, disrupts SOX10 binding to a fetal gut enhancer (CRE: RET+3), reduces RET expression and increases Hirschsprung disease (HSCR) risk 4-fold (Emison et al., 2010).
In this study, we demonstrate that RET loss-of-function is necessary, not sufficient, for clinical expression of HSCR based on the following observations in human patients, mouse models, human fetal gut tissue and cellular assays: (1) nearly every HSCR patient carries at least one non-coding RET deficiency allele, the risk arising largely from three common, non-coding, interacting RET alleles (rs2506030 at RET−7; rs7069590 at RET−5.5 and rs2435357 at RET+3: see methods for nomenclature explanation and Figure 1B for schematic representation of the position of each named RET CRE) with increased risk jointly specified by RET−5.5 and RET+3 with modulation by RET−7; (2) each risk allele resides within a Ret enhancer (CRE) active during mouse gut development, with RET−7, RET−5.5 and RET+3 being bound by Rarb, Gata2 and Sox10, respectively; (3) enhancer use is dynamic with all three enhancers active early in gut development but only RET+3 active later; (4) each risk variant significantly reduces its cognate enhancer activity through reduced binding of its respective TF, leading to reduced Ret gene expression; and, (5) in vitro and in vivo reduction of RET gene expression perturbs the entire RET GRN, including the genes that affect its production through TFs (SOX10, GATA2), activation (GDNF, GFRA1) and signal termination (CBL), through distinct and conserved feedback mechanisms. These results provide an explanation of how HSCR arises from amplification of the effects of multiple low-penetrance enhancer variants through coordinate dysregulation of the entire RET GRN, comprising unlinked but functionally related genes.
Figure 1. Genomic map and biological activity of enhancers within the RET locus.

(A) A 350 kb genomic segment annotated with topologically associated domains (TAD) in 9 human cell lines with experimentally tested (mouse Neuro2a cells) and ENCODE-predicted (DNaseI Hypersensitivity, DHS) enhancers, and H3K4me1 and H3K27ac marks, from a 108 day human fetal intestine. All enhancers lie within a core 225 kb TAD common to all cell lines. (B) Fine-mapping of a 153 kb sub-region containing all identifiable enhancers, 38 HSCR-associated SNPs and known transcription factor (TF) ChIP-seq sites in human SK-N-SH cells. The 8 SNPs in color disrupt a TF binding site and lie within an enhancer. (C) in vitro luciferase assays in Neuro2A cells showing enhancer activity in wildtype and risk allele constructs: red elements at RET−7 (containing rs2506030), RET−5.5 (containing rs7069590) and RET+3 (containing rs2435357) demonstrate statistically significant allelic differences in enhancer activity; green elements do not. (D) Luciferase assays of multiple-enhancer constructs in Neuro2a cells (Table 1) demonstrating an exponential relationship between loss of enhancer activity and increasing risk of HSCR from risk allele dosage. The risk alleles in each haplotype are marked in red. The error bars represent standard errors of the mean (** P<0.001).
RESULTS
Screening multiple RET CREs for enhancer loss-of-function variants
Given the existence of one loss-of-function enhancer allele at RET leading to HSCR (Emison et al., 2010; Emison et al., 2005), we hypothesized that should other loss-of-function RET enhancer variants exist they will also lead to HSCR. Thus, we searched a 153 Kb region upstream of RET for all known and predicted RET enhancers, a region associated in our prior genome-wide association study (GWAS)(Jiang et al., 2015), and part of a 343 kb TAD (chr10:43,300,994–43,643,327) evident in human ES cells and eight other cell lines (Dixon et al., 2012; Rao et al., 2014) with a common core of 225 kb (chr10:43,375,000–43,600,000) (Figure 1A). We identified 16 unique CREs: 5 predicted using DNase I hypersensitivity (DHS), H3K4me1 and H3K27ac sites from a 108 day human fetal intestine (Bernstein et al., 2010), and 13 we previously identified from mammalian sequence conservation followed by experimental verification in the mouse neuroblastoma cell line Neuro2A (Grice et al., 2005)(Figure 1A). In parallel, we screened all known common (>10% allele frequency) single nucleotide polymorphisms (SNPs) within this locus (Abecasis et al., 2012) for HSCR association (Figure 1B). Of 146 variants, 38 were associated (P≤ 5×10−8) either by direct genotyping or imputation based on a prior GWAS (Jiang et al., 2015) (Table S1).
Association of an individual variant can arise from causality or linkage disequilibrium (LD) with a causal variant; thus, we restricted attention to eight SNPs in CREs that overlapped known TF bound regions assessed in the human neuroblastoma cell line SK-N-SH (Figure 1B, Table S1) (Consortium, 2012). These variants were tested for differential luciferase activity by cloning 1kb fragments containing either the reference or the variant allele into the pGL4.23 vector with a minimal TATA-box of the mouse β-globin gene transfected into Ret-expressing Neuro2A cells. All 8 CREs acted as enhancers, five with similar activity for both alleles (Figure 1C); however, CREs containing three variants – allele G at rs2506030 (RET−7), allele T at rs7069590 (RET−5.5) and allele T at rs2435357 (RET+3) - showed statistically significant reduction in reporter activity (Figure 1C). In support of their potential disease role, these variant alleles increased HSCR risk (see next section). The non-risk allele (A) at rs2506030 for RET−7 (chr10: 43,447,346–43,448,347) showed a 4.3-fold increased luciferase activity as compared to the control (basal promoter) while its risk allele (G) showed a 1.7-fold drop in activity compared to the non-risk allele (P=6×10−4). The corresponding values for rs7069590 in RET−5.5 (chr10: 43,552,395–43,553,394) were 5.8-fold increase and 1.4-fold decrease (P=1.6×10−6), and for rs2435357 at RET+3 (chr10: 43,581,812–43,582,711) were 12.7-fold increase and 1.9-fold decrease (P= 9×10−9). These three CREs are, therefore, potential enhancers, consistent with their electrophoretic mobility shift assay (EMSA) results, using a 20bp Cy5-labeled probe centered on each variant and using nuclear extracts from Neuro2A cells; further, the observed protein-DNA binding can be abrogated by a 10bp deletion containing the SNP (Figure S1).
Interactive effects of three RET enhancer variants in HSCR
Our forward genetic screen identified three putative functionally independent HSCR-associated enhancer variants among many other candidates that do not lie within known CREs: the previously discovered rs2435357 (Emison et al., 2005), one ~125kb upstream of RET (rs2506030) recently identified as a sentinel variant in GWAS (Jiang et al., 2015) and, a new variant~18kb upstream of RET (rs7069590). To examine their individual and joint effects, we genotyped them in 346 HSCR probands and 732 controls, all of European ancestry (Table 1, Table S2) to demonstrate three features. First, individual risk alleles are pervasive and have major effects, with frequency 41%, 76% and 25% in European controls increasing to 56%, 84% and 58% in HSCR cases, for rs2506030 (allele G), rs7069590 (allele T) and rs2435357 (allele T), respectively with highly significant odds ratios of 1.8 (P=4.46×10−11), 1.7 (P=4.36×10−6) and 4.1 (P=3.31×10−50), respectively. Second, most humans have at least one non-coding RET risk allele: homozygosity for the non-risk ACC haplotype is 1.7% in cases and 3.7% in controls. Third, there is wide variation in HSCR risk across variant haplotypes. We estimated the odds ratio for each haplotype relative to haplotype ACC that was free of any risk alleles: clearly, risk is elevated only for ATT (OR 3.13, 95% CI: 2.17–4.50, P=8.31×10−10) and GTT (OR 4.40, 95% CI: 3.26–5.94, P=3.62×10−22) haplotypes (Table 1). These haplotype effects are not the result of LD within the RET locus only since, in control samples, rs2506030, rs7069590 and rs2435357 are very weakly associated (all three pairwise r2 values ~0.08; Figure S2) but demonstrate significantly greater LD on haplotypes from affected individuals (rs2506030 - rs2435357: r2 = 0.16, P = 0.03 versus controls; rs7069590 - rs2435357: r2 = 0.19, P = 0.005 versus controls; rs2506030 - rs769590 in cases: r2 = 0.11, P = 0.4 versus controls). Haplotype effects are, therefore, also a consequence of epistasis between risk variants, chiefly between rs7069590 (allele T) and rs2435357 (allele T) with further modulation by rs2506030 (allele G) (Table 1). Since HSCR risk is sex-dependent (Badner et al., 1990; Emison et al., 2010) we looked at haplotype disease risk separately in males and females in our HSCR probands. Males show significant risk from both ATT (OR 3.06, 95% CI: 1.84–5.07, P=1.53×10−5) and GTT (OR 5.47, 95% CI: 3.53–8.48, P=2.91×10−14) haplotypes, as in the total data, but females show significant risk (OR 3.23, 95% CI: 1.91–5.46, P=1.17×10−5) from GTT haplotypes only (Figure S3). These results are consistent with a higher male risk effect but the big (2.6-fold) difference in sample size between male and female probands lowers the statistical power of testing for sex differences in risk.
Table 1. RET haplotype-specific risks of Hirschsprung disease (HSCR).
Haplotypes for three polymorphisms (rs2506030, rs7069590, rs2435357) located within the enhancers RET−7, RET−5.5 and RET+3, 124.6 kb upstream, 19.6kb upstream and 9.5kb downstream from the RET transcription start site, respectively, are shown; risk alleles are in bold. Also shown are the observed frequencies of haplotypes among 346 cases and 732 controls, the odds ratio with respect to the reference haplotype ACC that lacks any susceptibility allele, its 95% confidence interval (CI) and the statistical significance (P) of testing whether odds ratios differ from unity.
| Haplotype | Frequency in cases (n=346) | Frequency in controls (n=732) | Odds ratio (95% CI) | P | ||
|---|---|---|---|---|---|---|
| rs2506030 (RET−7) | rs7069590 (RET−5.5) | rs2435357 (RET+3) | ||||
| A | C | C | 0.12 | 0.20 | 1 (reference) | - |
| G | C | C | 0.03 | 0.04 | 1.39 (0.80–2.43) | 0.24 |
| A | T | C | 0.17 | 0.32 | 0.91 (0.66–1.25) | 0.54 |
| G | T | C | 0.11 | 0.20 | 0.96 (0.68–1.36) | 0.82 |
| A | T | T | 0.14 | 0.08 | 3.13 (2.17–4.50) | 8.31×10−10 |
| G | T | T | 0.42 | 0.16 | 4.40 (3.26–5.94) | 3.62×10−22 |
These results prompted us to assess whether these interactions could be recapitulated in vitro. We generated triple enhancer reporter constructs, containing the six polymorphic (frequency >1%) haplotypes by cloning 300bp regions centered on each SNP, so as to maintain the total construct size at ~1kb, and repeated our prior reporter assays. The two remaining haplotypes had extremely low frequencies in both cases and control and were not used for risk calculations: ACT had a frequency of 0.01 in cases and 0.006 in controls; GCT had a frequency of 0.0004 in cases and 0.0007 in controls. These results, in comparison to haplotype odds ratios (Table 1), demonstrate that there is significant loss of enhancer activity only for the disease haplotypes ATT and GTT (P=0.003 for log-log regression), while all non-risk haplotypes have enhancer activities similar to the reference ACC (Figure 1D).
RET enhancer usage is dynamic during gut development
To assess whether these elements can drive tissue-specific gene expression in vivo, we generated transient transgenic mice using a 1.3kb human DNA fragment for RET−7 (chr10: 43,447,274–43,448,627), a 2kb fragment for RET−5.5 (chr10: 43,551,864–43,553,915) and a 1kb fragment for RET+3 (chr10: 43,581,812–43,582,888) cloned into a lacZ vector followed by pronuclear injection into FVB embryos. Embryos from three independent lines were harvested at E11.5 and E12.5, two time points during the early migration and colonization of ENCCs in the mouse gut. Ret is largely expressed in ENCCs, the midbrain and the dorsal root ganglia (DRG) (Grice et al., 2005). RET−7 shows strong and consistent activity in the gut, DRG and the mid-brain region at E11.5, however, at E12.5, expression is restricted to the DRG and midbrain region only (Figure 2A, D). RET−5.5 shows analogous expression patterns at both time points in the gut and the DRG, however, midbrain expression is more evident at E12.5 (Figure 2B, E). In contrast, RET+3 shows activity both in the gut and the DRG at both time points (Figure 2C, F). Therefore, all three CREs are transcriptional enhancers of DRG and midbrain expression in development, although RET−5.5 may be inactive in the brain at E11.5. In contrast, while RET+3 is active throughout gut development till E14.5 (Grice et al., 2005), RET−7 and RET+2 are active enhancers only at earlier stages. Thus, the use of multiple RET enhancers is temporally dynamic and the larger genetic effect of rs2435357 (RET+3) may be owing to its effect throughout gut development from E11.5 to E14.5.
Figure 2. Tissue-specific enhancer activities of three RET cis-regulatory elements (CRE) during mouse development.

(A) Transgenic assays of the human wildtype element RET−7 in mouse embryos demonstrate lacZ-driven expression in the gut (black arrowhead) and the dorsal root ganglion (DRG, red arrow) at day E11.5 (A). Analogous assays of the wildtype elements RET−5.5 (B) and RET+3 (C) also show tissue-specific expression in the gut and DRG at E11.5. At E12.5, RET−7 (D) continues to drive expression in the DRG but gut expression is lost, as is also in RET−5.5 (E); RET+3 (F) continues to have strong activity at E12.5 in both the gut and DRG.
RARB, GATA2 and SOX10 are RET gut transcription factors
We scanned the RET−7 and RET−5.5 CRE sequences for TF biding sites (TFBS) from publically available motif databases (Bryne et al., 2008; Newburger and Bulyk, 2009; Wingender et al., 1996) using the software FIMO (MEME suite (Bailey et al., 2009)); the TF bound to RET+3 is known to be SOX10 (Emison et al., 2010). The best candidates were 5′ GACCTATTCC 3′ for RET−7 and 5′ TAATGCGATAG 3′ for RET−5.5, recognized by retinoic acid receptors (RAR) (P = 10−4) and the GATA family (P=3.3×10−4), respectively (SNP site in bold). We used mouse expression levels as a proxy to identify the specific RAR (RARA, RARB, RARG) and GATA (GATA1-5) bound using Taqman assays on total cDNA from mouse guts at E11.5, E12.5 and E14.5. In relative terms, Rara is constantly expressed throughout this developmental period, Rarg is equally expressed at E11.5 and E14.5 but increases at E12.5, while Rarb is expressed only at E11.5 (Figure 3A). Given RET−7 activity during mouse gut development (Figure 2), Rarb emerges as the prime candidate TF. Of the four Gata family genes expressed during this time, in relative terms, Gata2 and Gata3 are similar in being an early TF, Gata4 is expressed at a constant rate while Gata5 expression goes up temporally (Figure 3A). Given ChIP-seq evidence of GATA2 binding to the enhancer in SK-N-SH, we focused on it as the potential TF binding to RET−5.5. As a positive control, we showed that Sox10 gene expression is constant across E11.5-E14.5 (Figure 3A). To verify whether these enhancer regions were associated with open chromatin, a hallmark of CRE activity, we mapped all DNaseI hypersensitive (DHS) peaks from the ENCODE project for the human neuroblastoma cell line (SK-N-SH). The 3 CREs were DHS sites, additionally 5 more of our tested enhancers lie in open chromatin regions, providing further evidence of their enhancer activity in this cell line. There are many additional DHS peaks within the RET locus, highlighting many more potential RET CREs (Figure 3B).
Figure 3. Loss of enhancer activity at risk alleles and identification of cognate transcription factors (TF).

(A) Gene expression of putative TFs in the mouse gut at E11.5, E12.5 and E14.5 shows declining expression of Rarb, Gata2 and Gata3 but no change in Sox10; pair-wise comparisons are relative to E11.5. (B) Genomic map of the RET locus showing DHS sites with respect to enhancers in human SK-N-SH cells. ChIP in SK-N-SH with a RAR antibody shows enrichment of binding to RET−7, as compared to the background signal; the specificity of binding is shown by siRNA knockdown of RARB with concomitant reduced binding. Analogous assays for RET−5.5 and GATA2, and RET+3 and SOX10 show specific binding of these TFs to their cognate enhancers. (C) siRNA-mediated down-regulation of Sox10, Gata2, and Rarb expression in Neuro2A cells affects activity of wildtype but not risk alleles at enhancers, demonstrating specificity. All pairwise comparisons are with wildtype or risk alleles co-transfected with control siRNAs.
The error bars represent standard errors of the mean (*P<0.01, ** P<0.001).
For definitive confirmation, we conducted ChIP- qPCR in the human SK-N-SH cell line with specific or pan antibodies against SOX10, GATA2/3 and RARB. To test for specificity, we also conducted siRNA knockdown of each TF and repeated ChIP assays (Figure 3B). These experiments show that RET−7 strongly binds RARB (50-fold over control IgG) with a 4-fold drop in enrichment when RARB is knocked down (P= 5.6×10−5). Similarly, we detected enrichment in binding with GATA2 (26-fold) not GATA3 antibody, at RET−5.5, with a 2-fold drop in enrichment (P=0.03) when GATA2 is knocked down. This reduced binding is not as significant as compared to the other knockdowns, pointing to the possibility that other GATA proteins may compensate for GATA2 loss. As expected, SOX10 binding is enriched 23-fold followed by a 7- fold reduction when SOX10 is knocked down (P=6×10−3).
To assess if loss of the cognate TFs leads to a loss of enhancer activity, we performed luciferase assays in Neuro2A cells to assess in vitro enhancer activity of each variant allele independently transfected with Rarb, Gata2, Sox10 and Ret siRNA. This allowed independent assessment of transcript levels from loss of TF versus loss of Ret. For RET−7, reduced Rarb gene expression leads to 2-fold (P=4.3×10−5) loss of activity of the non-risk and 1.3 fold (P=4×10−2) loss of the risk alleles. The corresponding loss of activity from Gata2 is 1.7-fold (P=8×10−2) of the non-risk and 1.2-fold (P=0.04) of the risk allele at RET−5.5. Analogous experiments on RET+3 and Sox1o expression reduction showed 2.3-fold (P=4.0×10−4) and 1.6-fold (P=1.4×10−2) loss of enhancer activity for the non-risk and risk alleles, respectively (Figure 3C). These knockdown effects are stronger for the non-risk than risk alleles, suggesting that the enhancers with variants are compromised for occupancy.
To prove that the identified TFs do indeed control Ret expression, which is the only gene within the 153Kb region, we performed siRNA-mediated knockdown of each TF in Neuro2A cells and measured Ret and TF transcript levels using Taqman-based qPCR assays. These experiments showed Ret expression reduction by 6-fold (P=2.7×10−4), 2-fold (6×10−2) and 8-fold (P=3.8×10−4) for Rarb, Gata2 and Sox10 knockdown, respectively, proving that each TF can independently control Ret gene expression (Figure 4A). We achieved highly specific expression knockdown of Rarb (P=4.2×10−4), Gata2 (P=3.1×10−3), Sox10 (P = 2.2×10−5) and Ret (P = 1.1×10−5) by their respective siRNAs showing specificity of the effects (Figure 4B). Surprisingly, we also observed that Ret knockdown was accompanied by a 4.3-fold, and 1.6-fold statistically significant reduction in the expression of Sox10 (P=2×10−4) and Gata2 (P=8×10−2) respectively; Rarb was unaltered (Figure 4B). Thus given haplotype risk data (Table 1), the increased disease risk from the risk alleles at RET−5.5 and RET+3 can be attributed to reduced binding of the cognate TFs to their enhancers coupled with positive feedback from Ret on these two TFs. In other words, the enhancer variants have an amplifying effect, first by direct transcriptional loss of Ret and second by positive feedback of this Ret loss on expression of Sox10 and Gata2.
Figure 4. Loss-of-function of Ret and genes in its regulatory network (GRN).

(A) siRNA-mediated down regulation of Sox10, Gata2, and Rarb in Neuro2a cells attenuates Ret transcription, as does siRNA against Ret. (B) Additionally, loss of Ret expression leads to reduced expression of its transcription factors Sox10 and Gata2, but not Rarb, thus showing positive feedback. All pairwise comparisons are to control siRNA values. (C) Gene expression of the Ret GRN in the developing gut of wildtype and Ret null embryos show significant up-regulation of Gdnf and Gfra1 and down-regulation of Sox10 with Ret loss-of-function at E11.5 and E12.5; Cbl and Gata2 show loss of expression at E11.5 only; other components of canonical Ret downstream signaling, and Rarb, are unaffected by loss of Ret in vivo. All pairwise comparisons are between wildtype and null embryos at each stage. The error bars represent standard errors of the mean (*P<0.01, ** P<0.001).
Dysregulation of the RET GRN in HSCR mouse models
Molecular analyses in vitro can substantially differ from in vivo biology. Consequently, we assessed gene expression in vivo using Taqman assays in homozygote Ret wildtype versus null mice (Uesaka et al., 2008). We studied guts at E11.5 and E12.5, during the period when the three enhancers are active. In the Ret null gut, as compared to wildtype, Sox10 expression is reduced 4-fold at E11.5 (P=1.2×10−6) and 8-fold at E12.5 (P=0.3×10−6) (Figure 4C). Analogously, Gata2 is also reduced 2-fold (p=3×10−3) at E11.5 but is unaffected (P= 0.65) at E12.5. Rarb expression is unaffected both at E11.5 (P=0.64) and E12.5 (P=0.19), although its expression level is drastically reduced at the later time point. Thus, the in vivo results match in vitro analyses.
RET is an atypical receptor tyrosine kinase (RTK) whose abundance is regulated by three activities, its production (RET and its TFs), its activation into a phosphorylated dimer (requiring the ligand GDNF and co-receptor GFRA1) and termination of receptor signaling from proteosomal degradation (requiring the E3 ubiquitin ligase CBL) (Mulligan, 2014). Subsequently, RET transduces signals through the adaptor protein GRB10 to many downstream effectors such as P38A, ERK1 and AKT1 (Mulligan, 2014). Consequently, we asked how RET loss-of-function in vivo affects other genes within its GRN, using Taqman assays in homozygote Ret wildtype and null mice (Figure 4C). Interestingly, expression of Gdnf is significantly increased 1.5-fold at E11.5 (P=3×10−2) and 2-fold at E12.5 (P=6×10−4), as is the expression of Gfra1 at 3.7-fold at E11.5 (P=6.2×10−8) and 3.0-fold at E12.5 (P=3.0×10−5). Further, loss of Ret also leads to reduced Cbl expression: a 2-fold drop at E11.5 (P=3×10−3) but a non-significant decrease at E12.5 (p=0.6). In contrast, Grb10 remains unaffected (E11.5, P=0.26; E12.5, P=0.39). These effects are specific to the developmental Ret GRN and not to the basic RTK signaling because gene expression of its downstream effectors P38α (Mapk14) (E11.5, P=0.9; E12.5, P=0.5), Erk1 (E11.5, P=0.8; E12.5, P=0.5) and Akt1 (E11.5 P=0.5; E12.5, P=0.8) are unchanged (Figure 4C). Therefore, Ret regulates gene expression of its own TFs (positive feedback), ligand and co-receptor (negative feedback), and signal terminator (positive feedback). RET thus controls its own GRN and is thus the critical, rate-limiting step in enteric ganglionosis. This regulation in vivo must involve non-autonomous factors because Gdnf is not expressed by ENCCs but by the supporting mesenchymal cells.
Since loss or significant reduction of Ret signaling leads to aganglionois in mice and humans, we studied the global effect of such loss in the embryonic guts at E11.5 and E12.5 in both wildtype and Ret homozygous null mice. The effect of the loss of Ret transcripts is extremely severe at E11.5 when expression of 1,318 genes are significantly (q-value <0.01) altered between wildtype and Ret null embryonic guts. The effect is less severe at E12.5 when 516 genes are significantly (q-value<0.01) affected (Figure S4A). A particular feature of the data is that the complete loss of Ret signaling leads to significant (q-value <0.01) changes in the gene expression of many TFs at both developmental stages between wildtype and Ret null embryonic guts: 178 and 34 at E11.5 and E12.5, respectively (data not shown). The control of various TFs by Ret, presumably indirect, early in development affects the gene expression of many genes in enteric neurons but specifically affects the Ret GRN. Thus, both Gdnf (1.7 fold, q-value 0.0007 at E11.5 and 1.76 fold, q-value 0.002 at E12.5), and Gfra1 (1.8-fold, q-value 0.0007 at E11.5 and 1.6 fold, q-value 0.002 at E12.5) show increased expression, as also demonstrated by qPCR. As expected, Cbl showed 4-fold reduced expression at E11.5 (q-value 0.0007) but is unaffected at E12.5 (q-value 0.98). Gata2 also shows reduced expression at E11.5 (1.6 fold, q-value 0.0007) but remains unchanged at E12.5 (q-value 0.23). Finally, Sox10 has significant reduced expression at both E11.5 (3 fold, q-value 0.0007) and E12.5 (5 fold, q-value 0.0003) (Figure S4B). The other GRN genes (Grb10, P38α, Erk1, Akt1, Rarb), whose transcripts remained unaffected at both time points in development by qPCR, show no difference by RNA-seq (Figure S4B).
Since the effect of the risk allele at the Sox10 enhancer is the strongest in HSCR and loss of Ret activity affects Sox10 expression, we decided to investigate if disruption of the Ret GRN driven by reduced Sox10 expression in vivo is consistent with our results. We assayed Sox10 heterozygous mouse guts (Britsch et al., 2001) at E11.5 and E12.5, for expression of Ret GRN genes (Figure S5). Compared to wild type guts, the expression of Ret in Sox10 heterozygotes is significantly reduced by 1.6 fold at E11.5 (P=4×10−4) and 1.8 fold at E12.5 (P=2.5×10−6). Correspondingly, Gdnf expression is increased only at E11.5 by 1.4 fold (P=2×10−4) but unaffected at E12.5 (P=0.4). The expression of the co-receptor Gfra1 is significantly increased 2.3 fold at E11.5 (P=8×10−6) and 2 fold at E12.5 (6.6×10−6). Further, Sox10 affects Gata2 at both E11.5 (1.5 fold decrease, P=3×10−2), and E12.5 (1.6 fold decrease, P=2.6×10−2) but expression of Rarb is unaffected (E11.5, P=0.49; E12.5 P=0.22). Sox10 heterozygotes also show an effect on Cbl at E11.5 (1.6 fold decrease, P=0.03), with no changes in the Ret adaptor Grb10 (E11.5, P=0.26; E12.5, P=0.39). In contrast to Ret deficiency, Sox10 expression reduction affects P38α (Mapk14) at both E11.5 (1.4 fold decrease, P=3×10−2) and E12.5 (1.4 fold decrease, P=4×10−3), Erk1 only at E11.5 (1.4 fold decrease, P=2×10−2) but not later at E12.5 (P=0.93), and has no effect on Akt1 levels at E11.5 (P=0.57) but causes a 1.4 fold reduction at E12.5 (P=3×10−2). Thus, the effect of Sox10 deficiency on the Ret GRN is consistent with Ret loss-of-expression but has broader effects, as expected, since being a TF it has other ENS targets. The SOX10 enhancer variant is more specific since it affects genes only through RET loss-of-function.
The feedback regulation most relevant to HSCR is that of Ret on its three TFs Sox10, Gata2 and Rarb. We enquired how TF, enhancer and target gene activity were related: was it dose- or threshold-dependent? Consequently, we varied Ret expression by varying the concentration of Ret siRNA and studied both gene and protein expression of Rarb, Gata2, Sox10 and Ret in Neuro2A cells. We examined gene expression using Taqman assays on total cDNA and protein levels by Western blotting using specific antibodies (Figure 5A). Ret siRNA concentrations up to 15μM led up to 50% reduction in Ret gene expression (P = 2.5×10−5) but only a 1% decrease in Sox10 expression (P = 0.47), and virtually no change in expression of Rarb, and Gata2. When Ret siRNA concentration increased to 17μM and 25μM, Ret expression further decreased to 25% (P = 4.1×10−8) and ~0% (P = 7.8×10−11), respectively. In contrast, Sox10 expression levels decreased more slowly to 80% (P = 0.05) and 20% (P =2.0×10−9), respectively, while Gata2 decreased to 70% at 17μM (P=8×10−3) and 50% at 25 μM (P=5×10−3). These results are concordant with the detected protein levels of Ret, Sox10 and Gata2, suggesting that the effects are largely transcriptional (Figure 5B). Therefore, Ret enhancer variants that reduce its gene expression have persistent effects on Sox10 and Gata2 transcription, but with a lag, through some yet unknown mechanism. In the mouse, aganglionosis is seen only in RET null homozygotes, while heterozygotes of a Sox10 null mutation have the phenotype(Schuchardt et al., 1994; Southard-Smith et al., 1999). Thus, even small decreases in Sox10 protein can amplify the genetic effects of the primary loss-of-function effects of Ret.
Figure 5. Feedback between Ret and its transcription factors (TF).

(A) Ret gene expression in mouse Neuro2a cells with increasing doses of Ret siRNA (12–25 μM), with assays of transcript and protein levels of Ret, Sox10, Rarb and Gata2. Ret expression declines steadily followed by decreasing Sox10 expression only when Ret falls below 50% (17 μM); an analogous decline is observed for Gata2 but not Rarb. All pairwise comparisons are with transfections with control siRNAs. (B) Protein expression changes assessed by western blotting. The error bars represent standard errors of the mean (*P<0.01, ** P<0.001).
Dysregulation of the RET GRN in the human fetal gut
To assess the veracity of the above results and relevance for HSCR we collected 8 human fetal guts (dissected from stomach to large intestine) at Carnegie Stage 22 (CS22), corresponding to a developmental time when neuronal innervation of the gut is almost complete (Newgreen and Young, 2002; Wallace and Burns, 2005). These samples were genotyped for the three enhancer variants to determine their risk haplotypes classified as S (susceptible: ATT, GTT) or R (resistant: GTC, ATC, GCC, ACC) and comprised 2, 5 and 1 embryos with R/R, R/S and S/S genotypes (Figure 6A). This allowed assays of developmentally-relevant in vivo gene expression of the RET GRN by genotype using Taqman assays; we set the expression values of the reference RR genotype to unity (Figure 6B). These human data allow two significant and key inferences. First, gene expression of RET is 4 fold (P=5×10−8) and 16 fold (P=4×10−11) lower in R/S and S/S genotypes. Even in the general population, the S haplotype decreases RET expression sharply and does so in a non-additive manner. Second, for the R/S and S/S genotypes, respectively, gene expression of (1) GDNF is significantly increased by 1.7 fold (P=4×10−6) and 1.9 fold (P=3.3×10−6), (2) GFRA1 is increased by 1.5 fold (P= 3.8×10−6) and 1.8 fold (P= 2×10−6), and (3) CBL is decreased by 3 fold (P= 6×10−5) and 8 fold (P=3.4×10−7). Of the three TFs, SOX10 has a 4 fold (P= 3×10−6) and 13 fold (P= 1.2×10−8) decrease, GATA2 has a 5.4 fold (P=4.2×10−6) and a 13 fold (P= 3.1×10−9) decrease, but, RARB is unchanged. These results qualitatively recapitulate our observations in the Ret wildtype and null mouse embryos at the analogous developmental time although quantitative differences exist (Figure 4C). Therefore, the Ret GRN effects we deciphered in the mouse are highly conserved in the human with identical feedback effects. The fact that RET enhancer genotypes affect the expression of RET and other genes in its GRN, all unlinked to RET, suggests that these effects act through yet other CREs and enhancers.
Figure 6. Dysregulation of the RET gene regulatory network (GRN) in the human fetal gut.

(A) Combined genotypes of 3 enhancer variants in 8 fetal samples represented in terms of Hirschsprung disease (HSCR) resistant (R) and susceptible (S) haplotypes (Table 1). The risk alleles have been highlighted in red. (B) Average gene expression by genotype shows loss of RET expression by S haplotype dosage, and analogous effects on SOX10, GATA2 and CBL; GDNF and GFRA1 show the opposite effect, as in the mouse (Figure 4). All pairwise comparisons are with reference to the R/R haplotype. The error bars represent standard errors of the mean (*P<0.01, ** P<0.001).
DISCUSSION
RET is the major gene for HSCR but merely one of many susceptibility genes that add to the multifactorial risk of HSCR (Alves et al., 2013). Nevertheless, despite considerable genetic heterogeneity in HSCR, RET is the most critical disease gene since deleterious coding variants occur in 21%, intragenic deletions in 5% and enhancer variants in >98% of HSCR patients (Table 1) (Emison et al., 2010). Thus, each HSCR patient carries at least one RET deficiency allele. Since unaffected controls also harbor these common enhancer deficiency alleles, RET deficiency per se is necessary, not sufficient, for disease onset. This conclusion is consistent with the known role of Ret as one of the first and major proteins required for ENCC differentiation into enteric neurons (Mulligan, 2014; Schuchardt et al., 1994; Southard-Smith et al., 1999; Uesaka et al., 2008).
The disease significance of this study is that it uncovers two types of genomic interactions not normally considered in human genetics. The first involves differential enhancer use within the RET TAD and how this feature confers variable genetic risk to HSCR (Table 1). Variable enhancer use also explains why some human enhancer deficiency alleles and haplotypes have stronger risk effects than others (Figures 1, 2). Additionally, these enhancers and their cognate TFs are connected to Ret bi-directionally, once through direct effects on transcription and, subsequently, through target gene feedback onto its TF (Figure 4). This dual effect implies that enhancer variants of a target gene are equivalent to hypomorphic alleles of the corresponding TF but unlike the TF, that has many targets, have more specific effects. Our study suggests that the GWAS variant effects we detect in humans are an incomplete feature of their effect (Manolio et al., 2009; Welter et al., 2014), since they do not act alone but in combination with variants in other enhancers, and with use varying with cellular state (Andrey et al., 2013). Understanding the genetic effect of a non-coding variant on a gene or disease consequently requires knowledge of its multiple enhancers, their usage, the gene(s) they regulate and how the target gene(s) interacts with these enhancers.
The second type of genomic interaction connects the many genes within the Ret GRN that need to be coordinately regulated. Previous genetic and biochemical studies have shown how physiological levels of Ret are maintained by balance between its production, activation and degradation (Mulligan, 2014). Our studies here demonstrate that this balance is achieved not only through the effects of the TFs, ligand, co-receptor and signal terminating genes on Ret protein but also through Ret regulating these component genes (Figure 4). Here we identified at least seven genes, Rarb, Gata2, Sox10, Ret, Gdnf, Gfra1, Cbl, that comprise the Ret GRN with multiple positive and negative feedback mechanisms. Each of these genes can lead to HSCR: rare deleterious coding mutations in RET, SOX10 and GDNF are frequent in HSCR (Alves et al., 2013; Southard-Smith et al., 1999), and while GFRA1 coding mutations are absent in HSCR, mouse Gfra1 null mutants have aganglionosis (Tomac et al., 2000); finally, although CBL mutations are also absent in HSCR the related ubiquitin E3 ligase gene UBR4 has rare deleterious coding mutations in HSCR (Chakravarti, 2014). Our study now explains how mutations in any Ret GRN component leads to Ret deficiency and, in turn, how Ret deficiency can affect the functions of its entire GRN. Thus, HSCR is caused by dysregulation of the entire RET GRN irrespective of the source of the primary variant or mutation.
Our model of a coherent functional architecture of a complex disease centered on a rate-limiting gene and its GRN has four corollaries, three for complex disorders generally, and one for HSCR specifically. The first three corollaries are relevant to the tens of thousands of genetic variants discovered through GWAS (Welter et al., 2014). First, even larger numbers of undetected variants exist since upto ~50% of the phenotypic variance of studied traits can be explained by common variants alone (Yang et al., 2011). Given widespread non-coding polymorphisms (Abecasis et al., 2012), multiple CREs controlling each gene (Bernstein et al., 2010) and enrichment of trait association signals within CREs (Trynka et al., 2013), a comprehensive forward genetic screen for causal associations, such as we conducted for RET, may be highly effective for dissecting the regulatory biology for each trait. The existence of TADs that are relatively stable across cell types suggest a precise physical locus for such screens. Second, non-coding variants cannot, and should not, be viewed in isolation but only in the context of all such variants affecting a gene because their effects are transcriptionally integrated. This cellular integration is the purpose of the GRN and needs to be understood to elucidate disease mechanisms. Indeed, a systems-level view of the Ret GRN was necessary for us to understand the genetic risk conferred by RET haplotypes, an aspect unlikely to be specific to HSCR. Third, at any association locus, GWAS attempt to identify statistically independent genetic variants as causal factors of a disease because linkage disequilibrium (LD) within a locus does not allow the distinction between a true causal and an associated surrogate variant. However, there is no reason to believe that the distribution of causal enhancer variants within a TAD or locus is related to LD. Consequently, a functional approach, such as we used, may be more efficient for identifying causal variants within a association locus than mapping per se.
The fourth corollary, for HSCR, is that although, by implication, RARB, GATA2 and SOX10 are critical TFs for RET, there may be other TFs and additional variants involved. This likelihood is increased given the feedback interactions within the Ret GRN, in analogy with ‘shadow’ enhancers in Drosophila (Hong et al., 2008). Whether such feedback occurs directly or indirectly, is transcriptional or translational, is unknown. We do not believe that these are direct effects of Ret through its canonical signaling pathway, since the transcription of known downstream effectors (Akt1, Erk1) is unchanged. One possibility is the post-translational modification of Sox10 and Gata2 to retard their nuclear entry. Sox10 is a nucleo-cytoplasmic shuttle protein (Rehberg et al., 2002) and gives some credence to this hypothesis. Additionally, our results suggest crosstalk between ENCCs and mesenchymal cells, since Gdnf and Gfra1 are expressed in the gut mesenchyme, not in ENCCs. This points to much broader roles of Ret in the control of early processes that drive enteric neuron migration in the gut and potentially many other neural crest-derived structures.
The specific gut TFs we uncovered speaks to the underlying biology of ENS development and HSCR in three ways. First, Sox10, and Pax3 (acting earlier on ENCCs), are the major early TFs for neural crest cells to pursue different cell fates, while Ret and Ednrb are the two major signaling proteins that lead to their terminal differentiation into enteric neurons. This is consistent with coding mutations in each of these genes leading to high-penetrance HSCR (Carrasquillo et al., 2002; Schuchardt et al., 1994; Southard-Smith et al., 1999). Our studies now show the additional role of Rarb and Gata2 in ENS differentiation. Of these, the role of Rarb is consistent with an earlier observation that retinoic acid depletion leads to distal bowel aganglionosis, while retinoic acid treated explants produce abundant, densely distributed ENCCs in chains and increases Ret expression (Fu et al., 2010; Simkin et al., 2013). Second the role of Gata2, a major endothelial marker, lends credence to the role of endothelial cells in promoting proliferation and migration of enteric neurons (Nagy et al., 2009). It is possible that the reduced Ret expression leading to reduced Gata2 leads to reduced numbers of endothelial cells as well as enteric neurons. Third, the developmental hierarchy of Pax3, Sox10, Rarb, Gata2 and Ret action within the neural crest, and their narrowing developmental fields within the gut, shows why only Ret variants are restricted to the ENS whereas mutations in the other four TFs lead to syndromic disorders involving many organs, including the gut. Our results suggest that the major effects of RET in HSCR and in mouse Ret null mutants occur because of its additional collateral effect on its TFs Sox10, Gata2 and other members of its GRN.
A major remaining question is how widespread human susceptibility from common enhancer variants, a defining feature of all complex diseases, is converted to clinical disease (Kapoor et al., 2015). The hypotheses include stochastic effects on gene expression, additional variants in other genes dysregulating other GRNs, and environmental effects on gene expression. In addition, the effects of enhancer variants may be subject to epigenetic factors, owing to feedback interactions, implying that the usual small effects we measure in a GWAS is an average across many cellular states.
Identification of the causal regulatory factors in any complex human disease is fraught with the difficulty that although experimental systems are absolutely necessary to demonstrate causality they are surrogate systems that might not accurately reflect human physiology. Moreover, owing to widespread linkage disequilibrium between local polymorphisms it is hard to definitively ‘prove’ that the true causal variants have been uncovered except in such surrogate systems. The way out of this dilemma is to provide consistent and concordant evidence from multiple independent sources, as presented in this study.
Methods and Resources
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to the corresponding author Aravinda Chakravarti (Aravinda@jhmi.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient samples, controls and genotyping
Ascertainment of patients, their diagnosis and DNA isolation from whole blood used standard protocols (Kapoor et al., 2015); these studies were conducted with written informed consent approved by the Institutional Review Boards of Johns Hopkins University School of Medicine. Patients were classified by segment length of aganglionosis into three classes: short-segment (S-HSCR: aganglionosis up to the upper sigmoid colon), long-segment (L-HSCR: aganglionosis up to the splenic flexure) and total colonic aganglionosis (TCA). We used a primary sample of 356 probands (see below) comprising one affected individual per family and with the following distribution: 259/97 are male/female; 150/41/61/104 are S-HSCR/L-HSCR/TCA/unknown segment length; 248/108 are simplex/multiplex; 230/126 probands are isolated/had additional anomalies. Only study individuals self-identified as being of European ancestry were included. In addition, we used 757 controls from two sources: (i) 503 European-ancestry samples from the 1000 Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/) (Abecasis et al., 2012); (ii) 254 pseudo-controls created from the untransmitted alleles from genotyped parents of a child with HSCR.
Genotyping of the three RET SNPS (rs2506030, rs7069590 and rs2435357) was performed using specific TaqMan Human Pre-Designed genotyping assays following the manufacturer’s protocol (ThermoFisher Scientific). The assays IDs are C__26742714_10 for rs2506030, C__2046272_10 for rs7069590 and C__16017524_10 for rs2435357. The endpoint fluorescence measurements were performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems) and analyzed using Sequence Detection System Software v.2.1 (Applied Biosystems). After standard quality control, 346 probands and 732 (503 1000G and 229 trio) controls were analyzed.
Human fetal gut samples
Fetal gut tissues were obtained form the Human Developmental Biology Resource (www.hdbr.org) (Gerrelli et al., 2015), voluntarily donated by women undergoing termination of pregnancy following specific written consent. These studies were conducted with written informed consent approved by the Institutional Review Boards of Johns Hopkins University School of Medicine.
Transgenic mouse enhancer assays
To generate transgenic mice for each putative enhancer, 1–2kb human DNA fragments were cloned upstream into a Hsp lacZ vector. The sizes of the enhancer elements were kept as close as possible to the fragments tested in vitro (~1kb) except for RET−5.5, where a larger fragment (2kb) was used. The concentration of the linearized and purified DNA was determined fluorometrically and by agarose gel electrophoresis. The DNA was diluted to a concentration of 1.5 to 2 ng/μl and used for pronuclear injections of FVB embryos in an injection buffer (10 mM Tris, pH 7.5; 0.1 mM EDTA). Embryos were harvested at days E11.5 and E12.5 and dissected in cold 100 mM phosphate buffer pH 7.3, followed by 30 min of incubation with 4% paraformaldehyde at 4°C. The embryos’ heads were pu nctured with a 27 G needle to facilitate the penetration of the staining solution and washed three times for 30 min with wash buffer (2 mM MgCl2; 0.01% deoxycholate; 0.02% NP-40; 100 mM phosphate buffer, pH 7.3). Embryos were stained for 24 h at room temperature with freshly made staining solution (0.8 mg/ml X-gal; 4 mM potassium ferrocyanide; 4 mM potassium ferricyanide; 20 mM Tris, pH 7.5, in wash buffer). Stained embryos were rinsed 3 times in 100 mM phosphate buffer, pH 7.3, and postfixed in 4% paraformaldehyde. A domain of expression was considered positive only when observed in at least 3 independent transgenic embryos.
Ret and Sox10 null mice
Mice homozygous for a null Ret allele have been described previously (Uesaka et al., 2008). Homozygous Ret null mice are embryonic lethals by about E18.5, so Ret heterozygous mice were crossed to generate all possible genotypes (wildtype, heterozygotes and homozygotes). Genotyping was done from yolk sac DNA by using primers specific to the Ret locus for wildtype embryos (449bp PCR product) and CFP transgene (615 bp PCR product) for mutants (Key Resource Table). Embryonic guts at various developmental time points were dissected, and genotyped male embryonic gut selected for further analysis. The guts were washed in ice cold phosphate buffered saline and snap frozen in liquid nitrogen for RNA extraction. The Sox10lacZ mice have been described previously (Britsch et al., 2001) and embryos were collected as described for Ret null mice. Genotyping was performed on yolk sac DNA using primers specific to The Sox10 locus (506 bp PCR product) for wildtype embryos and lacZ transgene (364 bp PCR product) for mutants (Key Resource Table).
Genotyping for sex was done using primers mapping to Kdm5c/d genes, resulting in two 331 bp bands for X chromosome for females and an additional 301 bp Y chromosome band in males (Clapcote and Roder, 2005)
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of Johns Hopkins University (Protocol MO12M374) and were in accordance with Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines. All animals were fed a standard rodent chow ad libitum.
METHOD DETAILS
DNA and Chromatin Analysis
Nomenclature
There is no official or systematic nomenclature for non-coding elements and their sequence variants. In this paper, we have used the following system to facilitate description. Each polymorphism is referred to by its standard alias (rsID) with its alleles labeled as ‘risk’ and ‘non-risk’ alleles based on their odds ratios of disease: consequently, risk allele frequencies can have any value over the unit interval. Each cis-regulatory element (CRE) is defined with respect to the gene it transcriptionally affects (i.e. RET), with + or − signifying locations downstream or upstream of that gene’s transcriptional start site (TSS) respectively, and with the number representing the temporal order of discovery. Thus, the third conserved element transcriptionally affecting RET which is located downstream of the TSS in intron 1 is termed RET+3 (Emison et al., 2005; Grice et al., 2005). In the case of the non-integer RET−5.5, we accommodated finding a new element between two previously discovered CREs. Each CRE is of variable length, defined either by comparative sequence analysis or epigenomic marks, and a CRE is specifically called an enhancer when its biochemical activity has been demonstrated through in vitro and in vivo analyses (this paper). The latter biochemical analyses have used various sub-segments of the indicated CREs, but always centered on the polymorphic site that led to study of that CRE. Thus, RET+3 is an enhancer with a loss-of-function allele T at the polymorphism rs2435357 that increases Hirschsprung disease risk significantly.
Motif search
The 20bp sub-sequence for RET−7, CCAATGACCTATTCCAGTCT, and RET−5.5, ACATGAAATAATGCGATAGA centered at the SNPs of interest (rs2506030 and rs7069590) was scanned by the FIMO software with 890 known TF motifs available from three public motif databases, TRANSFAC, JASPAR and UniPROBE (Bailey et al., 2009; Bryne et al., 2008; Newburger and Bulyk, 2009; Wingender et al., 1996).
ChIP-seq peak calling
Three epigenomic data sets for the 108 day human fetal large intestine, histone H3K27ac ChIP-seq (GSM1058765), histone H3K4me1 ChIP-seq (GSM1058775), and DNaseI-seq (GSM817188), were downloaded from the NIH Roadmap Epigenomics Project. For the SK-N-SH cell line, DNaseI-seq data (GSM736559) were obtained from the ENCODE project. For each of the data sets, MACS software v1.4 (Zhang et al., 2008) with default setting was used to call “peaks” where the sequence reads were significantly enriched. With the default peak-calling threshold (P < 10−5), 51,771, 61,689, 66,930 and 52,534 genomic regions were identified in the GSM1058765, GSM1058775, GSM817188 and GSM736559 data sets, respectively.
Topologically Associating Domains (TADs)
We used published HiC data to map the TADs at the RET locus. The HESC data were from published sources (Dixon et al., 2012) and the other TADs were created using Juicebox (Rao et al., 2014) by mapping the normalized data for each cell type with 5kb resolution. The genomic coordinates (Hg19) for TADs around RET in each cell types are as follows: HESC (chr10:43,300,994–43,643,327), GM12878 (chr10:43,355,001–43,600,000), HeLa (chr10:43,355,001–43,605,000), HMEC (chr10:43,375,001–43,625,000), HUVEC (43,370,001–43,600,000), IMR90 (chr10:43,340,001–43,610,000), K562 (43,350,001–43,620,000), KBM-7 (43,355,001–43,610,000) and NHEK (chr10:43,360,001–43,595,000). A core 225 kb (chr10:43,375,000–43,600,000) domain is common to all cell types.
Cell lines
Neuro2a (ATCC CCL-131) and SK-N-SH (ATCC HTB-11) were purchased from ATCC and grown under standard conditions (DMEM/F12 + 10% FBS and 1% Penicillin Streptomycin).
Luciferase assays
400 ng of firefly luciferase vector (pGL4.23, Promega Corporation) containing a 1kb DNA sequence with the assayed SNP in the center and 2 ng of Renilla luciferase vector (transfection control) were transiently transfected into the mouse neuroblastoma cell line Neuro2A (5 – 6 × 104 cells/well) using 6 μl of FuGENE HD transfection reagent (Roche Diagnostic, USA) in 100μl of OPTI-MEM I medium (Invitrogen, USA). The cells were grown for 48 hours and luminescence measured using a Dual Luciferase Reporter Assay System on a Tecan multi-detection system luminometer, per the manufacturer’s instructions. All assays were performed in triplicate with independent readings in triplicate (n=9): the data presented are the means with their standard errors.
Electrophoretic mobility shift assays (EMSA)
Nuclear proteins were extracted from Neuro2A cells using the NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific). EMSAs were carried out using DNA probes modified with 5′ Cy5 labels (Integrated DNA Technologies). Equimolar amounts of complementary strands were mixed and heated to 95°C followed by gradual cooling to ambient temperature over at least 5 h to anneal probes. For binding studies, double-stranded DNA probes at 10 nM were mixed with 10 μg of nuclear proteins and 500 ng of Poly dI-dC (Sigma) in a buffer containing 40 mM Tris-HCl (pH 8.0), 0.4 mg/ml BSA, 200 μM ZnCl2, 400 mM KCl, 40% glycerol and 0.4% IGEPAL and incubated at 4°C in the dark for one hou r. The bound and unbound probes were subsequently run on a pre-run 6% 1X TBE polyacrylamide gel for ~30 min at 200 V and fluorescence detected using a Typhoon 9140 PhosphorImager (Amersham Biosciences). For the deletion construct, an oligonucleotide was designed lacking 10bp sequence containing the SNP. All assays were performed in triplicate. The following are the probe sequences we used: RET−7_A(Cy5-5′CCAATGACCTATTCCAGTCT-3′), RET−7_G (Cy5- Cy5-5′CCAATGACCTGTTCCAGTCT-3′), RET−7_deletion (Cy5-5′CCAATAGTCT-3′). RET−5.5_C (Cy5-5′-TGAAATAATGCGATAGATG-3′), RET−5.5_T (Cy5-5′-TGAAATAATGTGATAGATG-3′), RET−5.5_deletion (Cy5-5′-TGAAATGATGC-3′). RET+3_C (Cy5-5′-ACCCTTACACGGTCATCCAC-3′) RET+3_T(Cy5-5′-ACCCTTACATGGTCATCCAC-3′) and RET+3_deletion (Cy5-5′-ACCCTTCCAC-3′).
Gene expression Taqman assays
Total RNA was extracted from Neuro2A cells, individual male mouse embryonic guts at E11.5 and E12.5 and human fetal gut tissue at CS22 stage in development using TRIzol (Life Technologies, USA) and cleaned on RNeasy columns (Qiagen, USA). 1μg of total RNA was converted to cDNA using SuperScriptIII reverse transcriptase (Life Technologies, USA) and Oligo-dT primers. The diluted (1/10) total cDNA was subjected to Taqman gene expression (ThermoFisher Scientific) using transcript specific probes and primers (Table S3). Mouse β-actin (Actb) was used as an internal loading control for normalization. Five independent biological samples for mouse fetal gut at each stage or five independent wells for Neuro2a cells were used for RNA extraction and each assay was performed in triplicate (n=15); For human fetal gut each individual sample was assayed 3 times (n=3); the data presented are the means with their standard errors. Relative fold changes were calculated based on the 2ΔΔCt (threshold cycle) method. For siRNA experiments the 2ΔΔCt for control siRNA was set to unity; for measuring gene expression in mice guts, the 2ΔΔCt value for E11.5 wild type animals was set to unity. P values were calculated from pairwise 2-tailed t-tests.
Gene expression RNA-seq assays
Total RNA was extracted from 3 male mouse guts at E11.5 and E12.5. cDNA was prepared by oligo dT beads to select mRNA from the total RNA sample followed by heat fragmentation and cDNA synthesis from the RNA template, as part of the Illumina Tru Seq™ RNA Sample Preparation protocol. The resultant cDNA was used for library preparation (end repair, base ‘A’ addition, adapter ligation, and enrichment) using standard Illumina protocols. Libraries were run on a HiSeq 2000 using manufacturer’s protocols to a depth of 15 million reads per sample (75 base pairs, paired end) at the Broad Institute. The primary data were analyzed using the Broad Institute’s Picard Pipeline, which includes de-multiplexing, and data aggregation. The resultant BAM files were mapped to the mouse genome (assembly mm10/GRCm38) using Bowtie with its setting for paired end, non-strand specific library (Langmead et al., 2009). Successfully mapped reads were used to assemble transcripts and estimate their abundance using Cufflinks (Trapnell et al., 2012). The resultant data assigned Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values for each transcript and gene. The transcript file for each replicate were merged using Cuffmerge and analysed by Cuffdiff (Trapnell et al., 2012) to detect differentially expressed genes between wild type and Ret null samples at each stage. All data have been deposited in NCBI’s Gene Expression Omnibus and are accessible at GEO Series accession number GSE 84145.
siRNA assays
Ret (L-047013-00), Rarβ (L-040538-00), Sox10 (L-049957-01) and Gata2 (L-062114-00) SMARTpool siRNAs (a combination of 4 individual siRNA targeting each gene) along with ON-TARGETplus non-targeting siRNA (D-001810-10, negative control) (Dharmacon, USA)were transfected at concentration ranges from 12 to 25μM in Neuro2A cells at a density of 104–105 cells using FuGene HD Transfection reagent (Promega Corporation, USA) per the manufacturer’s instructions; negative control siRNAs were always transfected at 25μM concentration. Total RNA was extracted from the cells 48 hours post transfection and Taqman gene specific assays conducted, as previously described. Five independent transfections were used for each siRNA and each Taqman assay was performed in triplicate (n=15); the data presented are the means with their standard errors.
Western blot assays
Nuclear and cytoplasmic proteins were extracted from Neuro2A cells using the NE-PER nuclear and cytoplasmic extraction kit (ThermoFisher Scientific). 10μg of either nuclear extract (for the transcription factors and Histone H2A) or cytoplasmic extract (for Ret and Actb) were run on a gradient (4–12%) bis Tris polyacrylamide gel (Life Technologies, USA). The proteins were transferred to nitrocellulose membranes and incubated with the following antibodies: Ret (C31B4, Cell Signaling Technology) at 1:500 dilution, Sox10 (ab 155279, Abcam) at 1:1000 dilution, Rarβ (ab53161, Abcam) at 1:1000 dilution, Gata2 (ab22849, Abcam) at 1:1000 dilution, H2A (2578, Cell Signaling Technologies) at 1:2000 dilution and β-actin (4967S, Cell Signaling Technologies) at 1:1000 dilution. The blots were incubated with their respective HRP conjugated secondary antibodies followed by incubation for 5 minutes at room temperature with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and exposed to chemiluminescence film (GE healthcare).
Chromatin Immunoprecipitation–qPCR (ChIP-qPCR) assay
ChIP was performed three times independently for each antibody using 1×106 SK-N-SH cells for each transcription factor using the EZ-Magna ChIP kit (Millipore), as per the manufacturer’s instruction with the following modifications: the chromatin was sonicated with 30 seconds on and 30 seconds off for 10 cycles; sheared chromatin was pre-blocked with unconjugated beads for 4 hours and specific antibodies separately conjugated to the beads for 4 hours before IP was performed with the pre-blocked chromatin. The following antibodies were used: SOX10 (sc-17342X, Santa Cruz) 10μg, RAR (sc-773X, Santa Cruz) 10μg and GATA2 (ab22849, Abcam) 10μg. ChIP assays were also performed on cells 48 hours after transfection with the following siRNAs at 25μM to check for specificity of TF binding: SOX10 (L-017192-00), RARβ (L-003438-02) and GATA2 (L-009024-02) (Thermo Scientific). qPCR assays were performed using SYBR green (Life Technologies) using specific primers against the RET−7, RET−5.5 and RET+3 regions (Key Resource Table). The data were normalized to the input DNA and enrichment was calculated by fold excess over ChIP performed with specific IgG as background signal. All assays were done in triplicate for each independent ChIP (n=9).
QUANTIFICATION AND STATISTICAL ANALYSIS
Association tests
Genotype counts were obtained from HSCR cases or controls, including pseudo-controls that were generated from trio genotypes using PseudoCons (Cordell et al., 2004) (http://www.staff.ncl.ac.uk/richard.howey/pseudocons/index.html/). Genotypes at the three SNPs from all unrelated HSCR cases and controls were assessed separately for Hardy-Weinberg equilibrium (HWE) using standard tests (Kapoor et al., 2015). None deviated significantly from HWE (P>0.05), except at rs2435357 in HSCR cases where the significant deviation (P =6.3×10−12) reflects the high population association of this SNP with HSCR. Standard methods using χ2 statistics were used for calculation of odds ratios (OR), their upper and lower confidence limits and significance of their deviation from the null hypothesis of no association (OR = 1). Pairwise linkage disequilibrium or correlation measurements, r2, were compared between cases and controls using Fisher r-to-z transformation (Kapoor et al., 2015).
qPCR data
The number of samples (n) is mentioned in each experimental section represents either number of independent embryos used or the number of wells/plate from which cells were used for downstream RNA extraction (biological replicates). Also mentioned is the number of times the qPCR was done of each biological replicates, which constitutes technical replicates.
DATA AND SOFTWARE AVAILABILITY
All RNA seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible at GEO Series accession number GSE 84145.
Supplementary Material
EMSA using 20bp Cy5 labeled probes containing either the wild type or the risk allele for RET−7, RET−5.5 and RET+3 shows specific binding (black arrowheads) of both alleles with factor(s) present in Neuro2a nuclear extracts; binding is completely abrogated to probes containing a 10-bp deletion centered at each variant site.
Linkage disequilibrium (LD) between all 38 SNPs in Supplementary Table 1 was estimated using a previously described method (Gabriel et al., 2002). The 3 SNPs with allelic difference in enhancer activity (rs2506030, rs7069590 and rs2435357) are boxed.
A forest plot showing the haplotype odds ratio for three polymorphisms (rs2506030, rs7069590, rs2435357) located within the enhancers RET−7, RET−5.5 and RET+3, respectively in males and female among 346 cases and 503 controls (excluding 229 pseudo controls). The odds ratios are with respect to the reference haplotype ACC that lacks any susceptibility allele. The error bars are its 95% confidence interval (CI). The susceptibility alleles are marked in red.
(A) Scatter plot of log2FPKM values of genes expressed in Ret wildtype and null guts at E11.5 and E12.5. Genes in red have significant (q-value <0.01) expression difference between the states. (B) Bar plots showing the expression difference of the genes of the Ret GRN between wildtype and Ret null comparisons at each developmental stage. The error bars represent the confidence interval of FPKM (** q-value <0.001).
Gene expression of Ret pathway genes in the developing mouse gut at E11.5 and E12.5 in wild type and Sox10 heterozygote embryos show significant up-regulation of Gdnf and Gfra1 and down-regulation of Ret and Gata2 in Sox10 heterozygote embryos at both developmental stages; Cbl and Erk1 show loss of expression only at E11.5 while Rarb is unaffected. P38α also shows down-regulation at both stages while Akt1 shows down regulation only at E12.5. All pairwise comparisons are between wild type and null embryonic guts at each stage.
The error bars represent standard errors of the mean (* P<0.01, ** P<0.001).
Table S1 (Related to Figure 1 and table 1): HSCR-associated polymorphisms within the RET locus within putative enhancers. 38 SNPs (minor allele frequency ≥ 10%) at the RET locus showing genome-wide significant association (P≤ 5×10−8) with HSCR are shown from a recently published GWAS study (reference 17 in main text). SNPs marked in color disrupt a known transcription factor-binding site in the human SK-N-SH cell line (ENCODE ChIP-seq), of which the ones demonstrating in vitro allelic difference in enhancer activity are highlighted in red. The genomic region spanning 154,678 bp is characterized by four blocks of linkage disequilibrium (Supplementary Figure 1) defined by the 38 SNPs in 503 European ancestry reference samples (reference 21 in main text) as marked in gray; blocks 2 and 3 are demarcated by a thick line. Note that two polymorphisms, #14 and 23, show weak associations with all 36 other markers.
Table S2 (related to table 1): RET polymorphisms associated with HSCR. Genetic features of the three polymorphisms rs2506030, rs7069590 and rs2435357 that are located within the enhancers RET−7, RET−5.5 and RET+3, 124.6 kb upstream, 19.6kb upstream and 9.5kb downstream from the RET transcription start site, respectively, are shown. Also shown are the observed frequencies of the risk allele among 346 cases and 732 controls, the odds ratio of disease, its 95% confidence interval (CI) and its statistical significance (P value) on testing whether the odds ratio differs from unity.
Table S3 (related to STAR Methods): Taqman gene expression probes for mouse and human transcripts.
Table S4 (related to STAR Methods): Primers for ChIP-qPCR and for mouse genotyping
Acknowledgments
We wish to thank our patients, their families, referring physicians and genetic counselors for participating in the human genetic studies. We gratefully acknowledge the assistance of Dr. David Ginty (Harvard Medical School) for providing Ret mutant mice, Dr. William J Pavan (NHGRI, NIH) and Dr. Michael Wegner (Institut für Biochemie, Friedrich-Alexander-Universität) for providing Sox10 mutant mice, and Dr. Edward Rubin (Lawrence Berkeley National Laboratory) for helpful discussions. L.A.P. was supported by NIDCR FaceBase grant U01DE020060 and by NHGRI grants R01HG003988, and U54HG006997; the research was conducted at the E.O. Lawrence Berkeley National Laboratory and performed under Department of Energy Contract DE-AC02-05CH11231. The human embryonic and fetal material were provided by the Joint MRC/Wellcome Trust Human Developmental Biology Resource (www.hdbr.org), grant no. 099175/Z/12/Z. These studies were supported by an NIH MERIT Award HD28088 to AC.
Footnotes
Author Contributions
SC, AK and AC conceived and designed the study. SC conducted all in vitro and in vivo assays, AK all genotyping assays, and SC and SG the RNA-Seq experiments. JAA and LAP performed mouse transgenic enhancer assays, DRA helped with in vivo experiments, DL conducted ChIP-seq peak calling, and CB conducted family ascertainment and genetic counseling. SC, AK and AC wrote the manuscript; all authors were involved in manuscript revision.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U. An introduction to systems biology: design principles of biological circuits. Boca Raton, FL: Chapman & Hall/CRC; 2007. [Google Scholar]
- Alves MM, Sribudiani Y, Brouwer RW, Amiel J, Antinolo G, Borrego S, Ceccherini I, Chakravarti A, Fernandez RM, Garcia-Barcelo MM, et al. Contribution of rare and common variants determine complex diseases-Hirschsprung disease as a model. Dev Biol. 2013;382:320–329. doi: 10.1016/j.ydbio.2013.05.019. [DOI] [PubMed] [Google Scholar]
- Andrey G, Montavon T, Mascrez B, Gonzalez F, Noordermeer D, Leleu M, Trono D, Spitz F, Duboule D. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science. 2013;340:1234167. doi: 10.1126/science.1234167. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, Chakravarti A, Cohen JC, Hardy J. Mendelian disorders and multifactorial traits: the big divide or one for all? Nat Rev Genet. 2010;11:380–384. doi: 10.1038/nrg2793. [DOI] [PubMed] [Google Scholar]
- Badner JA, Sieber WK, Garver KL, Chakravarti A. A genetic study of Hirschsprung disease. Am J Hum Genet. 1990;46:568–580. [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic acids research. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nature biotechnology. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15:66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic acids research. 2008;36:D102–106. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo MM, McCallion AS, Puffenberger EG, Kashuk CS, Nouri N, Chakravarti A. Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet. 2002;32:237–244. doi: 10.1038/ng998. [DOI] [PubMed] [Google Scholar]
- Chakravarti A. 2013 William Allan Award: My multifactorial journey. Am J Hum Genet. 2014;94:326–333. doi: 10.1016/j.ajhg.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti AMAS, Lyonnet S. Hirschsprung Disease. In: Valle BALD, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. [Google Scholar]
- Clapcote SJ, Roder JC. Simplex PCR assay for sex determination in mice. Biotechniques. 2005;38:702, 704, 706. doi: 10.2144/05385BM05. [DOI] [PubMed] [Google Scholar]
- Consortium TEP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ, Barratt BJ, Clayton DG. Case/pseudocontrol analysis in genetic association studies: A unified framework for detection of genotype and haplotype associations, gene-gene and gene-environment interactions, and parent-of-origin effects. Genet Epidemiol. 2004;26:167–185. doi: 10.1002/gepi.10307. [DOI] [PubMed] [Google Scholar]
- Davidson EH. The regulatory genome: gene regulatory networks in development and evolution. Burlington, MA: Academic; 2006. New edn. [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll MC, Dobkin CS, Alter BP. Gamma delta beta-thalassemia due to a de novo mutation deleting the 5′ beta-globin gene activation-region hypersensitive sites. Proc Natl Acad Sci U S A. 1989;86:7470–7474. doi: 10.1073/pnas.86.19.7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emison ES, Garcia-Barcelo M, Grice EA, Lantieri F, Amiel J, Burzynski G, Fernandez RM, Hao L, Kashuk C, West K, et al. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am J Hum Genet. 2010;87:60–74. doi: 10.1016/j.ajhg.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, Lin S, Portnoy ME, Cutler DJ, Green ED, Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- Fu M, Sato Y, Lyons-Warren A, Zhang B, Kane MA, Napoli JL, Heuckeroth RO. Vitamin A facilitates enteric nervous system precursor migration by reducing Pten accumulation. Development. 2010;137:631–640. doi: 10.1242/dev.040550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Gerrelli D, Lisgo S, Copp AJ, Lindsay S. Enabling research with human embryonic and fetal tissue resources. Development. 2015;142:3073–3076. doi: 10.1242/dev.122820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB. Common genetic variation and human traits. The New England journal of medicine. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- Grice EA, Rochelle ES, Green ED, Chakravarti A, McCallion AS. Evaluation of the RET regulatory landscape reveals the biological relevance of a HSCR-implicated enhancer. Human molecular genetics. 2005;14:3837–3845. doi: 10.1093/hmg/ddi408. [DOI] [PubMed] [Google Scholar]
- Grosveld F, van Assendelft GB, Greaves DR, Kollias G. Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell. 1987;51:975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- Heanue TA, Pachnis V. Enteric nervous system development and Hirschsprung’s disease: advances in genetic and stem cell studies. Nature reviews Neuroscience. 2007;8:466–479. doi: 10.1038/nrn2137. [DOI] [PubMed] [Google Scholar]
- Hong JW, Hendrix DA, Levine MS. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Arnold S, Heanue T, Kilambi KP, Doan B, Kapoor A, Ling AY, Sosa MX, Guy M, Burzynski G, et al. Functional loss of semaphorin 3C and/or semaphorin 3D and their epistatic interaction with ret are critical to Hirschsprung disease liability. Am J Hum Genet. 2015;96:581–596. doi: 10.1016/j.ajhg.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Jiang Q, Chatterjee S, Chakraborty P, Sosa MX, Berrios C, Chakravarti A. Population variation in total genetic risk of Hirschsprung disease from common RET, SEMA3 and NRG1 susceptibility polymorphisms. Human molecular genetics. 2015;24:2997–3003. doi: 10.1093/hmg/ddv051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nature reviews Cancer. 2014;14:173–186. doi: 10.1038/nrc3680. [DOI] [PubMed] [Google Scholar]
- Nagy N, Mwizerwa O, Yaniv K, Carmel L, Pieretti-Vanmarcke R, Weinstein BM, Goldstein AM. Endothelial cells promote migration and proliferation of enteric neural crest cells via beta1 integrin signaling. Dev Biol. 2009;330:263–272. doi: 10.1016/j.ydbio.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic acids research. 2009;37:D77–82. doi: 10.1093/nar/gkn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgreen D, Young HM. Enteric nervous system: development and developmental disturbances--part 2. Pediatr Dev Pathol. 2002;5:329–349. doi: 10.1007/s10024-002-0002-4. [DOI] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehberg S, Lischka P, Glaser G, Stamminger T, Wegner M, Rosorius O. Sox10 is an active nucleocytoplasmic shuttle protein, and shuttling is crucial for Sox10-mediated transactivation. Mol Cell Biol. 2002;22:5826–5834. doi: 10.1128/MCB.22.16.5826-5834.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- Simkin JE, Zhang D, Rollo BN, Newgreen DF. Retinoic acid upregulates ret and induces chain migration and population expansion in vagal neural crest cells to colonise the embryonic gut. PLoS One. 2013;8:e64077. doi: 10.1371/journal.pone.0064077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southard-Smith EM, Angrist M, Ellison JS, Agarwala R, Baxevanis AD, Chakravarti A, Pavan WJ. The Sox10(Dom) mouse: modeling the genetic variation of Waardenburg-Shah (WS4) syndrome. Genome research. 1999;9:215–225. [PubMed] [Google Scholar]
- Tomac AC, Grinberg A, Huang SP, Nosrat C, Wang Y, Borlongan C, Lin SZ, Chiang YH, Olson L, Westphal H, et al. Glial cell line-derived neurotrophic factor receptor alpha1 availability regulates glial cell line-derived neurotrophic factor signaling: evidence from mice carrying one or two mutated alleles. Neuroscience. 2000;95:1011–1023. doi: 10.1016/s0306-4522(99)00503-5. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, Raychaudhuri S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesaka T, Nagashimada M, Yonemura S, Enomoto H. Diminished Ret expression compromises neuronal survival in the colon and causes intestinal aganglionosis in mice. J Clin Invest. 2008;118:1890–1898. doi: 10.1172/JCI34425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace AS, Burns AJ. Development of the enteric nervous system, smooth muscle and interstitial cells of Cajal in the human gastrointestinal tract. Cell Tissue Res. 2005;319:367–382. doi: 10.1007/s00441-004-1023-2. [DOI] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research. 2014;42:D1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knuppel R. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic acids research. 1996;24:238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, de Andrade M, Feenstra B, Feingold E, Hayes MG, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43:519–525. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
EMSA using 20bp Cy5 labeled probes containing either the wild type or the risk allele for RET−7, RET−5.5 and RET+3 shows specific binding (black arrowheads) of both alleles with factor(s) present in Neuro2a nuclear extracts; binding is completely abrogated to probes containing a 10-bp deletion centered at each variant site.
Linkage disequilibrium (LD) between all 38 SNPs in Supplementary Table 1 was estimated using a previously described method (Gabriel et al., 2002). The 3 SNPs with allelic difference in enhancer activity (rs2506030, rs7069590 and rs2435357) are boxed.
A forest plot showing the haplotype odds ratio for three polymorphisms (rs2506030, rs7069590, rs2435357) located within the enhancers RET−7, RET−5.5 and RET+3, respectively in males and female among 346 cases and 503 controls (excluding 229 pseudo controls). The odds ratios are with respect to the reference haplotype ACC that lacks any susceptibility allele. The error bars are its 95% confidence interval (CI). The susceptibility alleles are marked in red.
(A) Scatter plot of log2FPKM values of genes expressed in Ret wildtype and null guts at E11.5 and E12.5. Genes in red have significant (q-value <0.01) expression difference between the states. (B) Bar plots showing the expression difference of the genes of the Ret GRN between wildtype and Ret null comparisons at each developmental stage. The error bars represent the confidence interval of FPKM (** q-value <0.001).
Gene expression of Ret pathway genes in the developing mouse gut at E11.5 and E12.5 in wild type and Sox10 heterozygote embryos show significant up-regulation of Gdnf and Gfra1 and down-regulation of Ret and Gata2 in Sox10 heterozygote embryos at both developmental stages; Cbl and Erk1 show loss of expression only at E11.5 while Rarb is unaffected. P38α also shows down-regulation at both stages while Akt1 shows down regulation only at E12.5. All pairwise comparisons are between wild type and null embryonic guts at each stage.
The error bars represent standard errors of the mean (* P<0.01, ** P<0.001).
Table S1 (Related to Figure 1 and table 1): HSCR-associated polymorphisms within the RET locus within putative enhancers. 38 SNPs (minor allele frequency ≥ 10%) at the RET locus showing genome-wide significant association (P≤ 5×10−8) with HSCR are shown from a recently published GWAS study (reference 17 in main text). SNPs marked in color disrupt a known transcription factor-binding site in the human SK-N-SH cell line (ENCODE ChIP-seq), of which the ones demonstrating in vitro allelic difference in enhancer activity are highlighted in red. The genomic region spanning 154,678 bp is characterized by four blocks of linkage disequilibrium (Supplementary Figure 1) defined by the 38 SNPs in 503 European ancestry reference samples (reference 21 in main text) as marked in gray; blocks 2 and 3 are demarcated by a thick line. Note that two polymorphisms, #14 and 23, show weak associations with all 36 other markers.
Table S2 (related to table 1): RET polymorphisms associated with HSCR. Genetic features of the three polymorphisms rs2506030, rs7069590 and rs2435357 that are located within the enhancers RET−7, RET−5.5 and RET+3, 124.6 kb upstream, 19.6kb upstream and 9.5kb downstream from the RET transcription start site, respectively, are shown. Also shown are the observed frequencies of the risk allele among 346 cases and 732 controls, the odds ratio of disease, its 95% confidence interval (CI) and its statistical significance (P value) on testing whether the odds ratio differs from unity.
Table S3 (related to STAR Methods): Taqman gene expression probes for mouse and human transcripts.
Table S4 (related to STAR Methods): Primers for ChIP-qPCR and for mouse genotyping