

Abstract
Aim: Endoplasmic reticulum stress (ERS) and inflammation participate in cardiac fibrosis. Importantly, a novel paracrine/autocrine peptide intermedin1–53 (IMD1–53) in the heart inhibits myocardial fibrosis in rats. However, the mechanisms are yet to be fully elucidated.
Methods: Myocardial fibrosis in apolipoprotein E-deficient (ApoE -/-) mice and neonatal rat cardiac fibroblasts (CFs) were induced using homocysteine (Hcy).
Results: IMD1–53 inhibited myocardial fibrosis in vivo and in vitro. Picrosirius red staining showed that IMD1–53 reduced myocardial interstitial collagen deposition in ApoE-/- mice treated with Hcy and decreased the expression of myocardial collagen I and III, which was further verified in rat CFs. IMD1–53 attenuated myocardial hypertrophy, as shown by cardiomyocyte cross-sectional area, ratio of heart weight to body weight, and mRNA levels of atrial natriuretic peptide and brain natriuretic peptide. IMD1–53 inhibited the upregulation of ERS hallmarkers such as glucose-regulated protein 78 (GRP78), GRP94, activating transcription factor 6 (ATF6), ATF4, inositol-requiring enzyme 1α, spliced-X-box-binding protein-1, protein kinase receptor-like ER kinase, and eukaryotic translation initiation factor 2α in mouse myocardium and rat CFs treated with Hcy. In addition, IMD1–53 decreased the production of inflammatory factors such as tumor necrosis factor-α, monocyte chemotactic protein-1, interleukin-6 (IL-6), and IL-1β in the mouse myocardium and rat CFs treated with Hcy. Concurrently, IMD1–53 ameliorated the expression of nuclear factor-κB, transforming growth factor-β1, and c-Jun N-terminal kinase in the mouse myocardium and rat CFs treated with Hcy.
Conclusions: IMD potentially protects against myocardial fibrosis induced by Hcy in ApoE-/- mice, possibly via attenuating myocardial ERS and inflammation.
Keywords: Intermedin, Myocardial fibrosis, Endoplasmic reticulum stress, Inflammation, Homocysteine
Introduction
Epidemiological studies revealed that hypercholesterolemia or dyslipidemia was an independent determinant of increased left ventricular mass in patients1, 2). Hypercholesterolemia ApoE-/- mice showed age-dependent aortic stiffening3), cardiac hypertrophy3), and fibrosis4). Similar to the aging factor, homocysteine (Hcy) is another risk factor, which can lead to atherosclerotic vascular disease5, 6) and may accelerate the process of hypercholesterolemia-induced cardiac remodeling. Hcy is an amino acid-containing sulfydryl from methionine. The normal concentration of circulating Hcy is approximately 10 µmol/l, while the level of Hcy in hyperhomocysteinemia (HHcy) exceeds 15 µmol/l7). Plasma Hcy level was closely relative to left ventricular myocardial hypertrophy and left ventricle mass fraction8, 9). In addition, Hcy led to several heart morphological alterations including myocardial fibrosis and myocardial hypertrophy in vivo and in vitro7, 10–12).
Endoplasmic reticulum stress (ERS) is a subcellular process of ER homeostasis imbalance and function disorder13). Moderate ERS can enhance ER regulating ability and restore ER homeostasis13). However, prolonged and/or overwhelming severe ERS can induce cell apoptosis and organ damage14). Growing evidences from experimental and clinical research indicate that ERS is involved in several processes of cardiovascular diseases such as atherosclerosis, vascular calcification, myocardium ischemic-reperfusion injury, ischemic heart disease, cardiac fibrosis, and hypertrophy15–17). Inhibition of ERS with 4-phenyl butyric acid can attenuate myocardial fibrosis induced by isoprenaline or angiotensin II (Ang II)18, 19). Therefore, inhibiting ERS may provide cardiovascular protection.
Inflammation, a pathological response to various stimulators, is involved in myocardial fibrosis20, 21). Inflammatory cytokines have profibrotic effects by activating transforming growth factor-β1 (TGF-β1) signaling pathways in cardiac fibroblasts (CFs) and promoting CFs transforming into myofibroblasts22). Activated TGF-β1 can also upregulate collagen and fibronectin synthesis to promote extracellular matrix deposition23). Therefore, abrogation of inflammation might be a promising strategy for inhibiting myocardial fibrosis.
Circulating biologically active polypeptides and local autocrine/paracrine network imbalance in the heart are involved in the development of cardiovascular diseases24, 25). Intermedin (IMD) or adrenomedullin 2, a novel paracrine/autocrine biologically active polypeptide, is related to the calcitonin/calcitonin generelated peptide family. Human IMD gene is located on the distal arm of chromosome 22, and its prepropeptide is composed of 148 amino acids, named prepro-IMD. Prepro-IMD can yield IMD1–47 (prepro-IMD101—147), IMD8–47 (prepro-IMD108–147), and IMD1–53 (prepro-IMD93–145) by proteolytic cleavage and amidation26). IMD has the similar effects with adrenomedullin. Adrenomedullin receptor activity modifying protein (RAMP)2 system can maintain vascular integrity and homeostasis27). IMD can also maintain cardiovascular homeostasis via its calcitonin receptor-like receptor (CRLR)/RAMP complexes26). Our previous study showed that in CFs, endogenous IMD was significantly decreased in response to Ang II treatment, and administration of IMD1–53 suppressed Ang II-induced activation of CFs and hypertrophy of cardiomyocytes28, 29). In addition, IMD1–53 can protect against myocardial ischemia injury and vascular smooth muscle cell calcification by attenuating ERS30, 31). IMD can inhibit inflammation in diabetic and hyperlipidemia rats and rats with salt-sensitive hypertension32–34). However, whether IMD prevents myocardial fibrosis by inhibiting ERS and inflammation induced by Hcy in ApoE-/- mice has not been investigated. In the present study, we aimed to investigate the role and its possible mechanisms of IMD in myocardial fibrosis in ApoE-/- mice and rat CFs treated with Hcy.
Materials and Methods
Materials
All animal care and experimental protocols complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011) and were approved by the Animal Care Committee of Peking University Health Science Center. Male ApoE-/- mice (25 ± 1 g) were obtained from the Animal Center, Peking University Health Science Center (Beijing). Synthetic human IMD1–53 was from Phoenix Pharmaceuticals (Belmont, CA, USA). Antibodies for collagen I and III, glucose-regulated protein 78 (GRP78), GRP94, phosphorylated inositol requiring enzyme 1α (p-IRE1α) and IRE1α, activating transcription factor 6 (ATF6), ATF4, spliced-X-box-binding protein-1 (s-XBP-1), nuclear factor-κB (NF-κB) p65, and TGF-β1 were from Abcam PLC (Cambridge, UK). Antibodies for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), α-smooth muscle actin (α-SMA), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1α, monocyte chemotactic protein-1 (MCP-1), and all secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for phosphorylated protein kinase receptor-like ER kinase (p-PERK) and PERK, phosphorylated eukaryotic translation initiation factor 2α (p-eIF2α) and eIF2α, and phosphorylated c-Jun N-terminal kinase (p-JNK) and JNK were from Cell Signaling Technology (Danvers, MA, USA). Nitrocellulose membrane was from Hybond-C (Amersham Life Science, Buckinghamshire, UK), and the enhanced chemiluminescence (ECL) kit and Trizol reagent were from Applygen Technologies (Beijing). Deoxy ribonucleoside triphosphate was from Clontech (Palo Alto, CA, USA). Moloney murine leukemia virus (MMLV) reverse transcriptase, Taq DNA polymerase, Rnasin, and oligo (dT) were from Promega (Madison, WI, USA). Eva Green was from Biotium (Hayward, CA, USA). The sequences of oligonucleotide primers for real-time PCR amplification (Table 1) were synthesized by Beijing AuGCT DNA-SYN Biotechnology. Hcy and Hoechst 33342 were from Sigma (St. Louis, MO, USA). Other chemicals and reagents were of analytical grade.
Table 1. Primer sequences for real-time PCR.
| Gene | Upstream primer (5′-3′) | Downstream primer (5′-3′) | AnnT (°C) |
|---|---|---|---|
| ANP | CAGAGAGTGAGCCGAGACAG | AGCCCTTGGTGATGGAGAAG | 58 |
| BNP | TCTGGGACCACCTTTGAAGT | TGTTGTGGCAAGTTTGTGCT | 58 |
| Collagen I | ATCCTGCCGATGTCGCTAT | CCACAAGCGTGCTGTAGGT | 60 |
| Collagen III | CATGACTGTCCCACGTAAGCA | ATTCGCCTTCATTTGATCCCA | 58 |
| TNF-α | CGTCGTAGCAAACCACCAAG | GAGATAGCAAATCGGCTGACG | 60 |
| IL-6 | GCCTTCTTGGGACTGATGCT | TGCCATTGCACAACTCTTTTC | 60 |
| MCP-1 | AGGGACTGAGGCACTCCAGA | TGACGACGAGACTTCCAGACTACA | 58 |
| IL-1β | CTCACAAGCAGAGCACAAGC | TCCAGCCCATACTTTAGGAAGA | 58 |
| GAPDH | AGGGACTGAGGCACTCCAGA | TGACGACGAGACTTCCAGACTACA | 58 |
AnnT, Annealing temperature; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; TNF-α, tumor necrosis factor a; IL-6, interleukin 6; MCP-1, monocyte chemotactic protein 1; IL-1β, interleukin 1β.
Myocardial Fibrosis Model in ApoE-/- Mice
As described previously35, 36) with minor modification, 8-week-old male ApoE-/- mice (25 ± 1 g) were randomly divided into three groups: (1) control (n = 15), normal drinking water, or (2) Hcy (n = 18), Hcy (1.8 g/L dissolved in drinking water) for 6 weeks from the 18th week, or (3) Hcy plus IMD (n = 18), IMD1–53 (300 ng/kg/h dissolved in sterile saline) infused subcutaneously via Alzet mini-osmotic pumps (2004 and 2002, Cupertino, CA, USA) for 6 weeks at the same time as Hcy treatment as described earlier. All the ApoE-/- mice were given high fat diet (21% lard and 0.15% cholesterol) for 16 weeks from the eighth week. At the end of the experiment, all animals were killed by exsanguination, and hearts were quickly collected for further analysis.
Preparation of Primary Neonatal CFs and Hcy Treatment
Neonatal rat CFs were isolated from 1- to 2-dayold SD rats. Briefly, after being washed in Hank's balanced salt solution (HBSS), the rat myocardium was cut into pieces and digested in HBSS including trypsin (0.05%) and collagenase (0.055%). The supernatant was collected and added to high-glucose Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS). After centrifugation for 10 min at 1,000 rpm, the cell suspension was filtered with a sterile stainless steel cell cribble and plated at 37°C for 90 min to allow CFs to attach to culture dishes. Then the medium, which mostly contained cardiomyocytes, was decanted, and the purified CFs were cultured in fresh DMEM containing 10% FBS. Before reagent treatment, cells were starved with serum-free medium for 24 h. After incubation with IMD1–53 (10−7 mol/L) for 30 min, CFs were stimulated with Hcy (2 × 10−4 mol/L) for 24 h as described12).
Western Blot Analysis
Mouse whole heart or rat CFs were homogenized in lysis buffer. Equal amounts of protein samples were loaded and separated on 10% SDS-PAGE and then transferred to nitrocellulose membranes for 3 h at 4°C and 200 mA. After incubation in 5% nonfat milk for 1 h, membranes were incubated with the following primary antibodies: anti-GAPDH (1:2,000), anti-GRP78 and anti-GRP94 (both 1:3,000), anti-ATF6 (1:1,000), anti-ATF4 (1:3,000), anti-collagen I and III (both 1:4,000), anti-p-IRE1α and anti-IRE1α (both 1:500), anti-s-XBP-1 and anti-NF-κB (both 1:1,000), anti-TGF-β1 (1:500), anti-α-SMA (1:4,000), anti-TNF-α (1:500), anti-IL-6 and anti-IL-1β (both 1:500), anti-MCP-1 (1:500), anti-p-PERK and anti-PERK (both 1:500), anti-p-eIF2α (1:500), anti-eIF2α (1:1,000), and anti-p-JNK and anti-JNK (both 1:500) overnight at 4°C. After three washes for 5 min each in TBST (20 mmol/L Tris – HCl (pH 7.6), 150 mmol/L NaCl, and 0.1%Tween 20), membranes were incubated with secondary antibody (horseradish peroxidase-conjugated anti-mouse, anti-goat, or anti-rabbit IgG) for 1 h at room temperature (RT). The reaction was visualized by ECL. Protein levels were analyzed by use of NIH Image and normalized to that of GAPDH. All experiments were repeated at least three times.
Quantitative Real-Time PCR Analysis
Trizol reagent was used to extract total RNA from heart tissue. An amount of 2.0 µg RNA was reverse transcribed into cDNA with MMLV and oligo (dT) primer. The real-time PCR (7500 Fast Real-Time PCR System, Applied Biosystems, USA) was used to amplify cDNA. The amount of PCR product formed in each cycle was evaluated by Eva Green fluorescence. Relative quantification involved the 2−ΔΔCt method, with GAPDH as a reference. The primers for real-time PCR are in Table 1.
Immunofluorescence Assay of CFs
After a rinse with phosphate-buffered solution (PBS) three times, CFs were fixed with 4% paraformaldehyde at RT for 15 min, permeabilized with 0.1% TritonX-100 at RT for 10 min, sealed with 3% bovine serum albumin (BSA)/PBS at RT for 10 min, incubated with antibody α-SMA (1:100, diluted in 0.5% BSA/PBS) at 37°C for 1 h, then anti-rabbit IgG (1:500, diluted in 0.5% BSA/PBS) at 37°C for 1 h in the dark, then Hoechst 33342 (10 µg/ml, diluted in 0.5% BSA/PBS) at RT for 5 min in the dark, mounted with 50% glycerin, and observed under an immunofluorescence microscope.
Hematoxylin and Eosin and Picrosirius Red Staining
Mouse whole hearts were excised and fixed in 4% paraformaldehyde; embedded in paraffin; cut into 5-µm-thick sections; stained with hematoxylin for 15 min and with eosin for 3 min; underwent ethanol dehydration, xylene transparency, and neutral gum mounting; and observed under a microscope. Other slices were placed into 0.1% picrate Sirius-red dye solution for 1 min; then underwent ethanol dehydration, xylene transparency, and neutral gum mounting; and observed under a microscope.
Statistical Analysis
Graphpad software (GraphPad Prism v5.00 for Windows; GraphPad Software Inc., San Diego, CA, USA) was used for analyzing data, which were expressed as mean ± SD. Comparisons across two groups were analyzed by Student's t test. Comparisons among more than two groups were analyzed by oneway analysis of variance followed by Student–Newman–Keuls test. A P < 0.05 was considered significant.
Results
IMD1–53 Inhibited Myocardial Fibrosis In Vivo and In Vitro
Our previous in vitro experiments found that IMD inhibited CFs transforming into myofibroblasts induced by Ang II28). In the present study, we investigated whether IMD1–53 could inhibit myocardial fibrosis and hypertrophy in ApoE-/- mice and rat CFs with Hcy treatment. In vivo, picrosirius red staining showed that IMD1–53 treatment reduced collagen deposition in myocardial interstitial areas with Hcy treatment (Fig. 1a). Administration of IMD1–53 markedly reduced the mRNA levels of collagen I and III by 53.9% and 57.6% (both P < 0.05) and the protein levels by 43.8% (P < 0.01) and 54.4% (P < 0.05; Fig. 1b and c), respectively, compared with Hcy alone. In vitro, IMD1–53 pretreatment decreased the protein levels of collagen I, collagen III, and α-SMA by 54.1% (P < 0.05), 39.2%, and 25.5% (both P < 0.01; Fig. 1d), respectively, compared with Hcy alone. Moreover, immunofluorescence revealed that Hcy-treated CFs showed upregulated α-SMA expression, which was reduced with IMD1–53 treatment (Fig. 1e). Therefore, IMD1–53 directly suppressed CFs differentiating into myofibroblasts and myocardial fibrosis induced by Hcy.
Fig. 1.
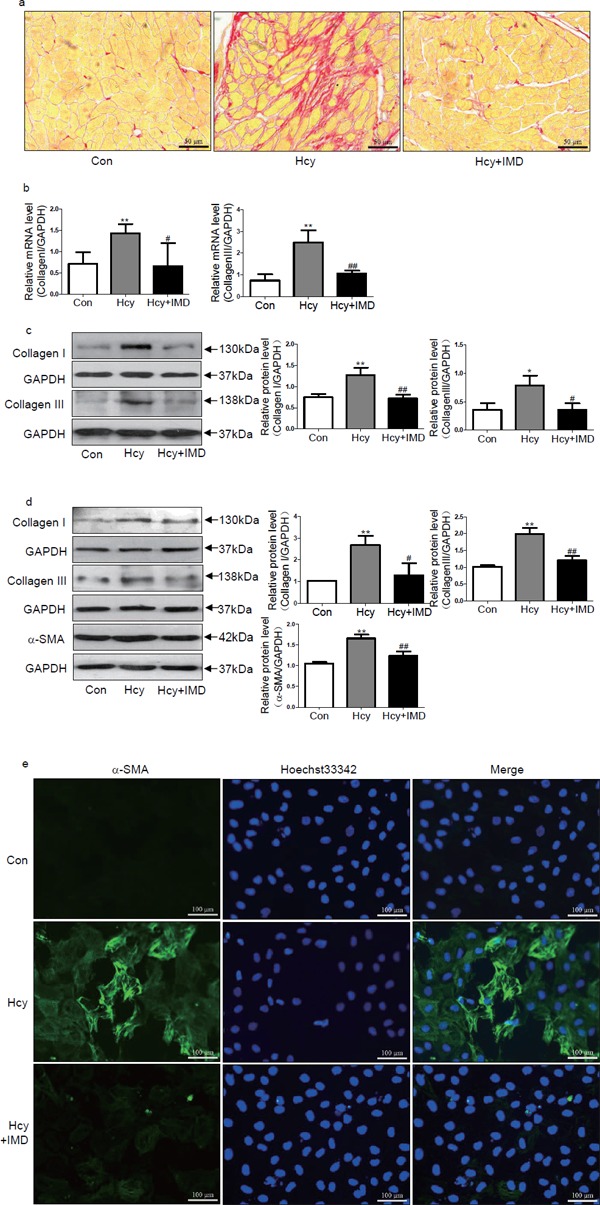
Intermedin1–53 (IMD1–53) inhibited myocardial fibrosis induced by homocysteine (Hcy) in vivo and in vitro. Picrosirius red staining (a) of myocardial interstitial collagen deposition. Bar, 50 µm. Quantitative real-time PCR analysis of mRNA levels (b) of collagen I and III in the myocardium of apolipoprotein E-deficient (ApoE-/-) mice in vivo. Results are relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Western blot analysis of protein levels (c) of collagen I and III in the myocardium of ApoE-/- mice in vivo. Western blot analysis of protein levels (d) of collagen I and III and α-smooth muscle actin (α-SMA) in rat cardiac fibroblasts (CFs) in vitro. GAPDH was a loading control. Data are mean ± SD, n = 3 in each group; *P < 0.05 and ** P < 0.01 versus Con. #P < 0.05 and ##P < 0.01 versus Hcy. (e) Immunofluorescence analysis of myofibroblasts differentiated from CFs with α-SMA antibody and Hoechst 33342 staining in Con, Hcy, Hcy + IMD groups. Bar, 100 µm.
H&E staining in mouse hearts in vivo revealed that IM1–53 attenuated the cross-sectional area of cardiomyocytes induced by Hcy (Fig. 2a). In addition, IMD1–53 infusion significantly reduced atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) mRNA expression and ratio of heart weight to body weight (HW/BW) by 32.4%, 50.2%, and 37.6% (all P < 0.01; Fig. 2b–d), respectively, with Hcy treatment. However, the functional parameters of apoE-/- mice such as systolic blood pressure and diastolic blood pressure have no significant difference among control, Hcy, and IMD1–53 treatment groups (Table 2).
Fig. 2.

Intermedin1–53 inhibited myocardial hypertrophy induced by homocysteine (Hcy) in vivo in ApoE-/- mice. H&E staining (a) in mouse myocardium. Bar, 50 µm. Quantitative real-time PCR analysis of mRNA levels of atrial natriuretic peptide (b) and brain natriuretic peptide (c) in mouse myocardium. Results are relative to glyceraldehyde-3-phosphate dehydrogenase. (d) Ratio of heart weight to body weight. Data are mean ± SD, at least n = 4 in each group; **P < 0.01 versus Con. ## P < 0.01 versus Hcy.
Table 2. The functional parameters of apolipoprotein E-deficient mice with control, homocysteine and intermedin1–53 treatment.
| Control | Hcy | Hcy + IMD | |
|---|---|---|---|
| HW/BW (mg/g) | 4.45 ± 0.23 | 6.59 ± 1.12** | 4.11 ± 0.68## |
| SBP (mmHg) | 112.33 ± 10.30 | 117.89 ± 6.49 | 117.78 ± 10.60 |
| DBP (mmHg) | 87.33 ± 10.04 | 84.33 ± 8.76 | 86.56 ± 10.65 |
| MBP (mmHg) | 95.67 ± 9.27 | 95.52 ± 5.97 | 96.96 ± 8.38 |
Data are shown as mean ± SD, n = 3 at least in each group.
P < 0.01 versus control,
P < 0.01 versus Hcy, respectively. HW/BW, heartweight/body weight; HR, heartrate; SBP, systolic blood pressure; DBP, diastolic blood pressure; MBP, mean blood pressure.
IMD1–53 Inhibited Myocardial ERS Induced by Hcy In Vivo and In Vitro
ERS is involved in cardiac hypertrophy and fibrosis13, 15). Our previous studies indicated that IMD1–53 can inhibit ERS and reduce myocardial ischemia injury and vascular calcification30, 31). Here we investigated whether IMD1–53 could reduce ERS in the ApoE-/- mice myocardium and in CFs induced by Hcy. In vivo, IMD1–53 treatment decreased the protein expression of the ERS markers GRP78, GRP94, ATF6, p-IRE1α, s-XBP-1, p-PERK, p-eIF2α, and ATF4 by 22.8%, 42.9%, 29.0%, 46.2%, 21.8%, 39.6% (all P < 0.05), 26.0%, and 34.5% (both P < 0.01; Fig. 3a and b), respectively, compared with Hcy alone. In vitro, IMD1–53 pretreatment significantly attenuated Hcy-upregulated GRP94, GRP78, p-PERK, p-IRE1α, s-XBP-1, ATF4, ATF6, and p-eIF2α protein expression in CFs by 26.1%, 36.0%, 32.4%, 42.7%, 45.7% (all P < 0.05), 34.7%, 42.0%, and 31.7% (all P < 0.01; Fig. 3c and d), respectively, compared with Hcy alone. Therefore, IMD can inhibit ERS in the ApoE-/- mouse myocardium and CFs induced by Hcy.
Fig. 3.
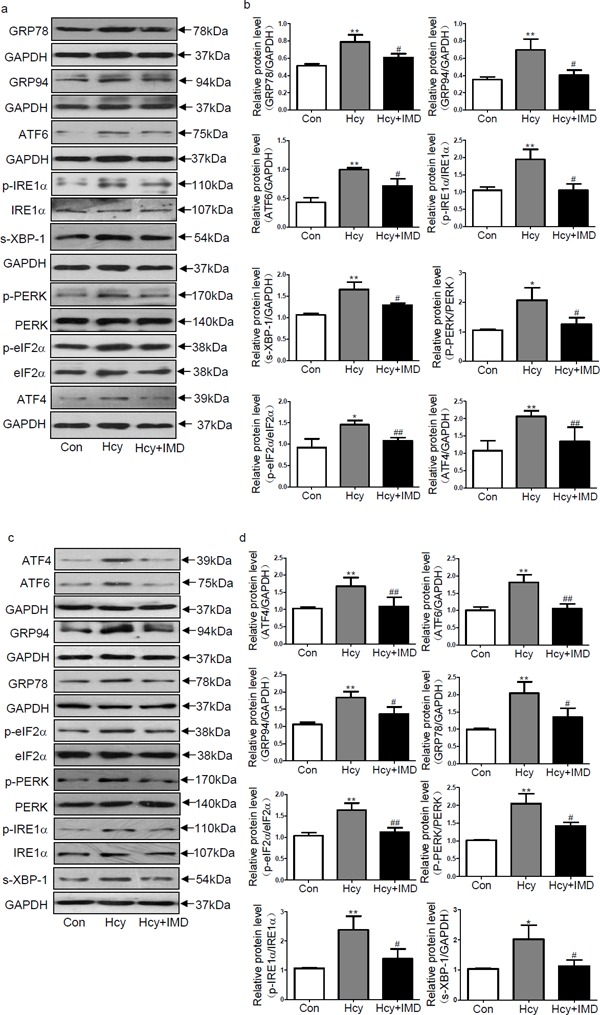
Intermedin1–53 attenuated endoplasmic reticulum stress (ERS) induced by homocysteine (Hcy) in vivo and in vitro. Western blot analysis of protein levels (a, b) of ERS hallmarkers such as glucose-regulated protein 78 (GRP78) and GRP94, activating transcription factor 6 (ATF6), phosphorylated inositol-requiring enzyme 1α (p-IRE1α), spliced-Xbox-binding protein-1 (s-XBP-1), phosphorylated protein kinase receptor-like ER kinase (p-PERK), phosphorylated eukaryotic translation initiation factor 2α (p-eIF2α) and ATF4 in the myocardium of ApoE-/- mice in vivo. Western blot analysis of protein levels (c, d) of ERS hallmarkers such as ATF6, ATF4, GRP94, GRP78, p-eIF2α, p-PERK, p-IRE1α, and s-XBP-1 in rat cardiac fibroblasts in vitro. Glyceraldehyde-3-phosphate dehydrogenase was a loading control. Data are mean ± SD, n = 3 in each group; *P < 0.05 and **P < 0.01 versus Con. #P < 0.05 and ##P < 0.01 versus Hcy.
IMD1–53 Inhibited Inflammation Induced by Hcy In Vivo and In Vitro
Compelling evidence affirmed that inflammation contributes to myocardial fibrosis and hypertrophy20, 21). Here, we investigated whether IMD1–53 could reduce inflammation in ApoE-/- mice and rat CFs induced by Hcy. In myocardium of ApoE-/- mice, administration of IMD1–53 reduced the mRNA levels of inflammation hallmarkers such as IL-6, TNF-α, MCP-1, and IL-1β in the mouse myocardium by 52.2% (P < 0.05), 56.0%, 79.1%, and 30.4% (all P < 0.01) (Fig. 4a), respectively, compared with Hcy alone. Meanwhile, IMD1–53 administration attenuated the protein levels of TNF-α, IL-6, MCP-1, IL-1β, NF-κB p65, p-JNK, and TGF-β1 by 44.0%, 37.1%, 42.1%, 39.8%, 33.6% (all P < 0.01), 28.1%, and 28.6% (both P < 0.05; Fig. 4b and c), respectively, compared with Hcy alone. In vitro, IMD1–53 treatment decreased TNF-α, MCP-1, IL-1β, NF-κB p65, p-JNK, TGF-β1, and IL-6 protein expression by 35.2%, 44.2%, 39.0%, 34.3%, 27.6%, 33.5% (all P < 0.05), and 47.3% (P < 0.01) (Fig. 4d and e), respectively, in CFs treated with Hcy compared with Hcy alone.
Fig. 4.
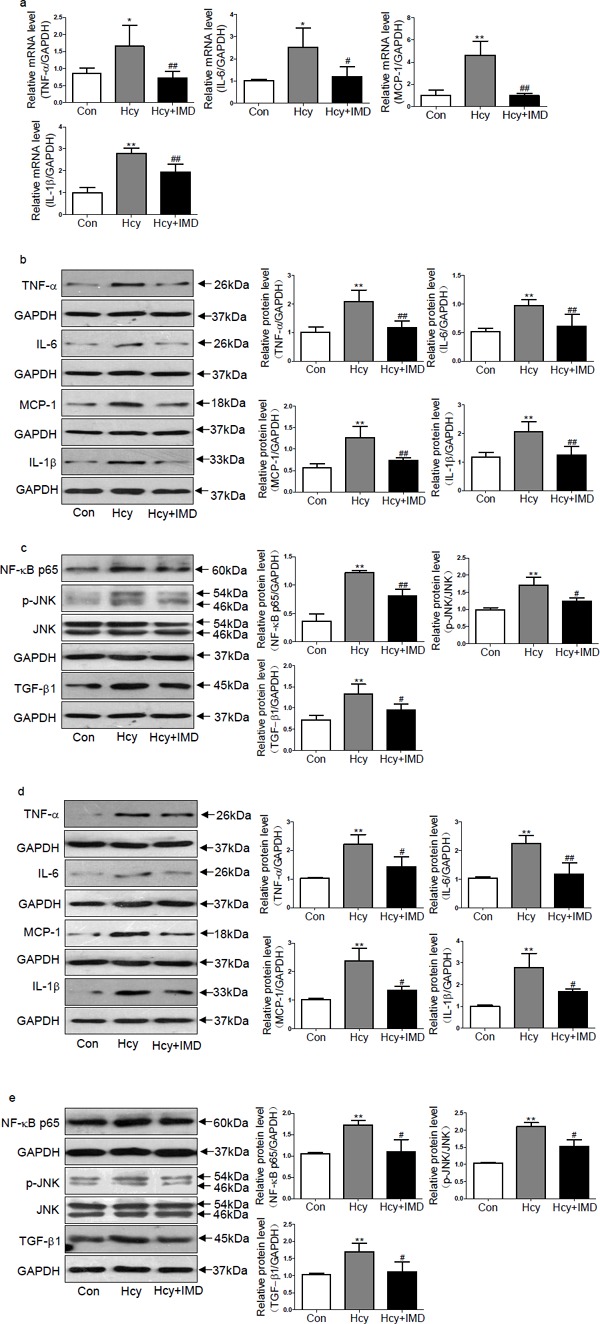
Intermedin1–53 attenuated homocysteine (Hcy)-induced inflammation in vivo and in vitro. Quantitative real-time PCR analysis of mRNA levels (a) of inflammation hallmarkers such as tumor necrosis factor-α (TNF-α), monocyte chemotactic protein-1 (MCP-1), interleukin-6 (IL-6) and IL-1β in the myocardium of ApoE-/- mice in vivo. Western blot analysis of protein levels (b, c) of TNF-α, IL-6, MCP-1, IL-1β, nuclear factor-κB (NF-κB) p65, phosphorylated c-Jun N-terminal kinase (p-JNK), and transforming growth factor β-1 (TGF-β1) in the myocardium of ApoE-/- mice in vivo. Western blot analysis of protein levels (d, e) of TNF-α, IL-6, MCP-1, IL-1β, NF-κB p65, p-JNK, and TGF-β1 in rat cardiac fibroblasts in vitro. Glyceraldehyde-3-phosphate dehydrogenase was a loading control. Data are mean ± SD, n = 3 in each group; *P < 0.05 and **P < 0.01 versus Con. #P < 0.05 and ## P < 0.01 versus Hcy.
Changes of Receptor System and Post-Receptor Pathways of IMD in Myocardial Fibrosis Induced by Hcy in ApoE-/- Mice
In this study, we first examined expression of IMD receptor system. It was found that Hcy increased protein expression of CRLR, RAMP2, and RAMP3 in apoE-/- mice myocardium by 120.2%, 92.2%, and 85.0% (all P < 0.05), respectively, compared with control group (Fig. 5a and b). However, protein levels of RAMP1 between control group and Hcy group revealed no significant differences (Fig. 5a and b). IMD1–53 supplementation increased the ratio of phosphorylated/total protein kinase A (PKA) and phosphorylated/total extracellular regulated protein kinase (ERK)1/2 in apoE-/- mice myocardium by 61.0% (P < 0.01) and 35.8% (P < 0.05) (Fig. 5c and d), respectively, compared with Hcy alone. However, the ratio of phosphorylated/total Akt between Hcy and Hcy + IMD group showed no significant differences (Fig. 5c and d).
Fig. 5.
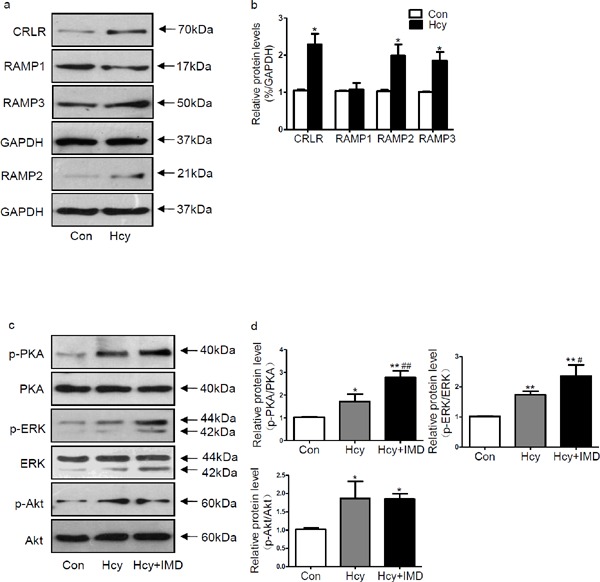
Western blot analysis of protein levels (a, b) of calcitonin receptor-like receptor, receptor activity modifying protein (RAMP)1, RAMP2, and RAMP3 in apoE-/- mice myocardium treated with homocysteine (Hcy). Glyceraldehyde-3-phosphate dehydrogenase was a loading control. Western blot analysis of protein levels (c, d) of phosphorylated Akt, total Akt, phosphorylated protein kinase A (PKA), total PKA, phosphorylated extracellular regulated protein kinase (ERK)1/2, and total ERK1/2 in apoE-/- mice myocardium. Data are mean ± SD, n = 3 in each group; *P < 0.05 and **P < 0.01 versus Con. #P < 0.05 and ##P < 0.01 versus Hcy.
Discussion
The major finding of the present study is that IMD protects against myocardial fibrosis by inhibiting myocardial ERS and inflammation induced by Hcy. IMD1–53 significantly attenuated myocardial interstitial collagen deposition induced by Hcy in ApoE-/- mice. IMD1–53 obviously inhibited Hcy-induced myocardial hypertrophy in vivo, as indicated by cardiomyocyte cross-sectional area, HW/BW ratio, and ANP and BNP levels. IMD1–53 inhibited the Hcy-induced upregulation of ERS markers such as GRP78, GRP94, ATF6, ATF4, and s-XBP-1 and ameliorated the Hcy-induced phosphorylation of IRE1α, PERK, and eIF2α in vivo and in vitro. Also, it decreased the Hcy-induced expression of inflammation markers such as TNF-α, MCP-1, IL-6, and IL-1β in vivo and in vitro. IMD1–53 also inhibited Hcy-activated NF-κB, TGF-β1, and JNK in vivo and in vitro.
Previous studies showed that hypercholesterolemia ApoE-/- mice exhibited age-dependent cardiac fibrosis4), hypertrophy3), and aortic stiffening3). Similar to the aging factor, Hcy can lead to atherosclerotic vascular disease5) and may accelerate the process of hypercholesterolemia-induced cardiac remodeling. In this study, we established cardiac fibrosis model in ApoE-/- mice with Hcy treatment for 6 weeks from the 18th week, which has the similar pathogenesis of cardiac fibrosis occurring in human hypercholesterolemia with HHcy. We found that ApoE-/- mice fed a high fat diet for 24 weeks without Hcy showed small amounts of collagen deposition and hypertrophic effects, which were consistent with previous report37). Moreover, Hcy-treated mice showed significantly increased collagen deposition and expression of fibrotic biomakers such as collagen I and II in myocardium, which indicates a fibrotic process. Meanwhile, Hcy had pro-hypertrophic effects, as indicated by cardiomyocyte cross-sectional area, HW/BW ratio, and ANP and BNP levels.
Previous studies have shown that biologically active polypeptides play vitol roles in promoting or resisting the development of cardiovascular diseases24, 25). The level of endogenous IMD, a novel biologically active protective polypeptide, was significantly decreased in response to Ang II stimuli28). In addition, exogenous IMD1–53 suppressed an Ang II-induced fibrotic response in CFs and Ang II-induced hypertrophic response in cardiomyocytes28, 29). Here, we discovered that Hcy increased protein expression of CRLR, RAMP2, and RAMP3 in apoE-/- mice myocardium, which were consistent with those reported by Nishikimi and Yang28, 38). In addition, compared with Hcy alone, IMD1–53 supplementation further activated cAMP/PKA and ERK1/2 post-receptor pathways, which were similar to those reported previously28, 39, 40). IMD1–53 also inhibited Hcy-induced upregulation of myocardial fibrotic biomakers such as collagen I and III and myocardial interstitial collagen deposition in ApoE-/- mice. It attenuated cardiomyocyte cross-sectional area, HW/BW, and mRNA levels of ANP and BNP in the myocardium of ApoE-/- mice treated with Hcy and directly attenuated collagen I and III synthesis in CFs and inhibited CFs transforming into myofibroblasts with Hcy treatment. Therefore, IMD1–53 conferred significant cardioprotection against myocardial fibrosis with Hcy treatment.
The ER, the factory of eukaryotic cells, has a variety of biological effects, such as regulating the synthesis and secretion of protein, lipids, cholesterol, glycans, and calcium homeostasis41–43). Many cellular stressors such as Hcy, viruses, metabolic factors, and hypoxia can injure fibroblasts44, 45), resulting in ERS. To maintain homeostasis, a cascade of pathways called adaptive unfolded protein response (UPR) was activated. Chaperone proteins such as GRP78 and GRP94 dissociated from ER transmembrane sensors, such as PERK, ATF6, and IRE1α, and combined with a lot of unfolded or misfolded proteins to promote the correct protein folding. Meanwhile, PERK, ATF6, and IRE1α were also initiated with chaperone protein dissociation to decrease the influx of nascent proteins into the ER, increase ER-associated protein degradation, and enhance the ER capacity to fold incoming proteins16). In the event of excessive and prolonged ERS, terminal UPR was activated, which can lead to apoptosis, inflammation, fibroblasts activation, and ultimately fibrosis development15, 16). Here we found that IMD1–53 inhibited Hcy-activated myocardial ERS in ApoE-/- mice, as shown by levels of ERS markers such as GRP78, GRP94, ATF6, s-XBP-1, ATF4, IRE1α, PERK, and eIF2α, which was further confirmed in CFs, which were in accordance with our previous study31). These data indicated that IMD1–53 may protect against myocardial fibrosis by attenuating ERS. However, further studies are required to determine which post-receptor signal transduction pathway mediates the anti-ERS effects of IMD.
Recent studies have confirmed that inflammation is another significant determinant of fibrotic remodeling20, 21, 46). Multiple risk factors such as Hcy can trigger infiltration of inflammatory cells and secretion of cytokines and chemokines, which can initiate TGF-β1 pathways23, 44). Activated TGF-β1 can induce myofibroblast formation and increase extracellular matrix deposition by upregulating collagen synthesis and inhibiting matrix degradation, which leads to myocardial fibrosis23). In addition, NF-κB and JNK pathways can be activated by Hcy7, 44). Here, we found that IMD1–53 inhibited the Hcy-increased levels of inflammatory molecules such as TNF-α, MCP-1, IL-6, and IL-1β in the ApoE-/- mouse myocardium treated with Hcy, which was further confirmed in CFs. Moreover, IMD1–53 treatment reversed Hcy-activated TGF-β1 expression in the ApoE-/- mouse myocardium and in CFs. These anti-inflammatory effects of IMD were identical to the other researches for diabetes, hyperlipidemia, and hypertension32, 34, 35). It was reported that NF-κB and JNK signal pathway played significant roles in proinflammatory process. Activated NF-κB can translocate into the nucleus to promote inflammatory gene expression and collagen synthesis43, 47). Phosphorylated JNK can activate activator protein 1, which can migrate to the nucleus to enhance TGF-β and collagen expression in CFs48). Here, results showed that IMD1–53 treatment reversed Hcy-activated NF-κB and JNK in the ApoE-/- mouse myocardium and in CFs. It has been confirmed that elevation of intracellular second messenger cAMP by adrenomedullin inhibited transcription factor NF-κB activation and reduce inflammatory adhesion molecule expression in lymphatic endothelial cells49, 50). The intracellular cAMP/PKA pathway involved in inhibiting the phosphorylation and subsequent degradation of inhibitory κB (IκB), leading to decrease of NF-κB nuclear translocation51, 52). Furthermore, in CFs, cAMP/PKA signaling activation by estrogen blocked myofibroblast formation and JNK activation by TGF-β153, 54). Here, results showed that IMD1–53 supplementation increased the phosphorylation of PKA and suppressed NF-κB and JNK activation compared with Hcy alone. Therefore, cAMP/PKA post-receptor pathway activation probably was involved in IMD suppressing NF-κB and JNK activation as well. Other report confirmed that ERK1/2 may induce phosphatases that effectively inhibit JNK activation via its dephosphorylation in hepatocyte55). Here, IMD1–53 supplementation increased ERK1/2 phosphorylation and decreased JNK phosphorylation compared with Hcy alone. Hence, ERK1/2 post-receptor pathways also probably involved in IMD suppressing JNK activation, which needed further investigation. All the above results suggest that IMD1–53 may protect against myocardial fibrosis via an antiinflammatory effect. However, the precise relationship between IMD1–53 and inflammation needs further investigation.
In summary, we found that myocardial paracrine/autocrine factor IMD protected against myocardial fibrosis via inhibiting ERS and inflammation in ApoE-/- mice treated with Hcy. The cardiac-protective peptide IMD may be a novel pharmacologic target to treat myocardial fibrosis occurring in hypercholesterolemia with HHcy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81170082, 91339203, 81670434 and 81270407 to Y.F. Qi).
Conflicts of Interest
None.
References
- 1). Lee TM, Chou TF, Tsai CH: Association of pravastatin and left ventricular mass in hypercholesterolemic patients: role of 8-isoprostaglandin f2alpha formation. J Cardiovasc Pharmacol, 2002; 40: 868-874 [DOI] [PubMed] [Google Scholar]
- 2). Sundström J, Lind L, Vessby B, Andrén B, Aro A, Lithell H: Dyslipidemia and an unfavorable fatty acid profile predict left ventricular hypertrophy 20 years later. Circulation, 2001; 103: 836-841 [DOI] [PubMed] [Google Scholar]
- 3). Wang YX: Cardiovascular functional phenotypes and pharmacological responses in apolipoprotein E deficient mice. Neurobiol Aging, 2005; 26: 309-316 [DOI] [PubMed] [Google Scholar]
- 4). Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R: ApoE deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb, 1994; 14: 133-140 [DOI] [PubMed] [Google Scholar]
- 5). Shenov V, Mehendale V, Prabhu K, Shetty R, Rao P: Correlation of serum homocysteine levels with the severity of coronary artery disease. Ind J Clin Biochem, 2014; 29: 339-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Kumakura H, Fujita K, Kanai H, Araki Y, Hojo Y, Kasama S, Iwasaki T, Ichikawa S, Nakashima K, Minami K: High-sensitivity C-reactive Protein, Lipoprotein (a) and Homocysteine are Risk Factors for Coronary Artery Disease in Japanese Patients with Peripheral Arterial Disease. J Atheroscler Thromb, 2015; 22: 344-354 [DOI] [PubMed] [Google Scholar]
- 7). Raaf L, Noll C, Cherifi Mel H, Samuel JL, Delcayre C, Delabar JM, Benazzoug Y, Janel N: Myocardial fibrosis and TGFB expression in hyperhomocysteinemic rats. Mol Cell Biochem, 2011; 347: 63-70 [DOI] [PubMed] [Google Scholar]
- 8). Peer M, Boaz M, Zipora M, Shargorodsky M: Determinants of left ventricular hypertrophy in hypertensive patients: identification of high-risk patients by metabolic, vascular, and inflammatory risk factors. Int J Angiol, 2013; 22: 223-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Kharlamova UV, Il'icheva OE: Effect of homocysteine on left ventricular structural and functional parameters in patients on programmed hemodialysis. Ter Arkh, 2013; 85: 90-93 [PubMed] [Google Scholar]
- 10). Pal Singh A, Kaur T, SinghDahiya R, Singh N, Singh Bedi PM: Ameliorative role of rosiglitazone in hyperhomocysteinemia-induced experimental cardiac hypertrophy. J Cardiovasc Pharmacol, 2010; 56: 53-59 [DOI] [PubMed] [Google Scholar]
- 11). Kassab S, Garadah T, Abu-Hijleh M, Golbahar J, Senok S, Wazir J, Gumaa K: The angiotensin type 1 receptor antagonist valsartan attenuates pathological ventricular hypertrophy induced by hyperhomocysteinemia in rats. J Renin Angiotensin Aldosterone Syst, 2006; 7: 206-211 [DOI] [PubMed] [Google Scholar]
- 12). Zhi H, Luptak I, Alreja G, Shi J, Guan J, Metes-Kosik N, Joseph J: Effects of direct Renin inhibition on myocardial fibrosis and cardiac fibroblast function. PLoS One, 2013; 8: e81612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Lenna S, Trojanowska M: The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr Opin Rheumatol, 2012; 24: 663-668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14). Szegezdi E, Logue SE, Gorman AM, Samali A: Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep, 2006; 7: 880-885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Tanjore H, Lawson WE, Blackwell TS: Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta, 2013; 1832: 940-947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Yoshida H: ER stress and diseases. FEBS J, 2007; 274: 630-658 [DOI] [PubMed] [Google Scholar]
- 17). Duan X, Zhou Y, Teng X, Tang C, Qi Y: Endoplasmic reticulum stress mediated apoptosis is activated in vascular calcification. Biochem Biophys Res Commun, 2009; 387: 694-699 [DOI] [PubMed] [Google Scholar]
- 18). Ayala P, Montenegro J, Vivar R, Letelier A, Urroz PA, Copaja M, Pivet D, Humeres C, Troncoso R, Vicencio JM, Lavandero S, Díaz-Araya G: Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp Mol Pathol, 2012; 92: 97-104 [DOI] [PubMed] [Google Scholar]
- 19). Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K: Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol, 2012; 32: 1652-1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Dobaczewski M, Frangogiannis NG: Chemokines and cardiac fibrosis. Front Biosci (Schol Ed), 2009; 1: 391-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Jia L, Li Y, Xiao C, Du J: Angiotensin II induces inflammation leading to cardiac remodeling. Front Biosci (Landmark Ed), 2012; 17: 221-231 [DOI] [PubMed] [Google Scholar]
- 22). Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, Qi Y, Du J: Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One, 2012; 7: e35144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Bujak M, Frangogiannis NG: The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res, 2007; 74: 184-195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Doroudgar S, Glembotski CC: The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol Med, 2011; 17: 207-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Miyahara Y, Ishibashi-Ueda H, Aizawa Y, Kangawa K, Nagaya N: Infusion of adrenomedullin improves acute myocarditis via attenuation of myocardial inflammation and edema. Cardiovasc Res, 2007; 76: 110-118 [DOI] [PubMed] [Google Scholar]
- 26). Ni X, Zhang J, Tang C, Qi Y: Intermedin/adrenomedullin2: an autocrine/paracrine factor in vascular homeostasis and disease. Sci China Life Sci, 2014; 57: 781-789 [DOI] [PubMed] [Google Scholar]
- 27). Koyama T, Sakurai T, Kamiyoshi A, Ichikawa-Shindo Y, Kawate H, Shindo T: Adrenomedullin-RAMP2 System in Vascular Endothelial Cells. J Atheroscler Thromb, 2015; 22: 647-653 [DOI] [PubMed] [Google Scholar]
- 28). Yang JH, Cai Y, Duan XH, Ma CG, Wang X, Tang CS, Qi YF: Intermedin1–53 inhibits rat cardiac fibroblast activation induced by angiotensin II. Regul Pept, 2009; 158: 19-25 [DOI] [PubMed] [Google Scholar]
- 29). Yang JH, Ma CG, Cai Y, Pan CS, Zhao J, Tang CS, Qi YF: Effect of intermedin1–53 on angiotensin II-induced hypertrophy in neonatal rat ventricular myocytes. J Cardiovasc Pharmacol, 2010; 56: 45-52 [DOI] [PubMed] [Google Scholar]
- 30). Chang JR, Duan XH, Zhang BH, Teng X, Zhou YB, Liu Y, Yu YR, Zhu Y, Tang CS, Qi YF: Intermedin1–53 attenuates vascular smooth muscle cell calcification by inhibiting endoplasmic reticulum stress via cyclic adenosine monophosphate/protein kinase A pathway. Exp Biol Med (Maywood), 2013; 238: 1136-1146 [DOI] [PubMed] [Google Scholar]
- 31). Teng X, Song J, Zhang G, Cai Y, Yuan F, Du J, Tang C, Qi Y: Inhibition of endoplasmic reticulum stress by intermedin(1–53) protects against myocardial injury through a PI3 kinase-Akt signaling pathway. J Mol Med (Berl), 2011; 89: 1195-1205 [DOI] [PubMed] [Google Scholar]
- 32). Hagiwara M, Bledsoe G, Yang ZR, Smith RS, Jr, Chao L, Chao J: Intermedin ameliorates vascular and renal injury by inhibition of oxidative stress. Am J Physiol Renal Physiol, 2008; 295: F1735-1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Li H, Bian Y, Zhang N, Guo J, Wang C, Lau WB, Xiao C: Intermedin protects against myocardial ischemia-reperfusion injury in diabetic rats. Cardiovasc Diabetol, 2013; 12: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34). Yang SM, Liu J, Li CX: Intermedin protects against myocardial ischemia-reperfusion injury in hyperlipidemia rats. Genet Mol Res, 2014; 13: 8309-8319 [DOI] [PubMed] [Google Scholar]
- 35). Li Y, Zhang H, Jiang C, Xu M, Pang Y, Feng J, Xiang X, Kong W, Xu G, Li Y, Wang X: Hyperhomocysteinemia promotes insulin resistance by inducing endoplasmic reticulum stress in adipose tissue. J Biol Chem, 2013; 288: 9583-9592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Zhou J, Møller J, Danielsen CC, Bentzon J, Ravn HB, Austin RC, Falk E: Dietary supplementation with methionine and homocysteine promotes early atherosclerosis but not plaque rupture in ApoE-deficient mice. Arterioscler Thromb Vasc Biol, 2001; 21: 1470-1476 [DOI] [PubMed] [Google Scholar]
- 37). Qin YW, Ye P, He JQ, Sheng L, Wang LY, Du J: Simvastatin inhibited cardiac hypertrophy and fibrosis in apolipoprotein E-deficient mice fed a “Western-style diet” by increasing PPAR α and γ expression and reducing TC, MMP-9, and Cat S levels. Acta Pharmacol Sin, 2010; 31: 1350-1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38). Nishikimi T, Tadokoro K, Akimoto K, Mori Y, Ishikawa Y, Ishimura K, Horio T, Kangawa K, Matsuoka H: Response of adrenomedullin system to cytokine in cardiac fibroblasts-role of adrenomedullin as an antifibrotic factor. Cardiovasc Res, 2005; 66: 104-113 [DOI] [PubMed] [Google Scholar]
- 39). Zhao L, Peng DQ, Zhang J, Song JQ, Teng X, Yu YR, Tang CS, Qi YF: Extracellular signal-regulated kinase 1/2 activation is involved in intermedin1–53 attenuating myocardial oxidative stress injury induced by ischemia/reperfusion. Peptides, 2012; 33: 329-335 [DOI] [PubMed] [Google Scholar]
- 40). Chen H, Wang X, Tong M, Wu D, Wu S, Chen J, Wang X, Wang X, Kang Y, Tang H, Tang C, Jiang W: Intermedin suppresses pressure overload cardiac hypertrophy through activation of autophagy. PLoS One, 2013; 8: e64757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41). Pah HL: Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev, 1999; 79: 683-701 [DOI] [PubMed] [Google Scholar]
- 42). Pavitt GD, Ron D: New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harb Perspect Biol, 2012; 4 pii: a012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Schröder M: Endoplasmic reticulum stress responses. Cell Mol Life Sci, 2008; 65: 862-894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Veeranki S, Tyagi SC: Defective homocysteine metabolism: potential implications for skeletal muscle malfunction. Int J Mol Sci, 2013; 14: 15074-15091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45). Ho YY, Lagares D, Tager AM, Kapoor M: Fibrosis--a lethal component of systemic sclerosis. Nat Rev Rheumatol, 2014; 10: 390-402 [DOI] [PubMed] [Google Scholar]
- 46). van Nieuwenhoven FA, Turner NA: The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vascul Pharmacol, 2013; 58: 182-188 [DOI] [PubMed] [Google Scholar]
- 47). Nattel S, Burstein B, Dobrev D: Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol, 2008; 1: 62-73 [DOI] [PubMed] [Google Scholar]
- 48). Thum T, Lorenzen JM: Cardiac fibrosis revisited by microRNA therapeutics. Circulation, 2012; 126: 800-802 [DOI] [PubMed] [Google Scholar]
- 49). Jin D, Otani K, Yamahara K, Ikeda T, Nagaya N, Kangawa K: Adrenomedullin reduces expression of adhesion molecules on lymphatic endothelial cells. Regul Pept, 2011; 166: 21-27 [DOI] [PubMed] [Google Scholar]
- 50). Parry GC, Mackman N: Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NFkappaB-mediated transcription. J Immunol, 1997; 159: 5450-5456 [PubMed] [Google Scholar]
- 51). Talero E, Di Paola R, Mazzon E, Esposito E, Motilva V, Cuzzocrea S: Anti-inflammatory effects of adrenomedullin on acute lung injury induced by Carrageenan in mice. Mediators Inflamm, 2012; 2012: 717851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52). Gonzalez-Rey E, Chorny A, Delgado M: Regulation of immune tolerance by anti-inflammatory neuropeptides. Nat Rev Immunol, 2007; 7: 52-63 [DOI] [PubMed] [Google Scholar]
- 53). Pedram A, Razandi M, O'Mahony F, Lubahn D, Levin ER: Estrogen receptor-beta prevents cardiac fibrosis. Mol Endocrinol, 2010; 24: 2152-2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54). Hammes SR, Levin ER: Minireview: Recent advances in extranuclear steroid receptor actions. Endocrinology, 2011; 152: 4489-4495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55). Schattenberg JM, Czaja MJ: Regulation of the effects of CYP2E1-induced oxidative stress by JNK signaling. Redox Biol, 2014; 3: 7-15 [DOI] [PMC free article] [PubMed] [Google Scholar]