

Abstract
Sulfonatocalix[4]arenes with an appended hydroxamic acid residue can detoxify VX and related V‐type neurotoxic organophosphonates with half‐lives down to 3 min in aqueous buffer at 37 °C and pH 7.4. The detoxification activity is attributed to the millimolar affinity of the calixarene moiety for the positively charged organophosphonates in combination with the correct arrangement of the hydroxamic acid group. The reaction involves phosphonylation of the hydroxamic acid followed by a Lossen rearrangement, thus rendering the mode of action stoichiometric rather than catalytic. Nevertheless, these calixarenes are currently the most efficient low‐molecular‐weight compounds for detoxifying persistent V‐type nerve agents under mild conditions. They thus represent lead structures for novel antidotes that allow treatment of poisonings by these highly toxic chemicals.
Keywords: calixarenes, chemical warfare agents, detoxification, hydroxamic acids, scavengers
With a percutaneous LD50 value of 10 mg/human, the organophosphonate (OP) O‐ethyl S‐[2‐(diisopropylamino)ethyl] methylphosphonothioate (VX; Figure 1) is one of the most toxic and most persistent chemical warfare agents.1 Analogues that likewise have the amino group in the side chain and the relatively stable phosphono–thioester bond are similarly harmful.
Figure 1.
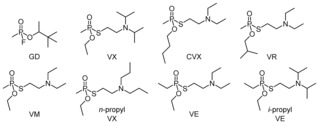
Structures of soman (GD) and the investigated V‐type nerve agents.
The toxicity of nerve agents is mainly related to their high propensity to covalently modify a serine residue in the active site of the enzyme acetylcholinesterase (AChE).2 In this form, AChE is unable to perform its function, namely to degrade the neurotransmitter acetylcholine, whose accumulation leads to severe toxic effects on the central and peripheral nervous system and ultimately to death.
A promising concept to treat poisonings by nerve agents involves the use of proteins, so called bioscavengers, that detoxify OPs before clinical signs occur.3 Bioscavengers have drawbacks, however, for example, low in vivo stability and immunogenicity, thus rendering synthetic scavengers attractive alternatives.4 With only a few exceptions,5 studies on synthetic scavengers have so far concentrated on cyclodextrin derivatives.6 Their mode of action is expected to resemble that of proteins, with an initial complexation step that positions the phosphorus atom of the nerve agent close to a substituent on the cyclodextrin ring to facilitate the reaction. Despite notable success in this context,6 cyclodextrins that detoxify V‐type nerve agents have so far remained elusive. A possible explanation could be that V‐type nerve agents are poor substrates for cyclodextrins because of their protonated side chain amino groups and, hence, polar nature at physiological pH values.7 If this assumption is correct, hosts for ammonium ions in water should be more promising scaffolds for scavengers for V‐type nerve agents. This idea is consistent with established design principles of supramolecular catalysts and reagents.8
Ajami and Rebek proposed the use of resorcinarene‐derived cavitands as OP scavengers.5c They described the synthesis of functionalized derivatives that could be used as a basis for such scavengers,9 but detoxification studies were not reported. Our approach involved the use of sulfonatocalix[4]arenes that are also strong cation binders in water.10, 11 These calixarenes were decorated with a substituent containing a hydroxamic acid, which is known to detoxify OPs.12, 13 Synthesis involved the coupling of monoamine 1 11a to carboxylic acids with a protected hydroxamic acid. Deprotection followed by chromatographic purification and ion exchange afforded the products as sodium salts. Scheme 1 shows the synthesis of 2 a as an example (for synthetic details, see the Supporting Information).
Scheme 1.
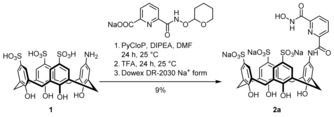
Synthesis of 2 a from 1 and the sodium salt of O‐tetrahydropyranyl (THP) protected 6‐(hydroxycarbamoyl)picolinic acid.
Of the compounds screened, 2 a and 2 b (Figure 2) exhibited highly promising properties. Analogues 2 c–e were then prepared as reference compounds. In addition, the sulfanilic acid derivatives 3 a–c were considered to study the extent to which the calixarene moiety contributes to the activity of 2 a and 2 b.
Figure 2.
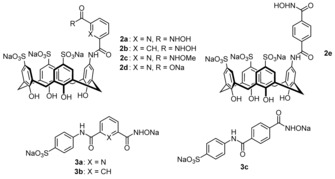
Structures of the investigated calixarenes and the corresponding reference compounds.
A previously established enzymatic in vitro assay was used to obtain quantitative information about the detoxification activities of the prepared compounds (for details, see the Supporting Information).14 Specifically, a nerve agent was incubated in Tris‐HCl buffer (0.1 m, pH 7.40) at 37 °C with an excess of a test compound. Aliquots of the solution were removed at regular time intervals to quantify their inhibitory effect on human AChE using a modified Ellman assay.14 AChE inhibition, described quantitatively by first‐order inhibition rate constants k obs, decreased during incubation because of scavenger‐mediated detoxification and spontaneous hydrolysis of the nerve agent. Fitting the time dependence of k obs to monoexponential decay curves yielded rate constants k that reflect how fast a nerve agent loses the ability to inhibit AChE (Figure 3). Correcting these rate constants for the rates of spontaneous hydrolysis, determined by performing the assay with the same OP in the absence of any scavenger, gave rate constants k detox, which were used to calculate the half‐lives t 1/2 of detoxification. Tris‐HCl buffer was chosen as the medium because we typically also consider G‐type nerve agents in the screening, whose spontaneous hydrolysis is too fast in phosphate buffer to allow reliable kinetic analyses.13, 14, 15 Selected measurements were also performed in phosphate buffer to assess the influence of the buffer on the detoxification of the V‐type nerve agents. It is worth noting that all of our detoxification studies involved the use of real nerve agents and not of less toxic simulants.
Figure 3.
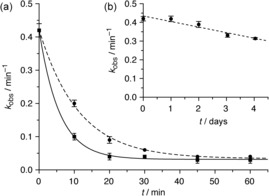
a) Decay curves of the first‐order inhibition rate constants k obs observed when VX (10 μm) was treated with 2 a (500 μm, solid line) or 2 b (500 μm, dashed line) in Tris‐HCl buffer (0.1 m, pH 7.40) at 37 °C. b) The rate of the spontaneous hydrolysis of VX under the same conditions in the absence of a scavenger. Note the different time scales.
Table 1 shows that the two calixarene‐based hydroxamic acids 2 a and 2 b mediate VX detoxification with half‐lives of about 5 min in Tris‐HCl buffer, which is approximately 3500 times faster than the spontaneous hydrolysis of VX under the same conditions. VX detoxification induced by 2 a is similarly fast in phosphate buffer. Analogues 2 c and 2 d, in which the hydroxamic acid group is methylated at the oxygen atom or replaced by a carboxylate group, respectively, are inactive. Shifting the hydroxamic acid group from the 3‐position of the aromatic substituent in 2 b to the 4‐position in 2 e also causes activity to disappear, and none of the reference compounds 3 a–c lacking the calixarene unit detoxify VX within the timeframe of the measurement. Notably, the activity of compounds 2 a and 2 b is not restricted to VX; structurally related V‐type nerve agents are also detoxified. The rates of detoxification of the methyl phosphonates in the presence of 2 a were almost equally fast, while the ethyl phosphonates VE and i‐propyl‐VE are detoxified significantly slower. In the case of 2 b, the V‐type nerve agents with linear alkyl groups in the side chains are detoxified less efficiently than VX, and going from the methyl phosphonates to the ethyl phosphonates causes another drop in activity. Equimolar mixtures of the sulfanilic acid 3 a and the calixarenes 1 or 2 c proved to be inactive, thus showing that the covalent linkage between the calixarene unit and the substituent in 2 a is required for effective detoxification.
Table 1.
Half‐lives t 1/2 [min] of the detoxification of V‐type nerve agents and soman (GD; see Table S1 for concentrations) mediated by 2 a–e and 3 a–c (500 μm) in Tris‐HCl buffer (0.1 m, pH 7.40) at 37 °C. Measurements were performed at least three times and results are given as means ± the standard deviation.
| VX | CVX | VR | VM | n‐propyl‐VX | VE | i‐propyl‐VE | GD | |
|---|---|---|---|---|---|---|---|---|
| 2 a | 3.7±0.4 | 2.8±0.1 | 3.4±0.6 | 2.9±0.5 | 2.4±0.2 | 15.1±0.4 | 17.0±0.6 | 17.7±0.9 |
| 2 a [a] | 2.9±0.2 | 2.2±0.1 | 2.3±0.1 | n.d.[b] | n.d. | n.d. | n.d. | 5.8±0.2 |
| 2 b | 7.5±0.3 | 25.0±0.7 | 28.6±0.4 | 20.3±0.4 | 25.3±0.8 | 105.2±4.7 | 40.5±1.4 | 49.5±3.4 |
| 2 c | >120[c] | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | >120 |
| 2 d | >120 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | >120 |
| 2 e | >120 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 91.2±6.8 |
| 3 a | >120 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 27.2±2.4 |
| 3 b | >120 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 46.6±2.8 |
| 3 c | >120 | >120 | >120 | >120 | >120 | >120 | >120 | 37.6±4.1 |
[a] Determined in phosphate buffer (0.1 m, pH 7.4). [b] Not determined. [c] Detoxification too slow to allow determination of a reliable half‐life.
For comparison, the effects of the prepared compounds on the detoxification of soman (GD) were also assessed. Table 1 shows that the O‐methylated hydroxamic acid 2 c also does not mediate the detoxification of GD, which suggests that the free hydroxamic OH group is required for the reaction. The carboxylate group in 2 d is presumably not sufficiently nucleophilic to have an effect. All the other compounds detoxify GD to some extent, irrespective of whether they contain a calixarene moiety or not. The half‐lives of GD detoxifications by 2 a and 3 a or by 2 b and 3 b are very similar. Thus, the detoxification of the neutral GD does not seem to benefit from the calixarene ring. By contrast, this ring is essential for the detoxification of the cationic V‐type nerve agents, as none of the sulfanilic acid derivatives exhibited notable activity. The lack of activity of the simple hydroxamic acids is consistent with the higher stability of phosphonothioates with respect to GD (the rate constants of spontaneous hydrolysis are 6.3×10−3 min−1 for GD and 5.2×10−5 min−1 for VX under the chosen conditions). The ability of 2 a and 2 b to overcompensate this stability difference and allow them to detoxify V‐type nerve agents even faster than GD, could, therefore, be an indication that the predicted interactions between the calixarenes and cationic OPs indeed facilitate detoxification.
To test this assumption, binding studies were performed by using VX and the inactive calixarene derivative 2 c as binding partners. Qualitative information about complex formation was obtained by comparing the 1H NMR spectrum of VX with that of VX in the presence of 2 equiv of 2 c in phosphate buffer (0.1 m in D2O, pD 7.81). Phosphate buffer was used to avoid signals from organic buffer molecules in the 1H NMR spectra. Figure 4 shows that the presence of 2 c has a pronounced effect on the resonances of the VX protons (for the effect of complexation on the receptor signals, see Figure S4). All the VX protons are shielded, with the strongest upfield shift of 1.3 ppm observed for the signals of the isopropyl‐CH3 groups. The shifts become smaller as the distance of the respective proton from the diisopropylamino group increases, but effects are even evident for the resonances of the ethyl‐CH3 and P‐CH3 protons. Another notable feature is the splitting of the isopropyl‐CH3 signal into two doublets, which indicates that the stabilization of the tetrahedral ammonium group of VX upon complexation causes the isopropyl‐CH3 groups to become diastereotopic.
Figure 4.
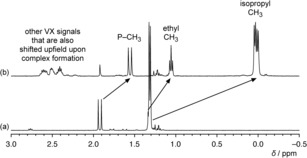
Sections of the 1H NMR spectra of a) VX (2.93 mm) in phosphate buffer (0.1 m in D2O, pD 7.81) and b) of a mixture of 2 c and VX in a 2:1 molar ratio. Characteristic shifts of the isopropyl, ethyl, and phosphonate methyl signals of VX are indicated.
The profound signal shifts observed in the 1H NMR spectrum are clear indications for the binding of VX to 2 c. Moreover, the extents of the shifts suggest that complex geometries are preferred, with the cationic ammonium group located inside the calixarene cavity. As a consequence, the phosphorus atom of the nerve agent should be preferentially oriented near the hydroxamic acid group of the scavenger, thus facilitating reaction. An NMR titration under the same conditions yielded a binding constant log K a of 4.11±0.12 for the complex between 2 c and VX, thus confirming the appreciable affinity of the sulfonatocalix[4]arene for positively charged nerve agents in water.
Efficient complexation of the nerve agents could, therefore, explain the detoxification activities of the calixarene‐based scavengers. The different activities observed for 2 a, 2 b, and 2 e may be related to differences in the nucleophilicities of their substituents or to structural aspects of the complexes formed. The former is less likely because benzohydroxamic acid and corresponding pyridine derivatives do not typically exhibit large differences in their pK a values or rates of reaction in the presence of OPs.5a,5b, 12a We therefore attribute the higher activity of 2 a with respect to 2 b, and also the different rates with which the investigated nerve agents are detoxified, to structural effects of the respective complexes. The loss of activity when moving the hydroxamic acid from the 3‐ to the 4‐position of the aromatic substituent may likewise be caused by the inability of the hydroxamic acid in the VX complex with 2 e to efficiently reach the phosphorous atom of the nerve agent.
Information about the pathways underlying the detoxification was obtained by following the reactions between VX and calixarenes 2 a and 2 b by 31P NMR spectroscopy and mass spectrometry. 31P NMR spectroscopy showed that the spontaneous hydrolysis of VX (2.93 mm) in Tris‐HCl buffer (0.1 m, pH 7.40) at 37 °C is associated with a progressive decrease in the VX signal. Concomitantly, signals appeared showing the formation of ethyl methylphosphonic acid (EMPA) and the toxic metabolite of VX, S‐(2‐(diisopropylamino)ethyl) methylphosphonothioate (EA‐2192). About 25 % of the VX was hydrolyzed after 24 h under these conditions, with the EMPA/EA‐2192 ratio amounting to 5.7:1.
Incubating VX (2.93 mm) in Tris‐HCl buffer (0.1 m, pH 7.40) at 37 °C with 1 equiv of 2 a led to a notable 78 % drop in VX concentration within the first hour of the experiment and a drop of 94 % when using 2 equiv of 2 a, thus confirming the high detoxification ability of this scavenger. The decrease in the VX concentration after 1 h was also significant in the case of 2 b, but somewhat smaller. VX degradation was complete after 24 h in the presence of 2 equiv of either calixarene. In contrast, the residual VX reached a plateau at 5 % and 20 % when only 1 equiv of 2 a and 2 b was present, respectively, which could be attributed to product inhibition, as the 2‐(diisopropylamino)ethanethiol group released during VX detoxification can be expected to bind to the calixarene ring with a similar affinity as VX. Importantly, scavenger‐induced VX degradation is associated only with the formation of EMPA, thus indicating that the calixarenes mediate the selective cleavage of the P−S bond of VX and efficiently suppress formation of the toxic metabolite EA‐2192. Since analogous results were obtained for 3 a and 3 c, the selectivity of the reaction seems to be characteristic for hydroxamic acids, as also observed in other studies,4 and not related to the calixarene moiety.
Mass spectrometry showed that VX degradation mediated by 2 a is associated with the formation of amine 4 and Tris adduct 5 shown in Scheme 2. Analogous products were observed when 2 b was reacted with VX. As a consequence, both scavengers are consumed during the reaction, thereby rendering their mode of action stoichiometric. The products observed indicate that the reaction includes a Lossen rearrangement, as also observed for the reaction of other organophosphates with hydroxamic acids.12 The isocyanates resulting from this rearrangement are either hydrolyzed or react with nucleophiles, in our experiments buffer molecules, to yield the corresponding adducts.
Scheme 2.
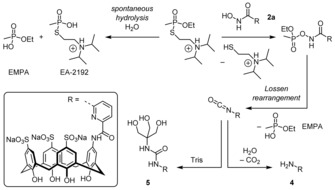
Reaction pathways of spontaneous VX hydrolysis in Tris‐HCl (Tris=tris(hydroxymethyl)aminomethane) buffer (0.1 m, pH 7.40, 37 °C) and VX detoxification mediated by 2 a.
Concluding, this study introduces a very promising concept for the development of synthetic scavengers for highly stable and persistent V‐type nerve agents. Calixarenes 2 a and 2 b mediate effective detoxification of VX and other V‐type nerve agents in aqueous buffer at 37 °C and pH 7.4 with so far unmatched activity for synthetic compounds. The observed high detoxification rates can be attributed to the combined presence of a recognition unit that binds to cationic nerve agents and a properly arranged nucleophilic group that mediates the selective cleavage of the P−S bond of the nerve agent. Relating the activities of 2 a and 2 b to those of bioscavengers, for example, on the basis of k cat/K M values, is difficult because the calixarenes are stoichiometric scavengers, while bioscavengers for VX act catalytically. Data compiled in a recent review show, however, that half‐lives of detoxification in the range of a few seconds are required for scavengers to be potentially useful in vivo.3b Thus, 2 a and 2 b cannot replace bioscavengers yet, but their activities might allow them to be used to supplement current nerve agent therapies by shortening the period of toxicologically relevant poison concentrations. In addition, they should allow decontamination of skin or sensitive equipment under much milder conditions than those normally associated with VX detoxification.1 Clearly, these calixarenes also represent highly promising lead structures for synthetic scavengers, whose activities can likely be improved in the future by fine‐tuning both the recognition unit and the reactive group.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by a contract with the German Armed Forces (E/UR2W/CF511/9A803). The generous funding is gratefully acknowledged. We also thank T. Kliemt for technical assistance.
C. Schneider, A. Bierwisch, M. Koller, F. Worek, S. Kubik, Angew. Chem. Int. Ed. 2016, 55, 12668.
References
- 1.
- 1a. Yang Y.-C., Baker J. A., Ward J. R., Chem. Rev. 1992, 92, 1729–1743; [Google Scholar]
- 1b. Talmage S. S., Watson A. P., Hauschild V., Munro N. B., King J., Curr. Org. Chem. 2007, 11, 285–298; [Google Scholar]
- 1c. Smith B. M., Chem. Soc. Rev. 2008, 37, 470–478; [DOI] [PubMed] [Google Scholar]
- 1d. Jang Y. J., Kim K., Tsay O. G., Atwood D. A., Churchill D. G., Chem. Rev. 2015, 115, PR1–PR76; [DOI] [PubMed] [Google Scholar]
- 1e. Yang Y.-C., Acc. Chem. Res. 1999, 32, 109–115. [Google Scholar]
- 2.
- 2a. Tuovinen K., Toxicology 2004, 196, 31–39; [DOI] [PubMed] [Google Scholar]
- 2b. Marrs T. C., Maynard R. L., Sidell F. R., Chemical Warfare Agents: Toxicology and Treatment, Wiley, Chichester, 2007. [Google Scholar]
- 3.
- 3a. Lenz D. E., Yeung D., Smith J. R., Sweeney R. E., Lumley L. A., Cerasoli D. M., Toxicology 2007, 233, 31–39; [DOI] [PubMed] [Google Scholar]
- 3b. Worek F., Thiermann H., Wille T., Toxicol. Lett. 2016, 244, 143–148; [DOI] [PubMed] [Google Scholar]
- 3c. Nachon F., Brazzolotto X., Trovaslet M., Masson P., Chem.-Biol. Interact. 2013, 206, 536–544. [DOI] [PubMed] [Google Scholar]
- 4. Sambrook M. R., Notman S., Chem. Soc. Rev. 2013, 42, 9251–9267. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Louise-Leriche L., Paǎunescu E., Saint-André G., Baati R., Romieu A., Wagner A., Renard P.-Y., Chem. Eur. J. 2010, 16, 3510–3523; [DOI] [PubMed] [Google Scholar]
- 5b. Saint-André G., Kliachyna M., Kodepelly S., Louise-Leriche L., Gillond E., Renard P.-Y., Nachon F., Baati R., Wagner A., Tetrahedron 2011, 67, 6352–6361; [Google Scholar]
- 5c. Ajami D., J. Rebek, Jr. , Org. Biomol. Chem. 2013, 11, 3936–3942; [DOI] [PubMed] [Google Scholar]
- 5d. Ruan Y., Taha H. A., Yoder R. J., Maslak V., Hadad C. M., Badjić J. D., J. Phys. Chem. B 2013, 117, 3240–3249; [DOI] [PubMed] [Google Scholar]
- 5e. Hiscock J. R., Sambrook M. R., Cranwell P. B., Watts P., Vincent J. C., Xuereb D. J., Wells N. J., Raja R., Gale P. A., Chem. Commun. 2014, 50, 6217–6220; [DOI] [PubMed] [Google Scholar]
- 5f. Dennison G. H., Sambrook M. R., Johnston M. R., Chem. Commun. 2014, 50, 195–197; [DOI] [PubMed] [Google Scholar]
- 5g. Chen S., Ruan Y., Brown J. D., Hadad C. M., Badjić J. D., J. Am. Chem. Soc. 2014, 136, 17337–17342; [DOI] [PubMed] [Google Scholar]
- 5h. Ruan Y., Dalkiliç E., Peterson P. W., Pandit A., Dastan A., Brown J. D., Polen S. M., Hadad C. M., Badjić J. D., Chem. Eur. J. 2014, 20, 4251–4256; [DOI] [PubMed] [Google Scholar]
- 5i. Hiscock J. R., Sambrook M. R., Wells N. J., Gale P. A., Chem. Sci. 2015, 6, 5680–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Letort S., Balieu S., Erb W., Gouhier G., Estour F., Beilstein J. Org. Chem. 2016, 12, 204–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rekharsky M. V., Inoue Y., Chem. Rev. 1998, 98, 1875–1918. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Breslow R., Dong S. D., Chem. Rev. 1998, 98, 1997–2011; [DOI] [PubMed] [Google Scholar]
- 8b. Sanders J. K. M., Chem. Eur. J. 1998, 4, 1378–1383; [Google Scholar]
- 8c. Molenveld P., Engbersen J. F. J., Reinhoudt D. N., Chem. Soc. Rev. 2000, 29, 75–86; [Google Scholar]
- 8d. Hooley R. J., J. Rebek, Jr. , Chem. Biol. 2009, 16, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Degardin M., Busseron E., Kim D.-A., Ajami D., J. Rebek, Jr. , Chem. Commun. 2012, 48, 11850–11852. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Perret F., Lazar A. N., Coleman A. W., Chem. Commun. 2006, 2425–2438; [DOI] [PubMed] [Google Scholar]
- 10b. Perret F., Coleman A. W., Chem. Commun. 2011, 47, 7303–7319; [DOI] [PubMed] [Google Scholar]
- 10c. Guo D.-S., Liu Y., Acc. Chem. Res. 2014, 47, 1925–1934. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Daze K. D., Ma M. C. F., Pineux F., Hof F., Org. Lett. 2012, 14, 1512–1515; [DOI] [PubMed] [Google Scholar]
- 11b. Hof F., Chem. Commun. 2016, 52, 10093–10108. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. B. E. Hackley, Jr. , Plapinger R., Stolberg M., Wagner-Jauregg T., J. Am. Chem. Soc. 1955, 77, 3651–3653; [Google Scholar]
- 12b. Stolberg M. A., Mosher W. A., J. Am. Chem. Soc. 1957, 79, 2618–2620; [Google Scholar]
- 12c. Swidler R., Plapinger R. E., Steinberg G. M., J. Am. Chem. Soc. 1959, 81, 3271–3274; [Google Scholar]
- 12d. Silva M., Mello R. S., Farrukh M. A., Venturini J., Bunton C. A., Milagre H. M. S., Eberlin M. N., Fiedler H. D., Nome F., J. Org. Chem. 2009, 74, 8254–8260. [DOI] [PubMed] [Google Scholar]
- 13. Brandhuber F., Zengerle M., Porwol L., Bierwisch A., Koller M., Reiter G., Worek F., Kubik S., Chem. Commun. 2013, 49, 3425–3427. [DOI] [PubMed] [Google Scholar]
- 14. Bierwisch A., Zengerle M., Thiermann H., Kubik S., Worek F., Toxicol. Lett. 2014, 224, 209–214. [DOI] [PubMed] [Google Scholar]
- 15. Zengerle M., Brandhuber F., Schneider C., Worek F., Reiter G., Kubik S., Beilstein J. Org. Chem. 2011, 7, 1543–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary